
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-metal-mediated N-oxyl radical (TEMPO)-induced acceptorless dehydrogenation of N-heterocycles via electrocatalysis†
Huiqing Hou‡
a,
Xinhua Ma‡a,
Yaling Yea,
Mei Wua,
Sunjie Shia,
Wenhe Zhengb,
Mei Lina,
Weiming Sun *a and
Fang Ke*a
*a and
Fang Ke*a
aSchool of Pharmacy, Fujian Provincial Key Laboratory of Natural Medicine Pharmacology, Fujian Medical University, Fuzhou 350004, China. E-mail: kefang@mail.fjmu.edu.cn; Fax: +86-591-22862016; Tel: +86-591-22862016
bThe First Affiliated Hospital of Fujian Medical University, Fuzhou 350004, China
First published on 16th February 2022
Abstract
The development of protocols for direct catalytic acceptorless dehydrogenation of N-heterocycles with metal-free catalysts holds the key to difficulties in green and sustainable chemistry. Herein, an N-oxyl radical (TEMPO) acting as an oxidant in combination with electrochemistry is used as a synthesis system under neutral conditions to produce N-heterocycles such as benzimidazole and quinazolinone. The key feature of this protocol is the utilization of the TEMPO system as an inexpensive and easy to handle radical surrogate that can effectively promote the dehydrogenation reaction. Mechanistic studies also suggest that oxidative TEMPOs redox catalytic cycle participates in the dehydrogenation of 2,3-dihydro heteroarenes.
Catalytic acceptorless dehydrogenation (CAD) is an important reaction in which hydrogen is liberated to change the substrate.1 In terms of atom-economy and waste generation, this approach has obvious advantages compared with traditional methods using stoichiometric amounts of oxidant or sacrificial hydrogen acceptors. In addition, acceptorless dehydrogenative coupling condensation of substituted alcohols with o-substituted anilines is an atom-economical and environmentally friendly method for the construction of new C–N bonds,2 which has recently emerged as an important process to afford many useful organic compounds such as imines, amides, amines, or nitrogen-containing heterocyclic compounds.3
Toward this direction, acceptorless dehydrogenation is mainly catalysed by transition metal catalysts by the liberation of hydrogen gas without the use of stoichiometric oxidants in the process.4 In 1981, an initial example of N-alkylation of alcohols by using a variety of amines via dehydrogenative coupling was showcased by Watanabe which required a noble metal catalyst ruthenium as RuCl2(PPh3)3 at a very high temperature of >200 °C.5 More recently, Fujita and coworkers reported the first catalytic system for the reversible dehydrogenation/hydrogenation of quinoline derivatives using an iridium-based complex in 2009.6 What is more, several of metal-free catalyst representatives for this important transformation of diamines and alcohols have been reported.7 In 2019, Malakar and co-workers demonstrated the metal-free 3-nitropyridine-catalyzed facile synthesis of 2-functionalized benzoxazoles, naphthoxazoles, benzothiazoles and benzimidazoles at 110 °C.7a In contrast, although significant advances have been made in metal- or metal-free-catalyzed acceptorless dehydrogenation, the use of metal complexes with multistep synthesized ligands or harsh catalytic conditions has restricted its applicability on large scales. From the standpoint of sustainable chemistry, the development of a new mild strategy via CAD from easily available starting materials is highly desirable.
Nitroxyl radicals are N,N-disubstituted NO radicals with an unpaired delocalized electron.8 These open-shell species have diverse reactivity and can undergo reactions such as a single-electron transfer (SET) to access three discrete oxidation states (i.e., TEMPO−/TEMPO/TEMPO+) or abstract hydrogen atoms from C–H/X–H bonds.9 Transition metals such as ruthenium, rhodium, palladium or copper with the assistance of co-catalyst TEMPO have been proved efficient co-catalysts, and have been utilized to CAD.10 More recently, TEMPO has been extensively used as a catalyst cooperating with electrocatalysis in CAD which is a green and atom-economical alternative.11 In general, the electrochemical reaction with electrons as the oxidizing/reducing agent can be performed at room temperature without the use of chemical oxidants, which makes it safe and low cost.12 Recently, Gong's group has developed an electrochemical dehydrogenation method for synthesis of quinazolin-4(3H)-ones under metal-free and oxidant-free conditions by combining acid-catalyzed annulation and TEMPO as the catalyst.11b Inspired by the Gong work, we envisaged electrocatalysis combined with nitroxyl radicals as a new strategy to direct catalytic acceptorless dehydrogenation of N-heterocycles under neutral conditions (Scheme 1).
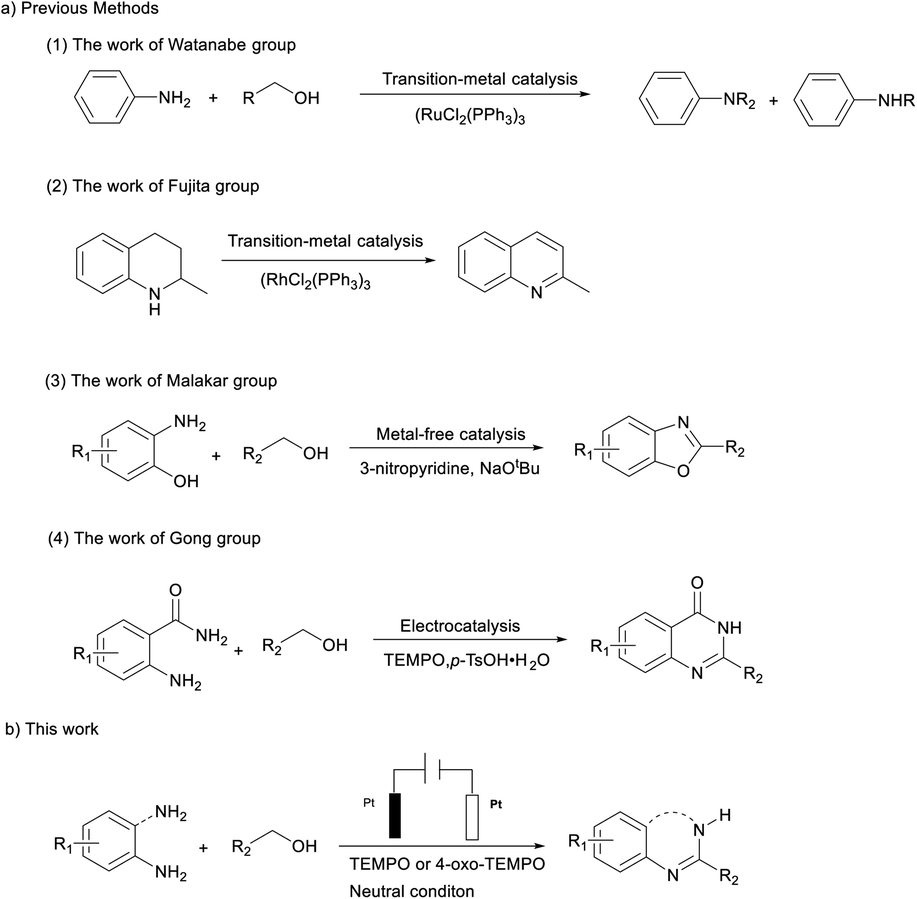 | ||
| Scheme 1 Methods for acceptorless dehydrogenative reactions. | ||
Furthermore, our previous reports on electrochemical synthesis show high reactivity for acceptorless dehydrogenation of primary alcohols to aldehydes.13 Therefore, simultaneous electron transfer and TEMPO-induced chemical reaction is a powerful tool to realize highly selective and relatively environmentally friendly CAD. Herein, we report such a reaction catalyzed by TEMPO to selectively generate nitrogen heterocycles under neutral conditions, with the evolution of H2 as the only byproduct.
In the preliminary studies, benzyl alcohol and o-phenylenediamine were selected as the substrates in MeCN/H2O solvent in the presence of the catalyst TEMPO (0.2 equiv.) and electrolyte tetrabutylammonium hexafluorophosphate (TBAPF6, 0.15 M) to produce a 93% yield of 3aa at constant current (80 mA) at room temperature. As illustrated in Table 1, entries 1–5 show that related nitroxyl derivatives were important catalysts for electrochemical CAD of the catalysts examined, but TEMPO exhibited the highest performance. This may be due to the introduction of an electron-donating group reducing the electron acquisition capacity of the TEMPO cation, leading to difficulty in dehydrogenation of 2,3-dihydro heteroarenes. A change of the electrolyte from TBAPF6 to tetrabutylammonium bromide (TBAB) or tetrabutylammonium iodide (TBAI) led to a decrease of the product yield (Table 1, entries 6 and 7). These results showed that the role of the halogen salts was not only as the electrolyte, but also as a redox mediator which competes to inhibit TEMPO oxidation. Further investigative experiments indicated that the product yield decreased when single or mixed solvents DMSO, DMF, THF, 1.4-dioxane and/or H2O were used instead of MeCN, indicating that MeCN/H2O was superior to the other solvents (Table 1, entries 8–12). The solvent effect may be related to the conductivity of the system. A decrease in electrical conductivity results in a decrease in yield. 1.4-Dioxane diminished the conductivity of the system drastically and hence did not yield 3aa. Moreover, organic compounds had poor solubility in H2O, leading to conductivity issues, and thus produced inferior yields of 3aa. We next turned our attention to investigating the effect of current on the yield. Decreased or trace desired produced was formed if the reaction was conducted at reduced or in the absence of current (Table 1, entries 13–15). When the reaction was conducted at lower constant current (40 and 60 mA), there was an obvious decrease in the yields (Table 1, entries 14 and 15). A decrease in current leads to a decrease in current density, which results in differing concentrations of electron oxidation products, inhibiting further oxidation of 2a. It was also noteworthy that when the Pt electrodes were replaced by graphite rods, the product yield was 31% (Table 1, entry 16). In addition, the discrepancy in reactivity between graphite electrode and Pt electrode may cause a difference in current density at the anode surface, which results in decreasing the rate of electron transfer as well. It was noted that increasing the time for reaction had little effect on the yield (Table 1, entry 17).
|
||
|---|---|---|
| Entry | Variations from the standard conditions | Yieldb (%) |
a Standard conditions: undivided cell, Pt anode (1.0 cm × 1.0 cm), Pt cathode (1.0 cm × 1.0 cm), 1a (0.5 mmol), 2a (0.8 mmol), TEMPO (0.1 mmol), TBAPF6 (0.15 M), CH3CN/H2O (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 3 mL), I = 80 mA at room temperature for 3 h.b Isolated yield. :2, 3 mL), I = 80 mA at room temperature for 3 h.b Isolated yield. |
||
| 1 | None | 93 |
| 2 | 4-Amino-TEMPO instead of TEMPO | 57 |
| 3 | 4-Hydroxy-TEMPO instead of TEMPO | 64 |
| 4 | 4-Oxo-TEMPO instead of TEMPO | 58 |
| 5 | No TEMPO | Trace |
| 6 | TBAB instead of TBAPF6 | 36 |
| 7 | TBAI instead of TBAPF6 | 23 |
| 8 | DMSO/H2O (v/v = 1:2) as solvent |
76 |
| 9 | DMF/H2O (v/v = 1:2) as solvent |
33 |
| 10 | THF/H2O (v/v = 1:2) as solvent |
46 |
| 11 | 1,4-Dioxane/H2O (v/v = 1:2) as solvent |
Trace |
| 12 | H2O as solvent | 73 |
| 13 | No current | Trace |
| 14 | 40 mA | 49 |
| 15 | 60 mA | 64 |
| 16 | C (+)/Pt (−) | 31 |
| 17 | 5 h | 92 |

With the optimal reaction conditions established, we next explored the generality of this protocol for the synthesis of N-heterocyclic compounds. As illustrated in Table 2, a variety of benzyl alcohols with electron-withdrawing or electron-donating groups were well tolerated and gave the corresponding products in good to excellent yields (3aa–3ak). Compared with electron-donating groups, electron-withdrawing groups (–NO2, –F, –Cl, –Br, –CF3) resulted in a relatively medium yield of 73–85% (Table 2, 3af–3ak). Heterocyclic alcohols (pyridinyl and thienyl) also proved to be good substrates for this process, giving good yields of benzazoles under similar conditions (3al and 3am). Notably, we were pleased to find an aliphatic alcohol (phenylethanol) could deliver the desired benzimidazole derivatives in a good yield of 68% (3an). An alcohol bearing a naphthyl group also participated in this reaction with high reactivity to afford the desired product in 73% yield (3ao). On the other hand, various substituted o-phenylenediamines were employed and the reaction proceeded successfully with a yield range of 79–90% (Table 2, 3ap–3at). Similar to the observations in for substituted alcohol compounds, o-phenylenediamine with electron-donating groups (–Me) gave relatively higher reactivity than those with electron-withdrawing (–Cl, and –NO2) ones (3ap–3as). Moreover, challenging substrates like pyridine-2,3-diamine also furnished the desired pteridin-4(3H)-one in 77% yield (Table 2, 3at).
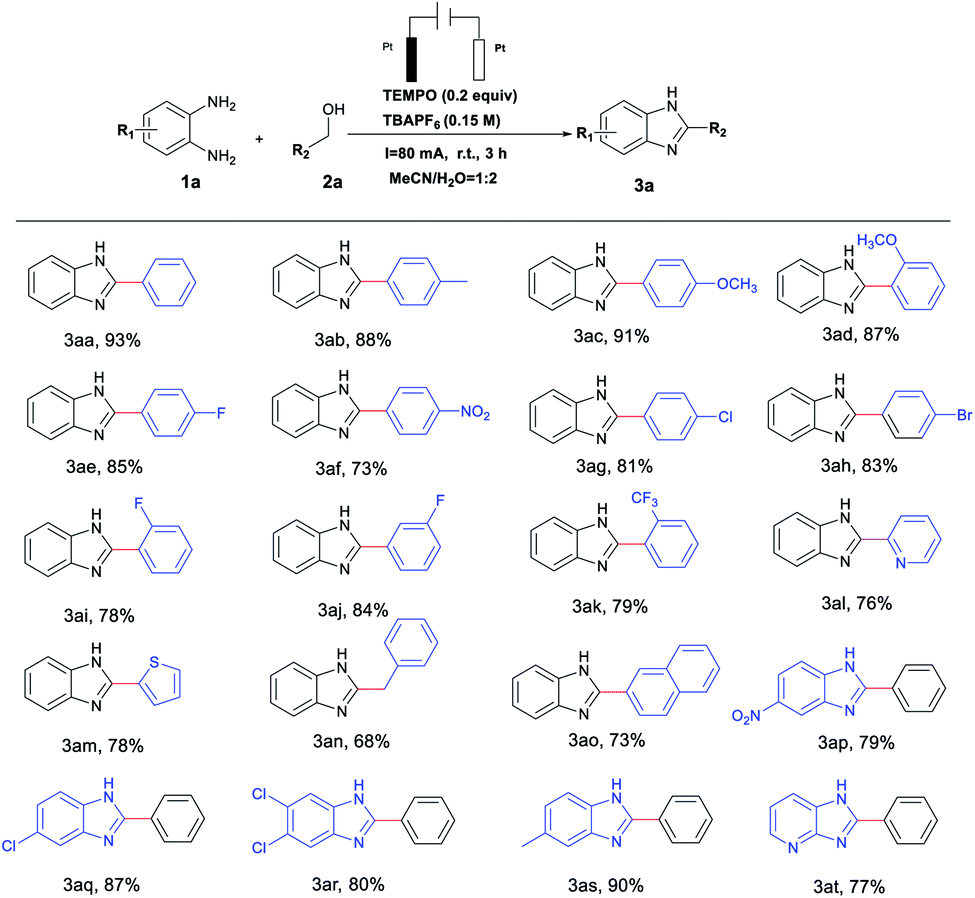
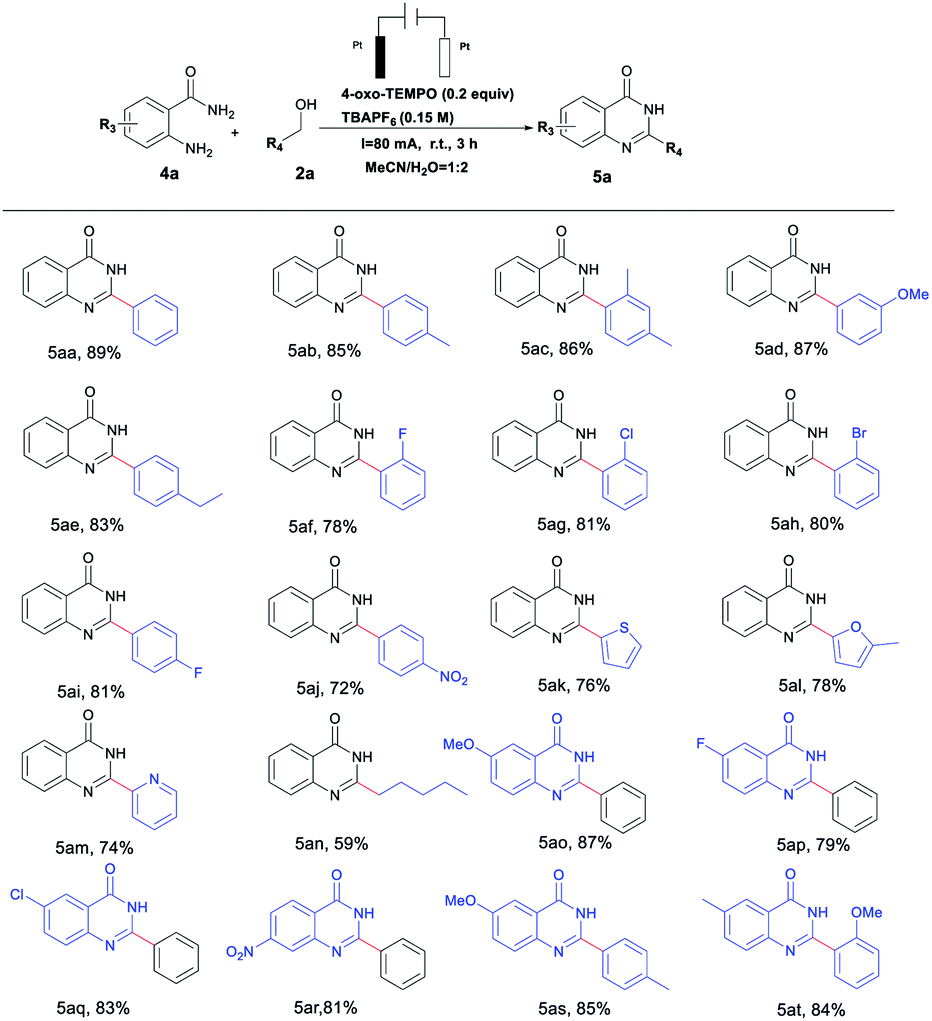
Interestingly, quinazolinone derivatives could also be reached in excellent yields by tuning reaction conditions to investigate use of 4-oxo-TEMPO as the catalyst, which are summarized in Table 3. It was observed that o-aminobenzamides or benzyl alcohols bearing various functional groups could be smoothly converted to the desired products in yields of 59–89%; not coincidentally, aminobenzamides or benzyl alcohols with electron-donating groups (–Me, –OMe or ethyl) gave relatively higher reactivity than those with electron-withdrawing (–F, –Cl or –Br) ones (Table 3, 5ab–5aj, 5ao–5at). In addition, heterocyclic alcohols and amides, and long-chain aliphatic alcohols were also well-tolerated in this catalytic system (Table 3, 5ak–5an).
In order to understand the reaction mechanism, several control experiments were carried out, as shown in Scheme 2. When 2a was subjected to the standard conditions, 75% and 72% yields of 6a were detected (Scheme 2(a)), while the yield of 6a was decreased to 22% or trace in the absence of catalyst or current (Scheme 2(b)). Comparing Scheme 2(c) and (d), we found that when 6a was added to the reaction system in the absence of catalyst, a decreased amount of 3aa was separated from the reaction mixture. This pretty obviously indicated that TEMPO still plays an important role after 1a and 6a are conjured to form intermediates. Furthermore, a similar scenario appears in the synthesis of quinazolinone compounds (Scheme 2(e) and (f)).
 | ||
| Scheme 2 Control experiments. | ||
When the radical inhibitor 2,6-di-tert-butyl-4-methylphenol (BHT) was added under standard conditions this transformation process was inhibited to a large degree (Scheme 3), which implies that the reaction goes through a radical process.
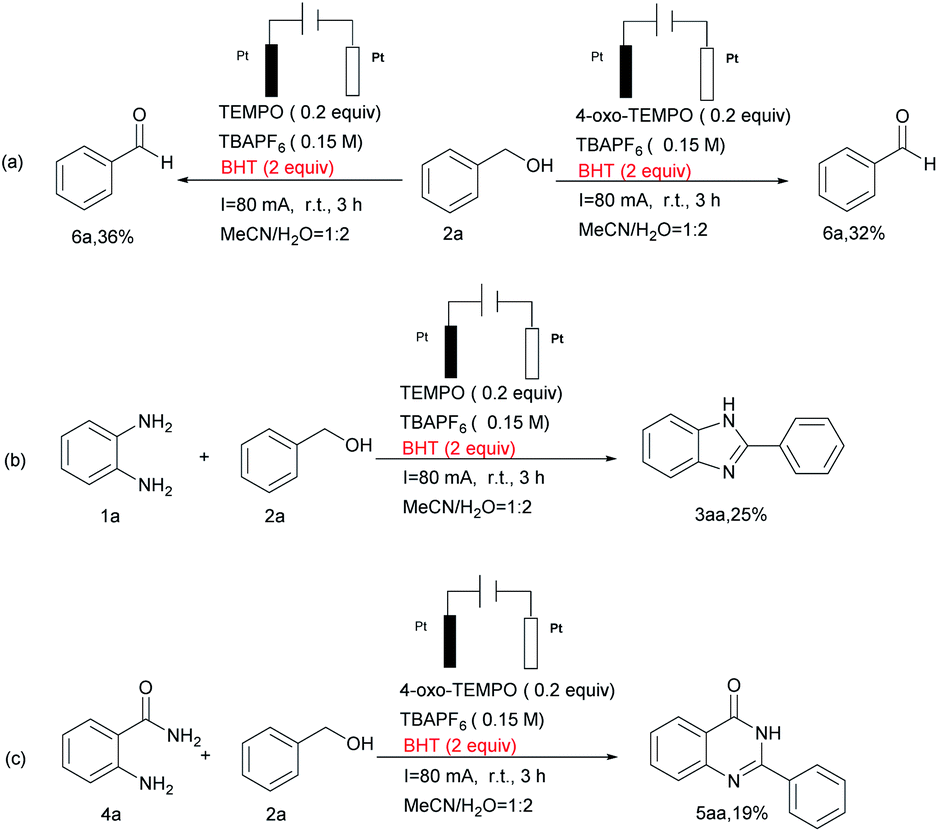 | ||
| Scheme 3 Radical trapping experiments. | ||
To gain an understanding of the reaction mechanism, cyclic voltammetry (CV) was then conducted (Fig. 1). By comparing CV curves a, and b (Fig. 1A), we observed that the TEMPO showed an oxidation peak at Ep = +1.50 V vs. SCE and a reduction peak at Ep = +0.34 V vs. SCE, while the peak changed slightly after the addition of 2a. These results indicate that both TEMPO and current together are required, to participate in the redox process of 2a. Furthermore, the mixture of 1a and 2a resulted in the appearance of a small curvilinear trend in comparison with that of curve d (Fig. 1B, curves c and d), which indicates that current and TEMPO act in coordination to promote further reactions of 1a and 2a. Furthermore, a similar curvilinear trend appears in the cyclic voltammetry of quinazolinones (ESI Fig. 1C and D†).
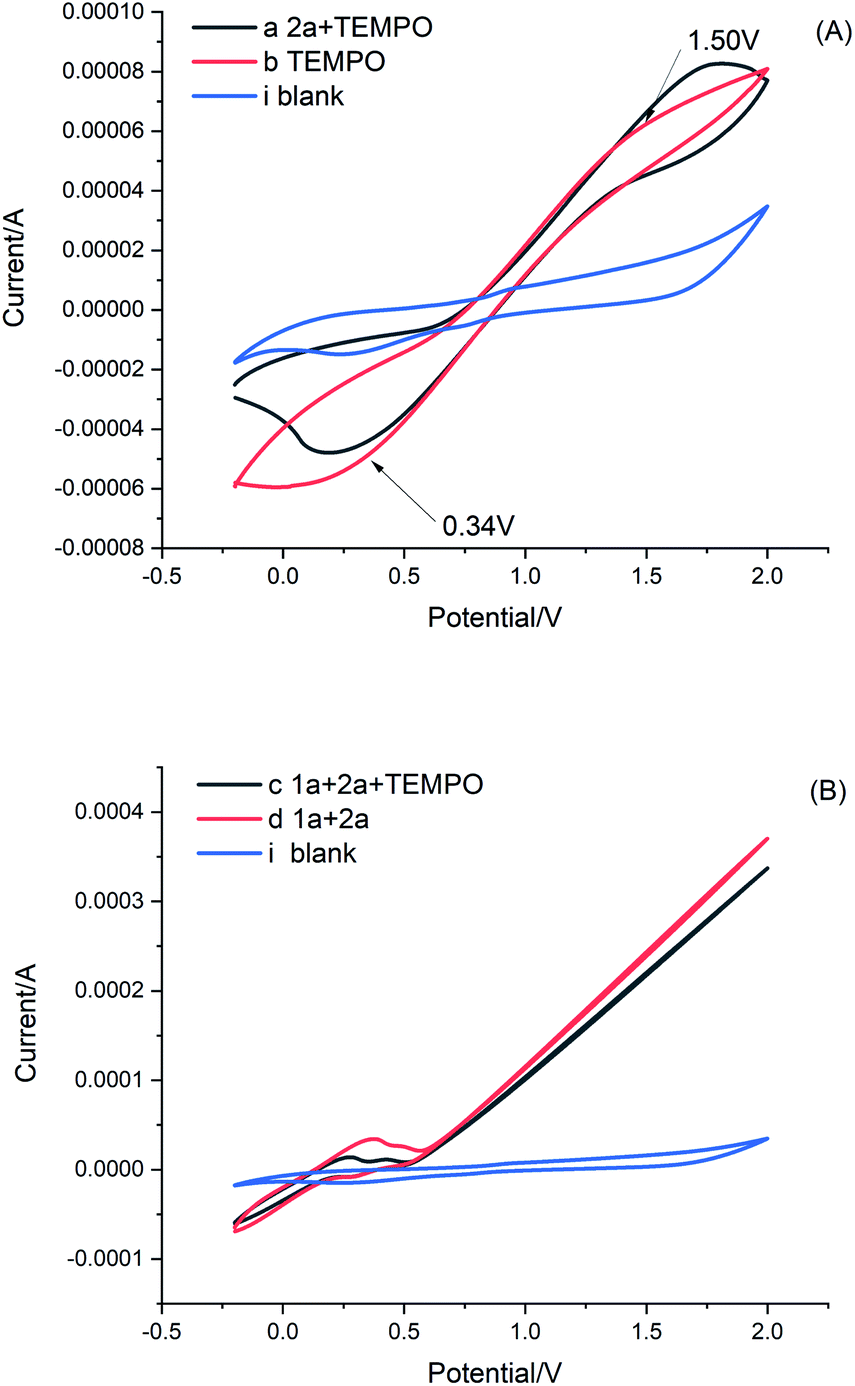 | ||
| Fig. 1 Cyclic voltammograms of reactants and their mixtures in 0.15 M TBAPF6/CH3CN:H2O (1:2) on a Pt disk working electrode (diameter: 3 mm) with a Pt wire and calomel electrode as the counter and reference electrode, respectively, at a scan rate of 0.1 V s−1. | ||
On the basis of the above results and the previous reports, a possible mechanism is provided in Scheme 4.14–17 We envisioned that the reaction proceeds initially through an oxidative redox cycle between TEMPO/4-oxo-TEMPO to TEMPO+/4-oxo-TEMPO+, which promotes the conversion of 2a to A.14 The consequent deprotonation of the radical cation A results in the formation of benzylic radical B. The following fast single-electron oxidation to C and its subsequent deprotonation produce the desired benzaldehyde (6a). In the meantime, water undergoes cathodic reduction to generate hydroxide accompanied by releasing hydrogen.15 Afterwards, as shown in pathway 1, 6a can readily react with 1a to obtain the imine D1, then imine D1 is converted to E1 which is oxidized (dehydrogenated) to afford the desired product 3aa via a radical-cation intermediate F1.16 Similarly, hexagonal nitrogen-containing heterocyclic ring 6a also works well in the same way as shown in pathway 2.17
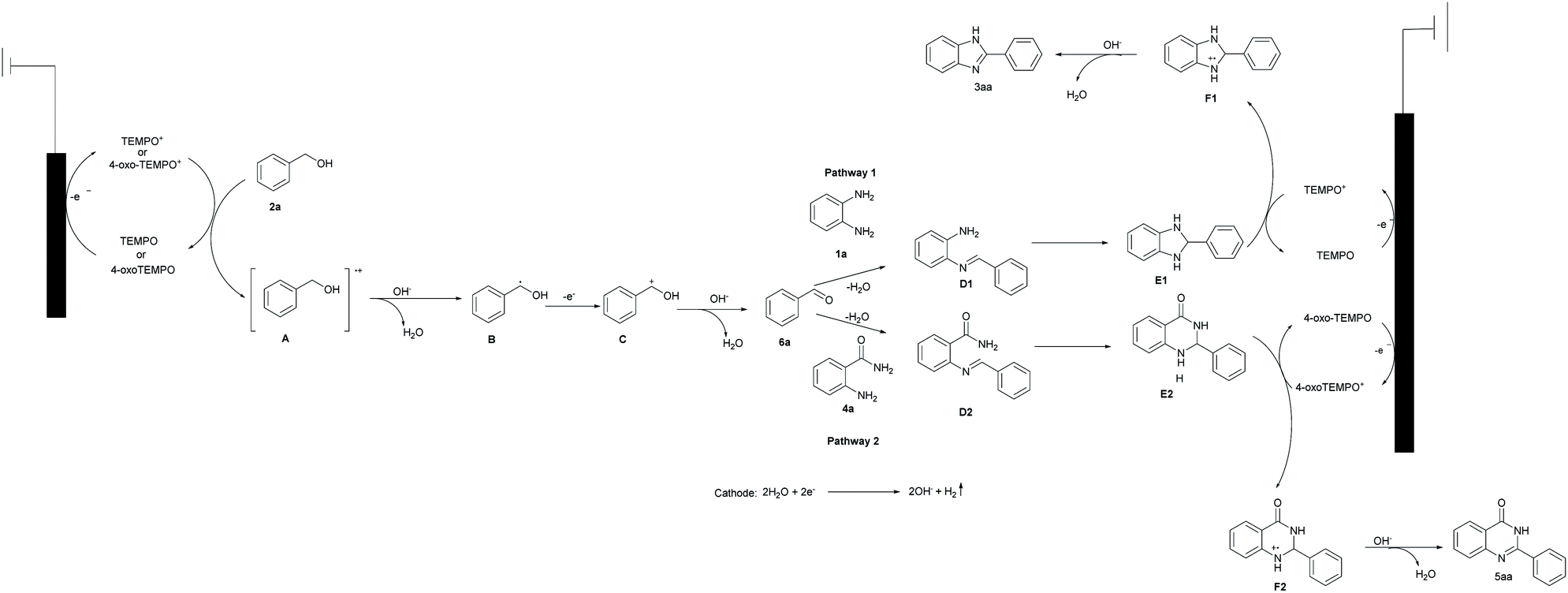 | ||
| Scheme 4 Proposed mechanism for this transformation. | ||
To gain a better understanding of the details of the reaction mechanism, density functional theory (DFT) calculations were performed, and the obtained energy profiles are plotted in Fig. 2. DFT results suggested the formation of the phenylmethanol radical B (˙C6H5CH2OH) and TEMPO+ via electron transfer from 2a to TEMPO+ with an activation energy barrier of +34.83 kcal mol−1. Similarly, 4-oxo-TEMPO can also promote the change from 2a to B with an energy barrier of +27.81 kcal mol−1. Afterwards, the resulting radical B will form 6a via single-electron oxidation by absorbing energy of +26.91 kcal mol−1 both in pathway 1 and in pathway 2, indicating that both 4-oxo-TEMPO+ and TEMPO+ can catalyze the transition from 2a to 6a. Subsequently, 6a can easily react with 1a and 4a to generate D1 and D2. And then, imine D is converted to radical-cation F via a loop-locked intermediate E under the catalysis of TEMPOs. Obviously, this step only has a very low energy barrier of 9.10 kcal mol−1 to overcome for D1, and no energy barrier for D2. Finally, the deprotonation of F1 and F2 yields the desired products 3aa and 5aa, along with releasing energy of −23.99 kcal mol−1 and −33.70 kcal mol−1, respectively. In total, this TEMPO catalysis system is suitable for the oxidative cyclization of nitrogen-containing heterocycles from the viewpoint of thermodynamics and chemical kinetics.
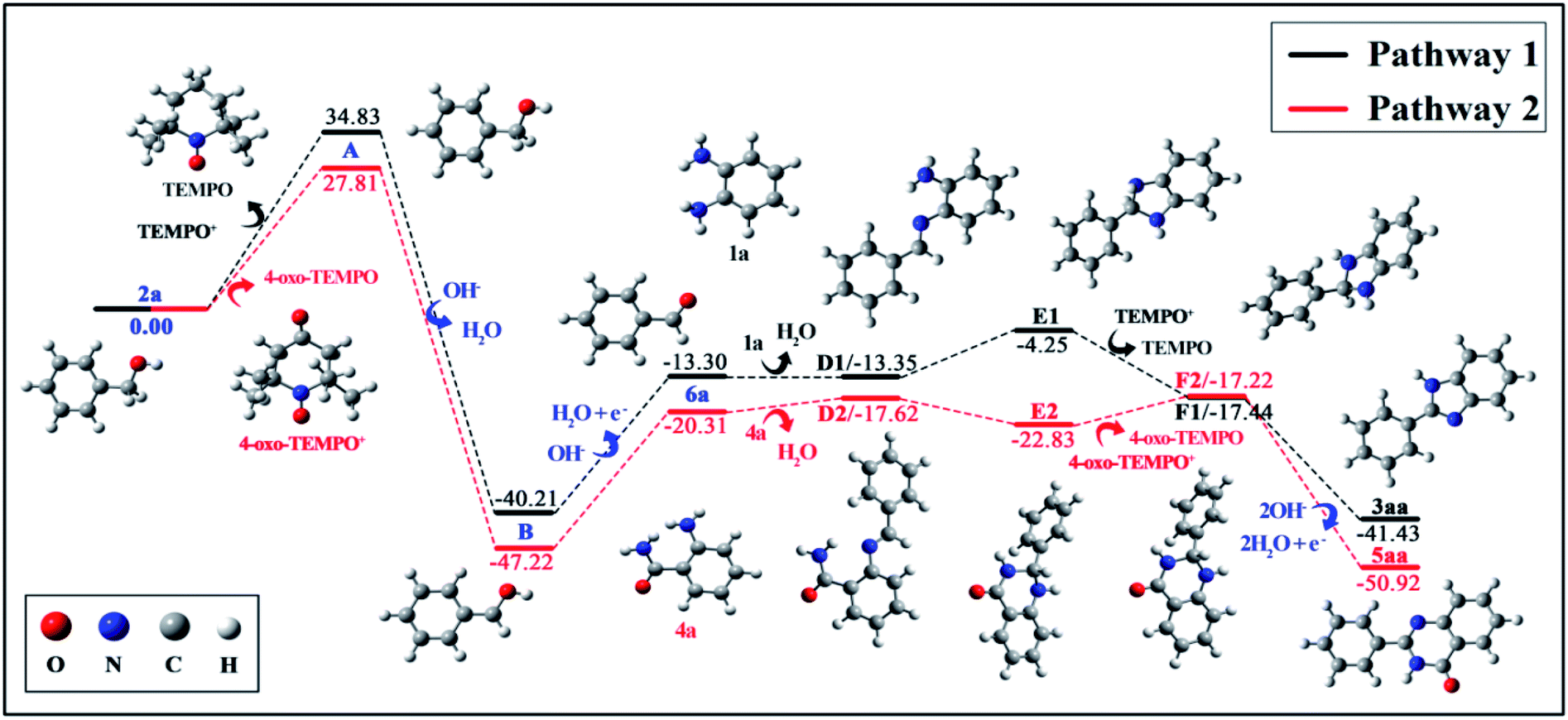 | ||
| Fig. 2 Computed energy profiles for the oxidative cyclization of nitrogen-containing heterocycles. | ||
Conclusions
In summary, an efficient electrocatalytic reaction via catalytic acceptorless dehydrogenation of a range of activated N-heterocycles has been developed using commercially available TEMPOs acting as oxidants under neutral conditions. Importantly, the present method employs a low-cost, environmentally friendly electrocatalyst and avoids using a metal catalyst. This catalysis system is of great value for oxidative cyclization of nitrogen-containing heterocycles from the viewpoint of green chemistry and organic synthesis. Moreover, DFT calculations were also performed to verify the mechanism that we proposed and to prove this system is suitable for the formation of N-heterocycles. Further synthetic application of this reaction for the efficient synthesis of various heterocyclic compounds is currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Fujian Provincial Foundation (Nos. 2020R1012-1, FJNMP-201902, FJNMP-2020004, 2019R11020034-1, 2020J01622, 2020J01627, and 2018QH2021).Notes and references
- (a) M. Zheng, J. Shi and T. Yuan, Angew Chem., Int. Ed., 2018, 57, 5487–5491 CrossRef CAS PubMed; (b) P. Ryabchuk, A. Agapova, C. Kreyenschulte, H. Lund, H. Junge, K. Junge and M. Beller, Chem. Commun., 2019, 55, 4969–4972 RSC; (c) T. liu, K. Wu, L. Wang and Z. Yu, Adv. Synth. Catal., 2019, 361, 3958–3964 CrossRef CAS; (d) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (e) P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed; (f) R. H. Crabtree, ACS Sustainable Chem. Eng., 2017, 5, 4491–4498 CrossRef CAS.
- (a) H. Song, B. Kang and S. H. Hong, ACS Catal., 2014, 4, 2889–2895 CrossRef CAS; (b) K.-N. T. Tseng, A. M. Rizzi and N. K. Szymcza, J. Am. Chem. Soc., 2013, 135, 16352–16355 CrossRef CAS PubMed; (c) S. Xu, L. M. Alhthlol, K. Paudel, E. Reinheimer, D. L. Tyer, D. K. Taylor, A. M. Smith, J. Holzmann, E. Lozano and K. Ding, Inorg. Chem., 2018, 57, 2394–2397 CrossRef CAS PubMed.
- (a) W. Zhao, P. Liu and F. Li, ChemCatChem, 2016, 8, 1523–1530 CrossRef CAS; (b) B. Saha, S. M. W. Rahaman, P. Daw, G. Sengupta and J. K. Bera, Chem.–Eur. J., 2014, 20, 6542–6551 CrossRef CAS PubMed; (c) S. K. Patra and J. K. Bera, Organometallics, 2007, 26, 2598–2603 CrossRef CAS.
- (a) E. Clot, O. Eisenstein and R. H. Crabtree, Chem. Commun., 2007, 2231–2233 RSC; (b) X. Cui, Y. Li, S. Bachmann, M. Scalone, A.-E. Surkus, K. Junge, C. Topf and M. Beller, J. Am. Chem. Soc., 2015, 137, 10652–10658 CrossRef CAS PubMed; (c) K. He, F. Tan, C. Zhou, G. Zhou, X. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 3080–3084 CrossRef CAS PubMed; (d) S. P. Midya, V. G. Landge, M. K. Sahoo, J. Rana and E. Balaraman, Chem. Commun., 2018, 54, 90–93 RSC; (e) M. Mastalir, G. Tomsu, E. Pittenauer, G. Allmaier and K. Kirchner, Org. Lett., 2016, 18, 3462–3465 CrossRef CAS PubMed; (f) G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Org. Lett., 2017, 19, 1080–1083 CrossRef CAS PubMed.
- Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667–2670 CrossRef CAS.
- R. Yamaguchi, C. Ikeda, Y. Takahashi and K. Fujita, J. Am. Chem. Soc., 2009, 131, 8410–8412 CrossRef CAS PubMed.
- (a) D. Kaldhi, N. Vodnala, R. Gujjarappa, S. Nayak, V. Ravichandiran, S. Gupta, G. K. Hazraa and C. C. Malakar, Tetrahedron Lett., 2019, 60, 223–229 CrossRef CAS; (b) G. M. Raghavendra, A. B. Ramesha, C. N. Revanna, K. N. Nandeesh, K. Mantelingu and K. S. Rangappa, Tetrahedron Lett., 2011, 52, 5571–5574 CrossRef CAS.
- (a) T. Vogler and A. Studer, Synthesis, 2008, 13, 1979–1993 Search PubMed; (b) H. Lv, R. D. Laishram, Y. Yang, J. Li, D. Xu, Y. Zhan, Y. Luo, Z. Su, S. More and B. Fan, Org. Biomol. Chem., 2020, 18, 3471–3474 RSC; (c) N. O. Balayeva, N. Zheng, R. Dillert and D. W. Bahnemann, ACS Catal., 2019, 9, 10694–10704 CrossRef CAS; (d) D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS; (e) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed.
- (a) J.-L. Zhan, M.-W. Wu, D. Wei, B.-Y. Wei, Y. Jiang, W. Yu and B. Han, ACS Catal., 2019, 9, 4179–4188 CrossRef CAS; (b) J. C. Siu, G. S. Sauer, A. Saha, R. L. Macey, N. Fu, T. Chauvire, K. M. Lancaster and S. Lin, J. Am. Chem. Soc., 2018, 140, 12511–12520 CrossRef CAS PubMed.
- (a) J. Yu, J. Xu and M. Lu, Appl. Organomet. Chem., 2013, 27, 606–610 CrossRef CAS; (b) Z.-G. Wang, X.-H. Cao, Y. Yang and M. Lu, Synth. Commun., 2015, 45, 1476–1483 CrossRef CAS.
- (a) Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS; (b) L. Cao, H. Huo, H. Zeng, Y. Yu, D. Lu and Y. Gong, Adv. Synth. Catal., 2018, 360, 4764–4773 CrossRef CAS; (c) Y.-L. Lai, J.-S. Ye and J.-M. Huang, Chem.–Eur. J., 2016, 22, 5425–5429 CrossRef CAS PubMed; (d) N. Sbei, A. V. Listratova, A. A. Titov and L. G. Voskressensky, Synthesis, 2019, 51, 2455–2473 CrossRef CAS.
- (a) P.-F. Zhang, H.-M. Lin, L.-W. Wang, Z.-Y. Mo, X.-J. Meng, H.-T. Tang and Y.-M. Pan, Green Chem., 2020, 22, 6334–6339 RSC; (b) P.-S. Gao, X.-J. Weng, Z.-H. Wang, C. Zheng, B. Sun, Z.-H. Chen, S.-L. You and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 15254–15259 CrossRef CAS PubMed; (c) J.-Y. Chen, H.-Y. Wu, Q.-W. Gui, S.-S. Yan, J. Deng, Y.-W. Lin, Z. Cao and W.-M. He, Chin. J. Catal., 2021, 42, 1445–1450 CrossRef CAS; (d) J. P. Barham and B. Konig, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (e) H. Ding, K. Xu and C.-C. Zeng, J. Catal., 2020, 381, 38–43 CrossRef CAS; (f) H.-T. Tang, J.-S. Jia and Y.-M. Pan, Org. Biomol. Chem., 2020, 18, 5315–5333 RSC; (g) Y. Wu, J.-Y. Chen, J. Ning, X. Jiang, J. Deng, Y. Deng, R. Xu and W.-M. He, Green Chem., 2021, 23, 3950–3954 RSC.
- (a) H. Hou, X. Ma, Y. Lin, J. Lin, W. Sun, L. Wang, X. Xu and F. Ke, RSC Adv., 2021, 11, 17721–17726 RSC; (b) Y. Hu, H. Hou, L. Yu, S. Zhou, X. Wu, W. Sun and F. Ke, RSC Adv., 2021, 11, 31650–31655 RSC; (c) L. Yang, H. Hou, L. Li, J. Wang, S. Zhou, M. Wu and F. Ke, Org. Biomol. Chem., 2021, 19, 998–1003 RSC.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- (a) D. Wang, P. Wang, S. Wang, Y.-H. Chen, H. Zhang and A. Lei, Nat. Commun., 2019, 10, 2796–2804 CrossRef PubMed; (b) R. Rajeev, B. Sharma, A. T. Mathew, L. Groege, Y. N. Sudhakar and A. Varghese, J. Electrochem. Soc., 2020, 167, 136508–136526 CrossRef CAS.
- (a) K. Tateyama, K. Wada, H. Miura, S. Hosokawa, R. Abe and M. Inoue, Catal. Sci. Technol., 2016, 6, 1677–1684 RSC; (b) A. Ziarati, A. Baadiei, G. M. Ziarani and H. Eskandarloo, Catal. Commun., 2017, 95, 77–82 CrossRef CAS; (c) Q. An, C. He, X. Fan, C. Hou, J. Zhao, Y. Liu, H. Liu, J. Ma, Z. Sun and W. Chu, ChemElectroChem, 2020, 7, 3969–3974 CrossRef CAS.
- (a) Y. Hu, S. Li, H. Li, Y. Li, J. Lin, C. Duanmu and B. Li, Org. Chem. Front., 2019, 6, 2744–2748 RSC; (b) M. Kumar, Richa, S. Sharma, V. Bhatt and N. Kumar, Adv. Synth. Catal., 2015, 357, 2862–2868 CrossRef CAS; (c) Q. Wang, M. Lv, J. Liu, Y. Lo, H. Cao, X. Zhang and Q. Xu, ChemSusChem, 2019, 12, 3043–3048 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08919f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |