
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron catalyzed C–C dehydrogenative coupling reaction: synthesis of arylquinones from quinones/hydroquinones†
Yu Dong *a,
Chun Xiea,
Jia Chena,
Ai Shena,
Qi-Qi Luoa,
Bing Hea,
Zhi-Fan Wanga,
Bo Changa,
Fan Yanga and
Zhi-Chuan Shib
*a,
Chun Xiea,
Jia Chena,
Ai Shena,
Qi-Qi Luoa,
Bing Hea,
Zhi-Fan Wanga,
Bo Changa,
Fan Yanga and
Zhi-Chuan Shib
aCollege of Chemistry and Life Science, Sichuan Provincial Key Laboratory for Structural Optimization and Application of Functional Molecules, Chengdu Normal University, Chengdu, 611130, P. R. China
bSouthwest Minzu University, Chengdu 610041, P. R. China
First published on 28th January 2022
Abstract
An atom-economical approach for the synthesis of arylquinones was achieved successfully via direct oxidative C–C dehydrogenative coupling reaction of quinones/hydroquinones with electron-rich arenes using an inexpensive Fe–I2–(NH4)2S2O8 system. The efficiency of this catalytic approach was established with a broad scope of substrates involving quinones and hydroquinones to give high yields (60–89%) of several arylated quinones. The present protocol is simple, practical, and shows good functional group tolerance.
Introduction
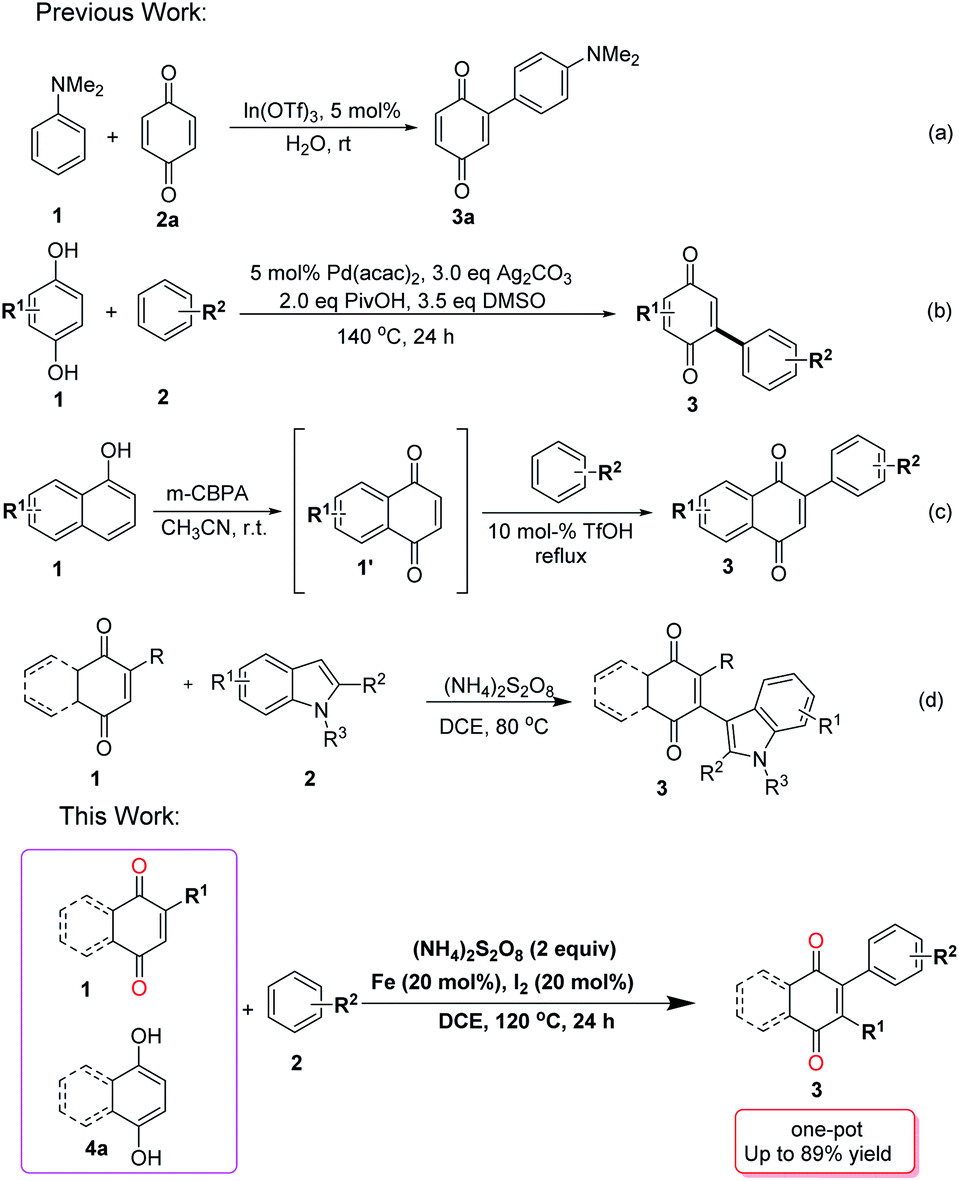
Quinone scaffolds are widely distributed in natural products and gained importance owing to their roles in biological systems,1 and were utilized as intermediates in materials,2 bioinspired synthetic products,3 and biosynthesis.4 They are also involved in many bioenergetic processes due to their electron transport properties.5 Of particular importance, aryl substituted quinones possess unique electronic properties,6 which have several applications in chemistry and many natural products. For example, Belamcanda chinensis, were used as a specific cyclooxygenase inhibitor, belamcandaquinones A and B isolated from the seed of a medicinal plant.7 Furthermore, aryl-substituted quinones are very useful in the dye industry owing to their important coloring properties.8 Thus, the synthesis of aryl substituted quinones has attracted significant attention and many methods have been developed.Several approaches were described for the synthesis of arylated quinones involving different types of starting materials. Utilized of prefunctionalized quinones and arenes requires the presence of expensive transition metal reagents. Examples include the transition-metal-catalyzed coupling of a stannylquinone with an arylhalide,9 haloquinone with a styrylstannane10 and halogenated quinones with boronic acids.11 Heck type arylation often comprises unfunctionalized quinones and pre-functionalized arenes. Commercially available, but costly arylboronic acids were used in such transformations, in the presence of metal reagents made of such as Ag, Rh, Ir, Pd, and Fe salts.12 Furthermore, several metal free methods using diazonium salt-hydrazine catalysts, diazonium salts, diaryliodonium salts, and arylhydrazine salts have also been described.13 On the other hand, oxidative C–H/C–H coupling assembling a diverse variety of complex aromatic systems was also employed in coupling of quinones and arenes. Such C–H/C–H coupling was carried out in the presence of metal salts such as In(OTf)3, Pd(OAc)2, Rh salts, Pd(TFA)2, and FeCl3.14
In general, the arylation of quinones compounds with carbon nucleophiles is achieved through Michael-type 1,4-conjugate addition reaction of electron-rich arenes15 or through the transition-metal-catalyzed coupling reactions. However, arene–quinone coupling have certain limitations, such as organometallic reagents generated from prefunctionalized starting material, aryl radicals obtained from prefunctionalized arenes and expensive transition metals. Hence, a 1,4-conjugate addition reaction followed by oxidation is a preferred route for the arylation reactions of quinones and, based on this concept, a few metal-catalyzed reactions were reported. Despite these advances, efforts still need to be made in terms of mild reaction conditions, inexpensive reagents, and wide substrate scope.16a
We have already reported the coupling of indoles and quinones in the absence of any catalyst,16c but herein we add a catalyst to expand the scope and perform the coupling not only with indoles but also with other arenes. In consideration of these important points and continuing our research interest on functionalized of quinone derivatives, we wish to report a sequential one-pot approach to arylated quinones through direct oxidative C–C dehydrogenative coupling reaction of quinones/hydroquinones with electron-rich arenes using an inexpensive Fe–I2–(NH4)2S2O8 system (Scheme 1).
 | ||
| Scheme 1 Approaches to arylated quinones compounds. | ||
Results and discussion
We selected the reaction of readily 1,4-naphthoquinone (1a) and N,N-dimethylaniline (2a) as the model reaction for optimizing the direct oxidative C–C dehydrogenative coupling reaction of quinones/hydroquinones with electron-rich arenes (see Table 1, as well as ESI†). Fortunately, under the reaction of Fe (10 mol%), oxidant (NH4)2S2O8 (2.0 equiv.) and DCE (2 mL), at 100 °C, for 24 h (Table 1, entry 1), the desired product 3a was obtained in 58% isolated yield. Other metal salts did not provide better results including FeSO4·7H2O, FeCl3, Zn, and Pd(OAc)2 also tested for the cross-coupling (entries 2–5, see ESI†). When the quantity of the catalyst (Fe) was increased to 20 mol% (entry 6), the yield of the product 3a increased (65%). Among the various additives screened, I2 was the most efficient (entries 8–10, see ESI†). Subsequently, the oxidant was screened. (NH4)2S2O8 was proved to be more efficient than others, such as K2S2O8 and Oxone (entries 11–12, see ESI†). Furthermore, when the temperature was raised to 120 °C, the yield increased to 89% (entry 14). It should be noted that increasing or decreasing the reaction temperature gave slightly inferior results, indicating that the transformation was sensitive to temperature (see ESI†). Based on these results, the best yield of 3a was obtained from the reaction of 2a (2 equiv.) and Fe (20 mol%), additive I2 (20 mol%), oxidant (NH4)2S2O8 (2 equiv.) in DCE at 120 °C under sealed tube.
|
|||||
|---|---|---|---|---|---|
| Entry | [Cat] (mol%) | Additive (mol%) | Oxidants (2 equiv.) | T °C | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.6 mmol, 2 equiv.), [Cat] (mol%), additive (20 mol%), oxidant (0.6 mmol, 2 equiv.), DCE (2 mL), 80–140 °C, sealed tube for 24 h. DCE stands for 1,2-dichloorethane.b Isolated yield. | |||||
| 1 | Fe (10) | — | (NH4)2S2O8 | 100 | 58 |
| 2 | FeSO4·7H2O (10) | — | (NH4)2S2O8 | 100 | 23 |
| 3 | FeCl3 (10) | — | (NH4)2S2O8 | 100 | 35 |
| 4 | Zn (10) | — | (NH4)2S2O8 | 100 | 41 |
| 5 | Pd(OAc)2 (10) | — | (NH4)2S2O8 | 100 | 34 |
| 6 | Fe (20) | — | (NH4)2S2O8 | 100 | 65 |
| 7 | Fe (30) | — | (NH4)2S2O8 | 100 | 53 |
| 8 | Fe (20) | CuBr | (NH4)2S2O8 | 100 | 24 |
| 9 | Fe (20) | CuI | (NH4)2S2O8 | 100 | 31 |
| 10 | Fe (20) | I2 | (NH4)2S2O8 | 100 | 76 |
| 11 | Fe (20) | I2 | K2S2O8 | 100 | 45 |
| 12 | Fe (20) | I2 | Oxone | 100 | 32 |
| 13 | Fe (20) | I2 | (NH4)2S2O8 | 80 | 41 |
| 14 | Fe (20) | I2 | (NH4)2S2O8 | 120 | 89 |
| 15 | Fe (20) | I2 | (NH4)2S2O8 | 140 | 70 |

With the optimized reaction conditions in hand, we next set out to explore the universality of this method (Table 2). Therefore, a series of electron-rich arenes with quinones, including anilines and indole derivatives, were subjected to this process. Subsequently, we evaluated the scope and the influence of the substituents on the electron-rich arenes moiety. A series of disubstituted anilines, such as dialkyl, dibenzyl, diallyl, and cycloalkylamine compounds, were examined and the desired arylated products obtained in high yields (3a–3j). The scale-up of the reaction was performed to reveal the applicability of this method. When 1,4-naphthoquinone 1a was scaled up by 5 mmol, 3a was isolated in 85% yield. When the benzyl group was attached to the N of the aniline, the product was obtained in excellent yield (3c). The exciting thing was that this protocol showed good compatibility with carbon–carbon double bonds (3d). Interestingly, the cyclic aniline compounds were capable of obtaining the corresponding products in good yields (3j). Monosubstituted aniline derivatives were converted into the respective products in moderate to good yields (3k and 3l). Although there is a possibility of NH-attack on quinones, we exclusively observed the C–C products. When we carried out the reaction with a meta-meta-substituted N,N-dimethylaniline as a substrate, we were also able to obtain an arylated product, albeit at a moderate yield (3m), wherein the methyl group could hamper the coupling. It was possible that the steric hindrance effect of the meta positions and led to a decrease in yield. Heterocyclic compounds, such as indoles (C-3) were successfully arylated under the optimized conditions (3n–3q). It is noteworthy that electron withdrawing indoles (2o) with a substituent at the 2-position also proved to be suitable coupling partners to provide the corresponding products 3o in 62% yield with a catalyst, but 3o was obtained in traces in the absence of Fe/iodine system.16c At the beginning, Fe and I2 gives FeI2 under the conditions of heating. FeI2 coordinates to the carbonyl oxygen of the 1,4-naphthoquinone, which increases the electrophilicity and assists 1,4-conjugative addition. Thus, Fe/iodine system can accelerate the reaction and increase the yield. We then proceeded to study the scope of quinone derivatives. We hypothesize that the sensitive chlorine at the β-position of naphthoquinone was compatible with the reaction conditions, which can easily be transformed further by cross-coupling reactions. Anthraquinone 1s was also employed, affording the corresponding product 3s in 83% yield.

Subsequently, the synthesis of arylated quinones for a broad substrate scope involved hydroquinone with various electron-rich arenes. As shown in Table 3, moderate to good yields of arylated quinones were obtained upon reacting 1,4 hydroquinone with various electron-rich arenes having disubstituted anilines, as in the case of dialkyl, dibenzyl and cycloalkylamine compounds. Monosubstituted aniline derivatives were converted into the respective products in moderate yields (3l). When we carried out the reaction with a meta-substituted N,N-dimethylaniline as a substrate, we were also able to obtain an arylated product, albeit at a moderate yield (3m). Heterocyclic compounds, such as indoles (C-3) were successfully arylated under the optimized conditions (3n and 3q).
| a Reaction conditions: 1a (0.3 mmol), 2 (0.6 mmol), Fe (20 mol%), I2 (20 mol%), (NH4)2S2O8 (2 equiv.), DCE (2 mL), 120 °C, sealed tube for 24 h. Isolated yield. Bn means benzyl. |
|---|
 |
Next, to gain understanding about the mechanism, control experiments were carried out (Scheme 2). The studies revealed that the radical scavenger BHT (2,6-di-tert-butyl-4-methylphenol) did not inhibit the reaction under standard conditions, ruling out the radical mechanism (Scheme 2a). The reaction between compounds 1a and 2a generated the coupling product 3a in a yield of 37% in the absence of (NH4)2S2O8 (Scheme 2b). The studies demonstrated the importance of (NH4)2S2O8. And the reaction of model compounds 1a and 2a generated the coupling product 3a without Fe and I2 in a yield of 34% (Scheme 2c). The corresponding product 3a is formed in 41% and 52% yield respectively in the presence of (NH4)2S2O8 with Fe or I2 (Scheme 2d and 2e), which showed that the Fe and I2 played a pivotal role in obtaining the desired product. The corresponding product 3a is formed in 67% yield in the presence of (NH4)2S2O8 with FeI2 (Scheme 2f).
 | ||
| Scheme 2 Control experiments. | ||
On the basis of this and previous reports,16 a possible reaction mechanism was proposed (Scheme 3). At the beginning, we hypothesize that Fe and I2 gives FeI2 under the conditions of heating. The reaction starts with the in situ oxidation of hydroquinone (4a) to 1,4-naphthoquinone (1a) over a (NH4)2S2O8 oxidant. Then, FeI2 coordinates to the carbonyl oxygen of the 1,4-naphthoquinone, which increases the electrophilicity and assists 1,4-conjugate addition. Due to nucleophilic nature of the electron-rich arenes 2a, it would be attracted to the electrophilic C-2 positions of naphthoquinone, and subsequently, 1,4-addition of the 2a to the 1,4-naphthoquinone afforded the intermediate B and aromatization by means of H abstraction. Finally, intermediate C undergoes (NH4)2S2O8 oxidation to give product 3a.
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have successfully demonstrated iron-mediated oxidative C–H/C–H cross-coupling reaction between quinones/hydroquinones and various electron-rich arenes to accomplish the arylated quinones using an inexpensive Fe–I2–(NH4)2S2O8 system. The significant aspects of our work allows modest functional group tolerance, including quinones and hydroquinones.Experimental section
To a solution of DCE (2 mL) was added 1 or 4a (0.3 mmol), 2 (0.8 mmol), Fe (0.06 mmol, 20 mol%), I2 (0.06 mmol, 20 mol%), and (NH4)2S2O8 (0.6 mmol, 2 equiv.). The reaction mixture was stirred at 120 °C for 24 h. After the completion of the reaction (monitored by TLC). The reaction was quenched with saturated salt water (2 mL) and the mixture was extracted with EtOAc (3 × 3 mL). The organic extracts were washed with brine, dried over Na2SO4, filtered and the solvent was removed in vacuo. The crude product was purified by silica gel column chromatography to give 3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the project supported by National undergraduate training program for innovation and entrepreneurship (Grant No. 202114389016 and 202114389005), the Foundation of Chengdu Normal University Talent Introduction Research Funding (2021YJRC202020) and Chengdu Normal University Project (2021CS21ZCY02).Notes and references
- (a) S. J. Gould, Chem. Rev., 1997, 97, 2499–2510 CrossRef CAS PubMed; (b) R. H. Thomson, Naturally Occurring Quinones IV, Blackie Academic & Professional, London, 1997 Search PubMed; (c) B. Zhang, G. Salituro, D. Szalkowski, Z. Li, Y. Zhang, I. Royo, D. Vilella, M. T. Díez, F. Pelaez and C. Ruby, Science, 1999, 284, 974–977 CrossRef CAS PubMed; (d) J.-K. Liu, Chem. Rev., 2006, 106, 2209–2223 CrossRef CAS PubMed.
- E. J. Son, J. H. Kim, K. Kim and C. B. Park, J. Mater. Chem. A, 2016, 4, 11179–11202 RSC.
- (a) J. L. Bolton and T. Dunlap, Chem. Res. Toxicol., 2017, 30, 13–37 Search PubMed; (b) N. El-Najjar, H. Gali-Muhtasib, R. A. Ketola, P. Vuorela, A. Urtti and H. Vuorela, Phytochem. Rev., 2011, 10, 353–370 CrossRef CAS; (c) G. G. Dias, A. King, F. De Moliner, M. Vendrell and E. N. da Silva Júnior, Chem. Soc. Rev., 2018, 47, 12–27 RSC.
- (a) M. Sugumaran and V. Semensi, J. Biol. Chem., 1991, 266, 6073–6078 CrossRef CAS PubMed; (b) J. P. Klinman and F. Bonnot, Chem. Rev., 2014, 114, 4343–4365 CrossRef CAS PubMed.
- B. Nowicka and J. Kruk, Biochim. Biophys. Acta, 2010, 1797, 1587–1605 CrossRef CAS PubMed.
- (a) I. CFR Ferreira, J. A Vaz, M. H. Vasconcelos and A. Martins, Anti-Cancer Agents Med. Chem., 2010, 10, 424–436 CrossRef PubMed; (b) C. Asche, Mini-Rev. Med. Chem., 2005, 5, 449–467 CrossRef CAS PubMed.
- (a) Y. Fukuyama, J. Okino and M. Kodama, Chem. Pharm. Bull., 1991, 39, 1877–1879 CrossRef CAS; (b) Y. Fukuyama, Y. Kiriyama, J. Okino and M. Kodama, Tetrahedron Lett., 1993, 34, 7633–7636 CrossRef CAS.
- (a) T. Bechtold, in Handbook of Natural Colorants, ed. T. Bechtold and R. Mussak, Wiley, New York, 2009, p. 151 CrossRef; (b) B. Nowicka and J. Kruk, Biochim. Biophys. Acta, 2010, 1797, 1587–1605 CrossRef CAS PubMed.
- T. P. Adarsh Krishna, S. Pandaram and A. Ilangovan, Org. Chem. Front., 2019, 6, 3244–3251 RSC.
- (a) N. Tamayo, A. M. Echavarren and M. C. Paredes, J. Org. Chem., 1991, 56, 6488–6491 CrossRef CAS; (b) Ó. d. Frutos, C. Atienza and A. M. Echavarren, Eur. J. Org. Chem., 2001, 2001, 163–171 CrossRef.
- (a) X. Gan, W. Jiang, W. Wang and L. Hu, Org. Lett., 2009, 11, 589–592 CrossRef CAS PubMed; (b) M. K. Hadden, S. A. Hill, J. Davenport, R. L. Matts and B. S. Blagg, Bioorg. Med. Chem., 2009, 17, 634–640 CrossRef CAS PubMed; (c) Z. Hassan, I. Ullah, I. Ali, R. A. Khera, I. Knepper, A. Ali, T. Patonay, A. Villinger and P. Langer, Tetrahedron, 2013, 69, 460–469 CrossRef CAS; (d) A. d. R. Louvis, N. A. Silva, F. S. Semaan, F. d. C. Da Silva, G. Saramago, L. C. de Souza, B. L. Ferreira, H. C. Castro, J. P. Salles and A. L. Souza, New J. Chem., 2016, 40, 7643–7656 RSC.
- (a) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. Del Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292–3295 CrossRef CAS PubMed; (b) D. Wang, B. Ge, L. Du, H. Miao and Y. Ding, Synlett, 2014, 25, 2895–2898 CrossRef CAS; (c) D. Wang, B. Ge, A. Ju, Y. Zhou, C. Xu and Y. Ding, J. Organomet. Chem., 2015, 780, 30–33 CrossRef CAS; (d) S. E. Walker, J. A. Jordan-Hore, D. G. Johnson, S. A. Macgregor and A. L. Lee, Angew. Chem., 2014, 126, 14096–14099 CrossRef; (e) A. Ortega, Á. Rincón, K. L. Jiménez-Aliaga, P. Bermejo-Bescós, S. Martín-Aragón, M. T. Molina and A. G. Csákÿ, Bioorg. Med. Chem. Lett., 2011, 21, 2183–2187 CrossRef CAS PubMed; (f) J. Wang, S. Wang, G. Wang, J. Zhang and X.-Q. Yu, Chem. Commun., 2012, 48, 11769–11771 RSC; (g) K. Komeyama, T. Kashihara and K. Takaki, Tetrahedron Lett., 2013, 54, 1084–1086 CrossRef CAS; (h) A. Deb, S. Manna, A. Maji, U. Dutta and D. Maiti, Eur. J. Org. Chem., 2013, 2013, 5251–5256 CrossRef CAS.
- (a) S. Shaaban, A. Jolit, D. Petkova and N. Maulide, Chem. Commun., 2015, 51, 13902–13905 RSC; (b) J. F. Bagli and P. L'Écuyer, Can. J. Chem., 1961, 39, 1037–1048 CrossRef CAS; (c) A. Honraedt, F. Le Callonnec, E. Le Grognec, V. Fernandez and F. o.-X. Felpin, J. Org. Chem., 2013, 78, 4604–4609 CrossRef CAS PubMed; (d) D. Wang, B. Ge, L. Li, J. Shan and Y. Ding, J. Org. Chem., 2014, 79, 8607–8613 CrossRef CAS PubMed; (e) P. Patil, A. Nimonkar and K. G. Akamanchi, J. Org. Chem., 2014, 79, 2331–2336 CrossRef CAS PubMed.
- (a) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed; (b) T. Itahara, J. Org. Chem., 1985, 50, 5546–5550 CrossRef CAS; (c) S. Zhang, F. Song, D. Zhao and J. You, Chem. Commun., 2013, 49, 4558–4560 RSC; (d) Y. Moon, Y. Jeong, D. Kook and S. Hong, Org. Biomol. Chem., 2015, 13, 3918–3923 RSC; (e) Z. She, Y. Shi, Y. Huang, Y. Cheng, F. Song and J. You, Chem. Commun., 2014, 50, 13914–13916 RSC; (f) H. B. Zhang, L. Liu, Y. J. Chen, D. Wang and C. J. Li, Adv. Synth. Catal., 2006, 348, 229–235 CrossRef CAS; (g) T. P. Adarsh Krishna, S. Pandaram and A. Ilangovan, Org. Chem. Front., 2019, 6, 3244–3251 RSC.
- (a) K. B. Jensen, J. Thorhauge, R. G. Hazell and K. A. Jørgensen, Angew. Chem., Int. Ed., 2001, 40, 160–163 CrossRef CAS; (b) R. Rasappan, M. Hager, A. Gissibl and O. Reiser, Org. Lett., 2006, 8, 6099–6102 CrossRef CAS PubMed; (c) B. Suchand, J. Krishna, K. Mritunjoy and G. Satyanarayana, RSC Adv., 2014, 4, 13941–13945 RSC.
- (a) J. H. Jiang, S. S. K. Boominathan, W. P. Hu, C. Y. Chen, J. K. Vandavasi, Y. T. Lin and J. J. Wang, Eur. J. Org. Chem., 2016, 2016, 2284–2289 CrossRef CAS; (b) M. C. Pirrung, K. Park and Z. Li, Org. Lett., 2001, 3, 365–367 CrossRef CAS PubMed; (c) Y. Dong, J.-X. Ye, Q.-Q. Luo, T. Mei, A. Shen, P. Huang, J. Chen, X. Zhang, C. Xie and Z.-C. Shi, Synlett, 2021, 32, 1772–1776 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08828a |
| This journal is © The Royal Society of Chemistry 2022 |