
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiurease screening of alkyl chain-linked thiourea derivatives: in vitro biological activities, molecular docking, and dynamic simulations studies†
Sana Yaqooba,
Abdul Hameed *ab,
Mahmood Ahmed*c,
Muhammad Imrand,
Muhammad Abdul Qadire,
Mahwish Ramzanf,
Numan Yousaff,
Jamshed Iqbalg and
Muhammad Muddassar*f
*ab,
Mahmood Ahmed*c,
Muhammad Imrand,
Muhammad Abdul Qadire,
Mahwish Ramzanf,
Numan Yousaff,
Jamshed Iqbalg and
Muhammad Muddassar*f
aH. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan. E-mail: abdul_hameed8@hotmail.com
bDepartment of Chemistry, University of Sahiwal, Sahiwal, Pakistan
cDepartment of Chemistry, Division of Science and Technology, University of Education, College Road, Lahore, Pakistan. E-mail: mahmoodresearchscholar@gmail.com
dKAM-School of Life Sciences, FC College (A Chartered University), Lahore, Pakistan
eSchool of Chemistry, University of the Punjab, Lahore, Pakistan
fDepartment of Biosciences, COMSATS University Islamabad, Park Road, Islamabad, Pakistan. E-mail: mmuddassar@comsats.edu.pk
gCenter for Advanced Drug Research, COMSATS Institute of Information Technology, Abbottabad 22060, Pakistan
First published on 23rd February 2022
Abstract
Urease has become an important therapeutic target because it stimulates the pathogenesis of many human health conditions, such as pyelonephritis, the development of urolithiasis, hepatic encephalopathy, peptic ulcers, gastritis and gastric cancer. A series of alkyl chain-linked thiourea derivatives were synthesized to screen for urease inhibition activity. Structure elucidation of these compounds was done by spectral studies, such as IR, 1H NMR and 13C NMR, and MS analysis. In vitro urease enzyme inhibition assay revealed that compound 3c was the most potent thiourea derivative among the series with IC50 values of 10.65 ± 0.45 μM, while compound 3g also exhibited good activity with an IC50 value of 15.19 ± 0.58 μM compared to standard thiourea with an IC50 value of 15.51 ± 0.11 μM. The other compounds in the series possessed moderate to weak urease inhibition activity with IC50 values ranging from 20.16 ± 0.48 to 60.11 ± 0.78 μM. The most potent compounds 3c and 3g were docked to jack bean urease (PDB ID: 4H9M) to evaluate their binding affinities and to find the plausible binding poses. The docked complexes were refined through 100 ns-long MD simulations. The simulation results revealed that the average RMSD of 3c was less than that of the 3g compound. Furthermore, the radius of gyration plots for both complexes showed that 3c and 3g docking predicted binding modes did not induce any conformational change in the urease structure.
1. Introduction
Urease (E.C.3.5.1.5) is a metal containing enzyme that is similar to amidohydrolases and is common in nature, being present in diverse varieties of microbes, animals, and plants, and it cleaves urea into carbon dioxide and ammonia.1,2 Urease provides an aid for the survival of Helicobacter pylori (H. pylori) in the stomach at low pH, which can stimulate the pathogenesis of many human health conditions, such as pyelonephritis, the development of urolithiasis, hepatic encephalopathy, peptic ulcer, gastritis and gastric cancer.3–5 Rapid diagnostic testing of urease is a precursor to diagnose H. pylori in the stomach because it depends on urease activity for its sustainability in the stomach at low pH.6,7 Urease from plants and bacteria have a common ancestral gene due to which they have no difference in sequence and have the same active sites, so jack bean urease was used here as a prototype for its study and characterization.8,9 Urease from jack beans has one catalytic subunit and two structural subunits, so it exists in trimeric form and the active site contains 840 amino acids and Ni2+ is present in the active site for its catalytic mechanism.10 Homohexameric molecules comprising six α subunits are present in urease from jack beans, whereas urease from bacteria comprises three different subunits, namely α, β, and γ, and despite the structural dissimilarity, the active site is always located on α subunits consisting of a binuclear nickel center.11,12Due to the association of urease with different bacterial infections, various types of urease inhibitors have been synthesized, such as Schiff bases-sulfonamides, phosphate derivatives, thiourea derivatives, hydroxamic acid, chelators of nickel atoms at the active site, thiolate compounds, analogs of barbituric acid (thiobarbiturates, barbiturates), and thiosemicarbazones.13–16 The structures of some good urease inhibitors previously reported are presented in Fig. 1. Thiourea has shown diverse pharmacological applications to serve as antioxidant, anti-inflammatory, anti-bacterial, antihypertensive, and anticancer agents.17 Keeping in view the importance of urea and the thiourea moiety, we synthesized a thiourea moiety, sandwiched between a short lipophilic chain and phenyl residue-based molecules to screen them as urease inhibitors to find leads as potential drug candidates. Moreover, the close resemblance of thiourea moiety-based molecules with the reference thiourea would be an additional benefit in finding a new urease inhibitor. Enzyme kinetics, molecular docking, and molecular dynamic (MD) simulation studies were also carried out, respectively, to get an insight into the inhibition mechanism and binding conformation of competitive inhibitors in the jack bean urease enzyme.
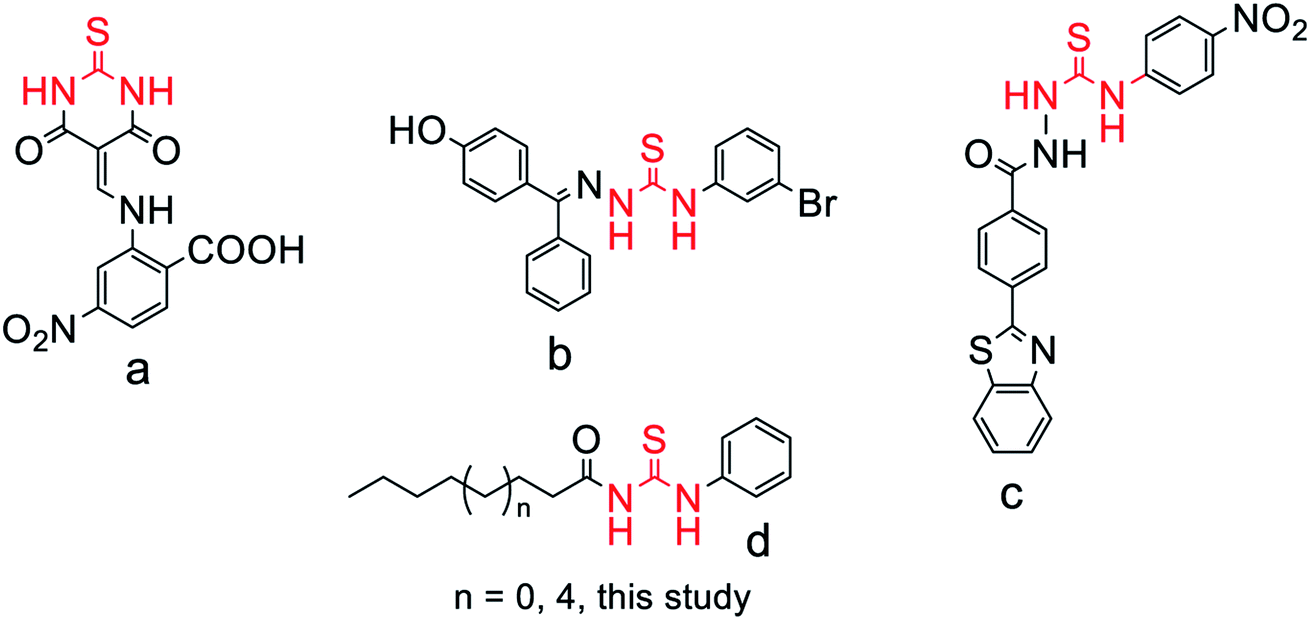 | ||
| Fig. 1 Previously identified scaffolds (a–c)14,18,19 and the structural framework (d) of target study as urease inhibitor. | ||
2. Experimental
All the reagents and solvents for the different reactions and purification were purchased from commercial suppliers (Falcon Scientific, Lahore, Hajvery Chemicals, Karachi and Central Scientific Store, Lahore, India) and were of analytical grade and used without further purification. High-purity water was prepared in our laboratory using the Milli-Q® water system, UK. The progress of the reactions was observed by thin layer chromatography (TLC), where, after spotting the pre-coated silica plates (Merck, Germany), the spots were visualized under ultraviolet (UV) light; while the Stuart® apparatus (Cole-Parmer, UK) was used to determine the melting points (mp) of all the products. IR (range, 4000–500 cm−1), 1HNMR (300–500 MHz), and 13C-NMR (125 MHz) spectra were recorded by an FTIR spectrophotometer and NMR spectrometer, respectively, from Bruker Technologies (USA) to elucidate the structures of the newly synthesized compounds, while the m/z ratio was determined by mass spectrometry (Finnigan MAT-321A, Germany). Chemical shifts (δ, in ppm) and the coupling constant (J, in Hz) were reported using DMSO-d6 as the solvent, whereas SiMe4 was used as the internal standard.2.1. General procedure for the synthesis of the thiourea-based molecules
Lauroyl chloride/caproyl chloride (5.0 mmol, 700 μL) were taken in an oven-dried round-bottom flask (25 mL), and then potassium thiocyanate solution (7.5 mmol, 729 mg in acetone) was added dropwise while stirring, and then the reaction mixture was refluxed at 50–60 °C for 1 h. The reaction mixture was placed at room temperature until it cooled down, and then substituted aniline (5.0 mmol, 840 mg in acetone) was added, and the mixture was refluxed again at 50–60 °C until the completion of the reaction, with the progress of the reactions observed by TLC (n-hexane, ethyl acetate as the solvent). Here, after spotting the pre-coated silica plates, the spots were visualized under ultraviolet (UV) light. After the completion of the reaction, the reaction mixture was ice cooled, and precipitates were formed, which were filtered and re-crystalized by employing absolute ethanol.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1614, 1535, 1326, 1165, 1068, 840, 720. EI-MS m/z (%), 402.1 (36.4), 219 (12.5), 198.1 (7.8), 184.1 (6.8); 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.69 (1H, s, NH), 11.54 (1H, s, NH), 7.91 (2H, d, J3′,2′/5′,6′ = 8.4 Hz, H-3′, H-5′), 7.76 (2H, d, J2′,3′/6′,5′ = 8.7 Hz, H-2′, H-6′), 2.45 (obscured by DMSO signal, H-2), 1.57 (2H, app q, J3 = 6.9 Hz, H-3), 1.23 (8H, m), 0.86 (3H, t, J12 = 6 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.07 (CS), 175.56 (CO), 141.5 (Ar–C), 134.8 (Ar–C), 125.74 (Ar–CH × 2), 125.69 (Ar–CH × 2), 124.4 (CF3), 35.72 (CH2), 31.2 (CH2), 28.9 (CH2 × 2), 28.7 (CH2), 28.6 (CH2 × 2), 28.4 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C20H29O1N2F3S1 (M): m/z 402.1953 found 402.1953.O), 1547, 1466, 1443, 1170, 1072, 906, 748, 611, 536. FAB (+ve) 357.1 (M + 1) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.41 (1H, s, NH), 11.58 (1H, s, NH), 7.88 (1H, app dd, J3′,4′ = 8.0 Hz, J3′,5′ = 1.2 Hz, H-3′), 7.71 (1H, app dd, J6′,5′ = 8.0 Hz, J6′,4′ = 1.2 Hz, H-6′), 7.43 (1H, td, J5′,(6′,4′) = 8.0 Hz, J5′,3′ = 1.2 Hz, H-5′), 7.24 (1H, td, J4′,(5′,3′) = 8.0 Hz, J4′,6′ = 1.2 Hz, H-4′), 2.49 (obscured by DMSO signal, H-2), 1.57 (2H, q, J3 = 12.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 6.0 Hz, H-8); 13C-NMR (100 MHz, DMSO-d6): δC 180.0 (CS), 175.6 (CO), 136.6 (Ar–C), 132.6 (Ar–CH), 128.6 (Ar–CH), 128.5 (Ar–CH), 127.7 (Ar–CH), 119.0 (Ar–C), 35.7 (CH2), 31.0 (CH2), 28.4 (CH2), 28.3 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H21O1N2Br1S (M): m/z 356.0558 found 356.056.O), 146 (100), 121 (26), 57 (50). EI-MS m/z (%), 306 (24.1), 291 (12.8), 306 (75), 179 (27.5), 165 (13.6), 164 (8.1), 127 (12.8). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.12 (1H, s, NH), 11.40 (1H, s, NH), 7.36 (1H, s, H-6′), 7.15 (1H, d, J3′,4′ = 8.0 Hz, H-3′), 7.01 (1H, d, J4′,3′ = 8.0 Hz, H-4′), 2.49 (obscured by DMSO signal, H-2), 2.25 (3H, s, CH3), 2.13 (3H, s, CH3), 1.55 (2H, q, J3 = 8.0 Hz, H-3), 1.26 (8H, m), 0.86 (3H, t, J8 = 8.0 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.6 (CS), 175.5 (CO), 136.4 (Ar–C), 135.2 (Ar–C), 130.2 (Ar–CH), 130.0 (Ar–C),127.6 (Ar–CH), 126.7 (Ar–CH), 35.68 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 20.5 (CH3), 17.1 (CH3), 13.9 (CH3); HREI-MS (M+ − I): calculated for C17H26O1N2S1 (M): m/z 306.1766 found 306.1777.O), 1606, 1511, 1463, 1343, 1251, 1158, 1027, 833, 742, 613, 437. EI-MS m/z (%), 308 (32.9), 181 (7.8), 167 (4.7), 128 (1.1), 122 (14.6), 108 (19.9). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.34 (1H, s, NH), 11.34 (1H, s, NH), 7.49 (2H, d, J2′,3′/6′,5′ = 8.0 Hz, H-6′, H-2′), 6.94 (2H, d, J3′,2′/5′,6′ = 8.8 Hz, H-3′, H-5′), 2.45 (2H, t, J2 = 14.0 Hz, H-2), 1.56 (2H, q, J3 = 12.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 6.0 Hz, H-8); 13C-NMR (100 MHz, DMSO-d6): δC 179.4 (CS), 175.9 (CO), 157.8 (Ar–C), 131.1 (Ar–C), 126.3 (Ar–CH × 2), 114.2 (Ar–CH × 2), 55.7 (–OCH3), 36.2 (CH2), 31.5 (CH2), 28.9 (CH2), 28.8 (CH2), 24.8 (CH2), 22.5 (CH2), 20.7 (CH2), 14.4 (CH3); HREI-MS (M+ − I): calculated for C16H24O2N2S (M): m/z 308.1588 found 308.155.O), 1546, 1185, 1373, 1039, 808, 701, 556, 531 m/z (%), 306 (75), 291 (78), 207 (10.3), 179 (27.5), 165 (35.5), 164 (19.4) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.12 (1H, s, NH), 11.40 (1H, s, NH), 7.36 (1H, s, H-2′), 7.15 (1H, d, J6′,5′ = 8.0 Hz, H-6′), 7.01 (1H, d, J5′,6′ = 8.0 Hz, H-5′), 2.46 (2H, t, J2 = 12.0 Hz, H-2), 2.25 (3H, s, CH3), 2.13 (3H, s, CH3), 1.57 (2H, q, J3 = 12.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 12.0 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.7 (CS), 175.5 (CO), 136.5 (Ar–C), 135.2 (Ar–C), 130.2 (Ar–CH), 130.0 (Ar–C), 127.6 (Ar–CH), 126.8 (Ar–CH), 35.7 (CH2), 31.0 (CH2), 28.4 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 20.5 (CH2), 17.1 (CH3 × 2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C17H26ON2S (M): m/z 306.1766 found 306.1769.O), 1541, 1219, 1166, 1093, 765, 697, 607, 512, 458. m/z (%), 306 (54), 291 (100), 179 (10.2), 165 (60.7), 120 (19.4), 105 (11.1) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 11.72 (1H, s, NH), 11.39 (1H, s, NH), 7.14–7.07 (3H, m, Ar–H), 2.49 (obscured by DMSO signal, H-2), 2.13 (6H, s, C), 1.58 (2H, q, J3 4.0 Hz, H-3), 1.28 (8H, m), 0.87 (3H, t, J8 = 6.0 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 180.1 (CS), 175.2 (CO), 136.0 (Ar–C), 134.9 (Ar–C × 2), 127.8 (Ar–CH), 127.3 (Ar–CH), 35.6 (CH2), 31.0 (CH2), 28.3 (CH2), 28.3 (CH2), 24.3 (CH2), 21.9 (CH2), 17.7 (CH3), 13.9 (CH3); HREI-MS (M+ − I): calculated for C17H26ON2S (M): m/z 306.1766 found 306.1770.O), 1558, 1488, 1336, 1265, 1152, 1017, 839, 727, 634, 561, 456. m/z (%), 353 (52), 226 (8.3), 212 (5.1), 196 (100), 181 (4.4), 127 (41.3) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 13.09 (1H, s, NH), 11.59 (1H, s, NH), 9.74 (1H, d, J6′,4 = 4.0 Hz, H-6′), 8.15 (1H, dd, J4′,3′ = 4.0 Hz, J4′,6′ = 8.0 Hz, H-4′), 7.34 (1H, d, J3′ = 8.0 Hz, H-3′), 4.01 (3H, s, OCH3), 2.49 (obscured by DMSO signal, H-2), 1.57 (2H, q, J3 = 12.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 8.0, Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 178.1 (CS), 175.6 (CO), 155.3 (Ar–C), 139.7 (Ar–C), 127.3 (Ar–C), 122.2 (Ar–CH), 117.0 (Ar–CH), 111.3 (Ar–CH), 57.2 (–OCH3), 35.6 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C16H23O4N3S (M): m/z 353.1409 found 353.1413.O), 1515, 1173, 784, 655, 615. FABP m/z (%), (M + I) 347.0 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 11.86 (1H, s, NH), 11.62 (1H, s, NH), 7.54 (2H, d, J3′,5′ = 8.0 Hz, H-3′, H-5′), 7.38 (1H, t, J4′ = 8.2 Hz, H-4′), 2.49 (obscured by DMSO signal, H-2), 1.58 (2H, q, H-3), 1.58 (2H, q, J3 = 8.0 Hz, H-3), 1.27 (8H, m), 0.87 (3H, t, J8 = 6.8, Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 180.8 (CS), 175.2 (CO), 134.1 (Ar–C × 2), 133.7 (Ar–C), 129.7 (Ar–CH × 2), 128.4 (Ar–CH), 35.6 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H21ON2Cl2S (M) m/z 347.0752 found 347.0746.O), 1525, 1472, 1234, 1164, 751, 782
O), 1614, 1535, 1326, 1165, 1068, 840, 720. EI-MS m/z (%), 402.1 (36.4), 219 (12.5), 198.1 (7.8), 184.1 (6.8); 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.69 (1H, s, NH), 11.54 (1H, s, NH), 7.91 (2H, d, J3′,2′/5′,6′ = 8.4 Hz, H-3′, H-5′), 7.76 (2H, d, J2′,3′/6′,5′ = 8.7 Hz, H-2′, H-6′), 2.45 (obscured by DMSO signal, H-2), 1.57 (2H, app q, J3 = 6.9 Hz, H-3), 1.23 (8H, m), 0.86 (3H, t, J12 = 6 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.07 (CS), 175.56 (CO), 141.5 (Ar–C), 134.8 (Ar–C), 125.74 (Ar–CH × 2), 125.69 (Ar–CH × 2), 124.4 (CF3), 35.72 (CH2), 31.2 (CH2), 28.9 (CH2 × 2), 28.7 (CH2), 28.6 (CH2 × 2), 28.4 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C20H29O1N2F3S1 (M): m/z 402.1953 found 402.1953.O), 1547, 1466, 1443, 1170, 1072, 906, 748, 611, 536. FAB (+ve) 357.1 (M + 1) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.41 (1H, s, NH), 11.58 (1H, s, NH), 7.88 (1H, app dd, J3′,4′ = 8.0 Hz, J3′,5′ = 1.2 Hz, H-3′), 7.71 (1H, app dd, J6′,5′ = 8.0 Hz, J6′,4′ = 1.2 Hz, H-6′), 7.43 (1H, td, J5′,(6′,4′) = 8.0 Hz, J5′,3′ = 1.2 Hz, H-5′), 7.24 (1H, td, J4′,(5′,3′) = 8.0 Hz, J4′,6′ = 1.2 Hz, H-4′), 2.49 (obscured by DMSO signal, H-2), 1.57 (2H, q, J3 = 12.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 6.0 Hz, H-8); 13C-NMR (100 MHz, DMSO-d6): δC 180.0 (CS), 175.6 (CO), 136.6 (Ar–C), 132.6 (Ar–CH), 128.6 (Ar–CH), 128.5 (Ar–CH), 127.7 (Ar–CH), 119.0 (Ar–C), 35.7 (CH2), 31.0 (CH2), 28.4 (CH2), 28.3 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H21O1N2Br1S (M): m/z 356.0558 found 356.056.O), 146 (100), 121 (26), 57 (50). EI-MS m/z (%), 306 (24.1), 291 (12.8), 306 (75), 179 (27.5), 165 (13.6), 164 (8.1), 127 (12.8). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.12 (1H, s, NH), 11.40 (1H, s, NH), 7.36 (1H, s, H-6′), 7.15 (1H, d, J3′,4′ = 8.0 Hz, H-3′), 7.01 (1H, d, J4′,3′ = 8.0 Hz, H-4′), 2.49 (obscured by DMSO signal, H-2), 2.25 (3H, s, CH3), 2.13 (3H, s, CH3), 1.55 (2H, q, J3 = 8.0 Hz, H-3), 1.26 (8H, m), 0.86 (3H, t, J8 = 8.0 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.6 (CS), 175.5 (CO), 136.4 (Ar–C), 135.2 (Ar–C), 130.2 (Ar–CH), 130.0 (Ar–C),127.6 (Ar–CH), 126.7 (Ar–CH), 35.68 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 20.5 (CH3), 17.1 (CH3), 13.9 (CH3); HREI-MS (M+ − I): calculated for C17H26O1N2S1 (M): m/z 306.1766 found 306.1777.O), 1606, 1511, 1463, 1343, 1251, 1158, 1027, 833, 742, 613, 437. EI-MS m/z (%), 308 (32.9), 181 (7.8), 167 (4.7), 128 (1.1), 122 (14.6), 108 (19.9). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.34 (1H, s, NH), 11.34 (1H, s, NH), 7.49 (2H, d, J2′,3′/6′,5′ = 8.0 Hz, H-6′, H-2′), 6.94 (2H, d, J3′,2′/5′,6′ = 8.8 Hz, H-3′, H-5′), 2.45 (2H, t, J2 = 14.0 Hz, H-2), 1.56 (2H, q, J3 = 12.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 6.0 Hz, H-8); 13C-NMR (100 MHz, DMSO-d6): δC 179.4 (CS), 175.9 (CO), 157.8 (Ar–C), 131.1 (Ar–C), 126.3 (Ar–CH × 2), 114.2 (Ar–CH × 2), 55.7 (–OCH3), 36.2 (CH2), 31.5 (CH2), 28.9 (CH2), 28.8 (CH2), 24.8 (CH2), 22.5 (CH2), 20.7 (CH2), 14.4 (CH3); HREI-MS (M+ − I): calculated for C16H24O2N2S (M): m/z 308.1588 found 308.155.O), 1546, 1185, 1373, 1039, 808, 701, 556, 531 m/z (%), 306 (75), 291 (78), 207 (10.3), 179 (27.5), 165 (35.5), 164 (19.4) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.12 (1H, s, NH), 11.40 (1H, s, NH), 7.36 (1H, s, H-2′), 7.15 (1H, d, J6′,5′ = 8.0 Hz, H-6′), 7.01 (1H, d, J5′,6′ = 8.0 Hz, H-5′), 2.46 (2H, t, J2 = 12.0 Hz, H-2), 2.25 (3H, s, CH3), 2.13 (3H, s, CH3), 1.57 (2H, q, J3 = 12.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 12.0 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.7 (CS), 175.5 (CO), 136.5 (Ar–C), 135.2 (Ar–C), 130.2 (Ar–CH), 130.0 (Ar–C), 127.6 (Ar–CH), 126.8 (Ar–CH), 35.7 (CH2), 31.0 (CH2), 28.4 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 20.5 (CH2), 17.1 (CH3 × 2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C17H26ON2S (M): m/z 306.1766 found 306.1769.O), 1541, 1219, 1166, 1093, 765, 697, 607, 512, 458. m/z (%), 306 (54), 291 (100), 179 (10.2), 165 (60.7), 120 (19.4), 105 (11.1) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 11.72 (1H, s, NH), 11.39 (1H, s, NH), 7.14–7.07 (3H, m, Ar–H), 2.49 (obscured by DMSO signal, H-2), 2.13 (6H, s, C), 1.58 (2H, q, J3 4.0 Hz, H-3), 1.28 (8H, m), 0.87 (3H, t, J8 = 6.0 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 180.1 (CS), 175.2 (CO), 136.0 (Ar–C), 134.9 (Ar–C × 2), 127.8 (Ar–CH), 127.3 (Ar–CH), 35.6 (CH2), 31.0 (CH2), 28.3 (CH2), 28.3 (CH2), 24.3 (CH2), 21.9 (CH2), 17.7 (CH3), 13.9 (CH3); HREI-MS (M+ − I): calculated for C17H26ON2S (M): m/z 306.1766 found 306.1770.O), 1558, 1488, 1336, 1265, 1152, 1017, 839, 727, 634, 561, 456. m/z (%), 353 (52), 226 (8.3), 212 (5.1), 196 (100), 181 (4.4), 127 (41.3) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 13.09 (1H, s, NH), 11.59 (1H, s, NH), 9.74 (1H, d, J6′,4 = 4.0 Hz, H-6′), 8.15 (1H, dd, J4′,3′ = 4.0 Hz, J4′,6′ = 8.0 Hz, H-4′), 7.34 (1H, d, J3′ = 8.0 Hz, H-3′), 4.01 (3H, s, OCH3), 2.49 (obscured by DMSO signal, H-2), 1.57 (2H, q, J3 = 12.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 8.0, Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 178.1 (CS), 175.6 (CO), 155.3 (Ar–C), 139.7 (Ar–C), 127.3 (Ar–C), 122.2 (Ar–CH), 117.0 (Ar–CH), 111.3 (Ar–CH), 57.2 (–OCH3), 35.6 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C16H23O4N3S (M): m/z 353.1409 found 353.1413.O), 1515, 1173, 784, 655, 615. FABP m/z (%), (M + I) 347.0 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 11.86 (1H, s, NH), 11.62 (1H, s, NH), 7.54 (2H, d, J3′,5′ = 8.0 Hz, H-3′, H-5′), 7.38 (1H, t, J4′ = 8.2 Hz, H-4′), 2.49 (obscured by DMSO signal, H-2), 1.58 (2H, q, H-3), 1.58 (2H, q, J3 = 8.0 Hz, H-3), 1.27 (8H, m), 0.87 (3H, t, J8 = 6.8, Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 180.8 (CS), 175.2 (CO), 134.1 (Ar–C × 2), 133.7 (Ar–C), 129.7 (Ar–CH × 2), 128.4 (Ar–CH), 35.6 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H21ON2Cl2S (M) m/z 347.0752 found 347.0746.O), 1525, 1472, 1234, 1164, 751, 782![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 545. m/z (%), 387.2 (M+ − I, 4.3), 277 (84.9), 200.0 (11.6), 186 (5.5), 109 (2.6) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.50 (1H, s, NH), 11.39 (1H, s, NH), 7.95 (1H, d, Ar–H), 7.42–7.19 (8H, m, Ar–H) 2.40 (2H, t, J2 = 7.6Hz, H-2), 1.52 (2H, q, J3 = 6.6 Hz, H-3), 1.24 (8H, m), 0.86 (3H, t, J8 = 6.8 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.8 (CS), 175.3 (CO), 138.5 (Ar–C), 134.3 (Ar–C × 2), 133.2 (Ar–CH), 129.7 (Ar–CH × 2), 129.4 (Ar–CH × 2), 128.3 (Ar–CH), 127.5 (Ar–CH), 127.4 (Ar–CH), 127.1 (Ar–CH), 35.6 (CH2), 31.0 (CH2), 28.9 (CH2 × 2), 24.3 (CH2), 22.0 (CH2), 13.89 (CH3); HREI-MS (M+ − I): calculated for C21H26O1N2S2 (M): m/z 386.1487 found 386.1478.O), 1546, 1467, 1374, 1262, 1176, 1094, 1072, 1020, 786, 724, 704, 608, 541. EI-MS m/z (%), 306 (47.5), 105 (14.9), 121 (100.0), 128 (2.1), 165 (65.5), 179 (15.2), 207 (13.2), 291 (66.6) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.07 (1H, s, NH), 11.404 (1H, s, NH), 7.25 (1H, m, Ar–H), 7.10 (2H, m, Ar–H), 2.49 (obscured by DMSO signal, H-2), 2.57 (3H, s, CH3), 2.06 (3H, s, CH3), 1.57 (2H, q, J3 = 4.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 8.0 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 180.1 (CS), 175.5 (CO), 137.1 (Ar–C), 136.5 (Ar–C), 132.2 (Ar–C), 128.4, 125.3, 124.7 (Ar–CH), 35.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 19.9 (C H3), 13.9 (CH3); 13.8 (CH3); HREI-MS (M+ − I): calculated for C17H26ON2S (M): m/z 306.1766 found 306.1755.O), 1535, 1515, 1149, 1021, 7537, 717. FABP m/z (%) (M + I) 405.1 1H-NMR (400 MHz, DMSO-d6): δH (ppm): 12.2 (1H, s, NH), 11.5 (1H, s, NH), 7.90 (1H, app dd, J6′,5′ = 8.0 Hz, J6′,4′ = 0.8 Hz, H-6′), 7.63 (1H, d, J3′,4′ = 8.0 Hz, H-3′), 7.44 (1H, app td, J5′(6′,4′) 8.0 Hz, J5′,3′ = 0.4 Hz, H-5′), 7.05 (1H, app td, J4′(3′,5′) = 7.6 Hz, J4′,6′) = 0.8 Hz, H-4′), 2.49 (obscured by DMSO signal, H-2), 1.56 (2H, q, J3 = 7.2 Hz, H-3), 1.28 (8H, m), 0.87 (3H, t, J = 6.6 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 180.2 (CS), 175.5 (CO), 140.0 (Ar–C), 138.8 (Ar–CH), 128.8 (Ar–CH), 128.6 (Ar–CH), 128.4 (Ar–CH), 97.1 (Ar–C), 35.7 (CH2), 31.0 (CH2), 28.3 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H21OI1N2S1 (M): m/z 404.0419 found 404.0410.O), 1542, 1325, 1170, 1130, 1067, 1020, 916, 722, 670, 617, 540. EI-MS m/z (%), 346 (81.9), 277 (2.0), 219 (30.8), 205 (3.7), 186 (5.8), 161 (100), 145 (19.0), 142 (6.2), 128 (2.0). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.683 (1H, s, NH), 11.54 (1H, s, NH), 7.90 (2H, d, J3′,2′/5′,6′ = 8.8 Hz, H-3′, H-5′), 8.03 (2H, d, J2′,3′/6′,5′ = 8.8 Hz, H-2′, H-6′), 2.09 (2H, t, J2 = 15.2 Hz, H-2), 1.57 (2H, app q, J3 = 14.4 Hz, H-3), 1.28–1.22 (8H, m), 0.86 (3H, t, J12 = 13.6 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.1 (CS), 175.6 (CO), 141.5 (C), 126.3 (C), 125.89/125.87 (C), 125.7 (Ar–CH), 125.7 (Ar–CH), 125.7 (Ar–CH), 124.4 (Ar–CH), 35.8 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C16H21O1N2F3S1(M): m/z 346.1327 found 346.1333.O), 1548, 1458, 1183, 1038, 746, 617, 456. EI-MS m/z (%), 294 (4.0), 167 (3.0), 152 (4.5), 127 (5.1), 108 (3.0), 91 (5.4). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.73 (1H, s, NH), 11.27 (1H, s, NH), 10.13 (1H, s, OH), 8.51 (1H, app dd, J6′,5′ = 8.0 Hz, J6′,4′ = 1.2 Hz, H-6′), 7.05 (1H, app td, J4′,(5′,3′) = 8.0 Hz, J4′,6′ = 1.2 Hz, H-4′), 6.91 (1H, app dd, J3′,4′ = 8.0 Hz, J3′,5′ = 1.2 Hz, H-3′), 76.81 (1H, td, J5′,(6′,4′) = 8.0 Hz, J5′,3′ = 1.2 Hz, H-5′), 1.56 (2H, q, J3 = 13.6 Hz, H-3), 1.26 (8H, m, H-3, H-4, H-5, H-6, H-7), 0.87 (3H, t, J8 = 6.8 Hz, H-8); 13C-NMR (125 MHz, DMSO-d6): δC 177.2 (CS), 175.1 (CO), 148.7 (Ar–C), 126.2 (Ar–CH), 125.9 (Ar–C), 122.9 (Ar–CH), 118.2 (Ar–CH), 114.9 (Ar–CH), 35.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H22O2N2S (M): m/z 294.1402 found 294.1399.O), 1545, 1163, 1060, 1023, 903, 835, 725, 536, 505, 433. EI-MS m/z (%), 306 (68.2), 179 (21.5), 165 (7.4), 128 (3.1), 121 (23.9), 106 (45.6). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.45 (1H, s, NH), 11.36 (1H, s, NH), 7.51 (2H, d, J2′,3′/6′,5′ = 8.0 Hz, H-6′, H-2′), 7.22 (2H, d, J3′,2′/5′,6′ = 8.0 Hz, H-3′, H-5′), 2.62 (2H, q, J1′′ = 7.6 Hz, H-1′′), 2.45 (2H, t, J2 = 14.4 Hz, H-2), 1.55 (2H, q, J3 = 10.8 Hz, H-3), 1.25 (8H, m, H-3, H-4, H-5, H-6, H-7), 1.19 (3H, t, J2′′ = 15.2 Hz, H-2′′), 0.87 (3H, t, J8 = 13.2 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 178.7 (CO), 175.5 (CS), 175.6 (CO), 141.8 (Ar–C), 135.4 (Ar–C), 127.8 (Ar–CH × 2), 124.1 (Ar–CH × 2), 35.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.7 (CH2), 24.9 (CH2), 22.0 (CH3), 15.5 (CH3), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H22O2N2S (M): m/z 306.1766 found 306.1777.O), 1586, 1538, 1472, 1404, 1163, 1032, 809, 731, 616. EI-MS m/z (%), 402 (3.9), 332 (2.9), 204 (26.9), 183 (91.5). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.49 (1H, s, NH), 11.53 (1H, s, NH), 8.06 (1H, d, J2′,6′ = 2.0 Hz, H-2′), 7.64 (1H, d, J5′,6′ = 8.8 Hz, H-5′), 7.57 (1H, app dd, J6′,5′ = 8.4 Hz, J6′,2′ = 2.4 Hz), 2.45 (2H, t, J2 = 14.4 Hz, H-2), 1.56 (2H, q, J3 = 13.2 Hz, H-3), 1.23 (8H, m), 0.87 (3H, t, J8 = 6.8 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.7 (CS), 175.8 (CO), 138.4 (Ar–C), 131.1 (Ar–C), 130.7 (Ar–CH), 128.5 (Ar–C), 126.4 (Ar–CH), 125.2 (Ar–CH), 36.1 (CH2), 31.7 (CH2), 30.9 (CH2), 29.4 (CH2), 29.4 (CH2), 29.2 (CH2), 29.1 (CH2), 28.8 (CH2), 24.6 (CH2), 24.5 (CH2), 14.3 (CH3); HREI-MS (M+ − I): calculated for C19H28ON2Cl2S (M): m/z 402.1299 found 402.1295.O), 1596, 1531, 1277, 1176, 1030, 837, 592, 484, 433. EI-MS m/z (%), 320 (11.1), 193 (5.2), 134 (10.9), 128 (16.4), 120 (13.2). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.75 (1H, s, NH), 11.52 (1H, s, NH), 7.98 (2H, d, J3′,2′/5′,6′ = 8.4 Hz, H-3′, H-5′), 7.87 (2H, d, J2′,3′/6′,5′ = 8.8 Hz, H-6′, H-2′), 2.56 (3H, s, –OCH3), 2.47 (2H, t, J2 = 16.0 Hz, H-2), 1.57 (2H, q, J3 = 13.6 Hz, H-3), 1.25 (8H, m, H-3, H-4, H-5, H-6, H-7), 0.87 (3H, t, J8 = 6.6 Hz, H-8); 13C-NMR (125 MHz, DMSO-d6): δC 196.8 (CO), 178.7 (CS), 175.6 (CO), 141.9 (Ar–C), 134.1 (Ar–C), 128.9 (Ar–CH × 2), 123.3 (Ar–CH × 2), 35.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 26.6 (CH2), 24.2 (CH2), 22.0 (CH3), 13.9 (CH3); HREI-MS (M+ − I): calculated for C20H24O2N2S1 (M): m/z 320.1558 found 320.1588.
545. m/z (%), 387.2 (M+ − I, 4.3), 277 (84.9), 200.0 (11.6), 186 (5.5), 109 (2.6) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.50 (1H, s, NH), 11.39 (1H, s, NH), 7.95 (1H, d, Ar–H), 7.42–7.19 (8H, m, Ar–H) 2.40 (2H, t, J2 = 7.6Hz, H-2), 1.52 (2H, q, J3 = 6.6 Hz, H-3), 1.24 (8H, m), 0.86 (3H, t, J8 = 6.8 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.8 (CS), 175.3 (CO), 138.5 (Ar–C), 134.3 (Ar–C × 2), 133.2 (Ar–CH), 129.7 (Ar–CH × 2), 129.4 (Ar–CH × 2), 128.3 (Ar–CH), 127.5 (Ar–CH), 127.4 (Ar–CH), 127.1 (Ar–CH), 35.6 (CH2), 31.0 (CH2), 28.9 (CH2 × 2), 24.3 (CH2), 22.0 (CH2), 13.89 (CH3); HREI-MS (M+ − I): calculated for C21H26O1N2S2 (M): m/z 386.1487 found 386.1478.O), 1546, 1467, 1374, 1262, 1176, 1094, 1072, 1020, 786, 724, 704, 608, 541. EI-MS m/z (%), 306 (47.5), 105 (14.9), 121 (100.0), 128 (2.1), 165 (65.5), 179 (15.2), 207 (13.2), 291 (66.6) 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.07 (1H, s, NH), 11.404 (1H, s, NH), 7.25 (1H, m, Ar–H), 7.10 (2H, m, Ar–H), 2.49 (obscured by DMSO signal, H-2), 2.57 (3H, s, CH3), 2.06 (3H, s, CH3), 1.57 (2H, q, J3 = 4.0 Hz, H-3), 1.26 (8H, m), 0.87 (3H, t, J8 = 8.0 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 180.1 (CS), 175.5 (CO), 137.1 (Ar–C), 136.5 (Ar–C), 132.2 (Ar–C), 128.4, 125.3, 124.7 (Ar–CH), 35.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 19.9 (C H3), 13.9 (CH3); 13.8 (CH3); HREI-MS (M+ − I): calculated for C17H26ON2S (M): m/z 306.1766 found 306.1755.O), 1535, 1515, 1149, 1021, 7537, 717. FABP m/z (%) (M + I) 405.1 1H-NMR (400 MHz, DMSO-d6): δH (ppm): 12.2 (1H, s, NH), 11.5 (1H, s, NH), 7.90 (1H, app dd, J6′,5′ = 8.0 Hz, J6′,4′ = 0.8 Hz, H-6′), 7.63 (1H, d, J3′,4′ = 8.0 Hz, H-3′), 7.44 (1H, app td, J5′(6′,4′) 8.0 Hz, J5′,3′ = 0.4 Hz, H-5′), 7.05 (1H, app td, J4′(3′,5′) = 7.6 Hz, J4′,6′) = 0.8 Hz, H-4′), 2.49 (obscured by DMSO signal, H-2), 1.56 (2H, q, J3 = 7.2 Hz, H-3), 1.28 (8H, m), 0.87 (3H, t, J = 6.6 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 180.2 (CS), 175.5 (CO), 140.0 (Ar–C), 138.8 (Ar–CH), 128.8 (Ar–CH), 128.6 (Ar–CH), 128.4 (Ar–CH), 97.1 (Ar–C), 35.7 (CH2), 31.0 (CH2), 28.3 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H21OI1N2S1 (M): m/z 404.0419 found 404.0410.O), 1542, 1325, 1170, 1130, 1067, 1020, 916, 722, 670, 617, 540. EI-MS m/z (%), 346 (81.9), 277 (2.0), 219 (30.8), 205 (3.7), 186 (5.8), 161 (100), 145 (19.0), 142 (6.2), 128 (2.0). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.683 (1H, s, NH), 11.54 (1H, s, NH), 7.90 (2H, d, J3′,2′/5′,6′ = 8.8 Hz, H-3′, H-5′), 8.03 (2H, d, J2′,3′/6′,5′ = 8.8 Hz, H-2′, H-6′), 2.09 (2H, t, J2 = 15.2 Hz, H-2), 1.57 (2H, app q, J3 = 14.4 Hz, H-3), 1.28–1.22 (8H, m), 0.86 (3H, t, J12 = 13.6 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.1 (CS), 175.6 (CO), 141.5 (C), 126.3 (C), 125.89/125.87 (C), 125.7 (Ar–CH), 125.7 (Ar–CH), 125.7 (Ar–CH), 124.4 (Ar–CH), 35.8 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.2 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C16H21O1N2F3S1(M): m/z 346.1327 found 346.1333.O), 1548, 1458, 1183, 1038, 746, 617, 456. EI-MS m/z (%), 294 (4.0), 167 (3.0), 152 (4.5), 127 (5.1), 108 (3.0), 91 (5.4). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.73 (1H, s, NH), 11.27 (1H, s, NH), 10.13 (1H, s, OH), 8.51 (1H, app dd, J6′,5′ = 8.0 Hz, J6′,4′ = 1.2 Hz, H-6′), 7.05 (1H, app td, J4′,(5′,3′) = 8.0 Hz, J4′,6′ = 1.2 Hz, H-4′), 6.91 (1H, app dd, J3′,4′ = 8.0 Hz, J3′,5′ = 1.2 Hz, H-3′), 76.81 (1H, td, J5′,(6′,4′) = 8.0 Hz, J5′,3′ = 1.2 Hz, H-5′), 1.56 (2H, q, J3 = 13.6 Hz, H-3), 1.26 (8H, m, H-3, H-4, H-5, H-6, H-7), 0.87 (3H, t, J8 = 6.8 Hz, H-8); 13C-NMR (125 MHz, DMSO-d6): δC 177.2 (CS), 175.1 (CO), 148.7 (Ar–C), 126.2 (Ar–CH), 125.9 (Ar–C), 122.9 (Ar–CH), 118.2 (Ar–CH), 114.9 (Ar–CH), 35.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 24.3 (CH2), 22.0 (CH2), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H22O2N2S (M): m/z 294.1402 found 294.1399.O), 1545, 1163, 1060, 1023, 903, 835, 725, 536, 505, 433. EI-MS m/z (%), 306 (68.2), 179 (21.5), 165 (7.4), 128 (3.1), 121 (23.9), 106 (45.6). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.45 (1H, s, NH), 11.36 (1H, s, NH), 7.51 (2H, d, J2′,3′/6′,5′ = 8.0 Hz, H-6′, H-2′), 7.22 (2H, d, J3′,2′/5′,6′ = 8.0 Hz, H-3′, H-5′), 2.62 (2H, q, J1′′ = 7.6 Hz, H-1′′), 2.45 (2H, t, J2 = 14.4 Hz, H-2), 1.55 (2H, q, J3 = 10.8 Hz, H-3), 1.25 (8H, m, H-3, H-4, H-5, H-6, H-7), 1.19 (3H, t, J2′′ = 15.2 Hz, H-2′′), 0.87 (3H, t, J8 = 13.2 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 178.7 (CO), 175.5 (CS), 175.6 (CO), 141.8 (Ar–C), 135.4 (Ar–C), 127.8 (Ar–CH × 2), 124.1 (Ar–CH × 2), 35.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 27.7 (CH2), 24.9 (CH2), 22.0 (CH3), 15.5 (CH3), 13.9 (CH3); HREI-MS (M+ − I): calculated for C15H22O2N2S (M): m/z 306.1766 found 306.1777.O), 1586, 1538, 1472, 1404, 1163, 1032, 809, 731, 616. EI-MS m/z (%), 402 (3.9), 332 (2.9), 204 (26.9), 183 (91.5). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.49 (1H, s, NH), 11.53 (1H, s, NH), 8.06 (1H, d, J2′,6′ = 2.0 Hz, H-2′), 7.64 (1H, d, J5′,6′ = 8.8 Hz, H-5′), 7.57 (1H, app dd, J6′,5′ = 8.4 Hz, J6′,2′ = 2.4 Hz), 2.45 (2H, t, J2 = 14.4 Hz, H-2), 1.56 (2H, q, J3 = 13.2 Hz, H-3), 1.23 (8H, m), 0.87 (3H, t, J8 = 6.8 Hz, H-8); 13C-NMR (75 MHz, DMSO-d6): δC 179.7 (CS), 175.8 (CO), 138.4 (Ar–C), 131.1 (Ar–C), 130.7 (Ar–CH), 128.5 (Ar–C), 126.4 (Ar–CH), 125.2 (Ar–CH), 36.1 (CH2), 31.7 (CH2), 30.9 (CH2), 29.4 (CH2), 29.4 (CH2), 29.2 (CH2), 29.1 (CH2), 28.8 (CH2), 24.6 (CH2), 24.5 (CH2), 14.3 (CH3); HREI-MS (M+ − I): calculated for C19H28ON2Cl2S (M): m/z 402.1299 found 402.1295.O), 1596, 1531, 1277, 1176, 1030, 837, 592, 484, 433. EI-MS m/z (%), 320 (11.1), 193 (5.2), 134 (10.9), 128 (16.4), 120 (13.2). 1H-NMR (400 MHz, DMSO-d6): δH (ppm) 12.75 (1H, s, NH), 11.52 (1H, s, NH), 7.98 (2H, d, J3′,2′/5′,6′ = 8.4 Hz, H-3′, H-5′), 7.87 (2H, d, J2′,3′/6′,5′ = 8.8 Hz, H-6′, H-2′), 2.56 (3H, s, –OCH3), 2.47 (2H, t, J2 = 16.0 Hz, H-2), 1.57 (2H, q, J3 = 13.6 Hz, H-3), 1.25 (8H, m, H-3, H-4, H-5, H-6, H-7), 0.87 (3H, t, J8 = 6.6 Hz, H-8); 13C-NMR (125 MHz, DMSO-d6): δC 196.8 (CO), 178.7 (CS), 175.6 (CO), 141.9 (Ar–C), 134.1 (Ar–C), 128.9 (Ar–CH × 2), 123.3 (Ar–CH × 2), 35.7 (CH2), 31.1 (CH2), 28.4 (CH2), 28.3 (CH2), 26.6 (CH2), 24.2 (CH2), 22.0 (CH3), 13.9 (CH3); HREI-MS (M+ − I): calculated for C20H24O2N2S1 (M): m/z 320.1558 found 320.1588.2.2. Urease inhibition assay
Antiurease activity was assessed by employing the indophenol method. Here, the synthesized compounds (inhibitors) were dissolved in dimethyl sulfoxide and serial dilution was made in the range of 1000–0.976 μM concentration, with thiourea used as the reference inhibitor of urease. The assays were performed in a 96-well plate, with incubation with phosphate buffer (10 μL, 50 mmol, K2HPO4), jack bean urease (20 μL, 5 units per mL) from Uni-Chem-UK, catalog no. U30550-2E, and inhibitor (20 μL) for 10 min at 37 °C. Then, urea was added as a substrate (40 μL, 20 mM) and the mixture was incubated again for 50 min at 37 °C. After incubation, the reaction mixture was placed at room temperature, and then phenol reagent (40 μL) was added, which was a mixture of phenol (1.0% w/v, in water) and sodium nitroprusside (0.005% w/v, in water) and alkali reagent (75 μL) comprising sodium hydroxide (0.5%, in water) and sodium hypochlorite (0.1% active chlorine, in water). Then, the well plate was placed at room temperature and a micro plate reader (LT-4500, Labtech International Ltd, UK) was employed to note the absorption of each well at 625 nm and the percentage urease inhibition was determined using the following formula. The blank was run in the same manner with water used instead of the inhibitor.21–24| % Urease inhibition = {1 − T/C} × 100 |
2.3. Molecular docking and dynamics studies
000 steps. After removing the clashes of systems, the solvation system was equilibrated at 300 K. Three additional equilibrations were run by increasing the temperature from 200 K to 250 K and 300 K to maintain the stability of the systems. Then the systems were subjected to 100 ns simulation in the production run. The MD trajectories were stored at every 2 ps during the production run. The analysis was carried out by the VMD and R package.3. Results and discussion
3.1. Chemistry
A series of thiourea-based molecules was prepared by a simple and efficient synthetic protocol by following the scheme explained in the experimental section with good to excellent yields. The different acyl derivatives were treated with potassium thiocyanate in dried acetone at 60–70 °C, which afforded the acyl isothiocynte. The intermediate obtained then was subsequently treated with various substituted aniline derivatives in the presence of dried acetone and refluxed again at 60–70 °C. The reaction mixture was monitored through thin-layered chromatographic analysis. The newly synthesized derivatives of thiourea 3a–3p (Scheme 1) were characterized by various spectroscopic techniques, including 1H-NMR, 13C-NMR, IR, and EIMS, and the details are shown in Section 2.1. | ||
| Scheme 1 Synthesis of the alkyl chain-linked thiourea derivatives (3a–3q). | ||
3.2. Urease inhibition
The successfully synthesized compounds were evaluated for their in vitro anti-urease activity. The enzyme (urease) inhibition data (Table 1) showed that all the compounds were active against urease. Thiourea was used as a reference in the urease inhibition assay with an IC50 value of 18.61 μM. The thiourea derivatives with 4′-bromo (3c) and 2,6-dimethyl (3g) substituents on the phenyl ring showed excellent activities against urease with IC50 values of 10.65 and 15.19 μM, respectively, compared to standard thiourea (IC50 = 18.61 μM). The other dimethyl substituents bearing thiourea derivatives (3f), (3k), and (3d) showed moderate to weak activity with IC50 values of 40.22, 46.17, and 60.11 μM, respectively.
|
||||||
|---|---|---|---|---|---|---|
| Compound | Phenyl substituents | IC50 (μM); mean ± SEM (% inhibition) | Vmax(app)a (μM min−1) | Km(app)b (mM) | Kic (μM) | Mode of inhibition |
| a Vmax(app) = maximum velocity that measures the rate of reaction of urease enzyme at 20 μM concentration of the inhibitor.b Km(app) = Michaelis–Menten constant that measures the affinity of urease for the substrate at 20 μM concentration of the inhibitor.c Ki (μM) = Inhibition constant derived from Lineweaver–Burk and Dixon plots.d Reference inhibitor of urease. | ||||||
| 3a | 2′-NO2, 4′-OCH3 | 31.09 ± 0.42 (95.2) | — | — | — | — |
| 3b | 4′-CF3 | 35.17 ± 0.46 (87.2) | — | — | — | — |
| 3c | 4′-Br | 10.65 ± 0.45 (96.3) | 6.91 | 4.17 | 1.52 | Competitive |
| 3d | 2′,5′-diCH3 | 60.11 ± 0.78 (61.3) | — | — | — | — |
| 3e | 4′-OCH3 | 35.17 ± 0.56 (71.2) | — | — | — | — |
| 3f | 3′,4′-diCH3 | 40.22 ± 0.45 (76.2) | — | — | — | — |
| 3g | 2′,6′-diCH3 | 15.19 ± 0.58 (96.3) | 15.63 | 6.09 | 9.28 | Competitive |
| 3h | 2′-OCH3, 5′-NO2 | 30.65 ± 0.75 (88.1) | — | — | — | — |
| 3i | 2′,3′-diCl | 22.03 ± 0.45 (91.4) | — | — | — | — |
| 3j | 2′-thiophenyl | 39.11 ± 0.72 (86.5) | — | — | — | — |
| 3k | 2′,3′-diCH3 | 46.17 ± 0.78 (85.8) | — | — | — | — |
| 3l | 2′-I | 31.07 ± 0.58 (88.7) | — | — | — | — |
| 3m | 4′-CF3 | 31.16 ± 0.25 (95.9) | — | — | — | — |
| 3n | 2′-OH | 35.58 ± 0.45 (87.4) | — | — | — | — |
| 3o | 4′-CH2CH3 | 42.78 ± 0.54 (87.4) | — | — | — | — |
| 3p | 3′,4′-diCl | 20.16 ± 0.48 (85.2) | — | — | — | — |
| 3q | 4′-Acetyl | 53.74 ± 0.58 (86.2) | — | — | — | — |
| Thiouread | — | 18.61 ± 0.11 (92.1) | 18.61 | 2.18 | 18.18 | Competitive |
The urease inhibition activity of other halogen-bearing thiourea derivatives (3i), (3p) with a dichloro substituent on the phenyl showed good activity with slightly higher IC50 values of 22.03 and 20.16 μM, compared to the reference thiourea (IC50 = 18.61 μM). While, in the case of the 2′-iodo substituent, the inhibitory activity was reduced with an IC50 value of 31.07 μM.

Moving toward other thiourea-based derivatives, the 4′-methoxyphenyl substituent-bearing derivative (3e) showed weak activity with an IC50 value of 35.17 μM, while the induction of the electron-withdrawing nitro group on the phenyl ring improved the inhibitory activity of the thiourea derivative with IC50 values of 31.09 μM (3a) and 30.65 μM (3h). Further, the derivatives (3m) and (3b) bearing the 4′-trifluoromethyl substituent also showed moderate to weak activity with IC50 values of 31.16 and 35.17 μM.
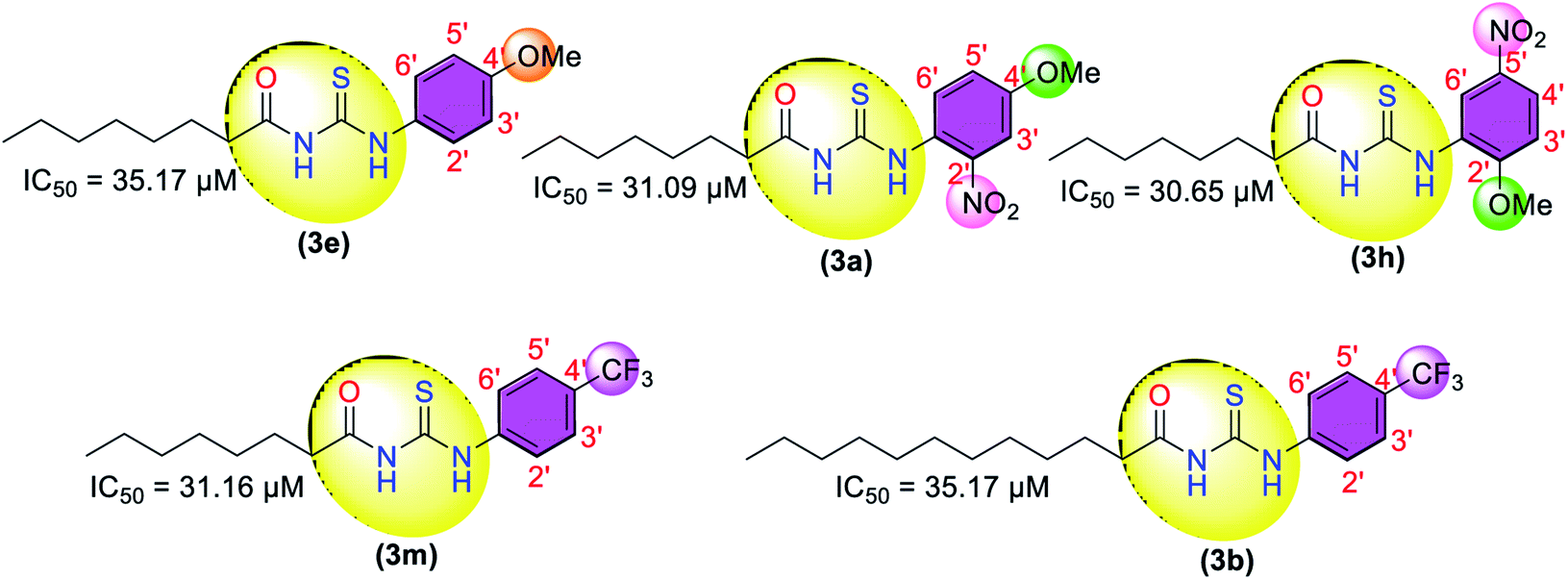
The other derivatives bearing 2′-thiophenyl (3j), 2′-hydroxy (3n), 4′-ethyl (3o), and 4′-acetyl (3q) also demonstrated inhibitory activities with IC50 values of 39.11, 35.58, 42.78, and 53.74 μM. The most potent compounds (3c) and (3g) could potentially serve as leads for the development of new urease inhibitors.
Kinetic studies were performed for the two most potent compounds 3c (IC50 = 10.65 ± 0.45 μM) and 3g (IC50 = 15.19 ± 0.58 μM) at five different concentrations, namely 0.0, 5.0, 10.0, 15.0, and 20.0 μM, along with four different conditions of urea (0.5, 1.0, 2.0, 4.0) as the substrate. The inhibition constant (Ki) as well as the inhibition mode of both inhibitors (3c and 3g) were determined by doing the enzymatic kinetics studies as mentioned above to evaluate whether the inhibitors were competitive, non-competitive, or mixed type.
Lineweaver–Burk plots were drawn to assess the mode of inhibition by evaluating the effect of the inhibitors (3c and 3g) on Vmax and Km. The effects of the inhibitors (compounds) on Vmax and Km were determined to assess the mode of inhibition by Lineweaver–Burk plots. The Km of urease enzyme increased while the Vmax was at 20 μM for the inhibitor (3c and 3g), which showed that both the inhibitors inhibited the enzyme in competitive ways. The Ki values for each inhibitor were determined by Lineweaver–Burk secondary plots (slope of each line vs. different concentrations of 3c and 3g) and were further confirmed by Dixon plot. It was concluded from the kinetic studies that both the compounds (3c and 3g) were competitive inhibitors with Ki values of 1.52 and 9.28 μM, respectively (Table 1). The enzymatic kinetics of the most active compounds are presented below in Fig. 2.
 | ||
| Fig. 2 Mode of inhibition exhibited by 3c and 3g as explained by (a) primary, (b) secondary Lineweaver–Burk and (c) Dixon plots. | ||
3.3. Molecular docking and simulation studies
The binding modes of the competitive inhibitors were predicted using molecular docking simulation studies. Both 3c and 3g compounds docked well in the binding site of the urease enzyme (as shown in Fig. 3a and c). It was also observed that both compounds made hydrogen bonding interactions with Arg439 and Ala440 residues of the backbone atoms of the urease enzyme (Fig. 3b and d). The long aliphatic tail of both compounds residing in the channel was present toward the solvent exposed site. To further validate the predicted binding mode of the compounds, MD simulations were performed for 100 ns time. The simulation trajectories were analyzed to measure both complexes structural stability by calculating the root mean square deviation (RMSD), root mean square fluctuations (RMSF), and radius of gyration (rGyr) over the whole simulation time. | ||
| Fig. 3 Predicted binding modes of 3c (A and B) and 3g (C and D) inhibitors, (A) Sticks model of the binding at the site surface of the urease enzyme, (B) Binding mode in the urease enzyme (cartoon structure) showing the hydrogen bonding (yellow dotted lines), (C) Sticks model of the binding site surface of the urease enzyme, (D) Sticks model of the binding site surface of the urease enzyme showing the hydrogen bonding interaction. Brown spheres represent nickel ions. | ||
The RMSD of the main chain was calculated from the trajectories of the urease-3c and 3g complexes. It was observed that both complexes remained at a ∼2–2.5 Å backbone RMS deviation until 30 ns, as shown in Fig. 4A and B. Between the ranges of 30 ns to 60 ns, urease bound with the 3c compound, and attainted an utmost RMSD value of ∼3.25 Å, while urease bound with 3g compound attained a maximum deviation of ∼3 Å. Irregular deviations in the RMSD of both complexes were observed during 30 to 60 ns and then the systems gained stability in the range of ∼2 to 2.5 Å. The average RMSD value of the urease-3g complex was 2.35 ± 0.29 Å, while urease-3c showed an average RMSD value of 2.37 ± 0.31 Å. The RMSD of the 3c and 3g compounds was also plotted,31 where both ligands remained in the range of ∼2 Å except for 3g, which showed a deviation of more than 2 Å from 80 to 100 ns. The RMSD graphs suggested that urease retained a stable conformation when bound to both 3c and 3g compounds throughout the simulation time.
 | ||
| Fig. 4 Molecular dynamics simulation analysis, (A and B) Calculated RMSD values of urease enzyme in complex with 3c and 3g compounds, (C) Comparison between the RMSF values of both complexes, (D) Compactness comparison of urease bound to 3c and 3g. | ||
To determine the dynamic behavior of the protein residues when bound to these compounds, RMSF values were calculated. The RMSF values of urease residues showed fluctuations from a range of ∼0.5–4 Å in the entire simulation period when bound with the 3c compound, while the RMSF of the urease-3g complex showed a maximum fluctuation of ∼3 Å (Fig. 4C). RMSF analysis revealed that urease showed more flexibility when bound to the 3c compound compared to the 3g compound at some frames. During the MD simulations, the main chain RMSF was determined over the trajectories and averaged over each residue for both complexes, which showed that the flexibility of amino acids was highest in the region 590–610. The start and end residues also showed high RMSF values as these were present on C and N terminals.
The radius of gyration provides an insight into the overall protein compactness over time. Fig. 4D shows the rGyr plots of both complexes for 100 ns-long simulation at 310 K. It shows that when 3c and 3g were bound with urease, the maximum rGyr of the complex reached ∼31.1 Å and ∼31.2 Å during 50–60 ns, respectively. The overall trajectories trend showed that rGyr remained similar for both complexes, except for minor deviations at the end of the simulations.
We also calculated the physicochemical properties of the synthesized compounds, as shown in Table 2. Most of the compounds were in acceptable octanol/water partition coefficient range, except for 3b, 3j, and 3p. Similarly, they showed good predicted cell permeability and an acceptable brain/blood partition coefficient. However, these compounds showed little higher predicted IC50 values for HERG K+ channels blockage.
| ID | MW | HBD | HBA | QPlogPo/w | QPlogHERG | QPPCaco2 | QPlogBB | QPlogKhsa | CNS |
|---|---|---|---|---|---|---|---|---|---|
| a MW = molecular weight, HBD = hydrogen bond donor, HBA = hydrogen bond acceptor, QPlogPo/w (−2.0 to 6.5) = predicted octanol/water partition coefficient, CNS (−2 to +2) = predicted central nervous system activity, QPlogHERG (<−5) = predicted IC50 value for blockage of HERG K+ channels, QPCaco2 (<25 poor, > 500 great) = predicted Caco2 cell permeability in nm s−1. QPlogBB (−3.0 to 1.2) = predicted brain/blood partition coefficient, QPlogKhsa (−1.5 to 1.5) = prediction of binding to human serum albumin. | |||||||||
| 3a | 353.435 | 1 | 4 | 4.489 | −5.545 | 479.455 | −1.358 | 0.715 | −2 |
| 3b | 402.517 | 1 | 2 | 7.508 | −6.365 | 2481.758 | −0.446 | 1.543 | 0 |
| 3c | 357.308 | 1 | 2 | 5.536 | −5.761 | 2483.855 | −0.219 | 0.906 | 0 |
| 3d | 306.465 | 1 | 2 | 5.638 | −5.613 | 3056.075 | −0.316 | 1.078 | 0 |
| 3e | 308.438 | 1 | 3 | 5.057 | −5.705 | 2485.241 | −0.467 | 0.774 | 0 |
| 3f | 306.465 | 1 | 2 | 5.553 | −5.603 | 2486.829 | −0.416 | 1.067 | 0 |
| 3g | 306.465 | 1 | 2 | 5.603 | −5.577 | 3520.209 | −0.247 | 1.042 | 0 |
| 3h | 353.435 | 1 | 4 | 4.403 | −5.618 | 324.484 | −1.584 | 0.734 | −2 |
| 3i | 347.302 | 1 | 2 | 5.904 | −5.604 | 3210.64 | 0.007 | 0.979 | 1 |
| 3j | 386.569 | 1 | 2 | 7.180 | −7.006 | 3141.839 | −0.377 | 1.497 | 0 |
| 3k | 306.465 | 1 | 2 | 5.596 | −5.635 | 2949.132 | −0.327 | 1.055 | 0 |
| 3l | 404.308 | 1 | 2 | 5.632 | −5.792 | 2909.121 | −0.142 | 0.923 | 0 |
| 3m | 346.41 | 1 | 2 | 5.947 | −5.756 | 2483.153 | −0.132 | 1.03 | 0 |
| 3n | 294.411 | 2 | 3 | 4.207 | −5.712 | 935.46 | −0.934 | 0.517 | −1 |
| 3o | 306.465 | 1 | 2 | 5.636 | −5.802 | 2487.485 | −0.484 | 1.047 | 0 |
| 3p | 403.409 | 1 | 2 | 7.463 | −6.241 | 3205.714 | −0.295 | 1.491 | 0 |
| 3q | 320.449 | 1 | 4 | 4.447 | −5.784 | 791.161 | −1.065 | 0.648 | −2 |
4. Conclusion
In conclusion, the urase inhibition activity of synthetic alkyl chain-conjugated thiourea derivaitives demonstrated N-((2′-bromophenyl)carbamothioyl)octanamide (3c) was the most active molecule among the series with an IC50 value of 10.65 ± 0.45 μM compared to standard thiourea (IC50 18.61 ± 0.11 μM). Further compound N-(2′,6′-dimethyl phenyl carbamothioyl)octanamide (3g) also exhibited good activity with an IC50 value of 18.61 ± 0.11 μM. The enzymatic kinetics and molecule docking studies showed that these compounds competitively bind with the urease enzyme, which is crucial for designing urease-targeted inhibitors. The complexes remained compact, and no large deviation of the urease backbone and compounds original poses were observed throughout the simulations. As a result, our findings suggest that this new family of urease inhibitors could be employed to design and assess prospective treatments for infections caused by urease-producing bacteria.Conflicts of interest
We wish to confirm that there are no known conflicts of interest associated with this publication.Acknowledgements
We thank Higher Education Commission of Pakistan for providing grant 8094/Balochistan/NRPU//R&D/HEC/2017 to purchase computational resources.References
- A. Hameed, K. M. Khan, S. T. Zehra, R. Ahmed, Z. Shafiq, S. M. Bakht, M. Yaqub, M. Hussain, A. d. l. V. de León and N. Furtmann, Bioorg. Chem., 2015, 61, 51–57 CrossRef CAS PubMed.
- W.-K. Shi, R.-C. Deng, P.-F. Wang, Q.-Q. Yue, Q. Liu, K.-L. Ding, M.-H. Yang, H.-Y. Zhang, S.-H. Gong and M. Deng, Bioorg. Med. Chem., 2016, 24, 4519–4527 CrossRef CAS PubMed.
- H. Mobley, M. D. Island and R. P. Hausinger, Microbiol. Mol. Biol. Rev., 1995, 59, 451–480 CrossRef CAS PubMed.
- A. Saeed, S.-u. Rehman, P. A. Channar, F. A. Larik, Q. Abbas, M. Hassan, H. Raza, U. Flörke and S.-Y. Seo, J. Taiwan Inst. Chem. Eng., 2017, 77, 54–63 CrossRef CAS.
- F. A. Larik, M. Faisal, A. Saeed, P. A. Channar, J. Korabecny, F. Jabeen, I. A. Mahar, M. A. Kazi, Q. Abbas and G. Murtaza, Bioorg. Chem., 2019, 86, 473–481 CrossRef CAS PubMed.
- K. Stingl, K. Altendorf and E. P. Bakker, Trends Microbiol., 2002, 10, 70–74 CrossRef CAS PubMed.
- P. Krishnamurthy, M. Parlow, J. B. Zitzer, N. B. Vakil, H. L. Mobley, M. Levy, S. H. Phadnis and B. E. Dunn, Infect. Immun., 1998, 66, 5060–5066 CrossRef CAS PubMed.
- K. Kappaun, A. R. Piovesan, C. R. Carlini and R. Ligabue-Braun, J. Adv. Res., 2018, 13, 3–17 CrossRef CAS PubMed.
- A. Hamad, M. A. Khan, I. Ahmad, R. Khalil, M. Khalid, U. Abbas, R. Azhar, J. Uddin, G. E.-S. Batiha and A. Khan, Sci. Rep., 2021, 11, 1–14 CrossRef PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, Structure, 1999, 7, 205–216 CrossRef CAS PubMed.
- M. W. Pinkse, C. S. Maier, J. I. Kim, B. H. Oh and A. J. Heck, J. Mass Spectrom., 2003, 38, 315–320 CrossRef CAS PubMed.
- D. J. Evans Jr, D. G. Evans, S. S. Kirkpatrick and D. Y. Graham, Microb. Pathog., 1991, 10, 15–26 CrossRef CAS.
- P. Kosikowska and Ł. Berlicki, Expert Opin. Ther. Pat., 2011, 21, 945–957 CrossRef CAS PubMed.
- A. Rauf, S. Shahzad, M. Bajda, M. Yar, F. Ahmed, N. Hussain, M. N. Akhtar, A. Khan and J. Jończyk, Bioorg. Med. Chem., 2015, 23, 6049–6058 CrossRef CAS PubMed.
- Y. Gull, N. Rasool, M. Noreen, A. Altaf, S. Musharraf, M. Zubair, F.-U.-H. Nasim, A. Yaqoob, V. DeFeo and M. Zia-Ul-Haq, Molecules, 2016, 21, 266 CrossRef PubMed.
- L. V. Modolo, A. X. de Souza, L. P. Horta, D. P. Araujo and A. de Fatima, J. Adv. Res., 2015, 6, 35–44 CrossRef CAS PubMed.
- B. Ali, K. M. Khan, S. Hussain, S. Hussain, M. Ashraf, M. Riaz, A. Wadood and S. Perveen, Bioorg. Chem., 2018, 79, 34–45 CrossRef CAS PubMed.
- M. Taha, N. H. Ismail, S. Imran, A. Wadood, F. Rahim, K. M. Khan and M. Riaz, Bioorg. Chem., 2016, 66, 80–87 CrossRef CAS PubMed.
- A. Arshia, A. Khan, K. M. Khan, S. M. Saad, N. I. Siddiqui, S. Javaid, S. Perveen and M. I. Choudhary, Med. Chem. Res., 2016, 25, 2666–2679 CrossRef CAS.
- S. Yaqoob, S. Rahim, A. M. Bhayo, M. R. Shah, A. Hameed and M. I. Malik, ChemistrySelect, 2019, 4, 10046–10053 CrossRef CAS.
- M. Ahmed, M. A. Qadir, A. Hameed, M. N. Arshad, A. M. Asiri and M. Muddassar, Biochem. Biophys. Res. Commun., 2017, 490, 434–440 CrossRef CAS PubMed.
- M. Imran, S. Waqar, K. Ogata, M. Ahmed, Z. Noreen, S. Javed, N. Bibi, H. Bokhari, A. Amjad and M. Muddassar, RSC Adv., 2020, 10, 16061–16070 RSC.
- M. Ahmed, M. Imran, M. Muddassar, R. Hussain, M. U. Khan, S. Ahmad, M. Y. Mehboob and S. Ashfaq, J. Mol. Struct., 2020, 1220, 128740 CrossRef CAS.
- S. A. A. S. Tirmazi, M. A. Qadir, M. Ahmed, M. Imran, R. Hussain, M. Sharif, M. Yousaf and M. Muddassar, J. Mol. Struct., 2021, 1235, 130226 CrossRef CAS.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- D. Elebeedy, I. Badawy, A. A. Elmaaty, M. M. Saleh, A. Kandeil, A. Ghanem, O. Kutkat, R. Alnajjar, A. I. Abd El Maksoud and A. A. Al-Karmalawy, Comput. Biol. Med., 2022, 105149 CrossRef PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. Cerutti, T. Cheatham, V. W. D. Cruzeiro, T. Darden and R. E. Duke, 2021.
- J. Zou, C. Tian and C. J. Simmerling, J. Comput.-Aided Mol. Des., 2019, 33, 1021–1029 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar and J. D. Madura, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo and T. Lee, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- A. Mahmoud, A. Mostafa, A. A. Al-Karmalawy, A. Zidan, H. S. Abulkhair, S. H. Mahmoud, M. Shehata, M. M. Elhefnawi and M. A. J. H. Ali, Heliyon, 2021, 7, e07962 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08694d |
| This journal is © The Royal Society of Chemistry 2022 |