
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-step formulation of magnetic nanoprobes for microRNA capture
Iveta Vilímová ,
Igor Chourpa,
Stéphanie David,
Martin Soucé and
Katel Hervé-Aubert*
,
Igor Chourpa,
Stéphanie David,
Martin Soucé and
Katel Hervé-Aubert*
EA6295 Nanomédicaments et Nanosondes, Université de Tours, Tours, France. E-mail: katel.herve@univ-tours.fr
First published on 2nd March 2022
Abstract
MicroRNAs (miRs) belong to a family of short non-coding endogenous RNAs. Their over-expression correlates with various pathologies: for instance, miRNA-155 (miR-155) is over-expressed upon the development of breast cancers. However, the detection of miRs as disease biomarkers suffers from insufficient sensitivity. In the present study, we propose a protocol for a rapid and efficient generation of magnetic nanoprobes able to capture miR-155, with the aim of increasing its concentration. As a nanoprobe precursor, we first synthesized superparamagnetic iron oxide nanoparticles (SPIONs) coated with covalently attached polyethylene glycol carrying a free biotin terminus (PEG-bi). Using streptavidin–biotin interactions, the nanoprobes were formulated by functionalizing the surface of the nanoparticles with the miR sequence (CmiR) complementary to the target miR-155 (TmiR). The two-step formulation was optimized and validated using several analytical techniques, in particular with Size-Exclusion High Performance Liquid Chromatography (SE-HPLC). Finally, the proof of the nanoprobe affinity to TmiR was made by demonstrating the TmiR capture on model solutions, with the estimated ratio of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22 TmiR:CmiR per nanoprobe. The nanoprobes were confirmed to be stable after incubation in serum.
:22 TmiR:CmiR per nanoprobe. The nanoprobes were confirmed to be stable after incubation in serum.
1. Introduction
MicroRNAs (miR) are small non-coding RNAs with lengths ranging from 19 to 23 nucleotides. MiRs act as regulators in post-transcriptional gene expression by targeting specific messenger RNAs. They play a major part in various biological processes, which are influenced by dysregulation in miR expression levels.1,2As such, miRs are considered as novel biomarkers for diagnostics and therapeutics of various diseases (e.g., numerous types of cancer, neurological and muscle degenerative diseases, cardiovascular disorders, etc.). A large part of the scientific research is focused on the atypical expression of miRs present in different types of cancer. Depending on their altered expression, miRs can be oncogenic (increased expression leading to inhibition of tumor suppressor genes and subsequent malignancy), or tumor-suppressing (obstructing the expression of oncogenes).3 The disruption of the balance between these two types impacts cellular development, leading to cancer development. As the expression levels of multiple miRs either increase or decrease compared to the healthy tissue, the focus is put on developing new methods with better sensitivity and detection limits.4,5
One of the popular analytical methods widely used for purity determination and batch-to-batch comparison of compounds (miRs included) in pharmaceutical use is Size Exclusion High Performance Liquid Chromatography (SE-HPLC). It is built on separating the mixture in a solution into smaller components. A liquid mobile phase passes through the column filled with particles of a certain size, i.e., stationary phase. As the solution is pumped through the column, the components of the solution interact differently with the surface of the particles of the stationary phase and are separated in the process, coming out of the column at different and specific retention times. Commercially available miRs are purified by HPLC, in order to eliminate shorter lengths species originating from the possible degradation. Precise determination of separation, isolation, and purification of nucleotides from biological samples still requires a lot of effort, and there are limits for qualitative analysis abilities by the means of quantification and identification.6–8
Beyond generally used methods for miR detection (qRT-PCR,9,10 Northern blotting,11 next-generation sequencing,12,13 microarrays,14,15 etc.), other approaches based on nanomaterials are becoming progressively more popular, either in combination with a well-known method or by developing altogether new ones.16
Methods and approaches of miR detection based on nanotechnologies offer an alternative path to a faster and less complicated detection process. Nanomaterials used can be in a form of nanoparticles, colloids, substrates, etc. They provide unique optical, electrical, and magnetic properties,17 and most importantly, high surface area, leaving room for a plethora of surface-active chemical groups, which can be utilized in the subsequent immobilization of target biomolecules. The sensing application and efficiency of nanomaterials have been demonstrated.18,19 Specific challenges include reproducibility, standardization, normalization, and data processing. Surprisingly, a significant part of the reported studies did not deal with natural bodily fluids (serum, blood, saliva, etc.) in the presence of possibly interfering biomolecules.20
One possible way of using nanoprobes is to capture on their surface the target miR (TmiR) present in the biological fluid. The TmiR capture efficiency and specificity can be enhanced by using the nanoprobe surface decorated with a complementary miR sequence21 (hereafter called CmiR), double/single-stranded DNA,22 or sequence of oligonucleotides.23 Thus, successful design, production, and use of the TmiR-specific nanoprobes depend on several important considerations such as (i) synthesis of the nanomaterial (core), (ii) surface modification (platform), and (iii) surface functionalization (specific activation).
Hybrid nanoprobes (inorganic core–organic shell) are of high interest since their utilization allows to take benefit from unique physical properties of the inorganic core while controlling the stability due to the organic shell.24
The core–shell nanoprobes chosen for this study are based on the hybrid nanoparticles developed by our group and made of superparamagnetic iron oxide nanoparticles (SPIONs) coated with polyethylene glycol (PEG).25
Our working hypothesis was that the TmiR present in biological samples can be efficiently captured on the surface of the nanoprobes functionalized with specific CmiR and then the TmiR can be reconcentrated by magnetic sorting (Fig. 1A), thus favoring their more sensitive detection. PEGylation of SPIONs is used for enhancement of their biocompatibility and colloidal stability in aqueous media while preserving relative surface neutrality. We have selected a PEG chain modified with N-succinimidyl ester (NHS) on one end, and with biotin on the other (here-after called PEG-bi), for further conjugation via streptavidin–biotin affinity interactions.26 Streptavidin- and biotin-modified materials are widely commercially available, for either the nanomaterial matrix or the biomolecule shell. Streptavidin-coated magnetic nanoparticles,27 microparticles, or beads are very popular: for instance, they were exploited for detection of miR-144 (ref. 28) (via fluorescence signal amplification), or miR-21 (ref. 29) (via liquid chromatography-electrospray ionization tandem mass spectrometry). In our study, we formulated the nanoprobes starting from the PEGylated SPIONs and using streptavidin as a linker between the biotinylated nanoparticle surface and biotinylated CmiR (Fig. 1B). The idea is that the PEGylated SPIONs play a role of a versatile nanoplatform that can be used to easily formulate nanoprobes decorated with various CmiRs and therefore adaptable to various TmiRs.
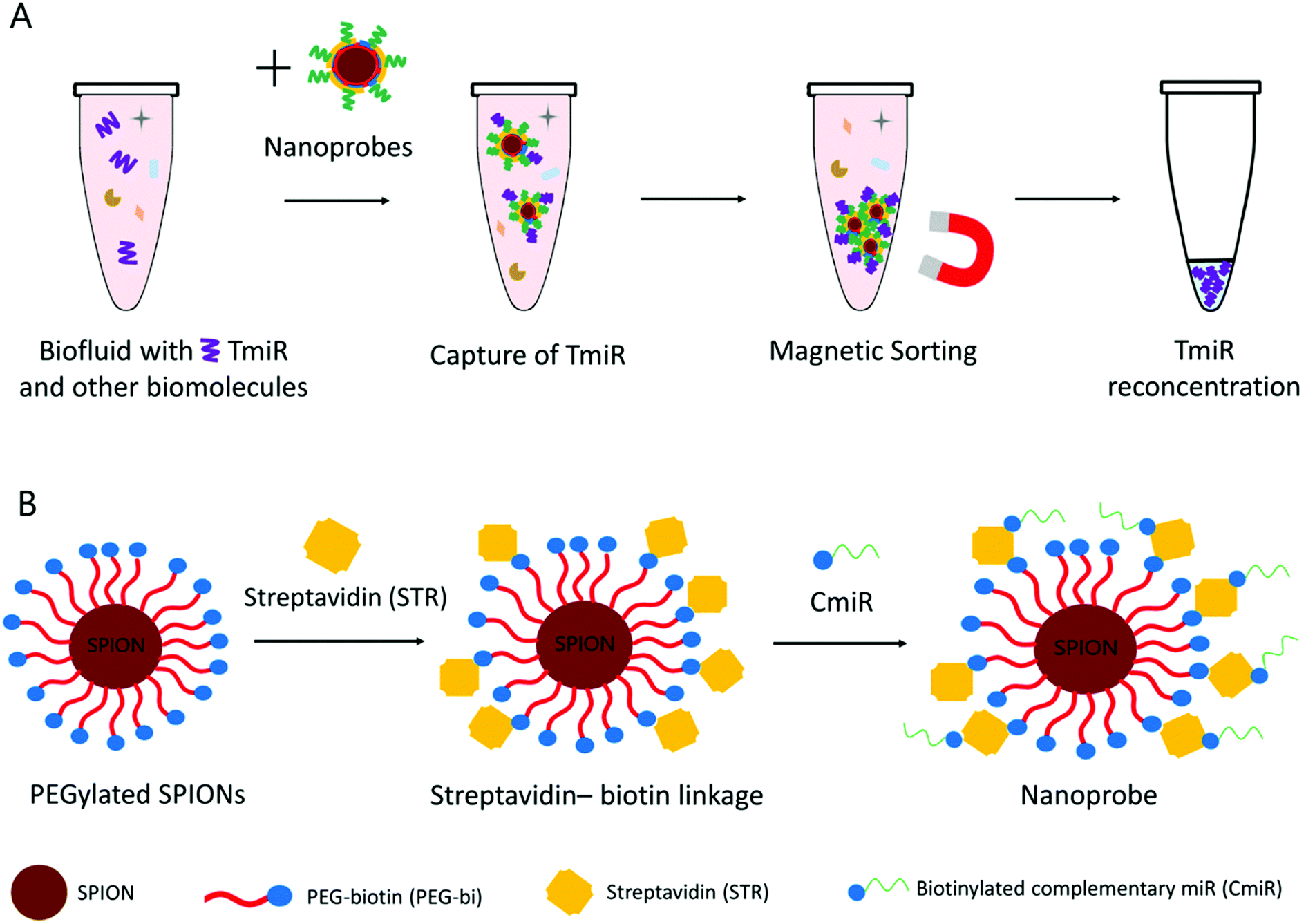 | ||
| Fig. 1 (A) TmiR capture and reconcentration in a biofluid. (B) Schematic representation of the nanoprobe formulation from the PEGylated SPIONs. | ||
After synthesis and characterization of SPION–PEG-bi nanoplatforms, we formulated and characterized several batches of the nanoprobes decorated with a model CmiR, specific to TmiR-155 upregulated in breast cancer cells and one of the most commonly reported miRs in body fluids.1,30
Several physicochemical characteristics of the nanosystems were established by means of a spectrum of analytical techniques: nanoparticle size (hydrodynamic diameter – DH by DLS-dynamic light scattering) and chemical composition (iron concentration by AAS – atomic absorption spectrophotometry, organic shell identification by IR spectrophotometry, miR concentrations by SE-HPLC). The results confirmed the successful synthesis of the SPION–PEG-bi nanoplatforms and allowed us to optimize the nanoprobes formulation in terms of their surface saturation with streptavidin (formulation step 1) and of their functionalization with CmiR (formulation step 2). The function of synthesized nanoprobes was confirmed by trying out their ability to capture TmiR-155, and their stability was tested by an incubation in serum.
2 Experimental
2.1 Materials
Deionized water (18 MΩ) was used in all experiments. α-Biotinyl-ω-NHS PEG, Mw 5000 Da, (NHS–PEG5000–biotin, PEG-bi) was purchased from Rapp Polymere (Tübingen, Germany). Streptavidin in lyophilized form, CmiR-155 modified with biotin ([Btn]-5′-ACCCCUAUCACGAUUAGCAUUAA-3′, 7674 g mol−1) and TmiR-155 (5′-UUAAUGCUAAUCGUGAUAGGGGU-3′, 7391 g mol−1) were purchased from Sigma-Aldrich (Saint Quentin-Fallavier, France). Standard miR solutions at required concentrations were prepared by adding RNAse-free deionized water.2.2 Synthesis of the nanoprobe precursors – SPIONs coated with PEG-bi (NP)
The synthesis of PEG-biotin-coated SPIONs was divided into 3 steps: (i) the coprecipitation of initial SPIONs, (ii) the silanization of SPION's surface, and (iii) the covering of silanized SPIONs with the PEG-bi layer.The synthesis of initial SPIONs by improved Massart's method and the silanization of SPIONs via surface amination with 3-aminopropyl-trimethoxysilane (APS) were performed according to the protocol published by our group.25 Afterwards, the surface of the particles was PEGylated by the following procedure.
A mixture of 1.6 mL of silanized SPIONs (0.14 mmol of iron) and 10 mL of DMSO was prepared, and the solution was dehydrated in a vacuum for 5 h with a rotary evaporator. Lyophilized NHS–PEG5000–biotin (220 mg, 0.04 mmol) was dissolved in 5 mL of dehydrated DMSO and added to the SPIONs mixture. The suspension was stirred for 24 h at the room temperature and afterward purified by dialysis (MWCO 1000 kDa) against water at the room temperature for 48 h.
2.3 Formulation of the nanoprobes
The formulation was made in two steps: (i) modification of the NP surface with streptavidin to result in NP–STR; and (ii) functionalization of the NP–STR with CmiR to result in NP-STR-CmiR.NP-STR1 (Fe/STR = 280 molar ratio) was generated by mixing 400 μL of NP suspension at an iron concentration of 350 mg L−1 with 500 μL of 1 mg mL−1 STR and 100 μL of PBS 10X. The other two batches, NP-STR2 (Fe/STR = 1400) and NP–STR3 (Fe/STR = 2100) were generated accordingly.
All the samples were shaken overnight at room temperature. The resulting NP–STR were purified by magnetic separation (protocol described in Section 2.6) in order to eliminate an excess of streptavidin.
The number of CmiR molecules per nanoparticle (N) was determined with the following formula:

2.4. TmiR capture by nanoprobes
The batch of nanoprobes NP–STR–CmiR5 (5 μM CmiR) was combined with the solution of TmiR to reach a 5 μM final concentration of TmiR and incubated for 2 hours. The resulting sample NP–STR–CmiR–TmiR was purified by magnetic separation (protocol in Section 2.6) in order to eliminate any free TmiR.2.5 Stability of nanoprobes
The batch of nanoprobes NP-STR-CmiR5 (5 μM CmiR) was combined with the solution of fetal bovine serum (FBS) (Eurobio, Les Ulis, France) in the ratio 1:1, and incubated at room temperature for 4 and 12 hours. The resulting samples (S4)-NP–STR–CmiR5 and (S12)-NP–STR–CmiR5 were purified by magnetic separation (protocol in the Section 2.6).
2.6 Magnetic separation
Magnetic separation (MS) was performed with MS columns (MACS® Column Technology, Miltenyi Biotec GmbH, Germany). The volume of the SPIONs suspension (up to 500 μL, depending on the initial volume of the sample) was applied into a MS column held in a magnetic MACS separator, and the separated liquid was collected. The column was then washed three times with the buffer (PBS 1X). The column was removed from the magnetic separator and flushed with the buffer (75% of the initial volume) resulting in a sample, utilized in the following synthetic procedures. MS was used for purification of the batches after each step of the synthesis, i.e., after saturation with STR, after functionalization with CmiR, and after TmiR capture.2.7 Analytical methods
All SE-HPLC measurements were performed in phosphate buffer mobile phase (0.15 mol L−1; the combination of KH2PO4 at 0.075 mol L−1 and Na2HPO4·2H2O at 0.075 mol L−1), with the flow rate 1.0 mL min−1, injection volume 10 μL, temperature 30 °C, duration 15 min and UV detection at 260 nm and 280 nm.
All samples for SE-HPLC measurements containing PEGylated SPIONs were magnetically separated, and the liquid from the sample was measured, in order to not inject a large concentration of magnetic nanoparticles into the column.
3 Results and discussion
3.1 Synthesis and characterization of the nanoprobe precursors
Coating nanosystems with neutral hydrophilic molecules such as PEG is important for biomedical applications, since it provides colloidal stability in aqueous media, independently from the pH (due to a sterical hindrance). In our study, the PEG chains were covalently attached to the silanized SPION surface, and the process involved three synthetic stages (Fig. 2), as described in the Experimental section. After the synthesis of initial SPIONs by coprecipitation of iron salts, the nanoparticles were silanized, i.e. coated with 3-aminopropyltrimethoxysilane (APS) molecules. The latter is bound to the hydroxyl groups abundant on the surface of SPIONs. The primary amines of APS remain available to form amide bonds with the activated esters of α-biotinyl-ω-NHS PEG. The resulting amide bond is well-known for its strength and stability.25 The PEGylated SPIONs were then extensively purified from the residual free PEG molecules by dialysis against water for 48 hours and stored in aqueous suspensions.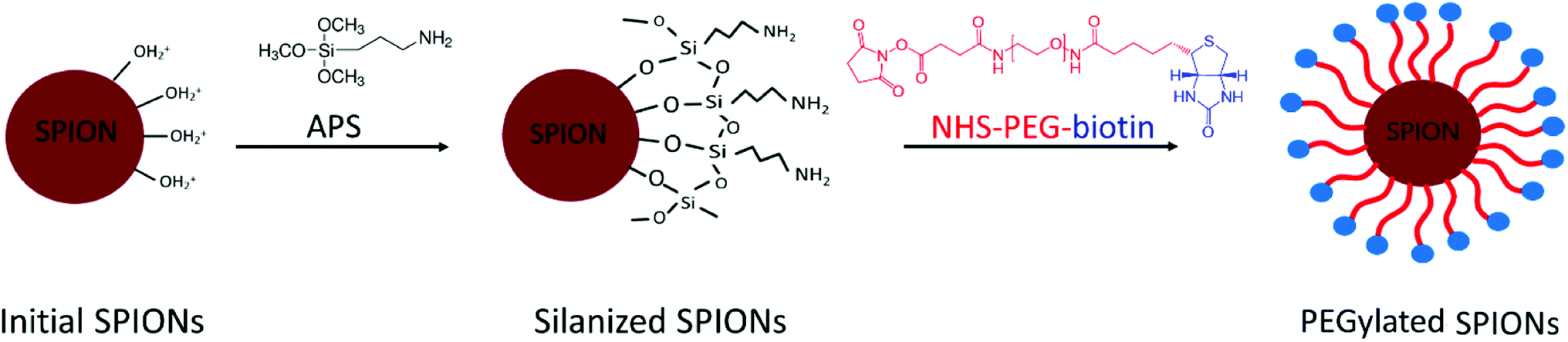 | ||
| Fig. 2 Schematic representation of the PEGylated SPIONs synthesis in 3 steps: (i) synthesis of the initial SPIONs; (ii) silanization of the SPIONs surface; (iii) PEGylation of the silanized SPIONs. | ||
The nanoparticles were characterized by a spectrum of analytical techniques: IR spectrometry to confirm the molecular composition and DLS to measure the NP DH and ZP. The iron concentration was determined by means of AAS and used for calculating the molar ratio in the next steps of the nanoprobe formulation.
Successful PEGylation of SPIONs was firstly confirmed by IR bands (Fig. 3A) characteristic of the PEG molecules: the strong bands at 3444 cm−1 (O–H stretching), 2921 cm−1 (C–H stretching of in CH2 and methylene groups), and 2853 cm−1 (C–H stretching). The amide bonds formed between PEG and silane were seen due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band at 1651 cm−1. More importantly, strong bands at 1099 cm−1 (O–H and C–O–H stretching, siloxane Si–O–SI stretching) characteristic for PEG molecules were observed and assigned to polycondensation of silanes.
O stretching band at 1651 cm−1. More importantly, strong bands at 1099 cm−1 (O–H and C–O–H stretching, siloxane Si–O–SI stretching) characteristic for PEG molecules were observed and assigned to polycondensation of silanes.
 | ||
| Fig. 3 Physicochemical properties of SPIONs. (A) IR spectra of PEGylated SPIONs, with the major bands labelled as follows: (1) 3444 cm−1; (2) 2921 cm−1; (3) 2853 cm−1; (4) 1651 cm−1; (5) 1099 cm−1. (B) Evolution of zeta potential (ZP) of initial SPIONs (red line), silanized SPIONs (grey line), and PEGylated SPIONs (yellow line) as a function of pH. | ||
Likewise, the successful formation of the polymeric shell was confirmed by the DLS results with the DH increase, from 36.9 ± 1.4 nm for initial SPIONs to 43.5 ± 3.4 nm after their silanization and 63.0 ± 4.0 nm after their PEGylation.
Similarly, ZP also changed after PEGylation: being positively charged for silanized SPIONs (18 ± 2.5 mV, pH 7) it became nearly neutral (−3.47 ± 0.97 mV, pH 7). The curves of ZP as a function of pH changed after the nanoparticle modification (Fig. 3B). The ZP of the initial SPIONs at lower pH is positive due to the presence of OH2+ on the surface of nanoparticles. The ZP decreases to nearly zero at pH 7 (isoelectric point), due to deprotonation of OH2+ to OH. At higher pH, the surface of SPIONs is covered with O− and the ZP is negative. For silanized SPIONs, the isoelectric point shifted closer to pH 9. As expected, the ZP of the PEGylated SPIONs was less sensitive to pH change and remained nearly zero. It can be concluded that the PEGylated SPIONs remained colloidally stable at neutral pH due to sterical hindrance.
3.2 Nanoprobe formulation and characterization
In our study, we have chosen a method of conjugation of miR to the PEGylated SPIONs via biotin (Bi)–streptavidin (STR) interaction, since the protocol is simple and cost-effective, requiring no additional reagents. Biotin-modified polymers and miRs are widely commercially available, and biotin insertion into the biomolecules is not intended to affect their properties or activity. Interaction between streptavidin and biotin is irreversible under physiologic conditions since the resulting bond is approaching in strength the covalent one and is unaffected by variation of temperature, pH, organic solvents, and denaturing agents. Furthermore, STR has four available binding sites, one in each of the four subunits of this protein, that allows each STR molecule to complex several molecules of biotin and thus of biotinylated molecules like PEG or miRs. Void of carbohydrate residues, STR has a low capability of forming non-specific bonds with other molecules. Once bound to the PEG-bi shell of the nanoparticles, STR should still have sites available for binding of biotinylated CmiR and thus enable the formation of nanoprobe described in Fig. 1.22,27First of all, the SE chromatograms were obtained for the ingredients taken separately (nanoparticles, STR, CmiR) then we analyzed their binary complexes (STR–CmiR), and finally the samples from nanoprobe synthesis steps. Fig. 4 presents chromatograms of PEGylated SPIONs, STR, CmiR, and of complex STR–CmiR. While all the chromatograms were also recorded with UV detection at 280 nm, CmiR has stronger absorbance at 260 nm, ergo herein mentioned results are obtained at this wavelength.
 | ||
| Fig. 4 SE-HPLC chromatograms. (A) PEGylated SPIONs (NP), tR: 4.4 min, (B) STR, tR: 8.2 min, (C) biotinylated CmiR, tR: 8.8 min, (D) STR–CmiR complex, tR: 6.8 and 8.8 min. | ||
PEGylated SPIONs had a relatively strong chromatographic peak at ca. 4.4 min (Fig. 4A). The peak of 1 g L−1 solution of streptavidin was observed at ca. 8.2 min (Fig. 4B). The strong peak of CmiR at concentration 5 μM was detected at ca. 8.8 min (Fig. 4C). The fact that the NP are eluted earlier than molecules is concomitant with their much bigger size. On the other hand, the close retention time about 8.2 and 8.8 min observed respectively for STR (Mw of ca. 55 kDa) and biotinylated CmiR (Mw of ca. 7.6 kDa) is much less expectable.
The formation of the complex STR–CmiR at molar ratio 3.78:1 was visible as the new peak rose at ca. 6.8 min (Fig. 4D) since the complex was larger than separate molecules. The uniform peak of the complex is in favor that all possible binding places on STR are probably occupied by the biotinylated CmiRs. In the mixed sample, the peak of free CmiR (ca. 8.8 min) decreased in intensity almost by a half (37.1 vs. 76.5 AU) compared to the free CmiR taken at the same concentration. The absence of the free STR peak (around 8.2 min) allows to suppose that, at the conditions used, all the STR molecules were bound to CmiR.
Formulation step 1 – modification of PEGylated SPIONs with STR. Aiming for a maximal coverage with STR at the biotin sites on the surface of the NP to achieve the best efficiency, we made the attempts with three Fe/STR molar ratios: 2100, 1400, and 280. At the lower Fe/STR molar ratio of 280 (sample NP–STR1), a weak peak of free STR was still present in the chromatograms (tR 8.2 min), while at higher Fe/STR molar ratio of 1400 and 2100 (the samples NP–STR2 and NP–STR3), no free STR peak was detected (Fig. 5). Thus, one could suppose that at the highest concentration (sample NP–STR1), the STR was able to saturate the available biotin sites.
 | ||
| Fig. 5 SE-HPLC analysis of the NP–STR (A): the full chromatograms of the samples: NP–STR1 (molar ratio Fe/STR = 280, red line, tR: 4.5, 6.9, 7.5 and 8.2 min), NP–STR2 (molar ratio Fe/STR = 1400, blue line, tR: 4.4, 6.3 min), NP–STR3 (molar ratio Fe/STR = 2100, black line, tR: 4.5 min and 6.3 min). (B) Zoom on the interval from 6 to 8.8 min with the overlayed peaks of the samples NP–STR1, NP–STR2, NP–STR3, and that of the STR–PEG-bi molecular complex (green dashed line). | ||
The weak peaks at 6.3, 6.5 and 6.9 min observed in those samples were possibly due to the presence of PEG-bi chains which were detached from the NP into solution after their complexation with the high molecular weight STR. Indeed, one can reasonably suppose that despite the purification via dialysis which was used to remove the excess of the free PEG-bi, it was still possible to desorb from the nanoparticles some minor fraction of the PEG-bi bound to STR. In agreement with this hypothesis, the peak of the molecular complex STR–PEG (Fig. 5B) had a close retention time of ca. 6.7 min. The 3 peaks at 6.3, 6.5 and 6.9 min might correspond to complex fractions with different STR binding sites. This result demonstrates the importance of the purification between the two formulation steps.
The presence of the free STR and the STR–PEG complexes in the NP–STR samples was undesirable since they might compete with STR-modified NP for the binding to the CmiR upon the next step of the nanoprobes formulation. Thus, we applied magnetic separation to purify the NP-STR and successfully eliminate the free STR and the PEG-bi–STR complexes.
Ideally, all the biotin sites on the PEGylated SPIONs surface would be occupied with STR. However, the steric hindrance of the large STR molecules can possibly prevent achieving the saturation of all the biotin sites since some of them are not accessible.
Since the sample NP–STR1 was the best covered with STR, the corresponding batches were purified from unbound species and used for the second formulation step.
Formulation step 2 – functionalization of the NP–STR with CmiR. Here again, our aim was to functionalize the NP–STR with as many CmiR as possible since it is supposed to favor the efficiency of the final nanoprobes. We started with the CmiR concentration of 5 μM and generated the samples NP–STR–CmiR5. As the formation of STR–CmiR complex was proved to be relatively fast, the 2 hours of incubation of the NP–STR with CmiR (under stirring, at the room temperature) was supposed long enough. The conjugation of both STR and CmiR did not markedly change the physicochemical properties of the respective batches (Table 1).
| DH [nm] | PDI | Zeta potential [mV] | |
|---|---|---|---|
| NP (PEGylated SPIONs) | 61.6 ± 2.0 | 0.20 ± 0.00 | 2.7 ± 0.1 |
| NP–STR1 | 67.0 ± 0.5 | 0.13 ± 0.01 | 3.1 ± 0.2 |
| NP–STR–CmiR5 | 71.2 ± 0.4 | 0.11 ± 0.01 | 2.5 ± 0.4 |
The batches of PEGylated SPIONs showed a comparable DH in both deionized water and in PBS 1X (Table 1): 61.6 ± 2.0 nm vs. 63.0 ± 4.0 nm. After the modification with STR, the DH in PBS 1X slightly increased to 67.0 ± 0.5 nm (Table 1), presumably due to the presence of STR molecules on the surface of the nanoparticles. Functionalization with CmiR led only to a minor increase of DH till 71.2 ± 0.4 nm. Zeta potential remained largely unchanged (Table 1), with values close to neutral. Interestingly, the PDI values diminished in going to the formulated nanoprobes compared to the PEGylated SPIONs. The PDI value of 0.11 is typically considered as characteristic of a monodisperse nanoparticle population. To summarize, the formulation steps did not decrease the colloidal stability of the nanoprobes.
The amount of CmiR bound to the nanoprobes was determined by means of the SE-HPLC analysis. The NP–STR–CmiR5 sample (Fig. 6) revealed 2 peaks: that of nanoparticles at ca. 4.4 min and that of the free CmiR at ca. 8.7 min. The latter decreased in the intensity down to 73.9% compared to the chromatogram of the pure CmiR. Thus, after incubation with 5 μM CmiR solution, about 26% of CmiR were bound to the nanoprobes (the concentration of the bound CmiR was 1.3 μM).
 | ||
| Fig. 6 SE-HPLC results for the second (final) step of the nanoprobes formulation. Typical chromatograms of NP–STR–CmiR5 (red curve, peaks at 4.4 and 8.7 min) compared to those of the pure CmiR at the same concentration of 5 μM (black curve, peak at ca. 8.8 min). | ||
Subsequently, we also prepared samples by incubating NP–STR with the lower and higher concentrations of CmiR: 1 μM (NP–STR–CmiR1) and 10 μM (NP–STR–CmiR10). According to the SE-HPLC data, at 1 μM and 10 μM of incubated CmiR, the nanoprobe-bound CmiR fraction was respectively 66% and 12% and corresponded to concentrations of 0.66 and 1.2 μM (Table 2). To summarize, using a 5 μM concentration of CmiR would be more advisable than 1 μM. In contrast, larger excess of 10 μM does not allow to increase the bound CmiR concentration and leads to use-less loss of the nucleic acid.
| Sample | Fraction of bound CmiR | Fraction of bound CmiR (concentration) | Number of bound CmiRs per NP |
|---|---|---|---|
| NP–STR–CmiR1 | 66% | 0.66 μM | 11 |
| NP–STR–CmiR5 | 26% | 1.3 μM | 22 |
| NP–STR–CmiR10 | 12% | 1.2 μM | 20 |
The estimated number of CmiR molecules grafted per nanoprobe was 11, 22 and 20 for the samples NP–STR–CmiR1, NP–STR–CmiR5, and NP–STR–CmiR10 respectively. To the best of our knowledge, very few publications reported the quantity of grafted CmiR31,32 or other oligonucleotides27 per nanoprobe or even the concentration of grafted CmiR.33,34 Although less explored in the literature, these values are important since they allow to evaluate both the conjugation method used and the expected efficiency of TmiR capture.35 It has been reported that only one CmiR per nanoprobe may be sufficient for TmiR detection.32
3.3 Proof of the nanoprobes affinity to TmiR-155
To determine the ability of nanoprobes to capture the target miR, the purified NP–STR–CmiR5 samples were incubated with the 5 μM solution of the TmiR-155, then were magnetically sorted. The chromatography data indicate that after the sorting, 21% (≈1 μM) of TmiR were bound to the nanoprobes, which corresponds to approximately 18 TmiRs per nanoprobe. This result is promising since it confirms that the nanoprobes were able to adsorb TmiR and they allowed to remove a significant fraction of it. The 18:22 molar ratio for TmiR:CmiR is also promising, while has not been considered as definitive, since it depends on the application protocols.
3.4 Proof of the stability of the nanoprobes in serum
To confirm the stability of the nanoprobes NP–STR–CmiR5 (5 μM CmiR), a new batch was prepared and combined with the solution of fetal bovine serum to be incubated for 4 and 12 hours, resulting in the samples (S4)-NP–STR–CmiR5 and (S12)-NP–STR–CmiR5 respectively. The sample (S4)-NP–STR–CmiR5 was visually monitored for signs of aggregation after 1, 2 and 3 hours, with no apparent change to the solution visible by the eye. We measured the physicochemical properties of the nanoprobes prior to the mixing with serum, and after the incubation and purification via magnetic separation (Table 3). The batch NP–STR–CmiR5 showed similar DH to the one after the incubation in the serum for both 4 h and 12 h, the size increased only slightly, 72.8 ± 3.3 nm vs. 78.2 ± 6.1 nm vs. 78.2 ± 1.7 nm, with a bigger standard deviation in the case of the sample (S4)-NP–STR–CmiR5. The polydispersity remained stable for all three batches. The zeta potential of the new batch NP–STR–CmiR5 shifted to the negative value of −7.1 ± 2.0 mV, and after the incubation showed no significant variation. The small changes in physicochemical properties could be explained by a small-scale absorption of proteins present in the serum to the surface of the nanoprobes. The nanoprobes however remain colloidally stable, with their properties not influenced even by a long incubation in the serum.| DH [nm] | PDI | Zeta potential [mV] | |
|---|---|---|---|
| NP–STR–CmiR5 | 72.8 ± 3.3 | 0.18 ± 0.01 | −7.1 ± 2.0 |
| (S4)-NP–STR–CmiR5 | 78.2 ± 6.1 | 0.19 ± 0.02 | −6.6 ± 1.0 |
| (S12)-NP–STR–CmiR5 | 78.2 ± 1.7 | 0.18 ± 0.01 | −4.7 ± 1.3 |
4. Conclusion
In this study, CmiR-functionalized magnetic nanoprobes were designed for TmiR-155 capture. As precursors of the nanoprobes, we synthesized SPIONs coated with covalently bound PEG biotinylated on its external terminus. Then, the precursors were used to formulate nanoprobes in two steps: (i) modification of the biotinylated external shell with streptavidin and (ii) functionalization of the STR-modified shell with biotinylated CmiR. Saturation of the surface of NP with streptavidin was achieved with molar ratio Fe/STR = 280. The corresponding batches of the NP–STR were then incubated with biotinylated CmiR (5 μM), and 26% of the latter were bound. The nanoprobes coated with 22 CmiR per nanoprobe were able to capture 18 TmiR per nanoprobe from the 5 μM TmiR solution. The nanoprobes were proved to be stable after an incubation in the serum. This result is promising, while in real samples the nanoprobes concentration has to be certainly used in excess compared to that of TmiR. The capture protocols optimization is a subject of an independent study we are starting.In close future, the developed nanoprobes will be also studied concerning their ability to capture the target miR (miR-155) in serum from patients and thus to monitor their concentration before and after an anti-cancer treatment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the ‘‘Ligue Nationale contre le Cancer” for their financial support (project NANOmiR) and especially to the local comities 53 and 56.References
- H.-Y. Loh, B. P. Norman, K.-S. Lai, N. M. A. N. A. Rahman, N. B. M. Alitheen and M. A. Osman, The Regulatory Role of MicroRNAs in Breast Cancer, Int. J. Mol. Sci., 2019, 20(19), 4940, DOI:10.3390/ijms20194940.
- P. Krishnan and S. Damaraju, The Challenges and Opportunities in the Clinical Application of Noncoding RNAs: The Road Map for MiRNAs and PiRNAs in Cancer Diagnostics and Prognostics, Int. J. Genomics, 2018, 2018(12), 1–18, DOI:10.1155/2018/5848046.
- A. A. Svoronos, D. M. Engelman and F. J. Slack, OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer, Cancer Res., 2016, 76(13), 3666–3670, DOI:10.1158/0008-5472.CAN-16-0359.
- R. D'Agata and G. Spoto, Advanced Methods for MicroRNA Biosensing: A Problem-Solving Perspective, Anal. Bioanal. Chem., 2019, 411(19), 4425–4444, DOI:10.1007/s00216-019-01621-8.
- T. Ouyang, Z. Liu, Z. Han and Q. Ge, MicroRNA Detection Specificity: Recent Advances and Future Perspective, Anal. Chem., 2019, 91(5), 3179–3186, DOI:10.1021/acs.analchem.8b05909.
- A. Kanavarioti, HPLC Methods for Purity Evaluation of Man-Made Single-Stranded RNAs, Sci. Rep., 2019, 9(1), 1–13, DOI:10.1038/s41598-018-37642-z.
- M. Kullolli, E. Knouf, M. Arampatzidou, M. Tewari and S. Pitteri, J. Am. Soc. Mass Spectrom., 2011, 72(2), 181–204, DOI:10.1007/s13361-013-0759-x.Intact.
- E. Boros, O. R. Pinkhasov and P. Caravan, Metabolite Profiling with HPLC-ICP-MS as a Tool for in vivo Characterization of Imaging Probes, EJNMMI Radiopharm. Chem., 2018, 3(1) DOI:10.1186/s41181-017-0037-5.
- E. M. Kroh, R. K. Parkin, P. S. Mitchell and M. Tewari, Analysis of Circulating MicroRNA Biomarkers in Plasma and Serum Using Quantitative Reverse Transcription-PCR (QRT-PCR), Methods, 2010, 50(4), 298–301, DOI:10.1016/j.ymeth.2010.01.032.
- M. Y. Pratama, L. Cavalletto, C. Tiribelli, L. Chemello and D. Pascut, Selection and Validation of MiR-1280 as a Suitable Endogenous Normalizer for QRT-PCR Analysis of Serum MicroRNA Expression in Hepatocellular Carcinoma, Sci. Rep., 2020, 10(1), 3128, DOI:10.1038/s41598-020-59682-0.
- É. Várallyay, J. Burgyán and Z. Havelda, MicroRNA Detection by Northern Blotting Using Locked Nucleic Acid Probes, Nat. Protoc., 2008, 3(2), 190–196, DOI:10.1038/nprot.2007.528.
- L. W. Lee, S. Zhang, A. Etheridge, L. Ma, D. Martin, D. Galas and K. Wang, Complexity of the MicroRNA Repertoire Revealed by Next-Generation Sequencing, Rna, 2010, 16(11), 2170–2180, DOI:10.1261/rna.2225110.
- A. D. Martinez-Gutierrez, O. M. Catalan, R. Vázquez-Romo, F. I. P. Reyes, A. Alvarado-Miranda, F. L. Medina, J. E. Bargallo-Rocha, L. T. O. Moreno, D. C. de León, L. A. Herrera, C. López-Camarillo, C. Pérez-Plasencia and A. D. Campos-Parra, MiRNA Profile Obtained by Next-generation Sequencing in Metastatic Breast Cancer Patients Is Able to Predict the Response to Systemic Treatments, Int. J. Mol. Med., 2019, 44(4), 1267–1280, DOI:10.3892/ijmm.2019.4292.
- M. Rusek, M. Michalska-Jakubus, M. Kowal, J. Bełtowski and D. Krasowska, A Novel MiRNA-4484 Is up-Regulated on Microarray and Associated with Increased MMP-21 Expression in Serum of Systemic Sclerosis Patients, Sci. Rep., 2019, 9(1), 1–13, DOI:10.1038/s41598-019-50695-y.
- J. E. Ideozu, X. Zhang, V. Rangaraj, S. McColley and H. Levy, Microarray Profiling Identifies Extracellular Circulating MiRNAs Dysregulated in Cystic Fibrosis, Sci. Rep., 2019, 9(1), 1–11, DOI:10.1038/s41598-019-51890-7.
- M. K. Masud, M. Umer, M. S. A. Hossain, Y. Yamauchi, N. T. Nguyen and M. J. A. Shiddiky, Nanoarchitecture Frameworks for Electrochemical MiRNA Detection, Trends Biochem. Sci., 2019, 44(5), 433–452, DOI:10.1016/j.tibs.2018.11.012.
- P. Christian, F. Von Der Kammer, M. Baalousha and T. Hofmann, Nanoparticles: Structure, Properties, Preparation and Behaviour in Environmental Media, Ecotoxicology, 2008, 17(5), 326–343, DOI:10.1007/s10646-008-0213-1.
- G. Barbillon, Latest Novelties on Plasmonic and Non-Plasmonic Nanomaterials for SERS Sensing, Nanomaterials, 2020, 10(6), 1–17, DOI:10.3390/nano10061200.
- L. Gloag, M. Mehdipour, D. Chen, R. D. Tilley and J. J. Gooding, Advances in the Application of Magnetic Nanoparticles for Sensing, Adv. Mater., 2019, 31(48), 1–26, DOI:10.1002/adma.201904385.
- I. Gessner, J. W. U. Fries, V. Brune and S. Mathur, Magnetic Nanoparticle-Based Amplification of MicroRNA Detection in Body Fluids for Early Disease Diagnosis, J. Mater. Chem. B, 2021, 9(1), 9–22, 10.1039/d0tb02165b.
- I. Gessner, X. Yu, C. Jüngst, A. Klimpel, L. Wang, T. Fischer, I. Neundorf, A. C. Schauss, M. Odenthal and S. Mathur, Selective Capture and Purification of MicroRNAs and Intracellular Proteins through Antisense-Vectorized Magnetic Nanobeads, Sci. Rep., 2019, 9(1), 1–10, DOI:10.1038/s41598-019-39575-7.
- B. Tian, Y. Han, E. Wetterskog, M. Donolato, M. F. Hansen, P. Svedlindh and M. Strömberg, MicroRNA Detection through DNAzyme-Mediated Disintegration of Magnetic Nanoparticle Assemblies, ACS Sens., 2018, 3(9), 1884–1891, DOI:10.1021/acssensors.8b00850.
- S. Grijalvo, A. Alagia, A. F. Jorge and R. Eritja, Covalent Strategies for Targeting Messenger and Non-Coding RNAs: An Updated Review on SiRNA, MiRNA and AntimiR Conjugates, Genes, 2018, 9(2), 74–111, DOI:10.3390/genes9020074.
- N. Knežević, I. Gadjanski and J. O. Durand, Magnetic Nanoarchitectures for Cancer Sensing, Imaging and Therapy, J. Mater. Chem. B, 2019, 7(1), 9–23, 10.1039/c8tb02741b.
- K. Hervé, L. Douziech-Eyrolles, E. Munnier, S. Cohen-Jonathan, M. Soucé, H. Marchais, P. Limelette, F. Warmont, M. L. Saboungi, P. Dubois and I. Chourpa, The Development of Stable Aqueous Suspensions of PEGylated SPIONs for Biomedical Applications, Nanotechnology, 2008, 19(46), 465608–465614, DOI:10.1088/0957-4484/19/46/465608.
- Y. Du and P. W. T. Pong, Multi-Binding Biotinylated Iron Oxide Nanoparticles as a Promising Versatile Material for Magnetic Biomedical Applications, Micro Nano Lett., 2018, 13(4), 415–420, DOI:10.1049/mnl.2017.0348.
- Y. Wang, P. D. Howes, E. Kim, C. D. Spicer, M. R. Thomas, Y. Lin, S. W. Crowder, I. J. Pence and M. M. Stevens, Duplex-Specific Nuclease-Amplified Detection of MicroRNA Using Compact Quantum Dot-DNA Conjugates, ACS Appl. Mater. Interfaces, 2018, 10(34), 28290–28300, DOI:10.1021/acsami.8b07250.
- M. Oishi, Comparative Study of DNA Circuit System-Based Proportional and Exponential Amplification Strategies for Enzyme-Free and Rapid Detection of MiRNA at Room Temperature, ACS Omega, 2018, 3(3), 3321–3329, DOI:10.1021/acsomega.7b01866.
- X. Li, J. Zhao, R. Xu, L. Pan and Y. M. Liu, Mass Spectrometric Quantification of MicroRNAs in Biological Samples Based on Multistage Signal Amplification, Analyst, 2020, 145(5), 1783–1788, 10.1039/c9an02064k.
- P. K. Lo, B. Wolfson, X. Zhou, N. Duru, R. Gernapudi and Q. Zhou, Noncoding RNAs in Breast Cancer, Briefings Funct. Genomics, 2016, 15(3), 200–221, DOI:10.1093/bfgp/elv055.
- G. Yammouri, H. Mohammadi and A. Amine, A Highly Sensitive Electrochemical Biosensor Based on Carbon Black and Gold Nanoparticles Modified Pencil Graphite Electrode for MicroRNA-21 Detection, Chem. Africa, 2019, 2(2), 291–300, DOI:10.1007/s42250-019-00058-x.
- M. Wang, W. Chen, L. Tang, R. Yan and P. Miao, Duplex-Specific Nuclease Assisted MiRNA Assay Based on Gold and Silver Nanoparticles Co-Decorated on Electrode Interface, Anal. Chim. Acta, 2020, 1107, 23–29, DOI:10.1016/j.aca.2020.01.041.
- H. N. Chan, S. L. Ho, D. He and H. W. Li, Direct and Sensitive Detection of Circulating MiRNA in Human Serum by Ligase-Mediated Amplification, Talanta, 2020, 206, 120217, DOI:10.1016/j.talanta.2019.120217.
- W. K. Wong, S. H. D. Wong and L. Bian, Long-Term Detection of Oncogenic MicroRNA in Living Human Cancer Cells by Gold@Polydopamine-Shell Nanoprobe, ACS Biomater. Sci. Eng., 2020, 6(7), 3778–3783, DOI:10.1021/acsbiomaterials.0c00633.
- H. Wang, H. Tang, C. Yang and Y. Li, Selective Single Molecule Nanopore Sensing of MicroRNA Using PNA Functionalized Magnetic Core–Shell Fe3O4-Au Nanoparticles, Anal. Chem., 2019, 91(12), 7965–7970, DOI:10.1021/acs.analchem.9b02025.
| This journal is © The Royal Society of Chemistry 2022 |