
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic energy levels of porphyrins are influenced by the local chemical environment†
Margaret Wolf‡
,
José J. Ortiz-Garcia‡ ,
Matthew J. Guberman-Pfeffer,
José A. Gascón and
Rebecca C. Quardokus*
,
Matthew J. Guberman-Pfeffer,
José A. Gascón and
Rebecca C. Quardokus*
Department of Chemistry, University of Connecticut, USA. E-mail: rebecca.quardokus@uconn.edu; Fax: +1-860-486-2981; Tel: +1-860-486-2844
First published on 6th January 2022
Abstract
Self-assembled islands of 5,10,15,20-tetrakis(pentafluoro-phenyl)porphyrin (2HTFPP) on Au(111) contain two bistable molecular species that differ by shifted electronic energy levels. Interactions with the underlying gold herringbone reconstruction and neighboring 2HTFPP molecules cause approximately 60% of molecules to have shifted electronic energy levels. We observed the packing density decrease from 0.64 ± 0.04 molecules per nm2 to 0.38 ± 0.03 molecules per nm2 after annealing to 200 °C. The molecules with shifted electronic energy levels show longer-range hexagonal packing or are adjacent to molecular vacancies, indicating that molecule–molecule and molecule–substrate interactions contribute to the shifted energies. Multilayers of porphyrins do not exhibit the same shifting of electronic energy levels which strongly suggests that molecule–substrate interactions play a critical role in stabilization of two electronic species of 2HTFPP on Au(111).
The packing of two-dimensional self-assembled monolayers is facilitated by molecule–surface and molecule–molecule interactions. At low coverage, molecules with repulsive intermolecular interactions can adsorb at preferred surface-adsorption sites,1–4 while at higher concentrations the preferred surface site may be sacrificed to increase density and maximize intermolecular interactions.3–6 Surface adsorption can change the conformational structure of a molecule to maximize these interactions resulting in a conformational shape not found in bulk crystals or solvated molecules.4,7–10 Rational manipulation of shape and local environment can fine tune the electronic properties of the adsorbate.
Porphyrins consist of four pyrrole-like moieties connected with methine bridges. The high level of conjugation, with 18 π electrons in the shortest cyclic path, allow porphyrins to strongly absorb light in the visible region, 400–700 nm.11–13 Porphyrins are known as the color of life molecule.14,15 Heme, an iron–porphyrin complex, is responsible for transporting oxygen through the bloodstream and gives red blood cells their bright color.16 Chlorophyll, a partially hydrogenated porphyrin, is responsible for the green color in plants and plays a critical role in photosynthesis.16 The strong interaction with light, overall chemical stability, and diversity through functionalization makes pyrrolic macrocycles ideal candidates for use in photovoltaics,17–21 chemical sensors,22–25 catalysis,26–28 and molecularly based devices.29–32
The existence of bistable switchable states is the first step towards use of porphyrins in applications of molecular memory storage or binary devices.33 Pyrrolic macrocycles have been shown to exhibit bistable switching behavior on surfaces including induced conformational switching,34–36 orientational flip-flopping,37–41 and tautomerization.42–46 Examples include subphthalocyanine arrays that adsorb with a shuttlecock shape and exhibit scanning tunneling microscopy (STM) induced reversible orientational switching on Cu(100),37 and current from the STM tip that has been used to induce hydrogen tautomerization in naphthalocyanine on an NaCl bilayer on Cu(111).42
Confinement to two dimensions through adsorption of 2HTFPP onto a surface creates a complex heterogeneous environment with influences from the substrate and neighboring molecules. In this study, we characterize the local density of states of 2HTFPP adsorbed on Au(111), and found that the local chemical environment can cause the energies of molecular orbitals to shift. This creates an environment where chemically identical molecules exist as two species on the surface.
The sample was prepared using an Au(111) substrate on mica (Phasis) that was cleaned through several rounds of annealing and argon sputtering. Images were collected at ultra-high vacuum and 77 K with a low-temperature scanning tunnelling microscope (LT-STM, Scienta Omicron) under constant current mode. The tips were mechanically etched 0.25 mm Pt80/Ir20 wire (NanoScience Instruments). The sample was prepared at room temperature by depositing 1 mM solution of 2HTFPP dissolved in dichloromethane via pulsed-solenoid valve in a vacuum chamber onto the clean gold or 2HTFPP dissolved in dimethylformamide via drop-casting and annealing to 200 °C.47
The electronic structure of 2HTFPP, neutral TFPP, and dianionic TFPP were calculated using density functional theory (DFT). The structures were optimised using the B3LYP48,49 density functional approximation and the 6-311+G(d) basis set. A harmonic vibrational frequency analysis confirmed the structure was in a local minimum on the ground state potential energy surface without imaginary frequencies. All calculations were performed on Gaussian 16 Rev. A.03.50 Simulated STM images were computed using the Tersoff and Hamann approximation51,52 as done by Kandel and co-workers.53,54 The tunneling current was modeled as a function of tip position, and the surface was modeled as a featureless and constant density of states to reproduce a constant current experiment. Through the Tersoff and Hamann approximation the tunneling current leads to information on the local density of states at a specific energy and a discrete location.
Pulse-depositing 2HTFPP in vacuum via a pulsed-solenoid valve resulted in ordered islands of the adsorbate, Fig. 1. The formation of close-packed islands, despite submonolayer coverage, indicates attractive intermolecular interactions between adsorbates. 2HTFPP has a packing density of 0.64 ± 0.04 molecules per nm2 with a 1-molecule unit cell and rectangular packing.
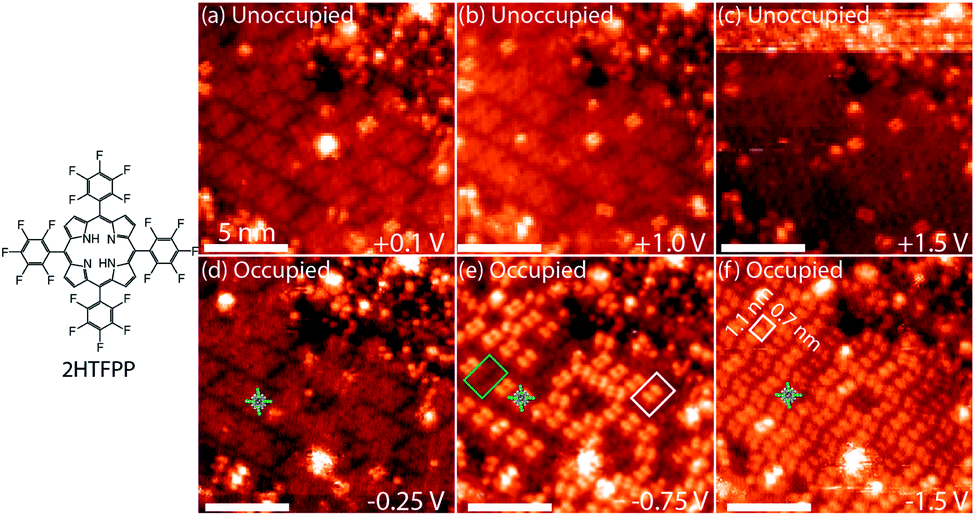 | ||
| Fig. 1 (a–c) Unoccupied and (d–f) occupied electron states of 2HTFPP. All scale bars are 5 nm. (e) Two bistable species, diffuse square outlined in green and a double-dot feature outlined in white. (f) 2HTFPP appears as double-dot features. The white box indicates the 1-molecule, rectangular unit cell. Images taken with a tunneling current of 5 pA. | ||
Changing the polarity of the bias voltage of the STM sample allows unoccupied (positive voltage) or occupied (negative voltage) molecular orbitals to be imaged along with the topography of the surface. The unoccupied electron states, Fig. 1a–c, have an even distribution of electron density across the molecule and appear as diffuse squares. As the magnitude of the bias voltage is increased, a larger number of electronic energy levels are included in the measurement. Increasing the magnitude of the negative bias voltage from −0.25 V to −1.5 V, Fig. 1d and f, results in a change in contrast from a diffuse square to a bright double-dot feature. Imaging at −0.75 V, an intermediate voltage between these two energy levels, we observe two species—a mixture of diffuse squares and double-dot features, Fig. 1e. These species are differentiated from one another by having the same ground state molecular orbitals at shifted energies. If all the molecules on the surface were equivalent, we would expect to image the same molecular orbitals of each molecule at each bias voltage. At −0.75 V we do not observe the same orbital shape for each molecule, but we observe influence of the HOMO−2 and HOMO−3 energy levels in approximately 60% of that adsorbed 2HTFPP. Interaction with the local chemical environment causes the energy levels to be shifted enough to be included at a bias voltage with a smaller magnitude.
We investigated the possibility that the two species were chemically different. If a gold atom formed a complex with the porphyrin, we would expect the displacement of the inner pyrrolic hydrogens. It was also possible that the presence of the STM tip was altering the adsorbates. Smykalla et al. reported that the STM tip can reversibly remove and replace the pyrrolic hydrogens which changes the shape and contrast of the porphyrin in the STM images.55 Deng and Hipps reported a small shift in orbital energies of 0.04 eV for vapor-deposited metallo-porphyrins on Au(111) due to the height of the tip.56 We observed a march larger shift of orbital energies and used DFT to calculate shapes of the occupied and unoccupied molecular orbitals of gas phase 2HTFPP, TFPP with the inner pyrrolic hydrogens removed, dianionic TFPP with the inner pyrrolic protons removed, and Au-TFPP with a gold atom coordinated to the porphyrin, Fig. 1 in ESI.† We used the electronic structure calculations to make theoretical STM images. We found that the double-dot feature was only present in the occupied orbitals of 2HTFPP, Fig. 2. Theoretical STM images of Au-TFPP, Fig. 2 in ESI,† does not show the double-dot feature. This suggests that intact 2HTFPP molecules make up the close-packed islands, and both detected species are chemically equivalent. The shift in electronic energy levels is due to the heterogeneous local chemical environment.
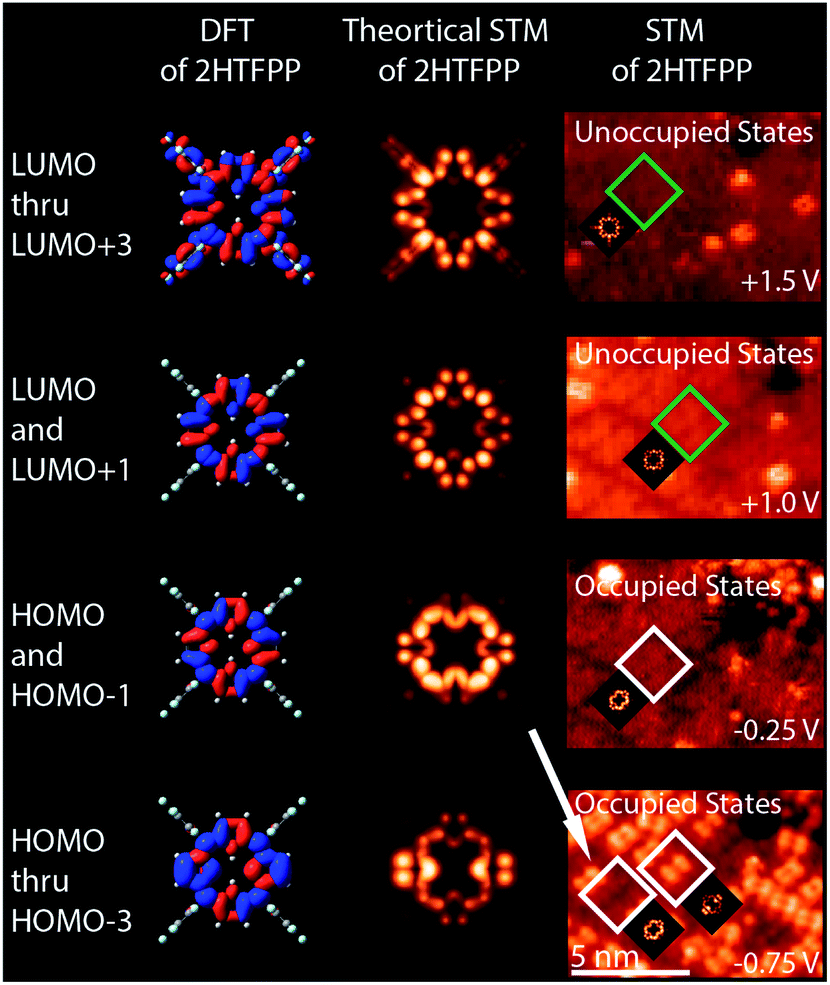 | ||
| Fig. 2 DFT, theoretical STM images, and comparison with experimental STM images of 2HTFPP. HOMO and HOMO−1 have a diffuse square shape, while HOMO thru HOMO−3 exhibit the double dot feature. Both of these shapes were experimentally observed at −0.75 V. | ||
The packing of adsorbates is a balance between molecule–molecule and molecule–substrate interactions. We observed various packing densities and unit cells that were dependent on sample preparation. Preparation with the pulsed-solenoid valve at room temperature created a 1-molecule unit cell with a packing density of 0.64 ± 0.04 molecules per nm2, Fig. 1. Annealing the sample to 200 °C resulted in a 1-molecule unit cell with a packing density of 0.38 ± 0.03 molecules per nm2, Fig. 4. Annealing altered both the adsorption sites and intermolecular ordering of 2HTFPP, and we no longer observed two electronic species on the surface. Annealing to 200 °C may remove excess solvent and provide the energy needed for the adsorbed species to leave the preferred adsorption site and maximize intermolecular interactions. This suggests that the local chemical environment plays a critical role in the creation of two bistable species. The surface of Au(111) undergoes herringbone reconstruction with 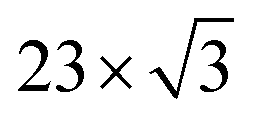 packing of the surface gold atoms.57,58 The surface consists of both face-centered cubic packing (fcc) and hexagonally-close packed (hcp) gold atoms with strained transition areas. The three different domains create a heterogeneous surface electronic structure.59 To investigate the relationship of species 1 and species 2 with the underlying substrate we characterized the images using Fast Fourier Transform (FFT). FFT converts an image from real space to frequency space and can reveal longer-range ordering of molecular assemblies.
packing of the surface gold atoms.57,58 The surface consists of both face-centered cubic packing (fcc) and hexagonally-close packed (hcp) gold atoms with strained transition areas. The three different domains create a heterogeneous surface electronic structure.59 To investigate the relationship of species 1 and species 2 with the underlying substrate we characterized the images using Fast Fourier Transform (FFT). FFT converts an image from real space to frequency space and can reveal longer-range ordering of molecular assemblies.
In order to focus the FFT on one species at a time, white circles were placed over each double dot feature, Fig. 3a and b. FFT were taken of the images with the circles to determine the packing of the adsorbates, Fig. 3c and d. Hexagonal ordering as well as longer-range rectangular packing can be seen in the FFT, Fig. 3c, of the locations of the double-dot features in the image showing two species of 2HTFPP, Fig. 3a. We observed rectangular packing of 2HTFPP when the magnitude of the bias voltage was large enough for all of the adsorbed porphyrins to exhibit bright double-dot features, Fig. 3b and d. The presences of hexagonal packing for one species, but not the overall self-assembled island suggests that the adsorption site on the underlying gold herringbone reconstruction is playing a role in the shift of electronic energy levels. We did not observe the two species after annealing to 200 °C, suggesting that solvent and intermolecular interactions may influence the location of the adsorption site on Au(111). We also observed that the double-dot features in Fig. 3a were more likely to exist around molecular defects and edges of the close-packed island suggesting that intermolecular interactions also play a role in the shift of the electronic energies of the molecular orbitals.
 | ||
| Fig. 3 (a) Circles placed over each 2HTFPP molecule with bright double dot features at −0.75 V and (b) −1.5 V (c) is FFT of (a) showing hexagonal order (d) is FFT of (b) showing rectangular order. | ||
Pulse deposition with a solenoid valve into vacuum has been shown to create meta-stable molecular motifs due to the rapid evaporation of solvent.53 We planned to directly compare room-temperature pulse deposition with drop-casting but were unable to resolve drop-cast porphyrins prior to annealing. Drop-casting followed by annealing resulted in an alternate packing of 0.21 molecules per nm2, Fig. 3 in ESI.† Despite the fluorinated phenyl group inverting the polarity of the ring, we observe π stacking of pentafluorophenyl rings after pulse-depositing and annealing to 200 °C, Fig. 4. Although this is unexpected, Blanchard et al. have previously observed face-to-face π stacking in the crystal structure of pentafluorophenyl substituted ferrocenes.60
 | ||
| Fig. 4 2HTFPP after annealing to 200 °C. The unit cell has dimensions of 1.6 nm × 1.6 nm and the monolayer has a packing density of 0.38 molecules per nm2. The bright double-dot features are doubly stacked porphyrins, the start of a second layer. The image was taken with a bias voltage of −10.0 mV and a tunneling current of 10 pA. | ||
Several molecules on top of the self-assembled island, the start of a second layer, appear with bright double-dot features, Fig. 4. We did not observe the coexistence of two electronic species in the second layer of 2HTFPP.
In conclusion, we observed two bistable species of 2HTFPP on Au(111) with non-degenerate electronic energy levels. DFT and theoretical STM images suggest that the two species are both 2HTFPP and not chemically altered through adsorption or interaction with the STM tip. The species with the affected electronic energy levels appear near molecular vacancies and exhibit longer-range hexagonal packing indicating that the two species are created through molecule–substrate and molecule–molecule interactions. Annealing to 200 °C decreases the packing density and alters the molecule–molecule and molecule–surface interactions. Adding a second layer of adsorbate only exhibits one electronic species which strongly suggests that the local chemical environment plays a critical role in the shift of the electronic energies of the molecular orbitals of 2HTFPP.
Author contributions
Margaret Wolf and José Ortiz-Garcia contributed equally to the collection and evaluation of experimental data. Margaret Wolf was involved in data curation, formal analysis, and writing of the manuscript. José Ortiz-Garcia was involved in data curation, formal analysis, DFT calculations, theoretical STM calculations, and writing of the manuscript. Matthew Guberman-Pfeffer and José Gascon were involved in DFT calculations and review and editing of the manuscript. Rebecca C. Quardokus was involved in conceptualization, data curation, formal analysis, supervision, and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Bischoff, K. Seufert, W. Auwärter, S. Joshi, S. Vijayaraghavan, D. Ecija, K. Diller, A. C. Papageorgiou, S. Fischer and F. Allegretti, et al., ACS Nano, 2013, 7, 3139–3149 CrossRef CAS PubMed.
- Y. Wang, J. Kröger, R. Berndt and W. Hofer, Angew. Chem., 2009, 121, 1287–1291 CrossRef.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- T. Lukasczyk, K. Flechtner, L. R. Merte, N. Jux, F. Maier, J. M. Gottfried and H.-P. Steinrück, J. Phys. Chem. C, 2007, 111, 3090–3098 CrossRef CAS.
- H. Peisert, J. Uihlein, F. Petraki and T. Chassé, J. Electron Spectrosc. Relat. Phenom., 2015, 204, 49–60 CrossRef CAS.
- D. Heim, K. Seufert, W. Auwärter, C. Aurisicchio, C. Fabbro, D. Bonifazi and J. V. Barth, Nano Lett., 2010, 10, 122–128 CrossRef CAS PubMed.
- Q. Zhang, X. Zheng, G. Kuang, W. Wang, L. Zhu, R. Pang, X. Shi, X. Shang, X. Huang and P. N. Liu, et al., J. Phys. Chem. Lett., 2017, 8, 1241–1247 CrossRef CAS PubMed.
- T. Yokoyama, S. Yokoyama, T. Kamikado and S. Mashiko, J. Chem. Phys., 2001, 115, 3814–3818 CrossRef CAS.
- J. Mielke, F. Hanke, M. V. Peters, S. Hecht, M. Persson and L. Grill, J. Am. Chem. Soc., 2015, 137, 1844–1849 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- S. Kumar, Y. K. Maurya, S. Kang, P. Chmielewski, T. Lis, J. Cybińska, D. Kim and M. Stepien, Org. Lett., 2020, 22, 7202–7207 CrossRef CAS PubMed.
- S. Shi, X. Wang, Y. Sun, S. Chen, X. Li, Y. Li and H. Wang, J. Mater. Chem., 2012, 22, 11006–11008 RSC.
- C. J. Kingsbury and M. O. Senge, Coord. Chem. Rev., 2021, 431, 213760 CrossRef CAS.
- L. R. Milgrom, The Colours of Life: An Introduction to the Chemistry of Porphyrins and Related Compounds, Oxford University Press, 1997 Search PubMed.
- S. Piña-Oviedo, C. Ortiz-Hidalgo and A. G. Ayala, Arch. Pathol. Lab. Med., 2017, 141, 445–462 CrossRef PubMed.
- M. V. Martinez-Diaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC.
- F. Jin, Z. Su, B. Chu, P. Cheng, J. Wang, H. Zhao, Y. Gao, X. Yan and W. Li, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- H. Imahori and S. Fukuzumi, Adv. Funct. Mater., 2004, 14, 525–536 CrossRef CAS.
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS.
- H. Yamada, H. Imahori, Y. Nishimura, I. Yamazaki, T. K. Ahn, S. K. Kim, D. Kim and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 9129–9139 CrossRef CAS PubMed.
- D. Gounden, N. Nombona and W. E. van Zyl, Coord. Chem. Rev., 2020, 420, 213359 CrossRef CAS.
- W. Göpel, Synth. Met., 1991, 41, 1087–1093 CrossRef.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517–2583 CrossRef CAS PubMed.
- Y. Chen and K. Wang, Photochem. Photobiol. Sci., 2013, 12, 2001–2007 CrossRef CAS PubMed.
- O. Nestler and K. Severin, Org. Lett., 2001, 3, 3907–3909 CrossRef CAS PubMed.
- H. Rao, C.-H. Lim, J. Bonin, G. M. Miyake and M. Robert, J. Am. Chem. Soc., 2018, 140, 17830–17834 CrossRef CAS PubMed.
- S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
- J. A. Mol, C. S. Lau, W. J. Lewis, H. Sadeghi, C. Roche, A. Cnossen, J. H. Warner, C. J. Lambert, H. L. Anderson and G. A. D. Briggs, Nanoscale, 2015, 7, 13181–13185 RSC.
- B. Limburg, J. O. Thomas, G. Holloway, H. Sadeghi, S. Sangtarash, I. C.-Y. Hou, J. Cremers, A. Narita, K. Müllen and C. J. Lambert, et al., Adv. Funct. Mater., 2018, 28, 1803629 CrossRef.
- Q. Wu, S. Hou, H. Sadeghi and C. J. Lambert, Nanoscale, 2018, 10, 6524–6530 RSC.
- M. L. Perrin, C. J. Verzijl, C. A. Martin, A. J. Shaikh, R. Eelkema, J. H. Van Esch, J. M. Van Ruitenbeek, J. M. Thijssen, H. S. Van Der Zant and D. Dulić, Nat. Nanotechnol., 2013, 8, 282–287 CrossRef CAS PubMed.
- C. Lent, P. Tougaw, W. Porod and G. Bernstein, Nanotechnology, 1993, 4, 49–57 CrossRef.
- Y. Wang, J. Kröger, R. Berndt and W. A. Hofer, J. Am. Chem. Soc., 2009, 131, 3639–3643 CrossRef CAS PubMed.
- T. Niu and A. Li, J. Phys. Chem. Lett., 2013, 4, 4095–4102 CrossRef CAS.
- Y.-S. Fu, J. Schwöbel, S.-W. Hla, A. Dilullo, G. Hoffmann, S. Klyatskaya, M. Ruben and R. Wiesendanger, Nano Lett., 2012, 12, 3931–3935 CrossRef CAS PubMed.
- H. Yanagi, K. Ikuta, H. Mukai and T. Shibutani, Nano Lett., 2002, 2, 951–955 CrossRef CAS.
- C. Kleint, Phys. Status Solidi A, 1979, 55, 447–456 CrossRef.
- J. Otsuki, Supramol. Chem., 2011, 23, 169–182 CrossRef CAS.
- V. Erokhin, S. Carrara, C. Paternolli, L. Valkova, S. Bernstorff and C. Nicolini, Appl. Surf. Sci., 2005, 245, 369–375 CrossRef CAS.
- H. Yanagi and K. Ikuta, Surf. Sci., 2005, 581, 9–16 CrossRef CAS.
- P. Liljeroth, J. Repp and G. Meyer, Science, 2007, 317, 1203–1206 CrossRef CAS PubMed.
- Z. Feng, S. Velari, C. Dri, A. Goldoni, M. Peressi and G. Comelli, J. Phys. Chem. C, 2020, 124, 11376–11382 CrossRef CAS.
- W. Auwärter, K. Seufert, F. Bischoff, D. Ecija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, Nat. Nanotechnol., 2012, 7, 41–46 CrossRef PubMed.
- H. Böckmann, S. Liu, J. Mielke, S. Gawinkowski, J. Waluk, L. Grill, M. Wolf and T. Kumagai, Nano Lett., 2016, 16, 1034–1041 CrossRef PubMed.
- J. N. Ladenthin, L. Grill, S. Gawinkowski, S. Liu, J. Waluk and T. Kumagai, ACS Nano, 2015, 9, 7287–7295 CrossRef CAS PubMed.
- M. Wolf, V. Hayes, C. R. Gerber, P. G. Quardokus, J. J. Ortiz-Garcia, C. Plummer and R. C. Quardokus, J. Vac. Sci. Technol., A, 2020, 38, 022413 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian-16 Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- J. Tersoff and D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 805 CrossRef CAS PubMed.
- J. Tersoff and D. Hamann, Phys. Rev. Lett., 1983, 50, 1998 CrossRef CAS.
- R. C. Quardokus, N. A. Wasio, J. A. Christie, K. W. Henderson, R. P. Forrest, C. S. Lent, S. A. Corcelli and S. A. Kandel, Chem. Commun., 2014, 50, 10229–10232 RSC.
- R. C. Quardokus, Y. Lu, N. A. Wasio, C. S. Lent, F. Justaud, C. Lapinte and S. A. Kandel, J. Am. Chem. Soc., 2012, 134, 1710–1714 CrossRef CAS PubMed.
- L. Smykalla, P. Shukrynau, C. Mende, T. Rüffer, H. Lang and M. Hietschold, Surf. Sci., 2014, 628, 92–97 CrossRef CAS.
- W. Deng and K. W. Hipps, J. Phys. Chem. B, 2003, 107, 10736–10740 CrossRef CAS.
- C. Wöll, S. Chiang, R. Wilson and P. Lippel, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 7988 CrossRef PubMed.
- J. V. Barth, H. Brune, G. Ertl and R. Behm, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9307 CrossRef CAS PubMed.
- F. Reinert and G. Nicolay, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 817–821 CrossRef CAS.
- M. D. Blanchard, R. P. Hughes, T. E. Concolino and A. L. Rheingold, Chem. Mater., 2000, 12, 1604–1610 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09116f |
| ‡ Authors contributed equally and share first-author accolades. |
| This journal is © The Royal Society of Chemistry 2022 |