
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccurate predictions of the electronic excited states of BODIPY based dye sensitizers using spin-component-scaled double-hybrid functionals: a TD-DFT benchmark study†
Qabas Alkhatiba,
Wissam Helal *a and
Ali Marashdehbc
*a and
Ali Marashdehbc
aDepartment of Chemistry, The University of Jordan, Amman 11 942, Jordan. E-mail: wissam.helal@ju.edu.jo
bDepartment of Chemistry, Al-Balqa Applied University, 19 117 Al-Salt, Jordan
cLeiden Institute of Chemistry, Gorlaeus Laboratories, Leiden University, P. O. Box 9502, 2300 RA Leiden, The Netherlands
First published on 11th January 2022
Abstract
The vertical excitation energies of 13 BODIPY based dye sensitizers are benchmarked by means of TD-DFT, using 36 functionals from different DFT rungs. Most TD-DFT results were found to overestimate the excitation energies, and show mean absolute error (MAE) values in the range 0.2–0.5 eV. The dispersion-corrected, spin-component-scaled, double-hybrid (DSD) functionals DSD-BLYP and DSD-PBEP86 were found to have the smallest MAE values of 0.083 eV and 0.106 eV, respectively, which is close to the range of average errors found in the more expensive coupled-cluster methods. Moreover, DSD-BLYP and DSD-PBEP86 functionals show excellent consistency and quality of results (standard deviation = 0.048 eV and 0.069 eV respectively). However, the range separated hybrid (RSH) and the range separated double hybrid (RSDH) functionals were found to provide the best predictability (linear determination coefficient R2 > 0.97 eV).
1. Introduction
The current environmental and energy dilemma has promoted substantial research and efforts in renewable resources of energy. Dye-sensitized solar cell (DSSC) photovoltaic systems are among the leading technologies in renewable energy and are therefore extensively explored.1–15 Boron dipyrromethene (BODIPY) chromophores are very promising in this regard, since they present sharp and intense absorption and emission spectra and excellent chemical and photochemical stabilities.16–18 Moreover, the optical properties of BODIPYs can be tuned by chemical modifications through a multitude of substitutions.19–23 Due to these remarkable optical and chemical properties, BODIPYs are widely used as light harvesting moieties in dye-sensitized solar cells (DSSC),24–31 heterojunction organic solar cells,32–34 and perovskite solar cells.35 In addition, BODIPY chromophores are also used in other diverse applications ranging from optoelectronics and OLEDs36,37 to bioimaging.38–40Theoretical modeling of dye sensitizers, which are the workhorse for all DSSC devices, can effectively aid the experimental research by providing basic design guidelines of new sensitizers and a deeper understanding of the photophysical and photochemical processes governing the functioning and the performance of solar cells.41–44 Of these fundamental photophysical processes, the absorption of sunlight by the dye sensitizer leading to the electronic excitation to an excited state, is the first and the most important step toward subsequent electron injection and dye and electrolyte regeneration. Therefore, being able to correctly model the optical absorption spectra of dye sensitizers is crucial.45 An ideal dye suitable for DSSCs should have a broad absorption range that covers both the visible and near infrared regions of the solar spectrum and its molar extinction coefficient must be high to enable efficient light harvesting.14,15 These and other optical properties can be calculated by most excited state computational methods.46–51 Typically, push–pull dyes designed with the “electron donor–π linker–electron acceptor” (D–π–A) conventional architecture, and many other variations such as the D–A–π–A architecture,52 are found to be efficient for photosensitized applications such as DSSCs.14,53
Despite many theoretical and computational efforts,54–70 the electronic molecular spectroscopy of BODIPYs is still not yet fully understood, and the accurate calculation of their excited-states is still a challenge for both theoretical and computational chemists. In general, conventional time dependent (TD) DFT is the preferred method for electronic excited states calculations on large compounds, since it is efficient and reasonably accurate, with errors of transition energies in the range 0.1–0.3 eV for many simple organic chromophores. Unfortunately, TD-DFT tends to highly overestimates the low-lying excited states (ES) of BODIPYs.54–58 Typically, the errors obtained with TD-DFT for BODIPYs are usually more than 0.3 eV.55,57 The failure of time-dependent regime with BODIPYs is mainly due to the presence of double excitations that play an important role in these compounds.55,61,71–73 Better results were obtained using the more expensive ab initio or combined ab initio and TD-DFT methods, such as combined TD-DFT and SOS-CIS(D),59–61 combined TD-DFT and Bethe–Salpeter formalism,62 spin-flip TD-DFT approach,63 and coupled-cluster methods.55,64–68 For instance, Brown and coworkers found that the mean absolute error (MAE) of the Laplace transformed local CC2 (LCC2*) and the DLPNO-STEOM-CCSD methods are around 0.1 eV.55,66 It should be emphasized, however, that these two accurate coupled-cluster methods, cannot be applied for reasonably large molecules.
It is our objective in this work to propose a density functional(s) that is reliable, accurate, simple to use, and relatively fast, for the calculations and predictions of the electronic absorption and other excited state properties of BODIPY sensitizers used in light harvesting technology such as DSSCs. In fact, the use of pure, hybrid, and range separated hybrid functionals to study the excited state properties of BODIPY dye sensitizers is not quantitatively or even qualitatively justified, despite their attractive computational speed for large molecules. In a recent benchmark study, where we have tested the double hybrid functionals on a set of relatively small BODIPY chromophores, we found that the dispersion-corrected, spin-component-scaled, double-hybrid (DSD) functionals DSD-BLYP and DSD-PBEP86 perform very well, with MAE values close to 0.1 eV.70 The average of errors found in that benchmark study using TD DH functionals fall in the range of errors of the more expensive coupled-cluster methods.55,66 In this work, we have decided to extend the benchmark of double hybrid functionals, on larger dyes that are designed on push–pull architectures and having electronic transitions that are characterized with significant long range charge transfer. The double hybrid functionals are not frequently used for the computational description of the optical properties of BODIPY and other dye sensitizers. Moreover, while there are numerous benchmark studies on organic dyes,74–76 benchmark studies on dyes sensitizers used in DSSCs are not frequent in the literature.41,77–80
Double hybrid (DH) functionals include a fraction of a perturbative second-order correlation part to the exchange–correlation (xc) functionals,81–83 and are increasingly used in TD-DFT calculations.84–91 Furthermore, time-dependent DH density functionals that include spin-component and spin-opposite scaling were recently proposed and they seems to be very promising for accurate calculations of excitation energies.92–94 We have investigated the performance of DH functionals for the prediction of the vertical excitations of a set of large BODIPY dye sensitizers. We have benchmarked the following DH functionals using TD-DFT: B2PLYP,81 B2GPPLYP,95 mPW2PLYP,96 the empirical dispersion-corrected, spin-component-scaled, double-hybrid (DSD) functionals (DSD-BLYP and DSD-PBEP86),97,98 and the range separated DH functionals ωB2PLYP and ωB2GPPLYP.87 In addition, we have tested the performance of TD-DFT using 29 other functionals from other rungs (see the Computational methods for details). Our benchmark set is composed of 13 BODIPY based dye sensitizer (see Fig. 1) that have been experimentally investigated as promising sensitizers for DSSC applications: B1,99 B2,100 B3,101 B4,102 B5,103 B6,104 B7,102 B8,105 B9,106 B10,107 B11,108 B12,109 and B13.110
 | ||
| Fig. 1 Molecular structure of BODIPY dyes considered in this study. | ||
2. Computational methods
All quantum chemical calculations have been carried out with ORCA 4.2.0 code.111,112 Molecular orbital isosurface densities have been visualized using Gabedit 2.4.8.113 The ground state (GS) equilibrium geometries of all molecules have been fully optimized without any symmetry restriction using DFT employing PBE0 functional114 and the Ahlrichs def2-TZVP115 basis set. PBE0 functional has been shown to produce accurate ground state geometries for BODIPY molecules.54 Geometry optimizations are repeated for all possible stable conformers of the molecules, and the subsequent excited state calculations correspond to the most stable conformer. Frequency calculations were carried out for all optimized geometries at the same level of theory. None of our optimized geometries shows any imaginary frequency values. We have also calculated coupled-cluster singles and doubles (CCSD)116,117 and the recently developed DLPNO-STEOM-CCSD method67,68,118 with def2-TZVP basis for compound B1.The lowest 20 singlet–singlet vertical electronic excitations on the optimized GS geometries were calculated by means of TD-DFT and the def2-TZVP basis set, without using the Tamm–Dancoff approximation (TDA), which is set as default in TD-DFT ORCA module. We have assessed the impact of solvent effects in all geometry optimizations and excited state calculations using the linear-response conductor-like polarizable continuum model (LR-CPCM),119 and solvents as indicated by the corresponding experimental results; see Table S1 in the ESI.† Furthermore, TD-DFT calculations in solvent were performed using the non-equilibrium regime, which is set as default for all ES calculations in ORCA.
All DFT and TD-DFT calculations were sped up with the resolution-of-the-identity (RI) approximation. The RIJCOSX procedure,120 a standard setting in ORCA which uses both the RI for Coulomb integrals (RI-J) and the chain-of-spheres approximation for exchange integrals (COSX),121 is employed with all functionals. The RI-MP2 (ref. 122) is also used for the DH and the range separated DH functionals. The def2/J auxiliary basis set123 were used with RIJCOSX, and the def2-TZVP/C124 with RI-DH. Converged SCF orbitals were obtained using the TightSCF setting in ORCA (energy change = 10−8 Eh). A multi-grid approach125 for the numerical quadrature integration was chosen, where the SCF iterations are done with a pruned grid of 40 radial shells (Gauss–Chebyshev) and 434 angular points (Lebedev434) per shell, whereas gradients and final energies are evaluated on a pruned grid of 45 radial shells and 590 angular points per shell.
TD-DFT calculations were performed using 36 functional of different rungs: generalized gradient approximation (GGA), meta-GGA (mGGA), global-hybrid GGA (GH-GGA), global-hybrid meta-GGA (GH-mGGA), range separated hybrid GGA (RSH-GGA), double hybrid GGA (DH-GGA), and range separated double hybrid GGA (RSDH-GGA). See Table 1 for more details. The exchange correlation energy of GH-GGA expression is:
| EGH-GGAxc = axEHFx + (1 − ax)EGGAx + EGGAc | (1) |
| EDH-GGAxc = (1 − ax)EDFTx + axEHFx + (1 − ac)EDFTc + acEMP2c | (2) |
| EDSD-DFTxc = (1 − ax)EDFTx + axEHFx + ccEDFTc + coEOS-MP2c+csESS-MP2c + Edisp | (3) |
| Functional | ax (%HF) | ac (%MP2) | cc | co | cs | ω bohr−1 | Exchange functional | Correlation functional | Year | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a List of abbreviations and symbols: GGA: generalized gradient approximation; mGGA: meta-GGA; GH: global hybrid; RSH: range separated hybrid; DH: double hybrid; RSDH: range separated double hybrid; ax: scale factor for exact (HF) exchange in GH-GGA [eqn (1)], GH-mGGA, RSH-GGA, DH-GGA [eqn (2)], and RSDH-GGA; ac second-order perturbative correlation for various DH-GGA [eqn (2)]; cc, co, and cs: scale factors of the DFT correlation, perturbative correlation contribution of opposite-spin electron pairs and that of same-spin electron pairs, respectively, for the DSD functionals [eqn (3)]; and ω: screening factor RS and functionals. | ||||||||||
| GGA | ||||||||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||||
| OLYP | OptX | LYP | 2001 | 131 and 132 | ||||||
| BLYP | B88 | LYP | 1988 | 131 and 133 | ||||||
| BP86 | B88 | P86 | 1988 | 133 and 134 | ||||||
| XLYP | B88 + PW91 | LYP | 2004 | 131 and 135 | ||||||
| PBE | PBE(X) | PBE(C) | 1996 | 136 | ||||||
| mPWPW | mPW91 | PW91 | 1998 | 137 | ||||||
| mPWLYP | mPW91 | LYP | 1998 | 131 and 137 | ||||||
| B97-D3 | RB97 | B97 | 2011 | 138 | ||||||
| MGGA | ||||||||||
|
||||||||||
| M06-L | M06-L(X) | M06-L(C) | 2006 | 139 | ||||||
| TPSS | TPSS | TPSS | 2003 | 140 | ||||||
| GH-GGA | ||||||||||
|
||||||||||
| O3LYP | 11.6 | OptX | LYP | 2001 | 131 and 132 | |||||
| B3LYP | 20 | B88 | LYP | 1993 | 141 and 142 | |||||
| B3P86 | 20 | B88 | P86 | 1993 | 134 and 141 | |||||
| X3LYP | 22 | B88 + PW91 | LYP | 2004 | 131 and 135 | |||||
| PBE0 | 25 | PBE(X) | PBE(C) | 1999 | 114 | |||||
| mPW1PW | 25 | mPW91 | PW91 | 1998 | 137 | |||||
| mPW1LYP | 25 | mPW91 | LYP | 1998 | 131 and 137 | |||||
| BH&HLYP | 50 | B88 | LYP | 1993 | 143 | |||||
| GH-mGGA | ||||||||||
|
||||||||||
| TPSSh | 10 | TPSS | TPSS | 2003 | 144 | |||||
| TPSS0 | 25 | TPSS | TPSS | 2005 | 145 | |||||
| M06 | 27 | M06(X) | M06(C) | 2008 | 146 | |||||
| M06-2x | 54 | M06-2X(X) | M06-2X(C) | 2008 | 146 | |||||
| RSH-GGA | ||||||||||
|
||||||||||
| LC-BLYP | 0–100 | 0.33 | B88 | LYP | 2004 | 147 | ||||
| CAM-B3LYP | 19–65 | 0.33 | B88 | LYP | 2004 | 148 | ||||
| ωB97 | 0–100 | 0.40 | ωB97 | B97 | 2008 | 149 | ||||
| ωB97X | 15.77–100 | 0.30 | ωB97X | B97 | 2008 | 149 | ||||
| ωB97X-D3 | 19.57–100 | 0.25 | ωB97X | B97 | 2013 | 150 | ||||
| ωB97X-D3(BJ) | 16.7–100 | 0.30 | ωB97X | B97 | 2018 | 151 | ||||
| ωB97X-V | 16.7–100 | 0.30 | ωB97X | B97 | 2014 | 152 | ||||
| DH-GGA | ||||||||||
|
||||||||||
| B2PLYP | 53 | 27 | B88 | LYP | 2006 | 81 | ||||
| B2GPPLYP | 65 | 36 | B88 | LYP | 2008 | 95 | ||||
| mPW2PLYP | 55 | 25 | mPW | LYP | 2006 | 96 | ||||
| DSD-BLYP | 69 | 54 | 46 | 37 | B88 | LYP | 2010 | 97 | ||
| DSD-PBEP86 | 70 | 43 | 53 | 25 | PBE | P86 | 2011 | 98 | ||
| RSDH-GGA | ||||||||||
|
||||||||||
| ωB2PLYP | 53 | 27 | 0.30 | ωB88 | LYP | 2019 | 87 | |||
| ωB2GPPLYP | 65 | 36 | 0.27 | ωB88 | LYP | 2019 | 87 | |||
3. Results and discussion
The BODIPY based dye sensitizers (B1–B13) investigated in this study are shown in Fig. 1. All dyes have been experimentally studied as potential successful sensitizers for DSSC applications.99–110 Our selected set of BODIPY based dye sensitizers represent different architectures and designs. First, the set of 13 BODIPY dyes represents three different designs:27,126 the horizontal design (B1–B7), the vertical design (B8–B11), and the fused design (B12 and B13). Second, many of our dyes (B1–B4, B6, B7, and B10) are based on the conventional so called D–π–A architecture, where an electron-donating (D) group is linked to a π spacer that is coupled in turn to an electron-withdrawing group (A); dye B5 is based on the D–A–π–A architecture,52 where an auxiliary acceptor is added before the π linker; dyes B8, B9, and B12 share the (D)2–π–A architecture. Dye B11 is based on a typical D–π–A, but the BODIPY unit here act as the light harvesting antenna and is linked to the acceptor; while dye B13 is based on a (D)2–π–(A)2 architecture or the so called “butterfly-shaped” BODIPY dye.110In order to enhance DSSC efficiency, long hydrophobic linear alkyl side chains are integrated into the dye sensitizer framework to prevent dye recombination with the semiconducting oxide electrode. Many molecules in our set contain these type of long alkyl chains: B2, B3, B5, B6, B11, and B13. In order to reduce the computational cost, the long linear alkyl chains in dyes B5, B6, and B13 are simply modeled by replacing them with methyl groups in all geometry and ES calculations (Fig. 1). The theoretical justification for such an approximation is that these outer linear alkyl chains are not involved in the electronic structure and optical properties for those highly conjugated chromophores. Furthermore, we have actually verified the effect of long alkyl side chain substitution with a methyl group on the calculated optical properties of one dye sensitizer, namely B2. We have found that replacing the alkyl chain C5H11 with a simple CH3 has almost no effect on the vertical energy of absorption. In particular, the computed vertical excitation energy of compound B2–C5H11 is found to be 0.015 eV lower than that B2–CH3. The differences in the oscillator strengths of B2–C5H11 and B2–CH3 are found to be insignificant as well: 1.47 and 1.52, respectively. In fact, this kind of approximation is also used in many other studies.127–129
Experimentally, the dye sensitizers and the electrolyte redox couple (such as I−/I3−) used as regenerator are typically dissolved in an organic solvent. Thus, all our geometry optimization and excited state calculations were performed using the implicit CPCM approximation in order to estimate the bulk solvent effects. The solvent used for each dye is the same as that used in the UV-vis absorption experiment, as indicated in Table S1 in the ESI.† The xyz coordinates of the optimized geometries in solvent for all dyes investigated in this work are reported in Table S43 in the ESI.†
The coupled-cluster T1 diagnostic test of Lee and Taylor130 was used, at the CCSD/def2-SVP level of theory, on the B1 dye sensitizer to check the reliability of using single reference methods for such compounds. A CCSD calculation produce the T1 diagnostic test which is based on the norm of the vector of single-excitation amplitudes from CCSD in a closed shell system,.130 If the T1 value is less than 0.02, the system is considered to be dominated by single reference wavefunction, otherwise, the system is considered to be of a multireference character. According to diagnostic test performed in this study, no multi-reference characteristics were found in the GS of B1 compound (T1 = 0.0139), which means that it is reliable to use a single reference method, such as TD-DFT, for describing the vertical ES. In fact, we did not calculate CCSD/def2-SVP for all compounds, since these calculations are very long for such large compounds. However, we believe that the other dyes will behave like the “parent” B1 dye, since they all share the same core, which is the “problematic” BODIPY fragment in all dye configurations.
The calculated vertical excitation energies do not take into consideration the vibronic effects, which are associated with all molecular electronic band spectra. Therefore, the experimental band maxima do not necessarily exactly match the vertical excitation energies that are obtained using conventional excited state methods, such as TD-DFT. Ideally, comparing 0–0 energies (E0–0) with the experimental absorption–fluorescence crossing point (AFCP) is more appropriate for excited state benchmarks.56–58 However, the calculation E0–0 needs the geometry optimization of the ES in solvent, and the subsequent calculation of ES Hessian in order to obtain ES vibrational frequencies in solvent, which is a very difficult task in general for moderate size molecules, and almost impossible for large molecules. In addition, accurate experimental AFCP values are rather uncommon for large molecules such dye sensitizers. It has been shown, however, that the use of the vertical approximation is justified in the case of BODIPY compounds.55,70 Considering the average large size of dye sensitizers in general and the large size of the dyes investigated in this study, we have decided to adopt the vertical excitation regime.46,55,66,70 Moreover, the practice of frequent calculations of large dyes in order to evaluate their potential use as light harvesting systems should be easy and straightforward. Thus it would be necessary to be able to calculate the optical properties of moderate to large size molecules using a simple protocol such as that of the vertical excitation energies, and not by using the complicated many-step protocol of finding E0–0 values. In fact, we are comfortable with comparing the vertical excitation energies directly with the experimental band maxima since the results of some of the functionals investigated in this study are very satisfactory, vide infra. Therefore, in this work, we compare the available experimental λmax values with the first dipole-allowed vertical excitation energies from the GS to a singlet ES.
TD-DFT vertical excitation energies typically produce errors in the range 0.2–0.3 eV for simple organic compounds,47,50 though sometimes “better” and “worse” predictions can be observed for particular families of chromophores.47 A TD-DFT functional is said to be accurate if the calculated results of electronic excitations are deviated from experiment by values equal to or less than 0.1 eV. The range separated hybrid (RSH) and the range separated double hybrid (RSDH) functionals are designed for excited state property calculations and should take into consideration the charge transfer present in the electronic transitions of the dye sensitizers. Therefore, RSH and RSDH functionals should in principle give the best performance among the whole set of functionals tested in this work. Interestingly, the functionals of these two rungs were found to produce the worst performance among all functionals; see Table 2 and Fig. 2 (the details of TD vertical excitations for all functionals and all dyes are reported in Tables S2–S6 in the ESI†). All RSH and RSDH functionals show mean absolute error (MAE) values of more than 0.4 eV. The two recent RSDH functionals ωB2PLYP and ωB2GPPLYP,87 that are optimized specifically for excited state properties and should in theory reproduce the correct asymptotic long-range behavior, have MAE values of 0.433 and 0.414 eV, respectively. For the case the ωB97 family of RSH functionals, the mean error becomes more than 0.5 eV. In fact, these findings are consistent with that of our recent benchmark study on small BODIPY chromophores, in which the range separated functionals (RSH and RSDH) were also among the worst performing ones with MAE values around 0.5 to 0.6 eV.70
| Functional | MAE | Max | Min | SD | R2 |
|---|---|---|---|---|---|
| GGA | |||||
|
|||||
| OLYP | 0.294 | 0.415 | −0.781 | 0.289 | 0.010 |
| BLYP | 0.319 | 0.396 | −0.836 | 0.314 | 0.001 |
| BP86 | 0.314 | 0.406 | −0.801 | 0.286 | 0.002 |
| XLYP | 0.315 | 0.395 | −0.869 | 0.317 | 0.002 |
| PBE | 0.308 | 0.407 | −0.782 | 0.280 | 0.003 |
| mPWPW | 0.308 | 0.406 | −0.785 | 0.289 | 0.003 |
| mPWLYP | 0.320 | 0.394 | −0.867 | 0.317 | 0.001 |
| B97-D3 | 0.310 | 0.411 | −0.840 | 0.314 | 0.002 |
| mGGA | |||||
|
|||||
| M06-L | 0.240 | 0.466 | −0.517 | 0.167 | 0.151 |
| TPSS | 0.172 | 0.453 | −0.299 | 0.126 | 0.519 |
| GH-GGA | |||||
|
|||||
| O3LYP | 0.196 | 0.412 | −0.582 | 0.164 | 0.612 |
| B3LYP | 0.244 | 0.554 | −0.370 | 0.176 | 0.594 |
| B3P86 | 0.228 | 0.566 | −0.066 | 0.184 | 0.721 |
| X3LYP | 0.259 | 0.582 | 0.041 | 0.175 | 0.820 |
| PBE0 | 0.336 | 0.645 | 0.100 | 0.167 | 0.857 |
| mPW1PW | 0.336 | 0.643 | 0.103 | 0.165 | 0.858 |
| mPW1LYP | 0.326 | 0.628 | 0.113 | 0.158 | 0.859 |
| BH&HLYP | 0.429 | 0.589 | 0.290 | 0.086 | 0.946 |
| GH-mGGA | |||||
|
|||||
| TPSSh | 0.233 | 0.631 | −0.124 | 0.183 | 0.579 |
| TPSS0 | 0.350 | 0.676 | 0.136 | 0.159 | 0.823 |
| M06 | 0.304 | 0.446 | −0.270 | 0.109 | 0.875 |
| M06-2x | 0.361 | 0.500 | 0.267 | 0.067 | 0.965 |
| RSH-GGA | |||||
|
|||||
| LC-BLYP | 0.469 | 0.541 | 0.384 | 0.048 | 0.975 |
| CAM-B3LYP | 0.415 | 0.522 | 0.318 | 0.060 | 0.979 |
| ωB97 | 0.518 | 0.608 | 0.416 | 0.056 | 0.966 |
| ωB97X | 0.510 | 0.592 | 0.413 | 0.053 | 0.972 |
| ωB97X-D3 | 0.496 | 0.571 | 0.396 | 0.053 | 0.976 |
| ωB97X-D3(BJ) | 0.532 | 0.615 | 0.432 | 0.054 | 0.972 |
| ωB97X-V | 0.532 | 0.615 | 0.432 | 0.054 | 0.972 |
| DH-GGA | |||||
|
|||||
| B2PLYP | 0.179 | 0.423 | −0.036 | 0.102 | 0.865 |
| B2GPPLYP | 0.280 | 0.409 | 0.207 | 0.053 | 0.972 |
| mPW2PLYP | 0.231 | 0.442 | 0.077 | 0.090 | 0.917 |
| DSD-BLYP | 0.083 | 0.114 | −0.175 | 0.048 | 0.886 |
| DSD-PBEP86 | 0.106 | 0.108 | −0.234 | 0.069 | 0.848 |
| RSDH-GGA | |||||
|
|||||
| ωB2PLYP | 0.433 | 0.500 | 0.358 | 0.043 | 0.979 |
| ωB2GPPLYP | 0.414 | 0.475 | 0.345 | 0.040 | 0.980 |
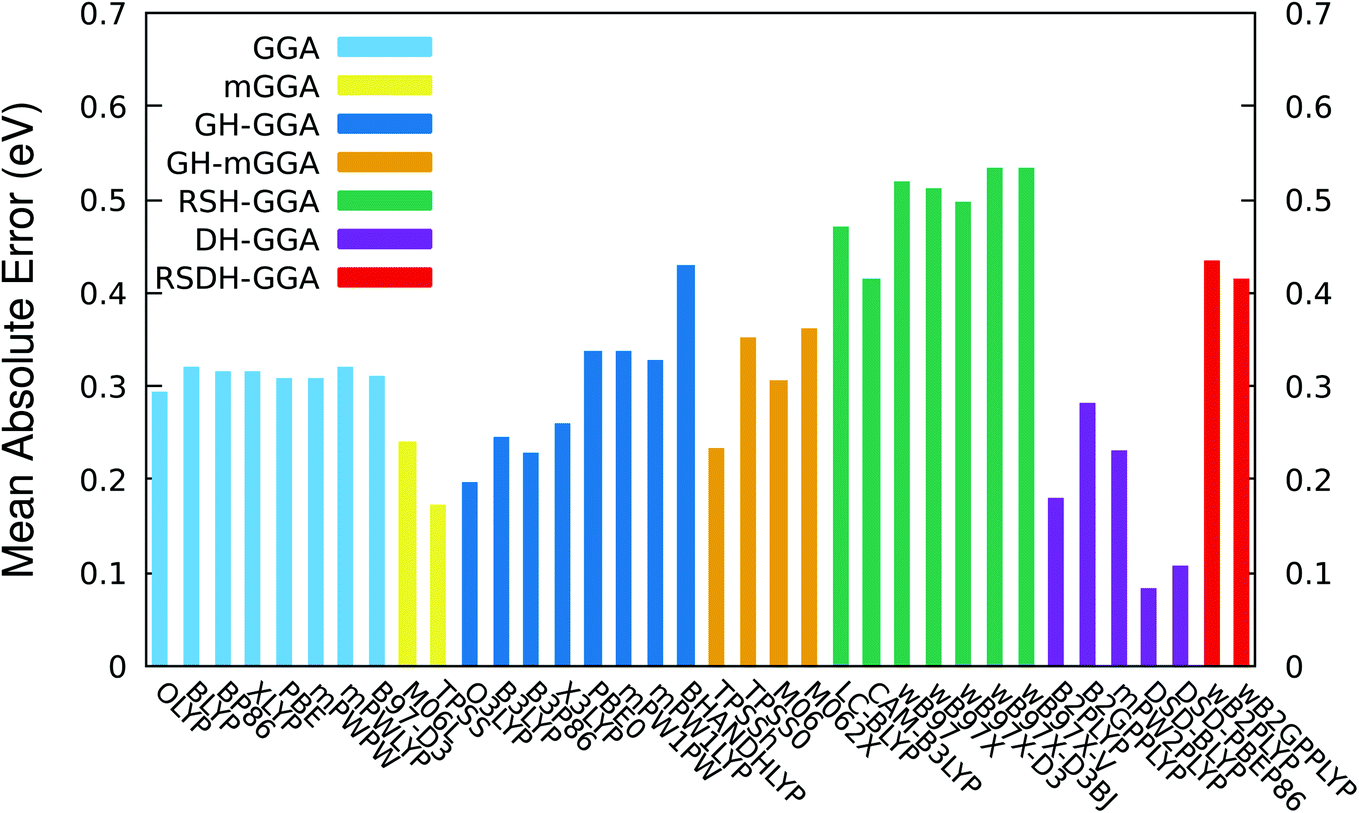 | ||
| Fig. 2 Histogram showing the mean absolute error (MAE) of TD-DFT excitation energies. | ||
The behavior of GGA, mGGA, GH-GGA, GH-mGGA and some DH functionals is unsatisfactory as well, with MAE values ranging in general between 0.2 and 0.4 eV. All pure GGA functionals show similar MAE values around 0.3 eV, regardless of the nature of the exchange or the correlation functional incorporated. Including the exact exchange HF component in the global hybrid functionals GH-GGA and GH-mGGA does not improve the quality of the results. In fact, an obvious deterioration of the values of the mean errors as a function of increasing the percentage of the HF exchange is observed; see Table 2 and Fig. 2 (in Table 2 and Fig. 2, the functionals of GH-GGA and GH-mGGA rungs are ordered in an ascendant manner according to their percentage of the HF exchange). For example, the MAE values of the O3LYP (ax = 11.6%) and the BH&HLYP (ax = 50%) are 0.196 and 0.429 eV, respectively. Indeed, the increase of calculated errors in the absorption energies obtained by the global hybrids as a function of the percentage of the HF exchange of BODIPY compounds is not surprising, as it was also revealed in our previous study for a set of smaller BODIPY chromophores.70
The two pure meta GGA functionals M06-L and TPSS show an improvement on their hybrid counterparts with MAE values of 0.240 and 0.172 eV, respectively. In fact, the two simple mGGA produce results comparable with that of some of the more demanding DH functionals: B2PLYP, B2GPPLYP, and mPW2PLYP. The original DH B2PLYP functional perform rather well, with an MAE value = 0.179 eV, in comparison with the B2GPPLYP and mPW2PLYP functionals with MAE values of 0.280 and 0.231 eV, respectively. Although the MAE values of the two DH functionals B2PLYP and B2GPPLYP (0.179 and 0.280 eV, respectively) are much lower than that of their range separated counterparts ωB2PLYP and ωB2GPPLYP (0.433 and 0.414 eV, respectively), it should be underlined that the percentages of HF exchange (ax scale parameter) and MP2 correlation (ac scale parameter) are the same for B2PLYP and ωB2PLYP (ax = 53% and ac = 27%) and for B2GPPLYP and ωB2GPPLYP (ax = 65% and ac = 36%). This may lead to the conclusion that the percentages of the HF exchange and MP2 correlation are irrelevant to the performance of the DH functionals toward electronic ES calculations.
The two dispersion-corrected, spin-component-scaled, double-hybrid (DSD) functionals, namely DSD-BLYP and DSD-PBEP86 produce the lowest MAE values among all functional investigated: 0.083 and 0.106 eV, respectively. It is therefore safe to conclude that these two empirical DH functionals developed by Martin and coworkers, reproduce “accurate” vertical excitation energies. This conclusion is especially true for the case of DSD-BLYP functional. The next lowest MAE values are almost double the small MAE value of the DSD-BLYP functional (0.179 eV for B2PLYP and 0.172 eV for TPSS). As a matter of fact, we have also obtained this remarkable performance of the two spin-component-scaled functionals in our recent study about another set of BODIPY model compounds which are smaller than the dye sensitizers studied in this work.70 It is interesting to note however, that the MAE values obtained using the two DSD functionals for the much larger and extended charge transfer dye sensitizers investigated in this work are slightly better than the smaller set of BODIPY chromophores studied previously.70 For instance, the MAE for DSD-BLYP functional is 0.083 eV for the larger dye sensitizers, and 0.119 eV for the significantly smaller size BODIPY set of chromophores.70 Not only the DSD functionals are the most accurate functionals among the large set of other functionals tested in this work, but those two empirical DH functionals are comparable with the excellent performance of the more expensive wavefunction based coupled-cluster methods in the gas phase, such as the LCC2* (ref. 55) (0.100 eV) and the DLPNO-STEOM-CCSD66 (0.114 eV). For comparison, we have calculated the lowest six excited states of dye B1 using the coupled-cluster DLPNO-STEOM-CCSD method. The vertical electronic excitation is found to be 2.239 eV compared with 2.220 for DSD-BLYP and 2.170 for DSD-PBEP86, while the experimental value is 2.321 eV (see Table S6†). It is worth to notice that the excellent mean absolute errors values produced by DSD-BLYP and DSD-PBEP86 is probably due to the incorporation of the spin-component-scaled (SCS) contributions to the MP2 energy in the correlation part of those functionals. The nature of the exchange and correlation functionals seems to be not relevant to the performance of these two double hybrids; see Table 1.
Moreover, the two DSDs show the best relative maximum error values (Max): 0.114 and 0.108 eV for DSD-BLYP and DSD-PBEP86, respectively; see Table 2 and Fig. S1† in the ESI. The next lowest Max values are that of pure GGA functionals (Max around 0.4 eV), which are almost four times higher than that of DSD-BLYP and DSD-PBEP86 functionals. All GH-GGA, GH-mGGA, RSH and RSDH functionals produce significantly higher maximum errors, reaching more than 0.6 eV for some of them. The relative minimum error (Min) of DSD-BLYP is −0.175 eV, and that for DSD-PBEP86 is −0.234 eV; see Table 2 and Fig. S2 in the ESI.† These two Min are, however, not the lowest among all the functionals. For instance, the following Min values are observed: −0.036 eV for the DH functional B2PLYP, and 0.077, −0.066 and 0.04 eV for the GH functionals mPW2PLYP, B3P86, and X3LYP, respectively. All Min values for all other functionals are greater than that of the DSDs, with the notable extreme deviation in producing too low excitation energies for some compounds in the case of pure GGAs where Min values are almost −0.8 eV, or the other flip of the coin in the case of the RSHs where the Min values are much higher in energy than the experimental data by an amount of at least +0.4 eV.
When it comes consistency and predictability, the two DSD functionals are again among the best functionals tested in this work. The standard deviation (SD) values of DSD-BLYP and DSD-PBEP86 are 0.048 and 0.069 eV, respectively (see Table 2 and Fig. S3 in the ESI†). In fact, the two RSDHs also show low SD results: 0.043 eV for ωB2PLYP and 0.040 eV for ωB2GPPLYP. Nevertheless, the linear determination coefficient (R2) values of DSD-BLYP (0.886) and DSD-PBEP86 (0.848) are far to be considered as satisfactory (see Table 2 and Fig. S4 in the ESI†). Actually the best R2 are found for the two RSDH functionals ωB2PLYP and for ωB2GPPLYP (0.979 and 0.980 eV, respectively), B2GPPLYP (0.972 eV), and all the RSH functionals (around 0.97 eV). Therefore, the RSH and RSDH functionals, together with the B2GPPLYP double hybrid functional, may all give accurate results if scaled properly.
It will be interesting to inspect the deviations from experiment of the two best performing DH functionals DSD-BLYP and DSD-PBEP86, with respect to the different BODIPY dye architectures and designs (see Table S6 in the ESI†). The mean of absolute errors of compounds B1–B7 that belong to the so called horizontal design, are 0.103 eV for DSD-BLYP and 0.154 eV for DSD-PBEP86, which is almost twice the mean of errors of compounds B8–B13 that belong to the vertical (B8–B11) and fused (B12 and B13) designs: 0.059 eV for DSD-BLYP and 0.051 eV for DSD-PBEP86. In all cases, we will not emphasize more on this conclusion since the difference in errors is insignificant, i.e. around 0.04 eV in the case of DSD-BLYP.
The analysis of the nature of the transitions and other excited state properties for all functionals and all dyes are presented in Tables S7–S42 in the ESI.† The results of all RSH, DH, and RSDH functionals, in addition to BH&HLYP and M06-2X functionals, show that the maxima of the absorption transition energies of all dyes correspond to the first dipole-allowed transition from the ground singlet state (S0) to the first singlet excited state (S1), i.e. an S0 → S1 transition. In addition, those transitions are found to be characterized by the electron excitations HOMO−1 → LUMO (mainly for B1–B7) or HOMO → LUMO (mainly for compounds B8–B13). In both cases, these two orbital excitations correspond to π → π* transitions; see Fig. S5 in the ESI† for the isosurface density plots of the frontier molecular orbitals involved in the main transitions of BODIPY dyes. On the other hand, the results of all other functionals (GGA, mGGA, most GH-GGA, and most GH-mGGA) erroneously predict that the transitions reach higher singlet states, e.g. S2, S3, etc, sometimes reaching S9 for B11 using pure GGAs and S7 for the same dye using mGGAs. The reason for this is that pure GGAs and mGGAs create unphysical low-lying states, sometime called ghost states. However, the promotion to those higher ESs are still characterized by HOMO−1 → LUMO or HOMO → LUMO excitations which also correspond to π → π* transitions. Finally, the set of BODIPY dye sensitizers investigated in our work were all tested experimentally to study their potential use in DSSCs. One of the key variables in determining the cell efficiency is the light harvesting efficiency (LHE) that is calculated using the oscillator strengths f obtained by TD-DFT excited state calculations: LHE = 1 − 10−f. The values of the oscillator strengths for all dyes using all functionals are reported in the ESI in Tables S7–S42.† It is noted that the functionals of the higher rungs (GH-GGA, GH-mGGA, RSH, DH, and RSDH) predict rather higher oscillator strengths values in general (f ∼1–1.5) for the dyes compared to the functionals of the lower rung such as GGA and mGGA (f ∼0.2–0.6). The calculated high values of oscillator strengths should in theory reproduce the high molar absorption coefficients that are typical to BODIPY dyes.
4. Conclusions
To summarize, we do not recommend the use of GH-GGA (B3LYP, PBE0, etc), GH-mGGA (TPSS0, M06-2X, etc), and RSH-GGA (CAM-B3LYP, ωB97X-D3, etc), by means of TD-DFT in order to study and predict the ES properties and especially the band maxima of dye sensitizers based on the BODIPY core. Although the aforementioned functionals are frequently used in ES calculations for BODIPY dyes by many researchers, with the B3LYP being the most abused, all these functionals suffer from at least one major problem: They all overestimate/underestimate excitation energies when compared to experimental results, with absolute average errors in the range 0.2 to 0.5 eV. Increasing the percentage of the exact HF exchange in the case of GH-GGA and GH-mGGA will not enhance the results, on the contrary it will worsen the situation. However, the RSH-GGA and RSDH-GGA functionals show high R2 values and therefore can be accurate if they are properly scaled. The mean absolute error values of pure GGA and mGGA functionals are lower than their hybrid or range separated hybrid counterparts (in the range 0.2–0.3 eV), but they actually suffer from creating unphysical low-lying ghost states, and thus will complicate and mislead the analysis and the transition assignments.The MAE of the dispersion-corrected, spin-component-scaled, double-hybrid (DSD) functionals DSD-BLYP and DSD-PBEP86 functionals are 0.083 eV and 0.106 eV, respectively, for the set of BODIPY based dye sensitizers investigated in this study. The error produced by these double hybrid spin-component-scaled functionals falls within the convention of chemical accuracy (≲0.1 eV) of a theoretical ES method. Moreover, the accuracy of TD ES calculations using the DSD functionals is remarkably comparable to the range of errors produced by the more expensive wavefunction coupled-cluster methods such as LCC2* and DLPNO-STEOM-CCSD. Indeed, the DSD-BLYP and DSD-PBEP86 functionals provide the lowest relative maximum error (0.114 and 0.108 eV, respectively) and among the lowest standard deviation values (0.048 and 0.069 eV, respectively) when compared to all other functionals benchmarked in this work.
We therefore recommend the use of the double hybrid functionals DSD-BLYP and DSD-PBEP86 for both the design of new BODIPY dyes, and for the analysis and band assignment purposes for such “tricky” dye sensitizers. As a matter of fact, TD-DFT with the DH functionals can be easily calculated routinely in most available recent codes. The calculations becomes reasonably fast even for large “real-life” molecules when using the resolution of identity (RI) approximation that is also incorporated in most recent codes.
Future work will concentrate on further extension of the results and conclusions of this study in two directions: first, benchmark more dye sensitizers from different families other than BODIPY dyes. Second, include more DH functionals with different fractions and types of spin-component-scaled contributions to test their performance in the prediction of the absorption spectra of different dye sensitizers.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support of this work by the Deanship of Scientific Research at the University of Jordan is gratefully acknowledged.References
- H. Anders, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Dye-sensitized solar cells, Chem. Rev., 2010, 110(11), 6595–6663 CrossRef PubMed , PMID: 20831177..
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Meso-substituted porphyrins for dye-sensitized solar cells, Chem. Rev., 2014, 114(24), 12330–12396 CrossRef CAS PubMed , PMID: 25495339..
- M. Ye, X. Wen, M. Wang, I. James, N. Zhang, C. Lin and Z. Lin, Recent advances in dye-sensitized solar cells: from photoanodes, sensitizers and electrolytes to counter electrodes, Mater. Today, 2015, 18(3), 155–162 CrossRef CAS.
- Y. Wu, W.-H. Zhu, S. M. Zakeeruddin and M. Grätzel, Insight into D–A–π–A structured sensitizers: A promising route to highly efficient and stable dye-sensitized solar cells, ACS Appl. Mater. Interfaces, 2015, 7(18), 9307–9318 CrossRef CAS PubMed , PMID: 25899976..
- J. Wang, K. Liu, L. Ma and X. Zhan, Triarylamine: Versatile platform for organic, dye-sensitized, and perovskite solar cells, Chem. Rev., 2016, 116(23), 14675–14725 CrossRef CAS PubMed , PMID: 27960267..
- N. Duvva, U. Chilakamarthi and L. Giribabu, Recent developments in tetrathiafulvalene and dithiafulvalene based metal-free organic sensitizers for dye-sensitized solar cells: a mini-review, Sustainable Energy Fuels, 2017, 1, 678–688 RSC.
- G. Richhariya, A. Kumar, P. Tekasakul and B. Gupta, Natural dyes for dye sensitized solar cell: A review, Renewable Sustainable Energy Rev., 2017, 69, 705–718 CrossRef CAS.
- G. D. Carlo, A. O. Biroli, F. Tessore, S. Caramori and M. Pizzotti, β-substituted ZnII porphyrins as dyes for DSSC: A possible approach to photovoltaic windows, Coord. Chem. Rev., 2018, 358, 153–177 CrossRef.
- A. Carella, F. Borbone and R. Centore, Research progress on photosensitizers for DSSC, Front. Chem., 2018, 6, 481 CrossRef CAS PubMed.
- K. Sharma, V. Sharma and S. S. Sharma, Dye-sensitized solar cells: Fundamentals and current status, Nanoscale Res. Lett., 2018, 13, 381 CrossRef PubMed.
- L. Zani, A. Dessi, D. Franchi, M. Calamante, G. Reginato and A. Mordini, Transition metal-catalyzed cross-coupling methodologies for the engineering of small molecules with applications in organic electronics and photovoltaics, Coord. Chem. Rev., 2019, 392, 177–236 CrossRef CAS.
- M. Urbani, M.-E. Ragoussi, M. K. Nazeeruddin and T. Torres, Phthalocyanines for dye-sensitized solar cells, Coord. Chem. Rev., 2019, 381, 1–64 CrossRef CAS.
- M. Urbani, G. d. l. Torre, M. K. Nazeeruddin and T. Torres, Phthalocyanines and porphyrinoid analogues as hole- and electron-transporting materials for perovskite solar cells, Chem. Soc. Rev., 2019, 48, 2738–2766 RSC.
- G. Boschloo, Improving the performance of dye-sensitized solar cells, Front. Chem., 2019, 7, 77 CrossRef CAS PubMed.
- Z. Yu, H. Anders and L. Sun, The application of transition metal complexes in hole-transporting layers for perovskite solar cells: Recent progress and future perspectives, Coord. Chem. Rev., 2020, 406, 213143 CrossRef CAS.
- A. Loudet and K. Burgess, BODIPY dyes and their derivatives: Syntheses and spectroscopic properties, Chem. Rev., 2007, 107(11), 4891–4932 CrossRef CAS PubMed.
- B. N. Alexander and A. J. Bard, Electrochemistry and electrogenerated chemiluminescence of BODIPY dyes, Acc. Chem. Res., 2012, 45(11), 1844–1853 CrossRef PubMed , PMID: 22515199..
- J. Bañuelos, BODIPY dye, the most versatile fluorophore ever?, Chem. Rec., 2016, 16(1), 335–348 CrossRef PubMed.
- A. Bessette and S. H. Garry, Design, synthesis and photophysical studies of dipyrromethene-based materials: insights into their applications in organic photovoltaic devices, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Structural modification strategies for the rational design of red/nir region BODIPYs, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- G. Yuan and D. F. O'Shea, Azadipyrromethenes: from traditional dye chemistry to leading edge applications, Chem. Soc. Rev., 2016, 45, 3846–3864 RSC.
- L. Vellanki, R. Sharma and M. Ravikanth, Functionalized boron-dipyrromethenes and their applications, Rep. Org. Chem., 2016, 6, 1–24 Search PubMed.
- W. Sheng, F. Lv, B. Tang, E. Hao and L. Jiao, Toward the most versatile fluorophore: Direct functionalization of BODIPY dyes via regioselective C–H bond activation, Chin. Chem. Lett., 2019, 30(10), 1825–1833 CrossRef CAS.
- R. Ziessel and A. Harriman, Artificial light-harvesting antennae: electronic energy transfer by way of molecular funnels, Chem. Commun., 2011, 47, 611–631 RSC.
- S. P. Singh and T. Gayathri, Evolution of BODIPY dyes as potential sensitizers for dye-sensitized solar cells, Eur. J. Org. Chem., 2014, 2014(22), 4689–4707 CrossRef CAS.
- M. Mao and Q.-H. Song, The structure-property relationships of D-π-A BODIPY dyes for dye-sensitized solar cells, Chem. Rec., 2016, 16(2), 719–733 CrossRef CAS PubMed.
- H. Klfout, S. Adam, M. Elkhalifa and H. He, BODIPYs for dye-sensitized solar cells, ACS Appl. Mater. Interfaces, 2017, 9(46), 39873–39889 CrossRef CAS PubMed , PMID: 29072443..
- H. Liu, L. Liu, Y. Fu, E. Liu and B. Xue, Theoretical design of D–π–A–A sensitizers with narrow band gap and broad spectral response based on boron dipyrromethene for dye-sensitized solar cells, J. Chem. Inf. Model., 2019, 59(5), 2248–2256 CrossRef CAS PubMed , PMID: 30908031..
- Y. Higashino, S. Erten-Ela and Y. Kubo, π-Expanded dibenzo-BODIPY with near-infrared light absorption: Investigation of photosensitizing properties of nio-based p-type dye-sensitized solar cells, Dyes Pigm., 2019, 170, 107613 CrossRef CAS.
- E. A. Yildiz, G. Sevinc, H. Gul Yaglioglu and M. Hayvali, Strategies towards enhancing the efficiency of BODIPY dyes in dye sensitized solar cells, J. Photochem. Photobiol., A, 2019, 375, 148–157 CrossRef CAS.
- E. A. Yildiz, G. Sevinc, H. Gul Yaglioglu and M. Hayvali, The effect of molecular structure and ultrafast electron injection dynamics on the efficiency of BODIPY sensitized solar cells, Opt. Mater., 2019, 91, 50–57 CrossRef CAS.
- A. Aguiar, J. Farinhas, W. da Silva, M. E. Ghica, C. M. A. Brett, J. Morgado and A. J. F. N. Sobral, Synthesis, characterization and application of meso-substituted fluorinated boron dipyrromethenes (BODIPYs) with different styryl groups in organic photovoltaic cells, Dyes Pigm., 2019, 168, 103–110 CrossRef CAS.
- G. Thumuganti, V. Gupta and S. P. Singh, New dithienosilole- and dithienogermole-based BODIPY for solar cell applications, New J. Chem., 2019, 43, 8735–8740 RSC.
- J. Yang, C. H. Devillers, P. Fleurat-Lessard, H. Jiang, S. Wang, C. P. Gros, G. Gupta, G. D. Sharma and H. Xu, Carbazole-based green and blue-BODIPY dyads and triads as donors for bulk heterojunction organic solar cells, Dalton Trans., 2020, 49, 5606–5617 RSC.
- A. Ortiz, Triarylamine-BODIPY derivatives: A promising building block as hole transporting materials for efficient perovskite solar cells, Dyes Pigm., 2019, 171, 107690 CrossRef CAS.
- M. Poddar and R. Misra, Recent advances of BODIPY based derivatives for optoelectronic applications, Coord. Chem. Rev., 2020, 421, 213462 CrossRef CAS.
- B. Maria Squeo and M. Pasini, BODIPY platform: a tunable tool for green to NIR OLEDs, Supramol. Chem., 2020, 32(1), 56–70 CrossRef.
- M. Liu, S. Ma, M. She, J. Chen, Z. Wang, P. Liu, S. Zhang and J. Li, Structural modification of BODIPY: Improve its applicability, Chin. Chem. Lett., 2019, 30(10), 1815–1824 CrossRef CAS.
- A. Turksoy, D. Yildiz and E. U. Akkaya, Photosensitization and controlled photosensitization with BODIPY dyes, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef CAS.
- J. Yang, H. Jiang, N. Desbois, G. Zhu, C. P. Gros, Y. Fang, F. Bolze, S. Wang and H.-J. Xu, Synthesis, spectroscopic characterization, one and two-photon absorption properties, and electrochemistry of truxene π-expanded BODIPYs dyes, Dyes Pigm., 2020, 176, 108183 CrossRef CAS.
- M. Pastore, S. Fantacci and F. De Angelis, Modeling excited states and alignment of energy levels in dye-sensitized solar cells: Successes, failures, and challenges, J. Phys. Chem. C, 2013, 117(8), 3685–3700 CrossRef CAS.
- T. Le Bahers, T. Pauporte, P. P. Laine, F. Labat, C. Adamo and I. Ciofini, Modeling dye-sensitized solar cells: From theory to experiment, J. Phys. Chem. Lett., 2013, 4(6), 1044–1050 CrossRef CAS PubMed , PMID: 26291375..
- M. Pastore, T. Etienne and F. De Angelis, Structural and electronic properties of dye-sensitized TiO2 for solar cell applications: from single molecules to self-assembled monolayers, J. Mater. Chem. C, 2016, 4, 4346–4373 RSC.
- M. Pastore, First principle modelling of materials and processes in dye-sensitized photoanodes for solar energy and solar fuels, Computation, 2017, 5(1), 1 RSC.
- F. De Angelis, S. Fantacci and A. Sgamellotti, An integrated computational tool for the study of the optical properties of nanoscale devices: application to solar cells and molecular wires, Theor. Chem. Acc., 2007, 117, 1093–1104 Search PubMed.
- A. V. Marenich, C. J. Cramer, D. G. Truhlar, C. A. Guido, B. Mennucci, G. Scalmani and M. J. Frisch, Practical computation of electronic excitation in solution: vertical excitation model, Chem. Sci., 2011, 2, 2143–2161 RSC.
- A. D. Laurent and D. Jacquemin, TD-DFT benchmarks: A review, Int. J. Quantum Chem., 2013, 113(17), 2019–2039 CrossRef CAS.
- F. Santoro and D. Jacquemin, Going beyond the vertical approximation with time-dependent density functional theory, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2016, 6(5), 460–486 CrossRef CAS.
- P.-F. Loos and D. Jacquemin, Evaluating 0–0 energies with theoretical tools: A short review, ChemPhotoChem, 2019, 3(9), 684–696 CrossRef CAS.
- P.-F. Loos, S. Anthony and D. Jacquemin, The quest for highly accurate excitation energies: A computational perspective, J. Phys. Chem. Lett., 2020, 11(6), 2374–2383 CrossRef CAS PubMed , PMID: 32125872..
- R. . Izsák, Single-reference coupled cluster methods for computing excitation energies in large molecules: The efficiency and accuracy of approximations, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2020, 10(3), e1445 Search PubMed.
- Y. Wu and W. Zhu, Organic sensitizers from D–π–A to D–A–π–A: effect of the internal electron-withdrawing units on molecular absorption, energy levels and photovoltaic performances, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC.
- G. Garcia, C. Adamo and I. Ciofini, Evaluating push–pull dye efficiency using TD-DFT and charge transfer indices, Phys. Chem. Chem. Phys., 2013, 15, 20210–20219 RSC.
- B. Le Guennic, O. Maury and D. Jacquemin, Aza-boron-dipyrromethene dyes: TD-DFT benchmarks, spectral analysis and design of original near-IR structures, Phys. Chem. Chem. Phys., 2012, 14, 157–164 RSC.
- M. R. Momeni and A. Brown, Why do TD-DFT excitation energies of BODIPY/aza-BODIPY families largely deviate from experiment? answers from electron correlated and multireference methods, J. Chem. Theory Comput., 2015, 11(6), 2619–2632 CrossRef CAS PubMed.
- S. Chibani, B. Le Guennic, A. Charaf-Eddin, O. Maury, C. Andraud and D. Jacquemin, On the computation of adiabatic energies in aza-boron-dipyrromethene dyes, J. Chem. Theory Comput., 2012, 8(9), 3303–3313 CrossRef CAS PubMed.
- S. Chibani, B. Le Guennic, A. Charaf-Eddin, A. D. Laurent and D. Jacquemin, Revisiting the optical signatures of BODIPY with ab initio tools, Chem. Sci., 2013, 4, 1950–1963 RSC.
- S. Chibani, A. Charaf-Eddin, B. Mennucci, B. Le Guennic and D. Jacquemin, Optical signatures of obo fluorophores: A theoretical analysis, J. Chem. Theory Comput., 2014, 10(2), 805–815 CrossRef CAS PubMed , PMID: 26580054..
- S. Chibani, A. D. Laurent, B. Le Guennic and D. Jacquemin, Improving the accuracy of excited-state simulations of BODIPY and aza-BODIPY dyes with a joint SOS-CIS(D) and TD-DFT approach, J. Chem. Theory Comput., 2014, 10(10), 4574–4582 CrossRef CAS PubMed , PMID: 26588151..
- A. Charaf-Eddin, B. Le Guennic and D. Jacquemin, Excited-states of BODIPY–cyanines: ultimate TD-DFT challenges?, RSC Adv., 2014, 4, 49449–49456 RSC.
- B. Le Guennic and D. Jacquemin, Taking up the cyanine challenge with quantum tools, Acc. Chem. Res., 2015, 48(3), 530–537 CrossRef CAS PubMed.
- B. Paul, S. Chibani, B. Le Guennic, I. Duchemin, X. Blase and D. Jacquemin, Combining the Bethe–Salpeter formalism with time-dependent DFT excited-state forces to describe optical signatures: NBO fluoroborates as working examples, J. Chem. Theory Comput., 2014, 10(10), 4548–4556 CrossRef PubMed , PMID: 26588148..
- O. Qi, P. Qian and Z. Shuai, Toward quantitative prediction of fluorescence quantum efficiency by combining direct vibrational conversion and surface crossing: BODIPYs as an example, J. Phys. Chem. Lett., 2020, 11(18), 7790–7797 CrossRef PubMed , PMID: 32787317..
- I. K. Petrushenko and K. B. Petrushenko, Effect of meso-substituents on the electronic transitions of BODIPY dyes: DFT and RI-CC2 study, Spectrochim. Acta, Part A, 2015, 138, 623–627 CrossRef CAS PubMed.
- F. de Jong, M. Feldt, J. Feldt and J. N. Harvey, Modelling absorption and emission of a meso-aniline-BODIPY based dye with molecular mechanics, Phys. Chem. Chem. Phys., 2018, 20, 14537–14544 RSC.
- M. Feldt and A. Brown, Assessment of local coupled cluster methods for excited states of BODIPY/aza-BODIPY families, J. Comput. Chem., 2021, 42(3), 144–155 CrossRef CAS PubMed.
- R. Berraud-Pache, N. Frank, G. Bistoni and R. Izsak, Computational design of near-infrared fluorescent organic dyes using an accurate new wave function approach, J. Phys. Chem. Lett., 2019, 10(17), 4822–4828 CrossRef CAS PubMed , PMID: 31386375..
- R. Berraud-Pache, N. Frank, G. Bistoni and R. Izsák, Unveiling the photophysical properties of boron-dipyrromethene dyes using a new accurate excited state coupled cluster method, J. Chem. Theory Comput., 2020, 16(1), 564–575 CrossRef PubMed , PMID: 31765141..
- A. Schlachter, A. Fleury, K. Tanner, S. Armand, B. Habermeyer, R. Guilard and P. D. Harvey, The TDDFT excitation energies of the BODIPYs; the DFT and TDDFT challenge continues, Molecules, 2021, 26(6) CrossRef CAS PubMed.
- W. Helal, Q. Alkhatib and M. Gharaibeh, Can time-dependent double hybrid density functionals accurately predict electronic excitation energies of BODIPY compounds?, Comput. Theor. Chem., 2022, 1207, 113531 CrossRef CAS.
- S. Grimme and F. Neese, Double-hybrid density functional theory for excited electronic states of molecules, J. Chem. Phys., 2007, 127(15), 154116 CrossRef PubMed.
- B. Moore and J. Autschbach, Longest-wavelength electronic excitations of linear cyanines: The role of electron delocalization and of approximations in time-dependent density functional theory, J. Chem. Theory Comput., 2013, 9(11), 4991–5003 CrossRef PubMed , PMID: 26583416..
- R. R. Valiev, A. N. Sinelnikov, Y. V. Aksenova, R. T. Kuznetsova, M. B. Berezin, A. S. Semeikin and V. N. Cherepanov, The computational and experimental investigations of photophysical and spectroscopic properties of BF2 dipyrromethene complexes, Spectrochim. Acta, Part A, 2014, 117, 323–329 CrossRef CAS PubMed.
- D. Jacquemin, E. A. Perpete, O. A. Vydrov, G. E. Scuseria and C. Adamo, Assessment of long-range corrected functionals performance for n → π* transitions in organic dyes, J. Chem. Phys., 2007, 127(9), 094102 CrossRef PubMed.
- D. Jacquemin, E. A. Perpete, G. E. Scuseria, I. Ciofini and C. Adamo, TD-DFT performance for the visible absorption spectra of organic dyes: Conventional versus long-range hybrids, J. Chem. Theory Comput., 2008, 4(1), 123–135 CrossRef CAS PubMed , PMID: 26619986..
- D. Jacquemin, E. A. Perpète, I. Ciofini and C. Adamo, Accurate simulation of optical properties in dyes, Acc. Chem. Res., 2009, 42(2), 326–334 CrossRef CAS PubMed , PMID: 19113946..
- M. Pastore, E. Mosconi, F. De Angelis and M. Gratzel, A computational investigation of organic dyes for dye-sensitized solar cells: Benchmark, strategies, and open issues, J. Phys. Chem. C, 2010, 114(15), 7205–7212 CrossRef CAS.
- M. Pastore, S. Fantacci and F. De Angelis, Ab initio determination of ground and excited state oxidation potentials of organic chromophores for dye-sensitized solar cells, J. Phys. Chem. C, 2010, 114(51), 22742–22750 CrossRef CAS.
- C. Bernini, L. Zani, M. Calamante, G. Reginato, A. Mordini, M. Taddei, R. Basosi and A. Sinicropi, Excited state geometries and vertical emission energies of solvated dyes for DSSC: A PCM/TD-DFT benchmark study, J. Chem. Theory Comput., 2014, 10(9), 3925–3933 CrossRef CAS PubMed , PMID: 26588536..
- H. Unal, D. Gunceler and E. Mete, A study of the density functional methods on the photoabsorption of BODIPY dyes, J. Photochem. Photobiol., A, 2014, 278, 14–18 CrossRef CAS.
- S. Grimme, Semiempirical hybrid density functional with perturbative second-order correlation, J. Chem. Phys., 2006, 124(3), 034108 CrossRef PubMed.
- L. Goerigk and S. Grimme, Double-hybrid density functionals, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2014, 4(6), 576–600 CrossRef CAS.
- J. M. L. Martin and G. Santra, Empirical double-hybrid density functional theory: A ‘third way’ in between WFT and DFT, Isr. J. Chem., 2020, 60(8–9), 787–804 CrossRef CAS.
- L. Goerigk, M. Jonas and S. Grimme, Computation of accurate excitation energies for large organic molecules with double-hybrid density functionals, Phys. Chem. Chem. Phys., 2009, 11, 4611–4620 RSC.
- L. Goerigk and S. Grimme, Double-hybrid density functionals provide a balanced description of excited 1La and 1Lb states in polycyclic aromatic hydrocarbons, J. Chem. Theory Comput., 2011, 7(10), 3272–3277 CrossRef CAS PubMed , PMID: 26598161..
- E. Bremond, M. Savarese, A. Jose Perez-Jimenez, J. C. Sancho-Garcia and C. Adamo, Speed-up of the excited-state benchmarking: Double-hybrid density functionals as test cases, J. Chem. Theory Comput., 2017, 13(11), 5539–5551 CrossRef CAS PubMed , PMID: 28976749..
- M. Casanova-Páez, M. B. Dardis and L. Goerigk, ωB2PLYP and ωb2gpplyp: The first two double-hybrid density functionals with long-range correction optimized for excitation energies, J. Chem. Theory Comput., 2019, 15(9), 4735–4744 CrossRef PubMed , PMID: 31298850..
- D. Mester and M. Kallay, Combined density functional and algebraic-diagrammatic construction approach for accurate excitation energies and transition moments, J. Chem. Theory Comput., 2019, 15(8), 4440–4453 CrossRef CAS PubMed , PMID: 31265275..
- A. Ottochian, C. Morgillo, I. Ciofini, M. J. Frisch, G. Scalmani and C. Adamo, Double hybrids and time-dependent density functional theory: An implementation and benchmark on charge transfer excited states, J. Comput. Chem., 2020, 41(13), 1242–1251 CrossRef CAS PubMed.
- D. Mester and M. Kallay, A simple range-separated double-hybrid density functional theory for excited states, J. Chem. Theory Comput., 2021, 17(2), 927–942 CrossRef CAS PubMed , PMID: 33400872..
- L. Goerigk and M. Casanova-Paez, The trip to the density functional theory zoo continues: Making a case for time-dependent double hybrids for excited-state problems, Aust. J. Chem., 2021, 74(1), 3–15 CrossRef CAS.
- S. Tobias and L. Goerigk, Time-dependent double-hybrid density functionals with spin-component and spin-opposite scaling, J. Chem. Theory Comput., 2017, 13(9), 4307–4323 CrossRef PubMed , PMID: 28763220..
- D. Mester and M. Kallay, Spin-scaled range-separated double-hybrid density functional theory for excited states, J. Chem. Theory Comput., 2021, 17(7), 4211–4224 CrossRef CAS PubMed , PMID: 34152771..
- M. Casanova-Paez and L. Goerigk, Time-dependent long-range-corrected double-hybrid density functionals with spin-component and spin-opposite scaling: A comprehensive analysis of singlet–singlet and singlet–triplet excitation energies, J. Chem. Theory Comput., 2021, 17(8), 5165–5186 CrossRef CAS PubMed , PMID: 34291643..
- A. Karton, A. Tarnopolsky, J.-F. Lamere, G. C. Schatz and J. M. L. Martin, Highly accurate first-principles benchmark data sets for the parametrization and validation of density functional and other approximate methods. Derivation of a robust, generally applicable, double-hybrid functional for thermochemistry and thermochemical kinetics, J. Phys. Chem. A, 2008, 112(50), 12868–12886 CrossRef CAS PubMed.
- S. Tobias and S. Grimme, Towards chemical accuracy for the thermodynamics of large molecules: new hybrid density functionals including non-local correlation effects, Phys. Chem. Chem. Phys., 2006, 8, 4398–4401 RSC.
- S. Kozuch, D. Gruzman and J. M. L. Martin, DSD-BLYP: A general purpose double hybrid density functional including spin component scaling and dispersion correction, J. Phys. Chem. C, 2010, 114(48), 20801–20808 CrossRef CAS.
- S. Kozuch and J. M. L. Martin, DSD-PBEP86: in search of the best double-hybrid DFT with spin-component scaled MP2 and dispersion corrections, Phys. Chem. Chem. Phys., 20104–20107, 13, 2011 Search PubMed.
- J.-B. Wang, X.-Q. Fang, X. Pan, S.-Y. Dai and Q.-H. Song, New 2, 6-modified BODIPY sensitizers for dye-sensitized solar cells, Chem.–Asian J., 2012, 7(4), 696–700 CrossRef CAS PubMed.
- M. Mao, Q.-S. Li, X.-L. Zhang, G.-H. Wu, C.-G. Dai, Y. Ding, S.-Y. Dai and Q.-H. Song, Effects of donors of BODIPY dyes on the performance of dye-sensitized solar cells, Dyes Pigm., 2017, 141, 148–160 CrossRef CAS.
- M. Mao, X.-L. Zhang, X.-Q. Fang, G.-H. Wu, Y. Ding, X.-L. Liu, S.-Y. Dai and Q.-H. Song, 2,6-conjugated BODIPY sensitizers for high-performance dye-sensitized solar cells, Org. Electron., 2014, 15(9), 2079–2090 CrossRef CAS.
- S.-C. Yeh, Li-J. Wang, H.-M. Yang, Yu-H. Dai, C.-W. Lin, C.-T. Chen and R.-J. Jeng, Structure-property relationship study of donor and acceptor 2,6-disubstituted BODIPY derivatives for high performance dye-sensitized solar cells, Chem.–Eur. J., 2017, 23(59), 14747–14759 CrossRef CAS PubMed.
- M. Mao, J.-B. Wang, Zu-F. Xiao, S.-Y. Dai and Q.-H. Song, New 2,6-modified BODIPY sensitizers for dye-sensitized solar cells, Dyes Pigm., 2012, 94(2), 224–232 CrossRef CAS.
- W.-J. Shi, T. Kinoshita and D. K. P. Ng, Ethynyl-linked donor-π-acceptor boron dipyrromethenes for panchromatic dye-sensitized solar cells, Asian J. Org. Chem., 2017, 6(6), 758–767 CrossRef CAS.
- S. Kolemen, O. Altan Bozdemir, Y. Cakmak, G. Barin, S. Erten-Ela, M. Marszalek, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and E. U. Akkaya, Optimization of distyryl-BODIPY chromophores for efficient panchromatic sensitization in dye sensitized solar cells, Chem. Sci., 2011, 2, 949–954 RSC.
- J. Zhang, F. Lu, S. Qi, Y. Zhao, K. Wang, B. Zhang and Y. Feng, Influence of various electron-donating triarylamine groups in BODIPY sensitizers on the performance of dye-sensitized solar cells, Dyes Pigm., 2016, 128, 296–303 CrossRef CAS.
- Y. Cakmak, S. Kolemen, M. Buyuktemiz, Y. Dede and S. Erten-Ela, Synthesis and dye sensitized solar cell applications of BODIPY derivatives with bis-dimethylfluorenyl amine donor groups, New J. Chem., 2015, 39, 4086–4092 RSC.
- Z. Lu, M. Liang, P. Dai, K. Miao, C. Zhang, Z. Sun and S. Xue, A strategy for enhancing the performance of borondipyrromethene dye-sensitized solar cells, J. Phys. Chem. C, 2016, 120(45), 25657–25667 CrossRef CAS.
- Y. Kubota, K. Kimura, J. Jin, K. Manseki, K. Funabiki and M. Matsui, Synthesis of near-infrared absorbing and fluorescing thiophene-fused BODIPY dyes with strong electron-donating groups and their application in dye-sensitised solar cells, New J. Chem., 2019, 43, 1156–1165 RSC.
- Y. Kubo, D. Eguchi, A. Matsumoto, R. Nishiyabu, H. Yakushiji, K. Shigaki and M. Kaneko, Boron–dibenzopyrromethene-based organic dyes for application in dye-sensitized solar cells, J. Mater. Chem. A, 2014, 2, 5204–5211 RSC.
- N. Frank, The orca program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2(1), 73–78 CrossRef.
- N. Frank, Software update: the orca program system, version 4.0, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8(1), e1327 Search PubMed.
- A.-R. Allouche, Gabedit—a graphical user interface for computational chemistry softwares, J. Comput. Chem., 2011, 32(1), 174–182 CrossRef CAS PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The pbe0 model, J. Chem. Phys., 1999, 110(13), 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- G. D. Purvis and R. J. Bartlett, A full coupled-cluster singles and doubles model: The inclusion of disconnected triples, J. Chem. Phys., 1982, 76(4), 1910–1918 CrossRef CAS.
- G. E. Scuseria, C. L. Janssen and H. F. Schaefer, An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations, J. Chem. Phys., 1988, 89(12), 7382–7387 CrossRef CAS.
- A. Sirohiwal, R. Berraud-Pache, N. Frank, R. Izsák and D. A. Pantazis, Accurate computation of the absorption spectrum of chlorophyll a with pair natural orbital coupled cluster methods, J. Phys. Chem. B, 2020, 124(40), 8761–8771 CrossRef CAS PubMed , PMID: 32930590..
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102(11), 1995–2001 CrossRef CAS.
- R. Izsák and N. Frank, An overlap fitted chain of spheres exchange method, J. Chem. Phys., 2011, 135(14), 144105 CrossRef PubMed.
- N. Frank, W. Frank, A. Hansen and U. Becker, Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange, Chem. Phys., 2009, 356(1), 98–109 Search PubMed.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, RI-MP2: optimized auxiliary basis sets and demonstration of efficiency, Chem. Phys. Lett., 1998, 294(1), 143–152 CrossRef CAS.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to RN, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms RB to RN, Theor. Chem. Acc., 2007, 117, 587–597 Search PubMed.
- T. Oliver and R. Ahlrichs, Efficient molecular numerical integration schemes, J. Chem. Phys., 1995, 102(1), 346–354 CrossRef.
- S. Xuan, N. Zhao, X. Ke, Z. Zhou, F. R. Fronczek, K. M. Kadish, K. M. Smith and M. G. H. Vicente, Synthesis and spectroscopic investigation of a series of push–pull boron dipyrromethenes (BODIPYs), J. Org. Chem., 2017, 82(5), 2545–2557 CrossRef CAS PubMed.
- L. R. Rutledge, S. M. McAfee and G. C. Welch, Design and computational characterization of non-fullerene acceptors for use in solution-processable solar cells, J. Phys. Chem. A, 2014, 118(36), 7939–7951 CrossRef CAS PubMed , PMID: 25111089..
- D. Jacquemin, I. Duchemin and X. Blase, 0–0 energies using hybrid schemes: Benchmarks of TD-DFT, CIS(D), ADC(2), CC2, and BSE/GW formalisms for 80 real-life compounds, J. Chem. Theory Comput., 2015, 11(11), 5340–5359 CrossRef CAS PubMed , PMID: 26574326..
- A. Ali, M. I. Rafiq, Z. Zhang, J. Cao, R. Geng, B. Zhou and W. Tang, TD-DFT benchmark for UV-visible spectra of fused-ring electron acceptors using global and range-separated hybrids, Phys. Chem. Chem. Phys., 2020, 22, 7864–7874 RSC.
- T. J. Lee and P. R. Taylor, A diagnostic for determining the quality of single-reference electron correlation methods, Int. J. Quantum Chem., 1989, 36(S23), 199–207 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- N. C. Handy and A. J. Cohen, Left-right correlation energy, Mol. Phys., 2001, 99(5), 403–412 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, Sep 1988, 38, 3098–3100 Search PubMed.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- X. Xu and W. A. Goddard, The X3LYP extended density functional for accurate descriptions of nonbond interactions, spin states, and thermochemical properties, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(9), 2673–2677 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., Oct 1996, 77, 3865–3868 Search PubMed.
- C. Adamo and V. Barone, Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The MPW and MPW1PW models, J. Chem. Phys., 1998, 108(2), 664–675 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125(19), 194101 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98(2), 1372–1377 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119(23), 12129–12137 CrossRef CAS.
- S. Grimme, Accurate calculation of the heats of formation for large main group compounds with spin-component scaled MP2 methods, J. Phys. Chem. A, 2005, 109(13), 3067–3077 CrossRef CAS PubMed , PMID: 16833631..
- Y. Zhao and D. G. Truhlar, The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120(1), 215–241 Search PubMed.
- Y. Tawada, T. Takao, S. Yanagisawa, T. Yanai and K. Hirao, A long-range-corrected time-dependent density functional theory, J. Chem. Phys., 2004, 120(18), 8425–8433 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393(1), 51–57 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128(8), 084106 CrossRef PubMed.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, Long-range corrected hybrid density functionals with improved dispersion corrections, J. Chem. Theory Comput., 2013, 9(1), 263–272 CrossRef CAS PubMed.
- A. Najibi and L. Goerigk, The nonlocal kernel in van der Waals density functionals as an additive correction: An extensive analysis with special emphasis on the B97M-V and ωB97M-V approaches, J. Chem. Theory Comput., 2018, 14(11), 5725–5738 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08795a |
| This journal is © The Royal Society of Chemistry 2022 |