
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability and fundamental properties of CuxO1−x as optoelectronic functional materials
Wei-Tao Fanab,
Zong-Yan Zhao *c and
Hong-Lie Shen*a
*c and
Hong-Lie Shen*a
aCollege of Materials Science and Technology, Jiangsu Key Laboratory of Materials and Technology for Energy Conversion, Nanjing University of Aeronautics & Astronautics, Nanjing 210016, P. R. China
bYcergy (Suzhou) Technology Co. Ltd, Suzhou 215121, P. R. China
cFaculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming 650093, P. R. China. E-mail: zzy@kust.edu.cn
First published on 28th January 2022
Abstract
Binary CuxO1−x compounds have some advantages as optoelectronic functional materials, but their further development has encountered some bottlenecks, such as inaccurate bandgap values and slow improvement of photoelectric conversion efficiency. In this work, all possible stoichiometric ratios and crystal structures of binary CuxO1−x compounds were comprehensively analyzed based on a high-throughput computing database. Stable and metastable phases with different stoichiometric ratios were obtained. Their stability in different chemical environments was further analyzed according to the component phase diagram and chemical potential phase diagram. The calculation results show that Cu, Cu2O and CuO have obvious advantages in thermodynamics. The comparison and analysis of crystal microstructure show that the stable phase of CuxO1−x compounds contains the following two motifs: planar square with Cu atoms as the center and four O atoms as the vertices; regular tetrahedron with O atoms as the center and four Cu atoms as the vertices. In different stoichiometric ratio regions, the electron transfer and interaction modes between Cu and O atoms are different. This effect causes energy differences between bonding and antibonding states, resulting in the different conductivity of binary CuxO1−x compounds: semi-metallic ferromagnetic, semiconducting, and metallicity. This is the root of the inconsistent and inaccurate bandgap values of CuxO1−x compounds. These compositional, structural, and property variations provide greater freedom and scope for the development of binary CuxO1−x compounds as optoelectronic functional materials.
1. Introduction
The key to the development of solar energy application technology lies in the development of efficient, broad-spectrum response, inexpensive and stable optoelectronic functional materials. Although traditional semiconductor oxide materials (such as Si, TiO2, ZnO, Fe2O3, etc.) have the advantages of abundant raw materials, high chemical stability, and environmental friendliness, etc., because most of them are wide bandgap semiconductors, such optoelectronic functional materials can only exhibit good optoelectronic conversion performance under UV excitation. In addition, due to the high recombination rate of photo-generated electron–hole pairs in the transport process, resulting in their low final quantum conversion efficiency. To improve the solar energy conversion efficiency of conventional oxide materials, researchers have tried to expand the spectral response range and suppress the photo-generated electron–hole recombination using material modification (e.g., ion doping, hetero-structure constructing, solid-solution design, etc.). In addition to material modification, the development of novel optoelectronic functional materials is also an essential solution. At present, novel optoelectronic functional materials that have been widely studied and researched include multi-metal oxides/nitrides/sulfides, layered compounds, and low-dimensional nanomaterials. However, due to the lack of sufficient and necessary theoretical guidance and reliable scientific data support, the progress of exploration work in this direction is not entirely satisfactory.In recent years, copper-based chalcogenide semiconductors have become a hot spot for research on new energy conversion materials due to their non-toxic, abundant in the earth's crust and environmentally friendly advantages, and have been widely developed and researched in solar cells, photocatalysis, lithium-ion batteries, thermoelectricity and other fields,1 among which Cu(In,Ga)Se2 has become a representative of absorber layer materials for thin-film solar cells;2 and Cu2ZnSnS4, which is developed based on Cu(In,Ga)Se2, is a representative of low-cost solar cell materials, and its solar energy conversion efficiency has exceeded 12.6% that is close to the standard for industrial application.3 In addition, other binary, ternary, and quaternary copper-based chalcogenides also exhibit excellent optoelectronic conversion performance, such as Cu2X (X = O, S, Se, Te), CuFeO2, CuAlS2, CuInS2, CuWO4, Cu2SnS3, Cu3MCh4 (M = V, Nb, Ta; Ch = S, Se, Te), CuZn2AS4 (A = Al, Ga, In), etc.4–11 Copper-based chalcogenides as optoelectronic functional materials have the following advantages: (1) these materials generally have direct bandgaps and much higher light absorption capacity than conventional optoelectronic materials such as Si, GaAs, and CdTe; (2) the composition of these materials is composed of adjacent similar elements in the periodic table, and because the crystal structures are the same or similar, so it is easy to obtain novel types of materials through element replacement and solid-solution design, providing a great degree of freedom for material design and optimization; (3) most copper-based chalcogenides with intrinsic defects can produce n-type or p-type conductivity, thus providing a convenient technical space for the construction of efficient heterojunction optoelectronic devices; (4) such materials can be well deposited on the surface of glass, plastic and other inexpensive substrates, which is conducive to batch production and reduce the cost of industrial production. These advantages make copper-based chalcogenides in the field of optoelectronic conversion technology has received widespread attention, and has shown potential market application prospects.
Among the copper-based chalcogenides, the binary copper-based oxides CuxO1−x have obvious development advantages and application markets, due to their simple and inexpensive composition, scalable production, non-toxicity, abundant sources, and long-term stability.12–16 Typical ones among them are copper oxide CuO and cuprous oxide Cu2O, which show bandgap values of about 1.2–2.1 eV, corresponding to the optimal bandgap values for ideal solar cells or photocatalytic materials, and are particularly suitable for solar energy-efficient applications such as solar cells and photocatalysis. Theoretical calculations show that the theoretical solar energy conversion efficiency of Cu2O can reach up to 18%,17 but the self-compensation effect makes it difficult to n-type Cu2O, thus making the preparation of p–n homo-junctions quite difficult, so the current maximum efficiency of Cu2O-based solar cells is only 2%.18 The performance of Cu2O thin films in the photochemical decomposition of water for hydrogen production is also of interest. Grätzel et al. designed a Cu2O/Ga2O3 photocathode with quantum conversion efficiency of up to 80% for solar decomposition of aquatic hydrogen, which can work stably for up to 100 hours with a TiO2 protective layer.19
The above-mentioned research progress has demonstrated that CuO and Cu2O are potential functional materials worth developing for optoelectronic applications, and further in-depth systematic studies are needed to improve the energy conversion efficiency. One important aspect is that the stoichiometric ratio (i.e., the value of x) of the binary copper-based oxides CuxO1−x varies significantly during preparation, especially sensitive to the oxygen partial pressure in the process conditions; and the heteromorphic effect is also prominent for a specific stoichiometric ratio. These two reasons make it very difficult to determine the fundamental physicochemical properties of CuxO1−x as optoelectronic functional materials, for example, the bandgap value of CuO oxide reported in the literature ranges from 1.2 eV to 1.9 eV. More importantly, the novel optoelectronic functional materials either have a special crystal structure, electronic structure, or can lead to a new perspective to recognize and understand the optoelectronic conversion process. In this regard, the binary CuxO1−x can be an ideal research object, enabling us to continuously understand the correlation and influence laws between the crystal structure, electronic structure, microstructure, optical properties, and stability of novel optoelectronic functional materials from a new perspective, to deeply analyze the corresponding optoelectronic conversion mechanism, and to provide research cases for the design and construction of efficient optoelectronic functional materials. On the other hand, at present, high-throughput computing based on density functional theory (DFT) has become a powerful tool to discover novel optoelectronic functional materials.20,21 For this purpose, this paper systematically analyzes the structural stability, crystal structure, electronic and optical properties of binary CuxO1−x compounds by DFT calculations based on high-throughput computation database.
2. Computational methods
In this paper, all DFT calculations are performed using the Quantum ATK package. The exchange–correlation energy is chosen from the meta-GGA method. The energy cutoff is set to 150 hartree, and the corresponding spread is set to 25 meV. 19 × 19 × 3 K-point grid is set by the Monkhorst–Pack method. In the PulayMixer algorithm, the energy convergence of the self-consistent field iterations is set to 0.0001 hartree. The LBFGS algorithm is chosen for geometry optimization, and the relevant convergence criteria are set to less than 0.05 eV Å−1 for stress, less than 0.1 GPa for strain, and less than 0.2 Å for maximum displacement. In the optimization process, the lattice basis vector, space group and Bravais lattice types are freely relaxed. The spin polarization effect was also considered in this work.3. Results and discussion
3.1 Phase diagram and stability
To comprehensively investigate all possible stoichiometric ratios and crystal structures of CuxO1−x, we first searched all possible binary CuxO1−x compounds in the Open Quantum Materials Database (OQMD), which is a general database for high-throughput calculations of materials.22,23 The obtained formation enthalpy (ΔH) is shown in Fig. 1(a) with the stoichiometric ratio x of the binary energy. It can be seen from the figure that the number of possible crystal structures of binary CuxO1−x compounds gradually increases, when the molar ratio of Cu to O is closed to 1 (i.e., x = 0.5). Thus, the paramorphism of CuO oxide is the most prominent in the OQMD, with the number of possible crystal structures reaching 31. In the ΔH − x relationship diagram, the convex hull line of binary CuxO1−x compounds can be obtained by connecting the lowest point of formation enthalpy, which is an important basis for judging the stability of binary CuxO1−x compounds. Based on this criterion, in addition to the common stable phases of tetragonal CuO (space group: no. 131, P42/mmc) and cubic Cu2O (space group: no. 224, Pn![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), binary CuxO1−x compounds have another stable phase: monoclinic CuO4 (space group: no. 10, P2/m). In addition to these three stable phases, binary CuxO1−x compounds with other stoichiometric ratios can also exist and be found under certain conditions. If ΔH < 0.06 eV per atom is used as a criterion for sub-stable phases, then four more metastable phases can be found as shown in red font in Fig. 1(a): cubic CuO2 (space group no. 205: Pa), rhombohedral Cu2O3 (space group no. 43, Fdd2), tetragonal Cu4O3 (space group no. 141, I41/amd), rhombohedral Cu8O (space group: no. 38, Amm2), and two sub-stable phases are not indicated in Fig. 1(a), which are tetragonal CuO (space group: no. 84, P42/m) and monoclinic CuO (space group: no. 15, C2/c). To systematically analyze the structure and properties of binary CuxO1−x compounds, we will take these seven compounds as the research objects and use the face-centered cubic phase of pure Cu as the reference in this work.
m), binary CuxO1−x compounds have another stable phase: monoclinic CuO4 (space group: no. 10, P2/m). In addition to these three stable phases, binary CuxO1−x compounds with other stoichiometric ratios can also exist and be found under certain conditions. If ΔH < 0.06 eV per atom is used as a criterion for sub-stable phases, then four more metastable phases can be found as shown in red font in Fig. 1(a): cubic CuO2 (space group no. 205: Pa), rhombohedral Cu2O3 (space group no. 43, Fdd2), tetragonal Cu4O3 (space group no. 141, I41/amd), rhombohedral Cu8O (space group: no. 38, Amm2), and two sub-stable phases are not indicated in Fig. 1(a), which are tetragonal CuO (space group: no. 84, P42/m) and monoclinic CuO (space group: no. 15, C2/c). To systematically analyze the structure and properties of binary CuxO1−x compounds, we will take these seven compounds as the research objects and use the face-centered cubic phase of pure Cu as the reference in this work.
 | ||
| Fig. 1 The DFT calculated: (a) enthalpy of generation (ΔH) versus stoichiometric ratio x; (b) chemical potential phase diagram; (c) grand potential when ΔμCu is zero; (d) p–T plot of the stable phase concerning oxygen partial pressure of binary CuxO1−x compounds. | ||
Since the compound formation is related to the chemical environment in the preparation process, we give the variation of the formation energy of different CuxO1−x compounds as the relative chemical potential of Cu and O changes in Fig. 1(b). It can be seen that as the increase of ΔμCu and ΔμO, the first to form in order are metallic Cu, Cu2O and CuO, and the other stable and metastable phases can only be formed in larger Cu and O chemical potential regions. This calculated result indicates that these stable and metastable phases need to be formed under specific synthesis conditions. So, to obtain these compounds, the synthesis process route needs to be carefully designed according to their formation conditions. Fig. 1(c) shows the giant potential (Ω) obtained according to the ΔμCu = 0 condition in Fig. 1(b), which expresses the phase transition process with the relative chemical potential of oxygen (ΔμO). In the oxygen-poor condition, metallic Cu is the stable phase; with the increase of ΔμO, the binary CuxO1−x compound forms the Cu2O phase; and in the oxygen-rich condition, CuO is the stable phase. The stable region of the Cu2O phase is relatively small, i.e., it is formed under relatively harsh conditions. On the phase partition line in this figure, there is a coexistence of two-phases. As the synthesis conditions (i.e., reaction temperature and oxygen partial pressure) change, binary CuxO1−x compound will undergo phase transition, as shown in Fig. 1(d). Under the same synthesis reaction temperature conditions, with the increase of oxygen partial pressure, binary CuxO1−x compounds start from Cu and go through two-phases transition processes, Cu/Cu2O and Cu2O/CuO, in order; while under the same oxygen partial pressure conditions, with the increase of reaction temperature, binary CuxO1−x compounds start from CuO and go through two-phases transition processes, CuO/Cu2O and Cu2O/Cu, in order.
3.2 Crystal structure properties
By structural optimization, we obtained the crystal structures of the stable and metastable phases of above-mentioned seven CuxO1−x compounds, as shown in Fig. 2. The detailed structural parameters corresponding to these crystal structures are listed in Table 1. The basic structural unit of tetragonal CuO is a planar square with one Cu atom at the center and four O atoms at the apex. However, the Cu atom is slightly off the center. These planar squares are first connected to form a two-dimensional plane, and then cross each other vertically to form a three-dimensional lattice. From another point of view, the tetrahedron with one O atom at the center and four Cu atoms at the apex can also be regarded as the basic structural unit of tetragonal CuO. In all CuxO1−x compounds considered in this work, CuO has the largest binding energy and formation energy, indicating it is the most thermodynamically stable. The most prominent structural feature of cubic Cu2O is the basic structural unit of tetrahedron with one O atom at the center and four Cu atoms at the apex. In other words, this basic structural unit can be considered as a linear dumbbell-like structure with Cu atoms at the center. The monoclinic CuO4 has a crystal constant of β = 91.565°, which means that this structure is rather close to the rhombohedral phase. Its prominent structural feature is the presence of two bonded O atoms between the Cu atoms. These four atoms coplanar form a chain-like linear structure. Furthermore, these chain-like linear structures are arranged parallel to each other along the c-axis. In the middle of these chain-like linear structures, two isolated O2-like molecules are distributed between each other. By comparing Fig. 2 and Table 1, it can be found that the crystal microstructure of the above three stable phases of CuxO1−x compound is relatively simpler and more symmetrical, and the interaction between Cu atom and O atom is relatively simple and unified. | ||
| Fig. 2 The model of crystal structure of binary CuxO1−x compounds in stable and metastable phases after structure optimization. | ||
| O2 (g) | CuO4 | CuO2 | Cu2O3 | CuO | Cu4O3 | Cu2O | Cu8O | Cu | ||
|---|---|---|---|---|---|---|---|---|---|---|
| a This is the population on O–O bond.b This is the O–O bond length.c This is the population on Cu–Cu bond.d This is the Cu–Cu bond length. | ||||||||||
| Crystal system | Molecule | Monoclinic | Cubic | Orthorhombic | Tetragonal | Tetragonal | Cubic | Orthorhombic | Cubic | |
| Symmetry | No. 10: P2/m | No. 205: Pa |
No. 43: Fdd2 | No. 131: P42/mmc | No. 141: I41/amd | No. 224: Pnm |
No. 38: Amm2 | No. 225: Fmm |
||
| Stability | Stable | Stable | Metastable | Metastable | Stable | Metastable | Stable | Metastable | Stable | |
| Lattice constants | — | a = 3.817 Å | a = b = c = 4.814 Å | a = 9.570 Å | a = b = 2.879 Å | a = b = 5.821 Å | a = b = c = 4.224 Å | a = 5.266 Å | a = b = c = 3.690 Å | |
| b = 3.060 Å | α = β = γ = 90° | b = 11.867 Å | c = 5.135 Å | c = 9.715 Å | α = β = γ = 90° | b = 6.063 Å | α = β = γ = 90° | |||
| c = 4.544 Å | c = 3.435 Å | α = β = γ = 90° | α = β = γ = 90° | c = 6.377 Å | ||||||
| α = γ = 90° | α = β = γ = 90° | α = β = γ = 90° | ||||||||
| β = 91.565° | ||||||||||
| Cell volume (Å3) | — | 53.059 | 111.580 | 390.053 | 42.558 | 329.221 | 75.375 | 203.590 | 50.238 | |
| Density (g cm−3) | — | 3.992 | 5.688 | 5.963 | 6.208 | 6.097 | 6.305 | 8.554 | 8.402 | |
| Binding energy (eV per atom) | 4.733 | 5.221 | 5.310 | 5.449 | 5.547 | 5.374 | 5.204 | 4.678 | 4.467 | |
| Formation energy (eV per atom) | — | 0.541 | 0.666 | 0.822 | 0.947 | 0.793 | 0.648 | 0.182 | — | |
| Mulliken population (e) | Cu | — | 0.98 | 0.76 | 0.83 | 0.61 | 0.33, 0.63 | 0.34 | 0.04, 0.09, 0.10 | 0 |
| O | 0 | −0.21, −0.29 | −0.36 | −0.54, −0.56 | −0.61 | −0.63, −0.65 | −0.68 | −0.63 | — | |
| Cu–O | 0.37a | 0.06, 0.15 | 0.15 | 0.44, 0.29, 0.37 | 0.54 | 0.26, 0.35, 0.48 | 0.33 | 0.22, 0.33 | 0.42c | |
| Bond length (Å) | Cu–O | 1.215b | 1.909, 2.069 | 2.374 | 1.799, 1.850, 1.870, 1.876 | 1.929 | 1.969, 1.896, 1.826 | 1.829 | 2.025, 1.898 | 2.609d |
| Bond angle (°) | O–Cu–O | — | 84.605, 95.395, 180 | 87.659, 92.341, 180 | 88.170, 88.099, 89.170, 94.005 | 83.450 | 82.185, 87.815, 180 | 180 | — | — |
| Cu–O–Cu | — | 95.395 | 108.655, 110.275 | 108.789, 112.518, 113.908 | 116.293 | 95.318, 114.002, 115.699 | 109.471 | 89.762, 90.238, 180 | — | |
The most prominent structural feature of cubic CuO2 is that the Cu atoms are still face-centered cubic as a sub-lattice, while the seven O2-like molecules are located at the midpoints of six sides and the body center. The structural features of the rhombohedral Cu2O3 are more complicated. The four O atoms outside the vertex are not coplanar with the Cu atoms, when viewed from the Cu atoms at the center. But the Cu atoms deviate from the coplanar of the four O atoms by only a small distance, so they can also be approximated as forming a planar rectangle. If viewed from the O atom at the center, there are two basic structural units in the rhombohedral Cu2O3, one is a tetrahedron with one O atom at the vertex and three Cu atoms as the bottom vertex, and the other is a chain-like linear structure with –Cu–O– interphase arrangement. These basic structural units intertwine to form a complex three-dimensional crystal lattice. The basic structural unit of tetragonal Cu4O3 is a square with one Cu atom at the center and four O atoms at the apex. These squares are intertwined and connected to form a three-dimensional lattice, through the linear dumbbell structure of O–Cu–O. The rhombohedral Cu8O has two basic structural units: an octahedron formed by six Cu atoms and a chain-like linear structure formed by –Cu–O–Cu–, which is arranged interdependently in parallel along the b-axis. As shown in Table 1, in the above four metastable phases of CuxO1−x compound, the interaction between Cu–O is more complex and inconsistent. Moreover, there are direct bonding and interaction between O–O or Cu–Cu. This leads to a slightly larger binding energy and formation energy, but the crystal structure is not the most stable.
Summarizing the crystal structures of the stable and metastable phases of above-mentioned binary CuxO1−x compounds, it can be found there are two stable structural units: one is a square with Cu atoms at the center and four O atoms at the apex; another is a tetrahedral structure with O atoms at the center and four Cu atoms at the apex, which is linearly connected by dumbbell-like O–Cu–O. The CuxO1−x compounds form a stable phase when the above basic structures dominate in the crystal structure. In the transitional form of these basic structural units, binary CuxO1−x compounds form a metastable phase. In addition, in the oxygen-rich stoichiometric ratio, CuxO1−x compounds contain a certain number of O2-like molecular configurations, while in the copper-rich stoichiometric ratio, CuxO1−x compounds contain a certain number of Cu-like clustered polyhedral configurations.
Analyzing the structural parameters in Table 1, it can be found that the density of binary CuxO1−x compounds increases gradually with the increase of the stoichiometric ratio x. However, their binding and formation energies increase first and then decrease. When x = 0.5, the binding energy of CuO is the largest, which indicates that the interaction between Cu and O atoms reaches the maximum. In other words, the reaction between metal Cu and oxygen O2 is most likely to produce such compounds. By analyzing the Mulliken population, it can be found that as the stoichiometric ratio x increases, the positive charge value of Cu atoms and the negative charge value on O atoms in binary CuxO1−x become smaller and smaller, while the charge number on the Cu–O bond increases and then decreases. These calculated results indicate that as the stoichiometric ratio x increases, the bonding between Cu and O atoms in binary CuxO1−x compounds is first dominated by ionic bonds, and then covalent bonds gradually take over. In the compound of CuO, the covalent bonds are reaching the strongest. Then, as the molar ratio of Cu atoms continues to increase, ionic bonds begin to gradually increase again. Moreover, a certain amount of metal bonding interactions appear in binary CuxO1−x compounds. The data on bond lengths and bond angles reflect the variation of the microstructure of the basic structural units in binary CuxO1−x compound with the stoichiometric ratio. The data in the table indicate that when these basic structural units tend to a regular configuration, the corresponding CuxO1−x compounds have larger binding and formation energies, and tend to form the stable phases.
3.3 Electronic structure
For optoelectronic functional materials, the most fundamental physical property is the band structure, which can directly give the type and value of the bandgap. It is extremely important for the screening of novel optoelectronic functional materials. Moreover, based on the band structure, the interaction process between electrons and photons can be deeply dissected, which is an important basis for understanding the intrinsic physical mechanism of optoelectronic functional materials. For binary CuxO1−x compounds, their most important fundamental physical parameter – the bandgap value – has been the most controversial issue reported in the literature. For example, the bandgap value of CuO has been reported in the literature with a rather wide range of values, 1.2–1.9 eV;24,25 the same is true for the case of Cu2O, whose bandgap value has been reported in the range of 2.0–2.2 eV.26 Therefore, a careful analytical study of this is very necessary.Fig. 3 presents the band structure diagrams of the stable or metastable phases of the above-mentioned eight binary CuxO1−x compounds. The first ones were found to range from x = 0.2 (CuO4) to x = 4/7 (Cu4O3), and the spins of these materials are unpaired, i.e., the spin-up electronic states do not completely overlap with the spin-down electronic states, showing a clear spin polarization phenomenon. More importantly, near the Fermi energy level (EF), the spin-up electronic states of the first 3 compounds (CuO4, CuO2, Cu2O3) have well-defined bandgaps, while the spin-down electronic states cross the Fermi energy level. Thus, these 3 compounds exhibit clear semimetallic ferromagnetic characteristics, which is a very attractive potential breakthrough direction for optoelectronic conversion applications. The other two compounds (CuO and Cu4O3) have well-defined indirect bandgap values of 1.67 and 0.85 eV, which exhibit semiconducting property. The remaining 3 compounds, on the other hand, exhibit a complete overlap between the spin-up electronic state and the spin-down electronic state, i.e., no spin-polarized features. Among them, Cu2O has a well-defined direct bandgap value of 1.13 eV and has a semiconducting property, while Cu8O and Cu have no bandgap and exhibit a distinct metallic property.
 | ||
| Fig. 3 Band structure and corresponding density of states of binary CuxO1−x compounds obtained from DFT calculations, the red line represents the spin-up electronic states and the blue line represents the spin-down electronic states. | ||
Secondly, it is important to note that the vibration of bandgap of these compounds is quite complex. The spin-up electronic state of CuO4 has a well-defined indirect bandgap of 7.45 eV. While, its spin-down electronic state has no bandgap, but has a pseudo bandgap above the Fermi level with a width of 4.11 eV. The spin-up electronic state of CuO2 has a well-defined indirect bandgap of 5.62 eV. While, its spin-down electronic state has a pseudo bandgap below and above the Fermi level with a width of 2.20 and 3.45 eV, respectively. The spin-up electronic state of Cu2O3 has a well-defined direct bandgap of 7.24 eV. While, its spin-down electronic state has a pseudo bandgap above the Fermi energy level with a width of 1.90 eV. In addition to the semiconductor bandgaps between the different spin electronic states mentioned above, the bandgaps between the different spin electronic states of CuO and Cu4O3 are 3.95 and 2.03 eV for the spin-up electronic states, and 2.57 and 0.85 eV for the spin-down electronic states, respectively. These complex bandgap features make the electron transition of binary CuxO1−x compounds with more complex processes, together with the polymorphism phenomenon discussed in Subsection 3.1. This may be the reason why it is experimentally difficult to determine the bandgap values of binary CuxO1−x compounds unambiguously.
Combined with the corresponding density of states (DOS) distribution, it can be found that the composition of band structure of CuxO1−x compounds shows a complex trend with the change of stoichiometric ratio. In the compound of CuO4, since the Cu atom loses more electrons, there is a large distance between the occupied and unoccupied states of its 3d electronic states. It is the reason why the spin-up electronic state of CuO4 has a wide bandgap value of 7.45 eV. In the energy range of −6 to 3 eV, there is obvious hybridization between the O-2p electronic state and the Cu-3d electronic state. The hybridized energy band between the spin-down states crosses the Fermi energy level, and thus becomes a semi-occupied state. It leads to a metallic electronic structure of CuO4. The electronic structure of CuO2 shows more obvious localization characteristics, where the hybridization between the O-2p electronic state and the Cu-3d electronic state occurs mainly in the energy range between −6 and −3 eV. At the Fermi energy level, an isolated half-full energy band is formed by the Cu-3d spin-down electronic state. It is the source of its metallic conductivity. In the compound of Cu2O3, the hybridization between the O-2p electronic state and the Cu-3d electronic state is more pronounced, and exhibits a distinct non-localization feature. At the top of the valence band, there are a small number of unoccupied states in the hybridized state, thus making Cu2O3 also metallic properties. As the value of x increases, the hybridized spin-down electronic states between the O-2p electronic state and the Cu-3d electronic state move further upward above the Fermi energy level. While, the hybridized states move downward below the Fermi energy level. Thus, CuO produces a well-defined wide bandgap. However, after the value of x is greater than 0.5, the above phenomenon changes in the opposite direction, because the number of Cu atoms losing electrons decreases further, leading to a decrease in the bandgaps of Cu4O3 and Cu2O. In the compound of Cu8O, the Cu atoms only lose a small number of electrons. The Cu atoms in the Cu clusters do not even lose electrons. At the same time, the O atoms only gain a small number of electrons. So, the electronic structure of Cu8O is extremely similar to that of metallic Cu as shown in Fig. 3. In Table 1, the Mulliken population of CuxO1−x compounds is also provided. These calculated results indicate that with the increase of stoichiometric ratio x, the number of electrons obtained by O atom is more and more, while the number of electrons lost by Cu atom is less and less. This means that ionic bonding interactions dominate in CuxO1−x compounds when the value of x is relatively small, while metal bonding interactions dominate in CuxO1−x compounds when the value of x is relatively large.
Combining the crystal structure and electronic structure calculations, it can be found that binary CuxO1−x compounds undergo semimetal–semiconductor–metal transitions with increasing values of stoichiometric ratio x. The main reason is that the different electron transfer and interaction modes between Cu and O atoms, which can cause the different composition and energy differences between the bonding and antibonding states. (1) When the value of x is small (x < 0.5), there are O2 molecule-like groups in binary CuxO1−x compounds, and a 2-coordinated structure is formed between Cu and O atoms. Therefore, the localization characteristics of the bonding and antibonding states are more prominent, resulting in the larger distance between them. So, although CuxO1−x exhibits metallicity, the pseudo bandgap between subbands is relatively larger. This electronic structure feature makes binary CuxO1−x compounds are expected to have potential applications in novel photoelectronic functional materials. (2) When the value of x is in the intermediate region (0.5 ≤ x ≤ 0.67), binary CuxO1−x compounds have tetrahedron as structural motifs. In these compounds, the bonding and antibonding states are in non-localization hybridized states mainly. Thus, they form semiconductors with well-defined moderate bandgap value, which is very suitable for photoelectronic conversion applications. (3) At large values of x (x > 0.67), there are metal-Cu-like nanoclusters in the crystal structure of binary CuxO1−x compounds. Furthermore, there are only very small amounts of electron transfer between Cu and O atoms. In these cases, the valence electrons of the Cu atoms become shared electrons and move throughout the crystal with an apparently non-localized near-free electron behavior, in which there is almost no pseudo bandgap. Thus, it makes binary CuxO1−x compound to show metallic properties once again, and have a clear advantage in conducting material applications.
3.4 Optical properties
According to band structure by DFT calculations, the corresponding optical properties can be further obtained based on the direct transition of electrons and the golden selection rule. Fig. 4 shows exactly the optical absorption spectra of seven binary CuxO1−x compounds. Due to their semi-metallic ferromagnetism, CuO4, CuO2, and Cu2O3 have strong light absorption in the UV region, while they show broadband absorption characteristics in the IR region, and the two light absorption peaks are independent of each other. As mentioned above CuO, Cu4O3, and Cu2O exhibit well-defined semiconducting features, so their light absorption curves also have well-defined absorption band edges accordingly. Cu8O has metallic properties, so it has strong light absorption in the whole UV-Visible-NIR region. For photoelectronic functional material applications, the calculated optical absorption spectra of binary CuxO1−x compounds are a good guide to explain some special phenomena found in experiments. For example, phenomena such as inaccurate bandgap values of Cu2O. It is expected that these calculated results will help clarify the difficulties and problems encountered in the optical measurements of binary CuxO1−x compounds.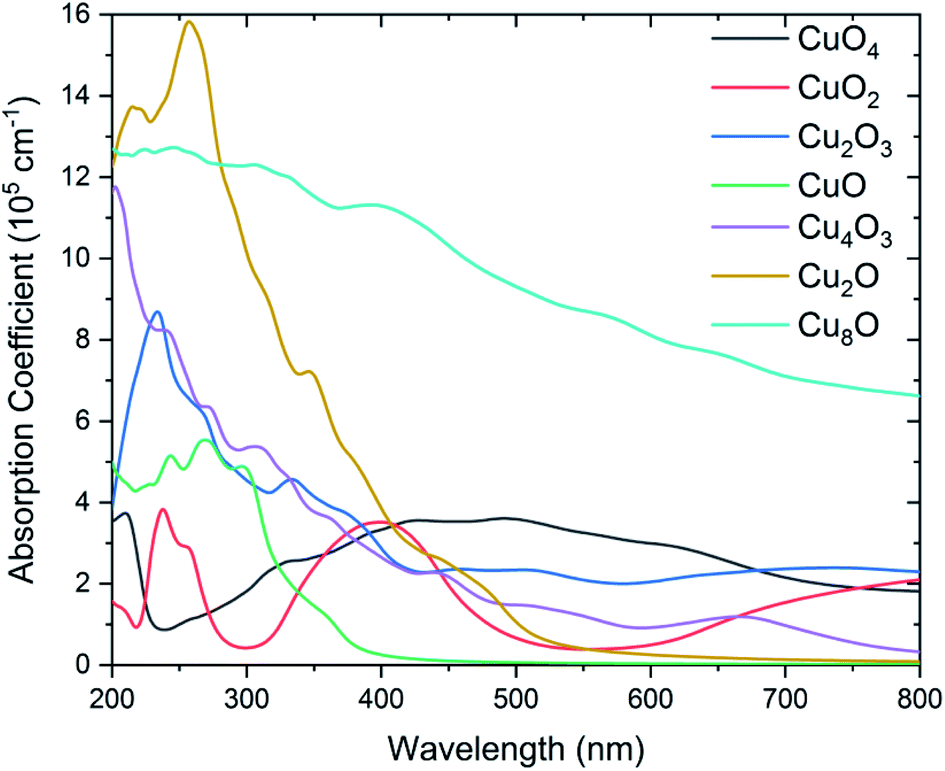 | ||
| Fig. 4 Optical absorption spectra of binary CuxO1−x compounds obtained by DFT calculations. | ||
4. Conclusions
In this paper, the possible stoichiometric ratios and crystal structures of binary CuxO1−x compounds are obtained from the material high-throughput calculation database. By comparing their formation enthalpies, stable phases (CuO4, CuO, Cu2O, Cu) and metastable phases (CuO2, Cu2O3, Cu4O3, Cu8O) with different stoichiometric ratios were obtained. Then, the stability of binary CuxO1−x compounds under different chemical environments is analyzed from the perspective of component phase diagram and chemical potential phase diagram. The results show that Cu, Cu2O and CuO have obvious advantages in thermodynamics. Moreover, with the change of reaction temperature and oxygen partial pressure in the preparation process conditions, binary CuxO1−x compounds will undergo two phase transition processes: Cu/Cu2O and Cu2O/CuO. Through the analysis of microstructure, it is found that the stable structural units of binary CuxO1−x compounds are: one is a square with Cu atom at the center and four O atoms at the apex; the other is a tetrahedral structure with O atom at the center and four Cu atoms at the apex. Moreover, the two basic structural units are linearly connected by dumbbell-like O–Cu–O. In terms of electronic structure, CuO4, CuO2, and Cu2O3 exhibit semimetallic ferromagnetism, CuO, Cu4O3, and Cu2O exhibit semiconducting features, and Cu8O and Cu exhibit definite metallicity. These complex electronic structures and bandgap variations are caused by the different electron transfer and interaction modes between Cu and O atoms, which result in different compositions and energy differences between bonding and antibonding states. This also makes the optical properties of binary CuxO1−x compounds show considerable complexity, and provides greater freedom and development space for their optoelectronic function applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (Grant No. 11964015).References
- Y. Zhao and C. Burda, Energy Environ. Sci., 2012, 5, 5564–5576 RSC.
- F.-J. Fan, L. Wu and S.-H. Yu, Energy Environ. Sci., 2014, 7, 190–208 RSC.
- W. Wang, M. T. Winkler, O. Gunawan, T. Gokmen, T. K. Todorov, Y. Zhu and D. B. Mitzi, Adv. Energy Mater., 2014, 4, 1301465 CrossRef.
- Y. Zhang, Y. Wang, L. Xi, R. Qiu, X. Shi, P. Zhang and W. Zhang, J. Chem. Phys., 2014, 140, 074702 CrossRef PubMed.
- F.-Q. Huang, M.-L. Liu and C. Yang, Sol. Energy Mater. Sol. Cells, 2011, 95, 2924–2927 CrossRef CAS.
- I. Kriegel, C. Jiang, J. Rodríguez-Fernández, R. D. Schaller, D. V. Talapin, E. da Como and J. Feldmann, J. Am. Chem. Soc., 2011, 134, 1583–1590 CrossRef PubMed.
- X.-J. Wu, X. Huang, X. Qi, H. Li, B. Li and H. Zhang, Angew. Chem., Int. Ed., 2014, 53, 8929–8933 CrossRef CAS PubMed.
- C. G. Read, Y. Park and K.-S. Choi, J. Phys. Chem. Lett., 2012, 1872–1876, DOI:10.1021/jz300709t.
- A. B. Kehoe, D. O. Scanlon and G. W. Watson, J. Mater. Chem. C, 2015, 3, 12236–12244 RSC.
- A. Ghosh, S. Palchoudhury, R. Thangavel, Z. Zhou, N. Naghibolashrafi, K. Ramasamy and A. Gupta, Chem. Commun., 2016, 52, 264–267 RSC.
- N. Guijarro, M. S. Prévot, X. Yu, X. A. Jeanbourquin, P. Bornoz, W. Bourée, M. Johnson, F. Le Formal and K. Sivula, Adv. Energy Mater., 2016, 6, 1501949 CrossRef.
- S. T. Omelchenko, Y. Tolstova, H. A. Atwater and N. S. Lewis, ACS Energy Lett., 2017, 431–437, DOI:10.1021/acsenergylett.6b00704.
- J. Türck, H.-J. Nonnenmacher, P. M. L. Connor, S. Siol, B. Siepchen, J. P. Heimfarth, A. Klein and W. Jaegermann, Prog. Photovolt.: Res. Appl., 2016, 24, 1229–1236 CrossRef.
- D. Hartung, F. Gather, P. Hering, C. Kandzia, D. Reppin, A. Polity, B. K. Meyer and P. J. Klar, Appl. Phys. Lett., 2015, 106, 253901 CrossRef.
- E. Asenath-Smith, J. M. Noble, R. Hovden, A. M. Uhl, A. DiCorato, Y.-Y. Kim, A. N. Kulak, F. C. Meldrum, L. F. Kourkoutis and L. A. Estroff, Chem. Mater., 2017, 29, 555–563 CrossRef CAS.
- W. Yuan, J. Yuan, J. Xie and C. M. Li, ACS Appl. Mater. Interfaces, 2016, 8, 6082–6092 CrossRef CAS PubMed.
- J. J. Loferski, J. Appl. Phys., 1956, 27, 777–784 CrossRef CAS.
- H. Raebiger, S. Lany and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 045209 CrossRef.
- L. Pan, J. H. Kim, M. T. Mayer, M.-K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- J. Yu, B. Zhang, X. Zhang, Y. Wang, K. Wu and M.-H. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 45023–45035 CrossRef CAS PubMed.
- B. Zhang, X. Zhang, J. Yu, Y. Wang, K. Wu and M.-H. Lee, Chem. Mater., 2020, 32, 6772–6779 CrossRef CAS.
- J. E. Saal, S. Kirklin, M. Aykol, B. Meredig and C. Wolverton, JOM, 2013, 65, 1501–1509 CrossRef CAS.
- S. Kirklin, J. E. Saal, B. Meredig, A. Thompson, J. W. Doak, M. Aykol, S. Rühl and C. Wolverton, npj Comput. Mater., 2015, 1, 15010 CrossRef CAS.
- L. Wang, K. Han, G. Song, X. Yang and M. Tao, Characterization of electro-deposited CuO as a low-cost material for high-efficiency solar cells, 2006 IEEE 4th World Conference on Photovoltaic Energy Conference, 2006, 130–133, DOI:10.1109/WCPEC.2006.279381.
- N. A. M. Shanid and M. A. Khadar, Thin Solid Films, 2008, 516, 6245–6252 CrossRef CAS.
- L. C. Caero, E. Hernández, F. Pedraza and F. Murrieta, Catal. Today, 2005, 107–108, 564–569 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |