
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrothermally synthesized poorly-crystalline binary oxides with ZrW2O8 composition: preparation, structural analysis, and catalytic activity for the alkylation of anisole with benzyl alcohol†
Takashi Yamamoto *ab,
Seina Heiuchib,
Maki Kondob,
Kei Tabusadania and
Atsushi Sakakic
*ab,
Seina Heiuchib,
Maki Kondob,
Kei Tabusadania and
Atsushi Sakakic
aDepartment of Science and Technology, Tokushima University, Tokushima 770-8506, Japan. E-mail: takashi-yamamoto.ias@tokushima-u.ac.jp
bDepartment of Mathematical and Material Sciences, Tokushima University, Tokushima 770-8502, Japan
cNichia Corporation, Anan, Tokushima 774-8601, Japan
First published on 28th January 2022
Abstract
Hydrothermally synthesized poorly-crystalline metastable Zr–W binary hydroxide (W/Zr = 2), after calcination, was confirmed to be a strong solid acid catalyst to promote the alkylation of anisole with benzyl alcohol. The preparation conditions, structure of the as-prepared catalysts and the calcined hydroxides were investigated using XRD, nitrogen adsorption isotherms, TG-DTA, and XANES/EXAFS techniques. The crystalline phase was controlled by the hydrochloric acid concentration used for preparing a mother gel, and 5–9 M HCl was suitable for preparing the active phase. The tungsten species exists as a six-valent WO6 distorted octahedron connected with the ZrO7 unit via corner-sharing linkages. The incompleteness of the network structure is suggested to be responsible for the solid acidity.
1. Introduction
ZrW2O8 is a compound appearing in the phase diagram of the ZrO2–WO3 system1 and is known as a negative thermal expansion material over a wide temperature range of 0.3–1050 K.2–4 It is thermodynamically stable between 1378 and 1530 K, and its low-temperature synthesis is possible by calcination of ZrW2O7(OH)2·2H2O crystals at ∼873 K.5 Generally, the highly crystalline ZrW2O7(OH)2 phase6 is synthesized during the hydrothermal treatment of a mother gel in acidic halide-containing media. The effects of HCl concentration used and/or the time of hydrothermal treatment on the crystalline phase have been examined.7–11 We have confirmed that a poorly crystalline phase (tentatively denoted as ZWO-I) forms before forming the well-crystalized ZrW2O7(OH)2·2H2O phase during hydrothermal treatment; the phase is stable after calcination at 873 K (Fig. S1†).12Tungsten–zirconium binary oxide catalysts developed by Hino and Arata,13,14 so-called tungstated-zirconia (WOx–ZrO2), have attracted considerable attention because of their strong acidity comparable to 100% H2SO4 and properties such as promoting the skeletal isomerization of light alkanes under mild conditions, easy preparation and handling, as well as high thermal stability of up to ∼1100 K.15–18 Although the origin of the acidic property and the structure of acid sites have been examined by many research groups,13,19–26 the problems remain unsolved to date. We presume that a mixed metal oxide forms at the interface between the loaded species and support, and a series of Zr–W binary oxides are candidates for a model catalyst, conventional tungstated-zirconia. Note that the W/Zr atomic ratio of conventional tungstated-zirconia (15 wt% as WO3) is <0.1, whereas that of ZrW2O8 is 2.
We reported that the as-mentioned poorly crystalline ZWO-I phase acts as a solid acid to promote the alkylation of anisole with benzyl alcohol, whereas the amorphous silica-alumina of the typical strong solid acid, highly crystalline ZrW2O8 and ZrW2O7(OH)2 are catalytically inert under the same reaction conditions.12 In this study, we closely examine the ZWO-I phase preparation condition, structure, and catalytic performance. Although we recorded W L-edge XAFS of the ZWO-I phase in a previous study, we did not perform Zr K-edge XAFS characterization or curve-fitting analysis at the W L3 edge. In the current solid acid catalyst, the Zr edge EXAFS analysis is expected to provide information about the boundary structure between the tungsten and zirconium species. The XRD pattern identical to the ZWO-I phase has been reported by Xing et al.,27 Ando et al.,28 and Lucas et al.29 Because of the wide lines in the powder X-ray diffraction patterns, the phase was identified as amorphous, and the precise structural characterization has not been performed. Ando et al. reported that the phase acts as a solid acid catalyst to promote the alkylation of anisole with benzyl alcohol.28 Because zirconium sulfate and sulfuric acid were used in their preparation, it is unknown whether sulfated zirconia30,31 of a typical strong solid acid exists on the surface or not. To our knowledge, there have been no other studies reporting that the ZWO-I phase itself promotes the acid-catalyzed reaction and detailed characterization.
2. Experimental
2.1. Preparation
Zr–W binary oxide precursor was prepared via the hydrothermal treatment of a mother gel at 453 K based on previous studies.12,27 In particular, 100 mL of the aqueous solution of 0.021 M (NH4)6H2W12O40 (ammonium metatungstate, Wako Chemicals) was added to 50 mL of 0.25 M ZrOCl2 (zirconium oxychloride, Nacalai, GR) at 333 K and stirred for 2 h. Then, 50 mL of 0–10 M hydrochloric acid (HCl) (Nacalai, GR) was added to the mixture and stirred for 5 h to prepare a mother gel. The mother gel was loaded into a Teflon bottle in an autoclave and statically heated at 453 K for 6–240 h. The white precipitate obtained was repeatedly washed with distilled water until Cl− was free based on the AgNO3 test. After drying at 383 K for 12 h, the as-synthesized binary hydroxide (ZWOH) was calcined at 673–1073 K for 3 h under air to form the corresponding binary oxide (ZWO). The samples prepared will be referred to hereafter as ZWOH_XM_Yh and ZWO_XM_Yh_Z where X, Y, and Z are the concentrations of the utilized HCl, time of hydrothermal treatment, and calcination temperature, respectively. Unless otherwise stated, the utilized HCl concentration, time of hydrothermal treatment, and calcination temperature were 6 M, 12 h, and 873 K, respectively.The reference catalysts used were tungstated-zirconia13,14 (home-made, reference JRC-WZ-1), sulfate-ion treated zirconium oxide (sulfated zirconia),30 amorphous silica-alumina (JRC-SAL-4, 12.6 wt% as Al2O3), and hexagonally ordered mesoporous silica FSM-16.32 Amorphous zirconium hydroxide Zr(OH)x was prepared by hydrolyzing a zirconium oxychloride aqueous solution at pH = 10 with an NH3 aqueous solution, followed by aging, washing, and drying at 383 K. Conventional tungstated-zirconia was prepared by impregnation of the (NH4)10H2W12O42 (ammonium paratungstate, Nacalai, GR) aqueous solution on Zr(OH)x at 353 K and drying, followed by calcination at 1073 K for 3 h.22 The loading amount was 15 wt% as WO3. Sulfated zirconia was prepared by impregnation of 0.5 M H2SO4 on Zr(OH)x at room temperature for 0.5 h; it was filtered, followed by calcination at 873 K for 3 h. FSM-16 was synthesized using a water glass (Fuji Silysia Chemical; SiO2: 25.3 wt%, Na2O: 7.56 wt%, Al: 54 ppm, Fe: 3.6 ppm, Ti: 2.3 ppm, Ca: 5.7 ppm) and hexadecyltrimethylammonium bromide (Kishida Chemical). The preparation procedure of FSM-16 and the acidic properties have been previously reported in detail.33–35 Monoclinic-ZrO2 was prepared by calcination of Zr(OH)x at 1573 K. In6WO12 was synthesized via a solid-state reaction by calcination of pelletized In2O3–WO3 mixture at 1273 K.
2.2. Characterization
XRD patterns were recorded using a Miniflex diffractometer (Rigaku) equipped with a Ni-filtered Cu Kα radiation source. The N2 adsorption isotherms at 77 K were measured using a BELSORP-mini (MicrotracBEL). Each sample was outgassed for 2 h before measurement at 573 K (ZWO) or room temperature (as-synthesized ZWOH). The Dollimore–Heal method was used to analyze the adsorption data to obtain the pore size distribution. A simultaneous thermogravimetric and differential thermal analysis (TG-DTA) was performed under air using a DTG-60 analyzer (Shimadzu) at a heating rate of 10 K min−1. The W L-edge X-ray absorption spectra were recorded using a laboratory-type spectrometer R-XAS Looper (Rigaku)36 in the transmission mode at room temperature. A curved Si(620) or Si(400) monochromator crystal was used in W L1-edge XANES and W L3-edge EXAFS experiments, respectively. Zr K-edge X-ray absorption spectra were recorded at the BL16B2 at SPring-8 using a Si(311) double-crystal monochromator. For XANES analysis, the background removal followed by normalization was performed using Igor Pro 6 program.37 The energy was not calibrated. The data reduction for the EXAFS analysis was performed using the REX2000 program.38 Fourier transforms were applied to EXAFS in the k-ranges of ∼3–11 Å−1 and 3–18 Å−1 at the W L3- and Zr K-edge, respectively. Because of the intrinsic contamination of Hf species in the used zirconium regent, the available k-range of W L3 edge EXAFS is limited to Hf L2-edge (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 739 eV). The FEFF8.4 program39 was used to calculate the phase shift and backscattering amplitudes.
739 eV). The FEFF8.4 program39 was used to calculate the phase shift and backscattering amplitudes.
2.3. Catalysis
The alkylation of benzyl alcohol with anisole was performed using a stirred batch reactor under dry N2 atmosphere typically at 393 K. Before the reaction, 100 mg of the catalyst was outgassed at 473 K for 2 h. The reactor was loaded with 0.64 mL of benzyl alcohol (6.2 mmol) and 10 mL of anisole (92 mmol). The product distributions were analyzed using a GC-14A gas chromatograph (Shimadzu) equipped with a flame ionization detector. n-Decane was used as an internal standard.3. Results
3.1. Structural analysis
 | ||
| Fig. 1 XRD patterns of the hydrothermally synthesized (a) binary hydroxide ZWOHs prepared at various HCl concentrations and (b) ZWOs calcined at 873 K. Hydrothermal treatment: 453 K, 12 h. ○: ZrW2O7(OH)2·2H2O, ●: ZWO-I, △: ZrW2O8, ◊: tungsten bronze. | ||
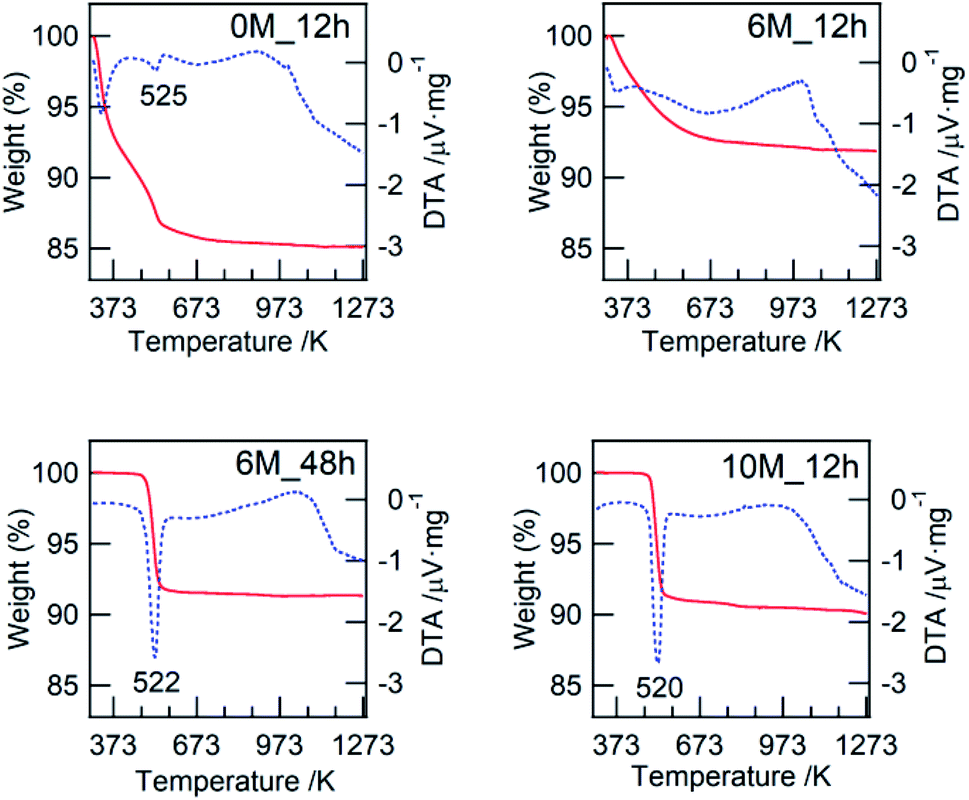 | ||
| Fig. 2 TG-DTA profiles of the typical as-synthesized binary hydroxide prepared with different HCl concentrations and hydrothermal treatment times. Solid curve: TG, dotted curve: DTA. | ||
 | ||
| Fig. 3 Nitrogen adsorption/desorption isotherms of calcined binary oxides ZWO_XM_12h_873 prepared with different HCl concentrations (X = 0–10). | ||
100 eV is known to be sensitive to the coordination symmetry of tungsten atom.43–45 Cubic ZrW2O8 (ref. 3 and 4) and In6WO12 (ref. 46) include distorted WO4 tetrahedron and regular WO6 octahedron, respectively. Both monoclinic WO3 and ZrW2O7(OH)2·2H2O6,47 contain distorted WO6 octahedrons. Fig. 4A shows the W L1 edge XANES spectra of various catalyst samples and reference compounds with different coordination symmetries. The intensity of the pre-edge peak for ZWO-I (ZWOH_6M_12h) and the calcined one was nearly the same as that for highly crystalline ZrW2O7(OH)2·2H2O (ZWOH_6M_48h), WO3, and conventional tungstated-zirconia catalysts. The apparent absorption edge position of the ZWO-I phase was higher than those of metallic tungsten and WIVO2 and was similar to that of WVIO3 (Fig. 4B).
 | ||
| Fig. 4 W L1-edge XANES spectra of (A) the hexavalent tungsten compounds and catalyst samples, and (B) comparison of the absorption edge. (a) ZWOH_6M_12h, (b) ZWO_6M_12h_873 (ZWO-I), (c) tungstated-zirconia (reference), (d) tungstated-zirconia (home-made), (e) ZrW2O7(OH)2·2H2O, (f) ZrW2O8, (g) WO3 and (h) In6WO12. | ||
Fig. 5 shows W L3 edge EXAFS spectra of catalyst samples and their radial structure functions (RSFs) obtained by Fourier transformation. Any distinct peaks were not confirmed at ∼3–4 Å in the RSFs of the ZWO-I phases. The calcination procedure at 873 K for the as-synthesized ZWO-I had little effect on the configuration of EXAFS and RSF, whereas calcination at 1073 K resulted in the transformation to nearly the same as those of WO3 (f). Despite their high crystallinity, certain peaks did not exist in their RSF longer than 2 Å for ZrW2O7(OH)2·2H2O (d), ZrW2O8 (e), and WO3 (f). We are unsure whether we will be able to perform a reliable curve-fitting analysis for the second coordination sphere of the current catalyst system at the W L3 edge. The curve-fitting analysis was then performed only on the first coordination sphere. The results are summarized in Table 1. Single-shell fitting was possible for ZrW2O8, whereas two-shell fittings were required for all the other samples. The evaluated EXAFS parameters were not so different from each other except for ZrW2O8.
 | ||
| Fig. 5 W L3-edge (A) EXAFS spectra and (B) their Fourier transforms. (a) ZWOH_6M_12h, (b) ZWO_6M_12h_873 (ZWO-I), (c) ZWO_6M_12h_1073, (d) ZrW2O7(OH)2·2H2O, (e) ZrW2O8, (f) WO3, (g) tungstated-zirconia (reference), and (h) tungstated-zirconia (home-made). | ||
| Catalyst | Shell | CNa | rb/Å | Δσc/Å |
|---|---|---|---|---|
| a Coordination number.b Interatomic distance.c Debye–Waller factor. | ||||
| ZWOH_6M_12h | W–O | 3.31 ± 0.09 | 1.787 ± 0.011 | 0.084 ± 0.039 |
| 1.71 ± 1.22 | 2.111 ± 0.033 | 0.097 ± 0.109 | ||
| ZWO_6M_12h_873 (ZWO-I) | W–O | 3.30 ± 0.96 | 1.782 ± 0.012 | 0.075 ± 0.044 |
| 1.41 ± 1.35 | 2.098 ± 0.047 | 0.097 ± 0.152 | ||
| ZWO_6M_12h_1073 | W–O | 4.20 ± 1.09 | 1.793 ± 0.009 | 0.099 ± 0.036 |
| 1.83 ± 0.91 | 2.125 ± 0.017 | 0.060 ± 0.083 | ||
| ZrW2O7(OH2)2 (ZWOH_6M_48h) | W–O | 2.55 ± 0.64 | 1.768 ± 0.006 | 0.076 ± 0.028 |
| 1.78 ± 0.12 | 2.115 ± 0.022 | 0.103 ± 0.074 | ||
| ZrW2O8 (ZWO_6M_48h_873) | W–O | 3.51 ± 0.56 | 1.779 ± 0.005 | 0.053 ± 0.027 |
| WO3 | W–O | 3.70 ± 0.84 | 1.779 ± 0.006 | 0.080 ± 0.028 |
| 2.12 ± 1.02 | 2.107 ± 0.016 | 0.079 ± 0.061 | ||
| WO3–ZrO2 (reference) | W–O | 2.86 ± 1.00 | 1.776 ± 0.020 | 0.081 ± 0.057 |
| 0.72 ± 1.02 | 2.053 ± 0.107 | 0.079 ± 0.261 | ||
| WO3–ZrO2 (home-made) | W–O | 3.27 ± 0.96 | 1.779 ± 0.015 | 0.105 ± 0.040 |
| 0.90 ± 1.07 | 2.093 ± 0.072 | 0.104 ± 0.183 | ||
 | ||
| Fig. 6 Zr K-edge (A) EXAFS spectra and (B) their Fourier transforms. (a) ZWOH_6M_12h, (b) ZWO_6M_12h_873 (ZWO-I), (c) ZWO_6M_12h_1073, (d) ZrW2O7(OH)2·2H2O, (e) ZrW2O8, (f) Zr(OH)x, (g) m-ZrO2, (h) tungstated-zirconia (reference), and (i) tungstated-zirconia (home-made). | ||
Table 2 summarizes the results of the curve-fitting analysis at the Zr K-edge. In cases for ZWO-I, the small peak at ∼3.5 Å in their RSFs is confirmed to be that of the Zr–W pair. The evaluated interatomic distance was comparable to that of the longer one of ZrW2O8 and ZrW2O7(OH)2. Unlike ZWO-I, the second coordination sphere of the two types of tungstated-zirconia was confirmed to be a Zr–Zr pair. The first coordination sphere at ∼1.8 Å could be fitted with a single Zr–O pair for ZrW2O8. Two shell fitting was required for all other catalyst samples, but significant differences in EXAFS parameters were not confirmed among them.
| Catalyst | Shell | CNa | rb/Å | Δσc/Å |
|---|---|---|---|---|
| a Coordination number.b Interatomic distance.c Debye–Waller factor.d Constrained to be the same. | ||||
| ZWOH_6M_12h | Zr–O | 4.11 ± 4.71 | 2.113 ± 0.065 | 0.082d ± 0.065 |
| 3.15 ± 3.85 | 2.233 ± 0.073 | 0.082d | ||
| Zr–W | 1.64 ± 0.16 | 3.941 ± 0.002 | 0.092 ± 0.004 | |
| ZWO_6M_12h_873 (ZWO-I) | Zr–O | 4.05 ± 4.69 | 2.108 ± 0.070 | 0.094d ± 0.070 |
| 3.53 ± 3.69 | 2.241 ± 0.072 | 0.094d | ||
| Zr–W | 2.39 ± 0.24 | 3.943 ± 0.003 | 0.107 ± 0.005 | |
| ZWO_6M_12h_1073 | Zr–O | 4.75 ± 2.32 | 2.111 ± 0.027 | 0.083d ± 0.038 |
| 3.13 ± 1.49 | 2.256 ± 0.044 | 0.083d | ||
| Zr–Zr | 4.06 ± 0.59 | 3.445 ± 0.003 | 0.081 ± 0.008 | |
| 3.68 ± 2.42 | 3.977 ± 0.014 | 0.114 ± 0.039 | ||
| ZrW2O7(OH2)2 (ZWOH_6M_48h) | Zr–O | 3.76 ± 3.19 | 2.096 ± 0.044 | 0.066d ± 0.059 |
| 4.06 ± 2.39 | 2.214 ± 0.038 | 0.066d | ||
| Zr–W | 2.67 ± 0.65 | 3.451 ± 0.004 | 0.073 ± 0.010 | |
| 3.06 ± 0.71 | 3.910 ± 0.003 | 0.066 ± 0.010 | ||
| ZrW2O8 (ZWO_6M_48h_873) | Zr–O | 6.49 ± 0.88 | 2.081 ± 0.005 | 0.079 ± 0.014 |
| Zr–W | 2.86 ± 1.09 | 3.720 ± 0.007 | 0.075 ± 0.018 | |
| 4.56 ± 1.11 | 3.900 ± 0.004 | 0.073 ± 0.011 | ||
| Zr(OH)x | Zr–O | 4.72 ± 2.19 | 2.121 ± 0.026 | 0.080d ± 0.036 |
| 1.89 ± 1.41 | 2.263 ± 0.050 | 0.080d | ||
| Zr–Zr | 3.59 ± 1.24 | 3.390 ± 0.021 | 0.117 ± 0.023 | |
| 0.21 ± 0.49 | 3.488 ± 0.013 | 0.063 ± 0.087 | ||
| m-ZrO2 | Zr–O | 4.40 ± 2.23 | 2.111 ± 0.027 | 0.078d ± 0.040 |
| 3.34 ± 1.43 | 2.252 ± 0.038 | 0.078d | ||
| Zr–Zr | 4.87 ± 0.68 | 3.450 ± 0.011 | 0.075 ± 0.008 | |
| 3.80 ± 2.38 | 3.981 ± 0.013 | 0.097 ± 0.036 | ||
| WO3–ZrO2 (reference) | Zr–O | 5.10 ± 2.67 | 2.119 ± 0.030 | 0.086d ± 0.040 |
| 3.25 ± 1.76 | 2.265 ± 0.049 | 0.086d | ||
| Zr–Zr | 4.47 ± 0.67 | 3.437 ± 0.003 | 0.083 ± 0.008 | |
| 4.82 ± 2.77 | 3.982 ± 0.013 | 0.104 ± 0.038 | ||
| WO3–ZrO2 (home-made) | Zr–O | 4.99 ± 1.54 | 2.127 ± 0.017 | 0.089d ± 0.026 |
| 2.44 ± 0.85 | 2.289 ± 0.041 | 0.089d | ||
| Zr–Zr | 6.61 ± 1.77 | 3.473 ± 0.013 | 0.094 ± 0.014 | |
| 6.83 ± 2.03 | 3.644 ± 0.018 | 0.101 ± 0.021 | ||
3.2. Catalysis
 | ||
| Fig. 7 Typical results of anisole alkylation with benzyl alcohol at 403 K. BOH: benzyl alcohol; DBE: dibenzyl ether; BA: benzyl anisoles. | ||
 | ||
| Fig. 8 HCl concentration dependency used for catalyst preparation on the reaction rate for anisole alkylation (circle) and surface area of a catalyst (triangle). Catalyst: ZWO_XM_12h_873, 0.1 g; benzyl alcohol: 0.64 mL; anisole: 10 mL; and reaction temperature: 403 K. | ||
Note that the liquid-phase alkylation of aromatics by solid acid catalysts has been examined using several models such as pseudo-first- or second-order kinetic equation and the Langmuir–Hinshelwood kinetic model.50 In this study, we confirm that equivalent HCl concentration dependency for the evaluated reaction rate could be obtained in case the pseudo-first-order kinetic equation model is applied.
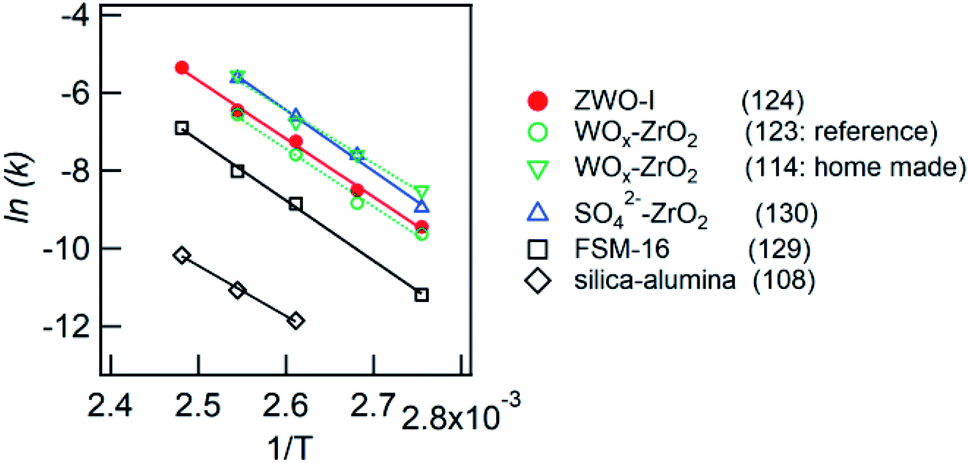 | ||
| Fig. 9 Arrhenius plot for anisole alkylation with benzyl alcohol. Catalyst: 0.1 g; benzyl alcohol: 0.64 mL; anisole: 10 mL. Values in parentheses show the activation energy (kJ mol−1). | ||
4. Discussion
4.1. Formation of poorly crystalline binary hydroxide ZWO-I phase
In this study, we confirmed that the poorly crystalline ZWO-I phase formed by the hydrothermal treatment of a mother gel for 12 h when 4–9 M HCl was added in their preparation. It has been reported that halide and hydronium ion concentrations are related to the reaction rate of crystallization and/or nucleation of a precursor of zirconium tungstate hydroxide.8,27 The present XRD patterns of the as-synthesized binary hydroxide with different hydrothermal treatment times (Fig. S1†) show that the ZWO-I phase is one of the metastable phases. A longer hydrothermal treatment would result in the transformation to a more stable ZrW2O7(OH)2·2H2O. This crystallization might occur until 12 h in the case 10 M HCl was used, as described in 3.1.1. ZWO-I phase was not formed even after 72 h of hydrothermal treatment at 453 K in case HCl concentration was <1 M. Then, we conclude that 6 M HCl is the best for forming the ZWO-I phase under this 12 h hydrothermal treatment at 453 K. The endothermic phenomena above 1000 K without weight loss were confirmed for all TG-DTA profiles. These phenomena were possibly attributed to the decomposition of a binary phase to WO3 and ZrO2. The similarity of the W L3 edge or Zr K-edge EXAFS spectra of ZWO_XM_1073 to the crystalline WO3 and ZrO2 supports this deduction.4.2. Active species
We confirmed that the calcined ZWO-I phase (ZWO_XM_873: Group B, X: 5–8) exhibited high activity for the anisole alkylation with benzyl alcohol. Calcined Group C and D catalysts (X: 0–3) promoted this reaction; however, the activities were lower than those of Group B. Although the ZWO-I phase was not confirmed in the XRD patterns of Group D (X: 0, 1), alkylation was promoted to some extent. This shows X-ray amorphous zirconium–tungsten binary oxides possess solid acidity, and the intrinsic activity is lower than that of the ZWO-I phase itself. Consequently, the highest activity per catalyst weight was achieved in cases where utilized HCl concentration for the catalyst preparation was 5–8 M. A series of Group-B ZWO-I catalysts (ZWO_XM_873, X = 5–9) have the equivalent level of surface area (80 ± 6 m2 g−1, n = 9). The surface area of Group A (ZWO_10M_873) was 8 m2 g−1, whereas highly crystalline ZrW2O7(OH)2·2H2O (ZWOH_6M_48–240h, ZWOH_10M_12h) and calcined ones (cubic ZrW2O8) were as small as ca. 1–2 m2 g−1. This suggests that a small number of poorly crystalline ZWO-I phase coexists accompanied by crystalline ZrW2O8 phase, and then alkylation was slightly promoted.As described in the Introduction section, XRD patterns similar to this ZWO-I phase have been reported by many research groups.27,28 Ando et al. reported that the calcined binary oxides (W/Zr = 1.4) at 673 K promoted the alkylation of anisole with benzyl alcohol. We believe that their catalysts are fundamentally similar to the present ZWO-I samples. Because no sulfate salts or sulfuric acid was used during our catalyst preparation procedure, we conclude this ZWO-I phase acts as a strong solid acid. The catalytic performance is suggested to be the same level as that of reference tungstated-zirconia.
The initial benzyl anisole production rate per ZWO-I weight was the same in cases where the catalyst amount was reduced to 1/10, as shown in Fig. S5.† The higher yield per ZWO-I weight was obtained at the higher substrate/catalyst ratio. It has been reported that the initial rate for the alkylation of toluene by benzyl alcohol over ion-exchanged clays exhibited roughly correlate with a number of Brønsted acid sites.52 We presume that the alkylation of anisole with benzyl alcohol is promoted by Brønsted acid sites on ZWO-I, as in a typical Friedel–Crafts reaction. This catalyst amount dependency suggests that the activation of benzyl alcohol by ZWO-I would control the reaction rate.
4.3. Structure of ZWO-I phase
As shown in the W L1 edge XANES spectra (Fig. 4), the pre-edge peak intensities of ZWO-I samples (ZWOH_6M_12h_as, ZWOH_6M_12h_873) were almost similar to those of ZrW2O7(OH)2 and WO3, which contain distorted WO6 units. The characteristic pre-edge peak intensity in W L1 edge XANES is well known to be sensitive to the coordination geometry.43–45 The similarity of the pre-edge peak intensity and the apparent absorption edge energy position suggest tungsten species in ZWO-I are distorted octahedral WVIO6.The RSFs of the ZWO-I phase at Zr K edge EXAFS afforded a single tiny peak, while distinct twin peaks could be observed in the RSF of ZrW2O8 and ZrW2O7(OH)2. Crystallographic data for ZrW2O8 (ref. 3 and 53) and ZrW2O7(OH)2 (ref. 6 and 47) show that two types of Zr–(O)–W pairs exist with different bond lengths. The shorter and longer bond lengths correspond to edge-sharing and corner-sharing Zr–O–W linkage between ZrOn and WOn polyhedron. The Zr–O–W bond length evaluated for ZWO-I phase samples was similar to the longer ones of the corner-sharing units. The EXAFS parameters of Zr–O pairs were similar to those for ZrW2O7(OH)2 and monoclinic ZrO2; the Zr–O polyhedron present in the two compounds is ZrO7. Distinct peaks at ∼2–4 Å were not confirmed in RSFs of ZWO-I samples and conventional tungstated-zirconia at the W L3 edge. We presume that the Zr–(O)–W linkage exists in the ZWO-I phase, but the narrow available k-ranges of W L3-edge EXAFS and/or the large Debye–Waller factor because of structural disorder might make it difficult to obtain their structural information. Unfortunately, we could not identify additional clear evidence around the local structure of W species in ZWO-I. Based on W L-edge and Zr K-edge XANES/EXAFS characterization, we propose that the Zr species in ZWO-I comprises ZrO7 polyhedron and distorted WO6 octahedron, each of which connects via a corner-sharing linkage. The ZWO-I phase was formed during the hydrothermal treatment of the mother gel before crystallization to ZrW2O7(OH)2. We considered that ZWO-I is a metastable phase having a local structure similar to ZrW2O7(OH)2, while the periodic structural network is incomplete where the edge-sharing polyhedral linkage is missing. The longer hydrothermal treatment would result in the formation of edge-sharing polyhedral units, leading to crystallization to ZrW2O7(OH)2. Moreover, the evaluated coordination number for the Zr–(O)–W pair of ZWO-I was slightly smaller than that of the longer one of highly crystalline ZrW2O7(OH)2.
We propose that only corner-sharing ZrO7–WO6 connections exist in the ZWO-I phase, and the incompleteness of the network would be responsible for generating strong acidity. The XRD pattern of the ZWO-I phase was not changed after the catalytic test at 363 K. Note that EXAFS characterization of an-synthesized and calcined ZWO-I samples demonstrated no discernible structural differences at the Zr K- and W L-edges. The calcination procedure significantly altered the pore structure and surface area (Fig. S6†). The activity enhancement behavior on calcination is a subject for additional investigation.
5. Conclusions
Hydrothermally synthesized poorly crystalline metastable Zr–W binary oxides (W/Zr = 2) act as strong solid acids to promote the anisole alkylation with benzyl alcohol. The catalytic performance was comparable to that of the reference conventional tungstated-zirconia. The HCl concentration used to prepare a mother gel controlled the crystalline phase (ZWO-I). The catalytically active phase was obtained in cases where HCl concentration was 5–8 M, and the hydrothermal treatment was performed at 453 K for 12 h. A higher HCl concentration and/or a longer hydrothermal treatment afforded a catalytically inert, highly crystalline ZrW2O7(OH)2·2H2O. The W species in ZWO-I existed as a six-valent WO6 distorted octahedron connected to the ZrO7 unit via corner-sharing linkage. The local structure was suggested to resemble that of ZrW2O7(OH)2, although edge-sharing Zr–O–W linkage was missing. The incompleteness of the network structure would be responsible for the strong solid acidity.Author contributions
Takashi Yamamoto: conceptualization, investigation, writing. Seina Heiuchi: investigation. Maki Kondo: investigation. Kei Tabusadani: validation. Atsushi Sakaki: synchrotron XAFS experiment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TY acknowledges to Professor M. Iwamoto (Waseda University) for providing a TG-DTA analyzer. Some of the experiments in Fig. S2 and S5† were carried out by T. Kataoka (Tokushima University). JRC catalysts were supplied by the Catalyst Society of Japan. The Zr K-edge XAFS experiments were performed with the approval of JASRI (Proposal No. 2018A5380 and 2018B5380). This work was partially supported by a research grant from Daiichi Kigenso Kagaku Kogyo Co., Ltd.References
- L. L. Y. Chang, M. G. Scroger and B. Phillips, J. Am. Ceram. Soc., 1967, 50, 211–215 CrossRef CAS.
- C. Martinek and F. A. Hummel, J. Am. Ceram. Soc., 1968, 51, 227–228 CrossRef CAS.
- J. S. O. Evans, T. A. Mary, T. Vogt, M. A. Subramanian and A. W. Sleight, Chem. Mater., 1996, 8, 2809–2823 CrossRef CAS.
- T. A. Mary, J. S. O. Evans, T. Vogt and A. W. Sleight, Science, 1996, 272, 90–92 CrossRef CAS.
- C. Closmann, A. W. Sleight and J. C. Haygarth, J. Solid State Chem., 1998, 139, 424–426 CrossRef CAS.
- M. S. Dadachov and R. M. Lambrecht, J. Mater. Chem., 1997, 7, 1867–1870 RSC.
- Q. F. Xing, X. R. Xing, R. B. Yu, L. Du, J. Meng, J. Luo, D. Wang and G. R. Liu, J. Cryst. Growth, 2005, 283, 208–214 CrossRef CAS.
- J. A. Colin, D. V. Camper, S. D. Gates, M. D. Simon, K. L. Witker and C. Lind, J. Solid State Chem., 2007, 180, 3504–3509 CrossRef CAS.
- L. C. Kozy, M. N. Tahir, C. Lind and W. Tremel, J. Mater. Chem., 2009, 19, 2760–2765 RSC.
- H. Wu, P. Badrinarayanan and M. R. Kessler, J. Am. Ceram. Soc., 2012, 95, 3643–3650 CrossRef CAS.
- A. I. Gubanov, E. S. Dedova, P. E. Plyusnin, E. Y. Filatov, T. Y. Kardash, S. V. Korenev and S. N. Kulkov, Thermochim. Acta, 2014, 597, 19–26 CrossRef CAS.
- T. Yamamoto, M. Kondo, T. Irie and N. Tanima, Adv. X-Ray Chem. Anal., Jpn., 2017, 48, 137–148 Search PubMed.
- K. Arata and M. Hino, Proc. 9th Int. Congr. Catal., 1988, pp. 1727–1734 Search PubMed.
- M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1988, 1259–1260 RSC.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- K. Arata, Appl. Catal., A, 1996, 146, 3–32 CrossRef CAS.
- H. Hattori and Y. Ono, Solid Acid Catalysis, Pan Stanford, Singapore, 2015 Search PubMed.
- W. Zhang, Z. Wang, J. Huang and Y. Jiang, Energy Fuels, 2021, 35, 9209–9227 CrossRef CAS.
- M. Scheithauer, R. K. Grasselli and H. Knözinger, Langmuir, 1998, 14, 3019–3029 CrossRef CAS.
- D. G. Barton, M. Shtein, R. D. Wilson, S. L. Soled and E. Iglesia, J. Phys. Chem. B, 1999, 103, 630–640 CrossRef CAS.
- N. Naito, N. Katada and M. Niwa, J. Phys. Chem. B, 1999, 103, 7206–7213 CrossRef CAS.
- T. Yamamoto, A. Orita and T. Tanaka, X-Ray Spectrom., 2008, 37, 226–231 CrossRef CAS.
- W. Zhou, E. I. Ross-Medgaarden, W. V. Knowles, M. S. Wong, I. E. Wachs and C. J. Kiely, Nat. Chem., 2009, 1, 722–728 CrossRef CAS PubMed.
- C. Thomas, J. Phys. Chem. C, 2011, 115, 2253–2256 CrossRef CAS.
- T. Yamamoto, A. Teramachi, A. Orita, A. Kurimoto, T. Motoi and T. Tanaka, J. Phys. Chem. C, 2016, 120, 19705–19713 CrossRef CAS.
- W. Zhou, N. Soultanidis, H. Xu, M. S. Wong, M. Neurock, C. J. Kiely and I. E. Wachs, ACS Catal., 2017, 7, 2181–2198 CrossRef CAS.
- X. R. Xing, Q. F. Xing, R. B. Yu, J. Meng, J. Chen and G. R. Liu, Phys. B, 2006, 371, 81–84 CrossRef CAS.
- M. Ando, A. Izumi, T. Murayama and W. Ueda, Abst. 108th CATSJ (Catalysis Society of Japan) Meetings, 2011, p. 1F11 Search PubMed.
- M. Lucas, K. Fabičovicová and P. Claus, ChemCatChem, 2018, 10, 612–618 CrossRef CAS.
- M. Hino, S. Kobayashi and K. Arata, J. Am. Chem. Soc., 1979, 101, 6439–6441 CrossRef CAS.
- X. Song and A. Sayari, Catal. Rev., 1996, 38, 329–412 CrossRef CAS.
- S. Inagaki, Y. Fukushima and K. Kuroda, J. Chem. Soc., Chem. Commun., 1993, 680–682 RSC.
- T. Yamamoto, T. Tanaka, T. Funabiki and S. Yoshida, J. Phys. Chem. B, 1998, 102, 5830–5839 CrossRef CAS.
- T. Yamamoto, T. Tanaka, S. Inagaki, T. Funabiki and S. Yoshida, J. Phys. Chem. B, 1999, 103, 6450–6456 CrossRef CAS.
- T. Yamamoto, S. Mori, T. Shishido, J. Kawai and T. Tanaka, Top. Catal., 2009, 52, 657–663 CrossRef CAS.
- T. Taguchi, J. Harada, A. Kiku, K. Tohji and K. Shinoda, J. Synchrotron Radiat., 2001, 8, 363–365 CrossRef CAS PubMed.
- T. Yamamoto, S. Mori, T. Kawaguchi, T. Tanaka, K. Nakanishi, T. Ohta and J. Kawai, J. Phys. Chem. C, 2008, 112, 328–331 CrossRef CAS.
- T. Taguchi, T. Ozawa and H. Yashiro, Phys. Scr., 2005, T115, 205–206 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- L. Bartha, A. B. Kiss and T. Szalay, Int. J. Refract. Met. Hard Mater., 1995, 13, 77–91 CrossRef CAS.
- G. Liu, S. Wang, Y. Nie, X. Sun, Y. Zhang and Y. Tang, J. Mater. Chem. A, 2013, 1, 10120–10129 RSC.
- P. Ou, X. Su, Y. Zeng, F. Zhang, J. Liu, C. Wang, S. Yang, H. Wang, Y. Wen and H. Zhao, CrystEngComm, 2021, 23, 1700–1703 RSC.
- J. A. Horsley, I. E. Wachs, J. M. Brown, G. H. Via and F. D. Hardcastle, J. Phys. Chem., 1987, 91, 4014–4020 CrossRef CAS.
- T. Yamamoto, X-Ray Spectrom., 2008, 37, 572–584 CrossRef CAS.
- S. Yamazoe, Y. Hitomi, T. Shishido and T. Tanaka, J. Phys. Chem. C, 2008, 112, 6869–6879 CrossRef CAS.
- D. Michel and A. Kahn, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1437–1441 CrossRef.
- Y. Cao, X. Deng, H. Ma, S. Wang and X. Zhao, Defect Diffus. Forum, 2006, 251–252, 21–26 CAS.
- K. Okumura, K. Yamashita, M. Hirano and M. Niwa, J. Catal., 2005, 234, 300–307 CrossRef CAS.
- A. Takagaki, D. Lu, J. N. Kondo, M. Hara, S. Hayashi and K. Domen, Chem. Mater., 2005, 17, 2487–2489 CrossRef CAS.
- P. Beltrame and G. Zuretti, Green Chem., 2004, 6, 7–13 RSC.
- H. Ishitani, H. Naito and M. Iwamoto, Catal. Lett., 2008, 120, 14–18 CrossRef CAS.
- T. Cseri, S. Békássy, F. Figueras and S. Rizner, J. Mol. Catal. A: Chem., 1995, 98, 101–107 CrossRef CAS.
- F. D. Vila, J. W. Spencer, J. J. Kas, J. J. Rehr and F. Bridges, Front. Chem., 2018, 6, 356 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08931e |
| This journal is © The Royal Society of Chemistry 2022 |