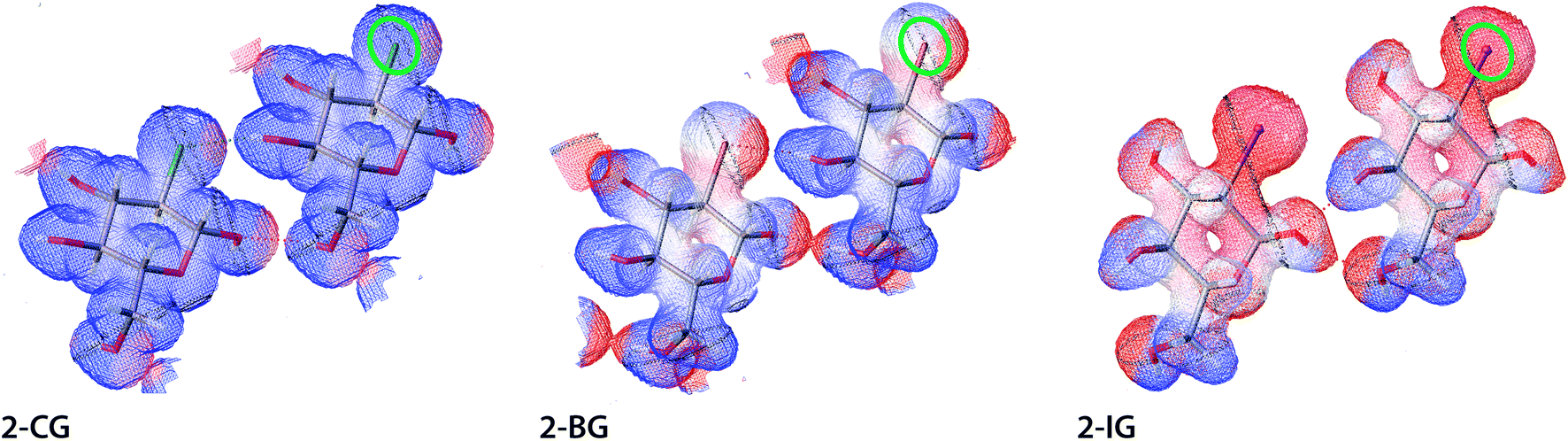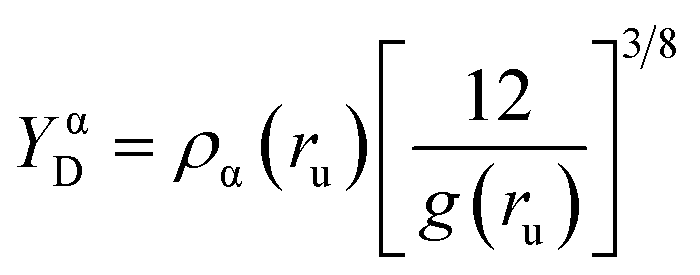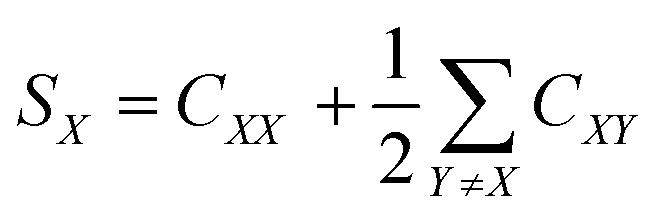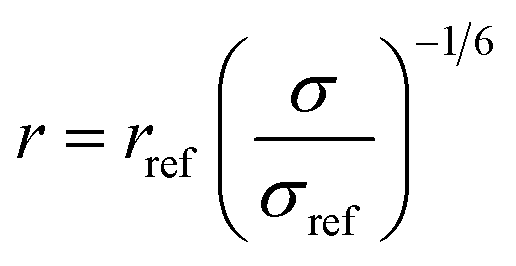DOI:
10.1039/D1RA08312K
(Paper)
RSC Adv., 2022,
12, 8345-8360
X-ray wavefunction refinement and comprehensive structural studies on bromo-substituted analogues of 2-deoxy-D-glucose in solid state and solution†
Received
12th November 2021
, Accepted 5th March 2022
First published on 15th March 2022
Abstract
The structural studies on two bromo-substituted derivatives of 2-deoxy-D-glucose (2-DG), namely 2-deoxy-2-bromo-D-glucose (2-BG) and 2-deoxy-2-bromo-D-mannose (2-BM) are described. 2-DG itself is an inhibitor of hexokinase, the first enzyme in the glycolysis process, playing a vital role in both cancer cell metabolism and viral replication in host cells. Because of that, 2-DG derivatives are considered as potential anti-cancer and anti-viral drugs. An X-ray quantum crystallography approach allowed us to obtain more accurate positions of hydrogen atoms by applying Hirshfeld atom refinement, providing a better description of hydrogen bonding even in the case of data from routine X-ray experiments. Obtained structures showed that the introduction of bromine at the C2 position in the pyranose ring has a minor influence on its conformation but still, it has a noticeable effect on the crystal structure. Bromine imposes the formation of a layered supramolecular landscape containing hydrogen bonds, which involves the bromine atom. Periodic DFT calculations of cohesive and interaction energies (at the B3LYP level of theory) have supported these findings and highlighted energetic changes upon bromine substitution. Based on molecular wavefunction from the refinement, we calculated the electrostatic potential, Laplacian, and ELI-D, and applied them to charge-density studies, which confirmed the geometry of hydrogen bonding and involvement of the bromine atom with these intermolecular interactions. NMR studies in the solution show that both compounds do not display significant differences in their anomeric equilibria compared to 2-DG, and the pyranose ring puckering is similar in both aqueous and solid state.
1. Introduction
A significant number of halogen-substituted carbohydrates have been synthesised over the period of 50 years, from simple monosaccharides1–3 to extended oligo- and polysaccharides.4 Many of these compounds are valuable tools in the synthetic chemistry of carbohydrates and related compounds.5,6 Significant synthetic effort was put into obtaining various fluorine derivatives3,7,8 since some of them found applications in medicine as imaging agents9 as well as probes to study protein–ligand interactions.10,11 Other halogenated sugars have not been extensively studied, mainly being used as simple intermediates in organic synthesis, and probably the most obscure class of them are bromine-substituted compounds. Most previously synthesised brominated carbohydrates are only applicable in synthesis, often stereo-controlled, of other mono and oligosaccharides12,13 or related compounds, including nucleotides.14 Nonetheless, some radiolabelled (76Br) glucose and mannose derivatives have been examined as potential tracers for sugar metabolism in the heart and brain.15 Since the structural scaffoldings of both mono- and disaccharides are composed of moieties inclined to create an intricate set of H-bonds between hydroxyl groups, a halogen substitution(s), gives an interesting opportunity for studies of halogen-mediated noncovalent interactions, including H-bonds and halogen bonds (XB) and the cross-play between them.16 Both of these interactions display similar features such as strength, range, and directionality. Some experimental and computational research focused on brominated compounds indicated that the relative strength of H-bonds and XBs varies depending upon the system. If both interactions coexist, they may display either competitivity or additivity.17–19
Currently, both C–H⋯F–C and C–H⋯Cl–C are classified as weak H-bonds where halogen acts as an acceptor of hydrogen, which is supported by both theoretical and experimental evidences.20–22 Unfortunately, structures of brominated organic compounds are often dominated by XBs and homo-halogen interactions, making it challenging to study relatively weak halogen-mediated H-bonds. Recently several publications focused on a more quantitative approach to Br-mediated intermolecular interactions, including charge-density analysis, confirmed that Br may act as both XB donor and H-bond acceptor. However, these studies were mostly limited to structures not heavily dependent upon H-bonds, such as derivatives of simple aromatic compounds.16,17,23 Thus, we are interested in investigating whether such X⋯H interactions are present in crystals forming a significant number of H-bonds, and if so, how prevalent they are. Another compelling aspect to examine is to investigate whether Cl, Br, and I substituents display supramolecular equivalence and a possible explanation for isostructurality among halogenated carbohydrates. The bromine chemistry is very similar to that of iodine, and the physical properties, including electronegativity or van der Waals radius are between values for chlorine and iodine. Additionally, degree of isostructurality between halogen moieties was reported, at least for certain classes of organic compounds.24 As we mentioned in our previous study, the number of available crystallographic structures of halogenated carbohydrates is very limited.25 This is especially evident in the case of brominated carbohydrates since there are only several structures deposited in the CCDC database, and almost all of them are synthetic intermediates with no resemblance to naturally occurring saccharides in terms of their structure and ability to form H-bonds. This is most likely due to challenges in preparation of high-quality crystals critical for X-ray studies. Instead of forming macroscopic crystals, most carbohydrates exist as amorphic solids or polycrystalline material composed of small and defected crystals. Underdeveloped structural chemistry of halogenated carbohydrates limits our understanding of their properties in solid state, including electronic properties and intermolecular interactions. Seeking a relatively simple yet attractive system to study Br-mediated interactions we focused on two brominated glucopyranoses: 2-deoxy-2-bromo-D-glucopyranose (2-BG) and 2-deoxy-2-bromo-D-mannopyranose (2-BM) since they resemble naturally occurring carbohydrates and may have clinical applications especially in cancer treatment.26 2-Deoxy-D-glucose (2-DG), their parental compound, is a synthetic analogue of D-glucose having 2′ hydroxyl group replaced by a hydrogen atom. Since D-glucose and D-mannose are C-2 epimers of both 2-BG and 2-BM are considered to direct analogues of 2-DG (Scheme 1). In order to achieve better understanding of the effects of bromine substitution on the crystal structure of simple carbohydrates, we also decided to include in this study two already known structures (Scheme S1†), namely β-D-fructofuranosyl α-D-glucopyranoside (sucrose) and its brominated derivative, β-D-fructofuranosyl α-D-4-bromo-glucopyranoside (4-BS).
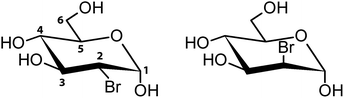 |
| | Scheme 1 Structural formulae of investigated compounds (depicted as α-anomers) with the numbering of carbon atoms. 2-BG (left) and 2-BM (right). | |
For many years the independent atom model (IAM) was a method of choice for crystallographic refinement. In this approach, it is assumed that atoms are separate and non-interacting entities of spherical electron density. However, recent development in both data collection and computational methods render IAM somewhat an obsolete method.27 These more sophisticated approaches to structure refinement takes into consideration atomic non-sphericity, thus providing a better characterisation of bonding in the crystal state. Non-spherical refinement could be based on pre-calculated multipole populations approximating the molecular wavefunction of the investigated system (Hansen–Coppens multipole model and its variations),28 or wavefunction computed for the system without any prior assumptions or pre-computed parameters (Hirshfeld atom refinement, HAR).29 In both cases, aspherical scattering factors are calculated using previously obtained non-spherical electron densities. However, only HAR provides easily interpretable information about chemical bonding. In this approach, the initial geometry derived from standard X-ray refinement is used to compute a molecular wavefunction, usually at the density functional theory (DFT) level of theory, which is later turned into a set of atomic density functions (called Hirshfeld atoms).30 These functions are immediately Fourier-transformed to obtain atomic form factors used in the further structural refinement resulting in an improved model. This procedure is then iterated until convergence is reached.31,32 Currently, HAR is regarded as the most accurate method of non-spherical refinement. This improved refinement is pivotal for hydrogen atoms, where the relative error introduced by a spherical core approximation is most severe. Furthermore, working on even moderate-quality data enables determination hydrogen atom positions at an accuracy level similar to neutron diffraction.32 Such refined structural information is useful for investigating thermodynamical properties of the crystal33 as well as intermolecular interactions.34 In this study, the crystal structures of both 2-BG and 2-BM have been established by X-ray crystallography using HAR to obtain models of improved quality when compared to the classical IAM approach. We described their supramolecular architecture focusing on analysis of the H-bond network. Next, we performed computational studies to unravel more details regarding the intermolecular interactions and energetic features of these crystals. Cohesive energies of the crystal lattice pinpointed the effect of bromine substitution on the global energetic landscape of these crystals. The Hirshfeld surface (HS) analysis provided the quantitative description of interatomic contacts in the structure, whereas energy frameworks showed the directionality and nature of these interactions. Furthermore, analysis of charge and density distribution in the crystals gave additional insight into the nature of intermolecular interaction and the quality of the structure themselves. Last but not least, high-resolution NMR spectral data unfolded their structural properties in the aqueous solution, including anomeric and conformational equilibria.
2. Results and discussion
2.1. Crystallization and crystal morphology
Both 2-BG and 2-BM were crystallised in ambient temperature from ethanol as a mixture of crystalline and amorphic, vitreous phases in ambient temperature. For both compounds, the crystalline phase was mostly composed of aggregates in which some monomeric single crystals were observed. Most of these crystals were of low quality, which renders them unsuitable for structural studies. However, upon closer examination we were capable of finding some crystals of quality suitable for this purpose. These crystals displayed an acicular or columnar habit, and most of them were macroscopically twinned. Our findings are in agreement with general opinion that crystallization of saccharides is usually difficult and time-consuming. In this particular case, the crystals appeared from the amorphous oil-liked phase after several months. Our previous attempts to crystallise 2-BG and 2-BM from water and several organic solvents (ethanol, isopropanol, acetonitrile) and their various mixtures were fruitless and always resulted in the syrup-like glassy phase (Fig. 1).
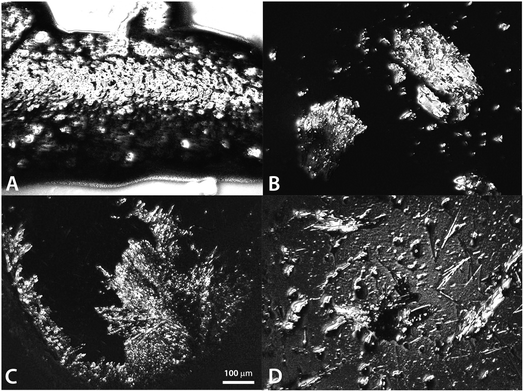 |
| | Fig. 1 General view on crystals of 2-BG (A) and (B) and 2-BM (C) and (D) (scale bar = 100 μm). | |
2.2. X-ray diffraction studies
Single-crystal X-ray analysis confirmed the identity of both studied compounds. 2-BG crystallise in the orthorhombic P212121 space group, and the asymmetric unit is composed of one molecule of the compound. On the other hand, 2-BM forms crystals in the monoclinic P21 space group, and its asymmetric unit contains two molecules. These molecules have very similar geometry except for the 1′OH group, which is located at equatorial position (anomer β) in one molecule and in axial position (anomer α) in the second one (Fig. S1†). Since the crystal of 2-BM used in the study display significant mosaicity, the quality of the model is lower than in the case of 2-BG, especially near the bromine atoms, where a toroidal residual electron density is observed (Fig. S2†), probably due to a partial disorder of this part of the molecule. All refinements parameters are summarised in ESI (Table S1†). Other parameters, including bond lengths, valence, and torsion angles, also can be found there (Tables S2–S7†). The non-spherical atom refinement was performed using NoSpherA2, the recent implementation of HAR in Olex2 program, at the B3LYP/def2-TZVPP level of theory. In this study, we also refined the existing structure of sucrose (available in the Olex2 as a learning set) using the same approach, and all refinement parameters for sucrose are included in ESI (Tables S1 and S8–S10†). For each structure, the application of HAR had a minor impact on global descriptors of refinement quality (Table 1), and only for sucrose, a more significant improvement in R1 and wR2 is observed. Importantly, in this refinement method the accuracy of hydrogen position is considerably better for all structures when compared to IAM, which almost always requires a number of structural constrains for H atoms. The improvement is most striking for sucrose structure, where the length of all C–H and O–H bonds is similar to neutron diffraction data, and no structural restrain or constrains was needed. Moreover, all hydrogen, except H8B, were refined anisotropically with sufficient quality (Fig. 2). Hydrogen positions in 2-BG were mostly reconstructed without additional constrains or restrains with exception for AFIX 13 and AFIX 0 for C2 atom, DFIX 1.09 (C5–H5) and DFIX 0.97 (O2–H2A and O3–H3A). Unfortunately, in the case of 2-BM, we were unable to obtain correct positions of hydrogens without using restrains on bond length. In contrast to some previous studies, we applied HAR to experimental data of average quality (or moderate-to-low in the case of 2-BM) typically obtained from routine X-ray experiments instead of sub-atomic high-resolution data.29,31,35,36 Our results show significant improvement of the accuracy of refinement of hydrogens for the each structure. Nonetheless, in the case of 2-BM, it was necessary to impose a set of restrains to achieve an appropriate length of X–H bonds. For the data collected from Mo radiation (λ = 0.71073 Å, sucrose structure), all hydrogen were refined as anisotropic atoms, but our attempts for aspherical hydrogen refinement for 2-BG were unsuccessful. Even though aspherical hydrogen refinement is possible for high-quality data set measured using Cu radiation (λ = 1.54184 Å) our data were not sufficiently robust to allow such a refinement.
Table 1 Comparison of global refinement quality in HAR and IAM approaches
| Compound |
2-BG |
2-BM |
Sucrose |
| HAR |
| R1 |
3.66% |
7.90% |
2.38% |
| wR1 |
8.38% |
22.85% |
4.10% |
| GooF |
0.948 |
1.050 |
1.093 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| IAM |
| R1 |
3.75% |
7.92% |
3.49% |
| wR1 |
8.35% |
22.34% |
8.87% |
| GooF |
1.020 |
1.167 |
1.052 |
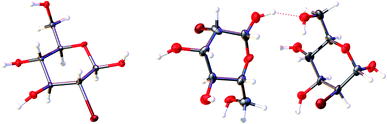 |
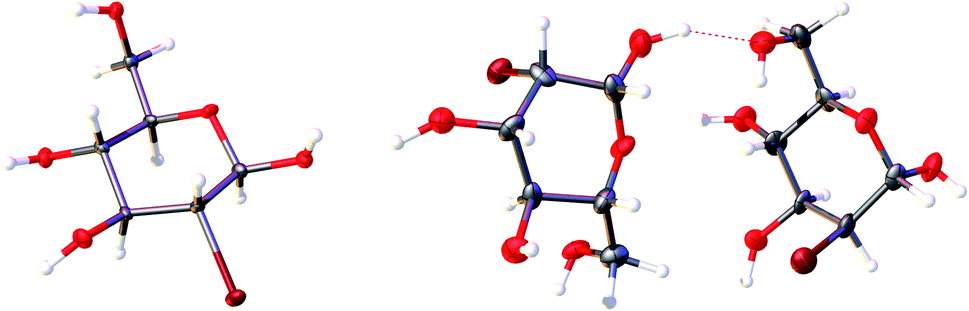
| | Fig. 2 The content of the asymmetric unit of the crystal lattice of investigated compounds. Atomic displacement ellipsoids are shown with a 50% probability level, and the H-atoms are shown as small spheres of arbitrary radius. A dashed line represents the O–H⋯H hydrogen bond. | |
2.3. Conformational analysis of the pyranose ring
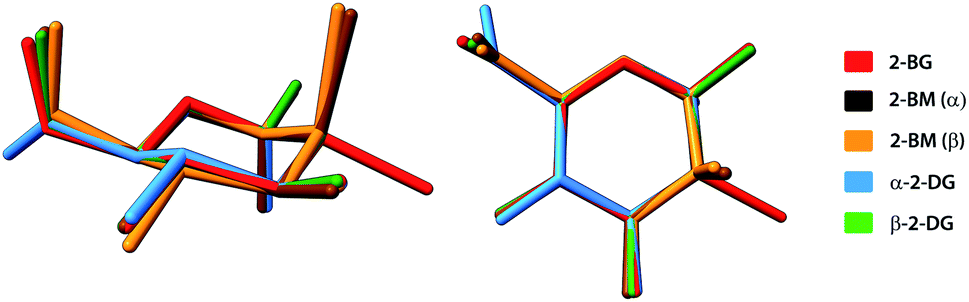
A typical way to describe the conformation of carbohydrates in their cyclic forms is to use Cremer–Pople (CP) parameters, which are generalised ring-puckering coordinates in a spherical coordinate system.37 The CP parameters provide measures to define the ring puckering quantitatively. In each structure, the carbohydrate molecule forms a pyranose ring existing in the chair 4C1 (D1) enantiomeric conformer, the predominant conformation for most carbohydrates either in solid-state or in aqueous solution. For all compounds, calculated CP parameters indicated only minor deviations from the ideal chair geometry (θ = 0°) and the most significant deviation is observed for the α-anomeric molecule of 2-BM (θ = 11.6°). For all 2-DG analogues, the geometry of pyranose displays high structural similarity to the parent compound (θ = 6.0° and 3.9° for the α and β anomers respectively). The structural alignment of these compounds is shown in Fig. 3. Similar observation can be made for 4-BS, indicating that bromine substitution does not alter the conformation of the pyranose ring. Slightly higher value of θ for α-2-BM may result from strain experienced by the molecule in a dense crystal packing, which is also reflected by a significant contribution of the repulsive term in the overall interaction energy (see Section 2.6 for details). These findings are in agreement with the previously published study on similar halogen-substituted analogues of 2-DG.25 The other CP parameters are summarised in Table S11.†
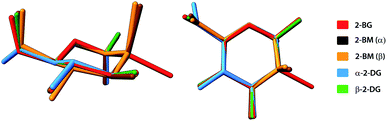 |
| | Fig. 3 Structural alignment of molecules of investigated halogen-derivatives compared to α- and β-anomers of their parent compound. H-atoms have been omitted for the sake of clarity. | |
2.4. Analysis of intermolecular interactions
In the crystal of the parent compound (2-DG), the adjacent molecules are held together by a network of strong O–H⋯O hydrogen bonds, and the crystal is additionally stabilised by the weaker intermolecular C–H⋯O contacts. The architecture of the crystal is slightly different for the α and β anomers of 2-DG but in both compounds the crystal network is dominated by a dense framework of H-bonds forming complex 3-dimensional assembly. Substituting the hydrogen atom in the C2 position in the pyranose ring by a bulky bromine atom alters the supramolecular landscape noticeably. These changes depend on the mutual arrangement of molecules enforcing particular schemes of intermolecular interactions in the crystal structure, and they manifest in different ways for 2-BG and 2-BM.
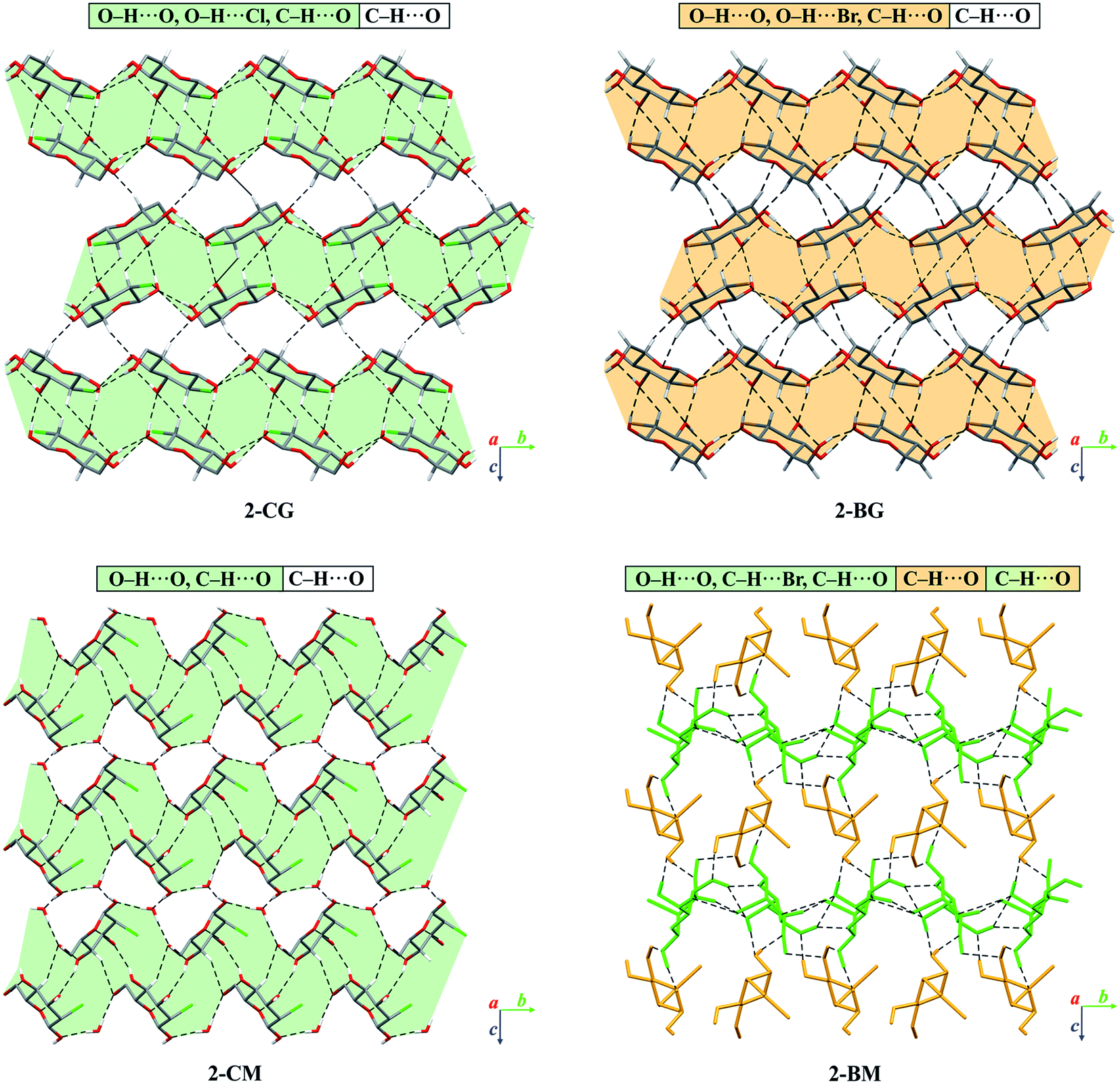
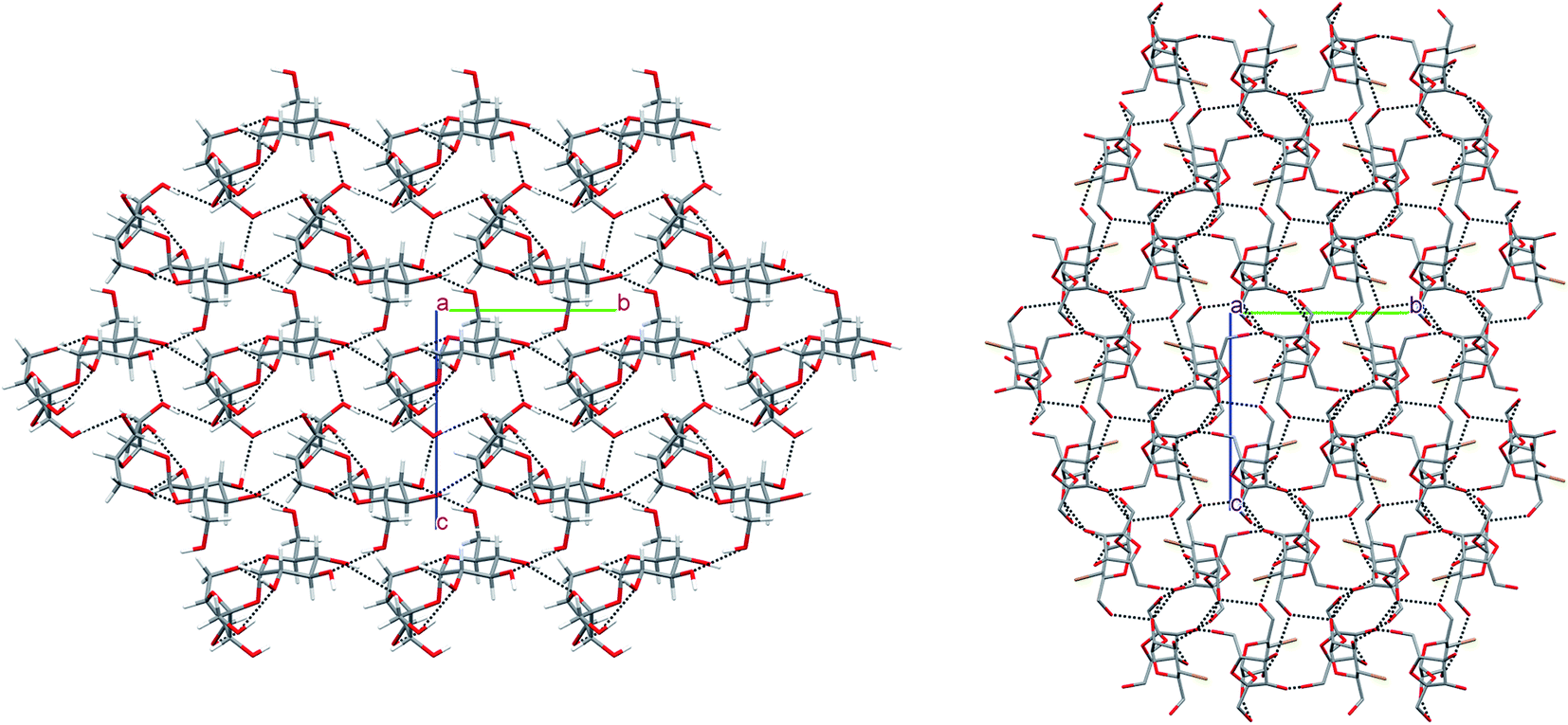
In the crystal of β-2-BG, molecules are organised in infinitive bilayers spreading along the (101) plane. Molecules within these supramolecular assemblies are held together by three strong O–H⋯O (d(D⋯A) = 2.717(5) − 2.810(5) Å; <(D–H⋯A) = 146(8) − 168(7)°) H-bonds and two weaker, O–H⋯Br (d(D⋯A) = 3.388(2) Å; <(D–H⋯A) = 168(5)°), C–H⋯O (d(D⋯A) = 3.270(6) Å; <(D–H⋯A) = 129.3(5)°) H-bonds. The total layered structure is stabilised only by two weak C–H⋯O H-bonds ((d(D⋯A) = 3.322(6) Å; <(D–H⋯A) = 123(4)°) and d(D⋯A) = 3.496(6) Å; <(D–H⋯A) = 165(3)°) involving the adjacent molecules from neighbouring bilayers. This supramolecular architecture bears a striking resemblance to crystal 2-deoxy-2-chloro-D-glucopyranose (2-CG), which is also organised in bilayers interacting via weak intermolecular C–H⋯O bonds (Fig. 4). In both crystals, the pattern of H-bonds and their connectivity is virtually the same, indicating that the replacement of Cl to Br atom preserves the structure of the H-bond network within the crystal even if the substituent has a higher van der Waals radius.25 Due to the presence of two different molecules in the asymmetric unit, the crystal lattice of 2-BM is more complicated. Both molecules form a dense 3-dimensional network of inter-and intramolecular H-bonds. Nevertheless, in the 2-BM crystal, it is possible to distinguish two distinctive layers creating the ABAB structural pattern, in which each layer is composed of either α-anomers (A) or β-anomers (B). Molecules in layer A interact with one another only through a weak C–H⋯O bond (d(D⋯A) = 3.521(17) Å; <(D–H⋯A) = 172(16)°), whereas molecules in layer B interacts with themselves via two O–H⋯O bonds and an O–H⋯Br bond (d(D⋯A) = 2.891(3) − 3.664(14) Å; <(D–H⋯A) = 130(9) − 143(−1)°). Both layers are held together by a dense network of strong O–H⋯O H-bonds (d(D⋯A) = 2.611(14) − 2.861(14) Å; <(D–H⋯A) = 104(13) − 177(−1)°). Structures of 2-BM and 2-deoxy-2-chloro-D-mannopyranose (2-CM) are more difficult to compare since 2-CM is a hydrate (CM:water = 1:1). Nonetheless both compounds form layered assembly connected via either water (in case of 2-CM) or α-2-BM molecules acting as a kind of “molecular glue”.25 We also analysed the structure of sucrose substituted at the C4 position by the bromine (4-BS) and compared it to the crystal (Fig. 5). It turned out that both compounds form a dense supramolecular assembly stabilised by a network of H-bonds. Sucrose molecules form a number of O–H⋯O (d(D⋯A) = 2.720(16) − 3.373(18) Å; <(D–H⋯A) = 121(3) − 173(19)°) as well as weaker C–H⋯O (d(D⋯A) = 2.903(2) − 3.679(16) Å; <(D–H⋯A) = 120(2) − 164(12)°), whereas 4-BS molecules also interact by a number of O–H⋯O (d(D⋯A) = 2.716(1) − 3.141(1) Å; <(D–H⋯A) = 123–169°) but interestingly, they do not form any C–H⋯O H-bonds. Instead, the bromine atom is involved in three C–H⋯Br (d(D⋯A) = 3.315(1) − 3.655(1) Å; <(D–H⋯A) = 103–150°) intermolecular contacts. The overall number of H-bonds in the crystal of 4-BS is smaller than in sucrose crystal, indicating less dense packing, which is also visible in the lower packing index for this compound (74.0% vs. 76.3%). To summarise, single H to Br substitution may have various effects on the carbohydrate crystals (Fig. 5). In the case of 2-DG derivatives, this modification disturbs the 3D molecular assembly present in structures of both α- and β-anomers of 2-DG as well as their fluorine-substituted derivatives, leading to layered structures loosely (2-BG) or tightly (2-BM) connected by H-bonds. Structural changes in 4-BS are more subtle since bulky Br atom destabilises some O–H⋯O interactions and removes any C–H⋯O making the crystal less packed the overall structure of sucrose is preserved. Also, due to its higher radius bromine interacts with a number of H atoms, forming an unusual triple-bifurcated system of H-bonds. The geometrical description of all H-bonds is presented in Tables S12–S15† as well as computed packing indexes for each compound (Table S16†).
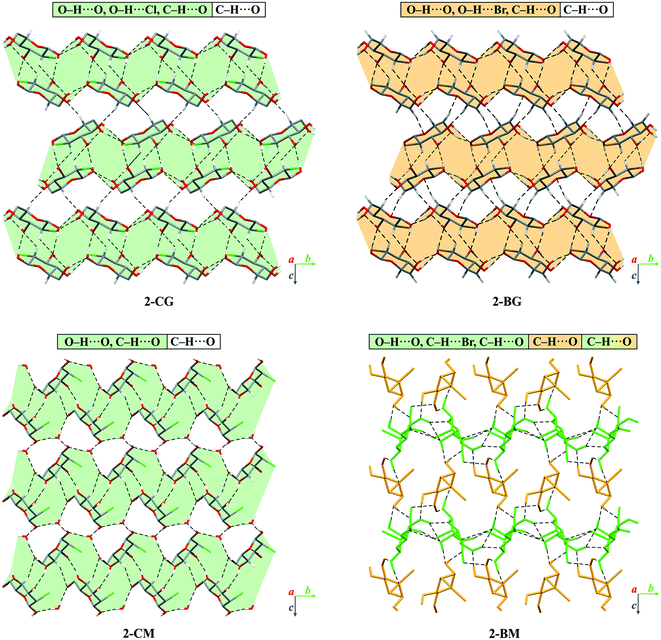 |
| | Fig. 4 Comparison of packing of molecules in the crystals of brominated derivatives of 2-DG with their chlorinated counterparts. The dashed lines represent hydrogen bonds. The H-atoms not participating in intermolecular hydrogen bonds have been omitted for the sake of clarity. Layered supramolecular entities in 2-CG, 2-CM and 2-BG have been highlighted in either light-orange (bromine) or light-green (chlorine). Different anomers of 2-BM were depicted as orange (α-anomer, layer A) or green (β-anomer, layer B). | |
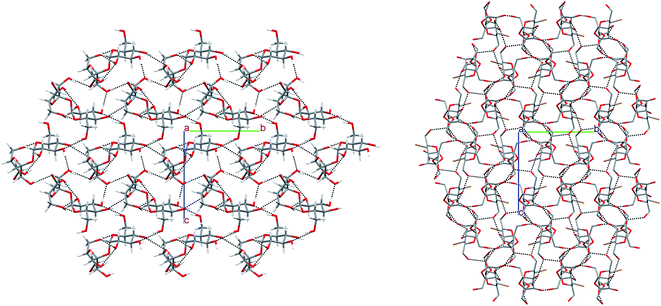 |
| | Fig. 5 Comparison of packing of molecules in the crystals of 4-BS and sucrose. The H-atoms not participating in intermolecular hydrogen bonds have been omitted for the sake of clarity. | |
2.5. Hirshfeld surface analysis
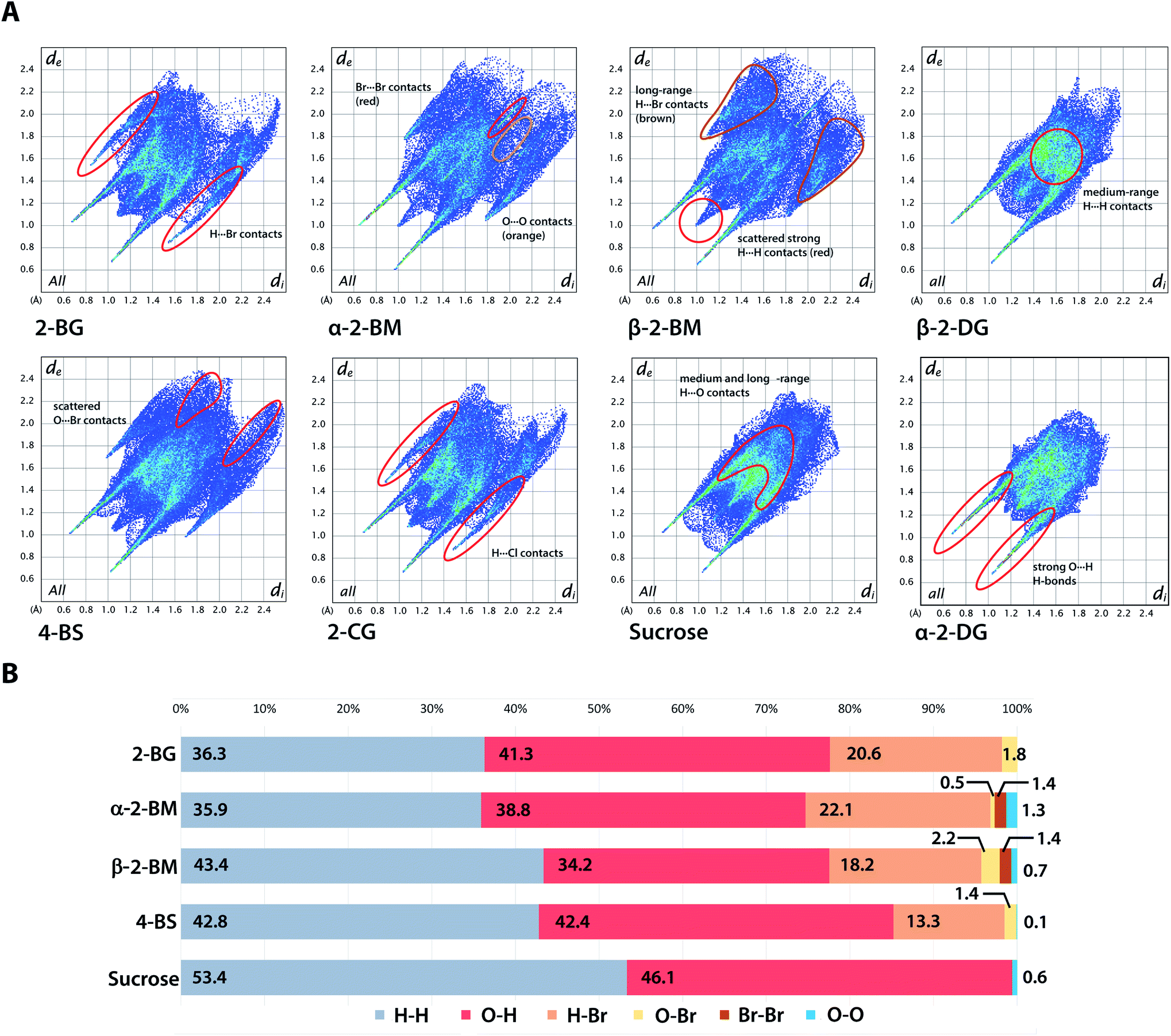
Hirshfeld surface (HS) of molecules in the crystal is defined as a surface where the contribution to the electron density of the crystal from the sum of spherical atoms of a given molecule (promolecule) have the same value as the contribution of adjacent molecules in the crystal (procrystal). HS analysis provides a quantitative description of intermolecular interactions in the crystal structures showing the contribution of each type of interatomic contact to the overall interaction pattern. The useful parameters that facilitate analysis of intermolecular interactions are di and de, which indicate the closest distance from a point on the HS to an atom nucleus located inside or outside the surface. These parameters can be applied to visualise intermolecular contact in the crystal using fingerprint plots describing the relation of di to de for each point on the HS, highlighting differences in the investigated crystals.38 Fingerprint plots for investigated brominated sugars and some related compounds are depicted in Fig. 6. The distributions of each type of atom–atom contacts and the HS itself for all studied compounds are shown in Fig. S3 and S4.† The most distinguishable feature of all studied compounds is the presence of diagonally-symmetric “spikes,” caused by the presence of strong O–H⋯O hydrogen bonds in the crystal structures (average estimated sum of di and de varies from 1.5 to 2.8 Å for all compounds). The position and shape of the spikes are virtually the same for each structure, indicating that introduction of bromine does not alter the global pattern of close O⋯H contacts, and similar results were found earlier for other halogenated sugars. The introduction of Br also has only a minor effect on the contribution of O⋯H contacts in 2-BG, which is similar to both 2-CG and 2-DG (41.3% vs. 41.1% vs. 43.8%). On the other hand, for 2-BM, the percentage of O⋯H contacts is reduced to 36.5%, averaging on both anomers. Due to different position in the supramolecular assembly a molecule of β-anomer participate in less O⋯H and Br⋯H contacts when compared to other investigated compounds, in favour of medium- to long-ranged H⋯H contacts likely caused by the more crowded molecular environment. Another visible effect of Br substitution is reduced contribution of H⋯H contacts (56–58% for 2-DG anomers and 53.4% for sucrose) to 36.3%, 39.7%, and 42.8% for 2-BG, 2-BM, and 4-BS respectively, on the favour of H⋯Br contacts. Nevertheless, the population of strongest H⋯H interactions is unaffected by that change, and even strengthened the in case of 2-BM. Percentage of H⋯Br contacts in 2-BG, 2-BM, and 4-BS is 20.6%, 20.2%, and 13.3%, respectively. The contribution of H⋯X contacts is virtually the same for 2-BG and both anomers in 2-BM as well as for 2-deoxy-2-iodo-D-glucopyranose (2-IG). For the chlorinated counterparts, H⋯X contribution is slightly smaller (below 20%) since the Cl atom is less bulky when compared to Br or I. Nonetheless, patters of H⋯Br and H⋯Cl contacts are very similar. Since the crystal structures of sucrose are mostly unaltered upon H to Br substitution, their HS surfaces are also more alike than in the case of 2-DG derivatives, which may explain the relatively low number of H⋯Br contacts. Despite these differences in the contribution of H⋯Br contacts, their pattern on the fingerprint plots (presence of “secondary spikes”) is very similar for all compounds since each of them is involved in the formation of either O–H⋯Br or C–H⋯Br H-bonds. Other types of atom–atom contacts, namely O⋯Br, Br⋯Br, and O⋯O, have only a marginal role in intermolecular contacts and are not to be discussed here. We also calculated the enrichment ratios (EXY) for interatomic contacts of significant contribution to HS (Table S17†). We excluded 2-BM since it contains two different molecules in the asymmetric unit to achieve a more straightforward comparison among investigated compounds. Due to the presence of strong H-bonds in all studied compounds, the EOH was higher than 1.0, and a similar trend manifests for H⋯Br contacts, which indicates that halogen is also preferred in contacts with H atoms since the capability to form H-bonds and dispersive interactions stronger than H⋯Br. These calculations support previously discussed findings regarding interatomic contacts on HS for studied compounds.
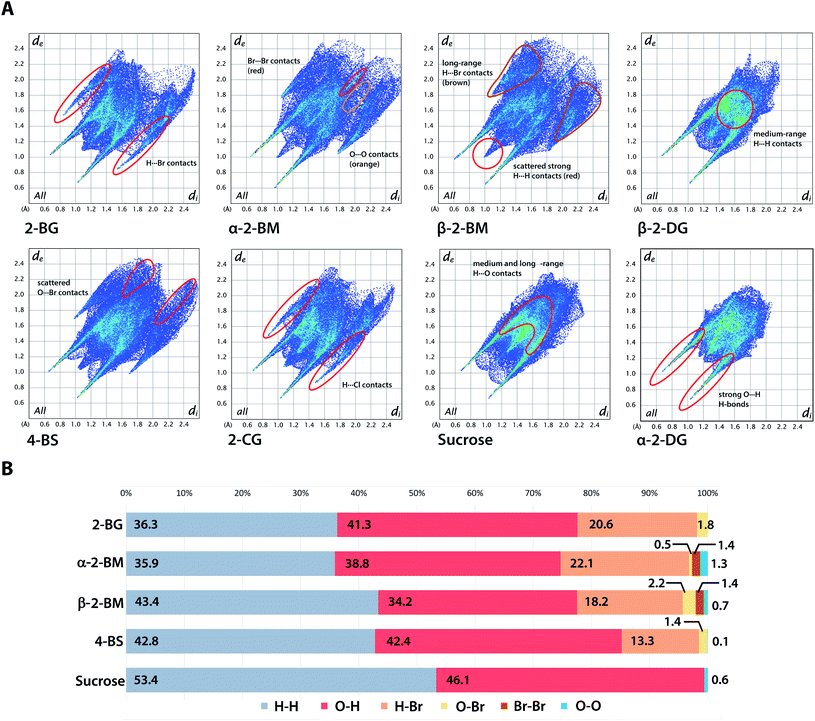 |
| | Fig. 6 Hirshfeld surface analysis of the crystal structures. (A) Two-dimensional fingerprint plots for the analysed crystals with localization of selected atom–atom interactions. (B) Contribution of each type of interatom contact to the overall Hirshfeld surface for chosen compounds. | |
2.6. Energetic features
The total cohesive energies for each compound are shown in Table 2. In the series of Gln derivatives, the energy slightly increases with the increasing size of the halogen substituents, which indicates that a steric hindrance introduced by bulkier substituents leads to slight destabilization of the lattice. Despite the high structural similarity between 2-BG and 2-CG, their difference in cohesive energy is more noticeable than in the case of 2-BG and 2-IG, which suggest that increasing van der Waals radius of the substituent destabilise the interlayer interactions to the point where they are virtually absent in the case of 2-IG. The structure of 2-BM bears some resemblance to 2-CM due to tighter binding between layers, which is also mirrored by higher cohesive energies for each mannose derivatives. In the case of sucrose and 4-BS, an opposite tendency is observed since bromine substitution increases the stability of the crystal. Both compounds have similar crystal structures but the bromine atom in 4-BS fits to empty space between adjacent molecules forming three relatively weak H-bonds, which may substantially contribute to the overall crystal stabilization than one strong H-bond present in this position in sucrose molecule. Moreover, the less densely packing of 4-BS crystal may reduce the number and strength of repulsive interaction within the crystals.
Table 2 Cohesive energies for the investigated compounds and their relatives. Brominated carbohydrates are highlighted in bold
| Compound |
Total energy (kJ mol−1) |
| α-2-DG |
−197.10 |
| β-2-DG |
−215.00 |
| β-2-CG |
−208.16 |
| β-2-BG |
−202.83 |
| β-2-CM |
−295.89 |
| 2-BM |
−215.04 |
| β-2-IG |
−199.70 |
| Sucrose |
−307.26 |
| 4-BS |
−338.14 |
Energy frameworks for all compounds are depicted in Fig. 7, and the energies of each molecular dimer are enlisted in Table S18.† Additionally, structures of dimers having the most significant contribution to overall lattice energy are depicted in Fig. S5.† The introduction of Br to 2-DG results in the formation of layered structures, which is also embodied in the connectivity of their energy frameworks. While α- and β-anomers of 2-DG have tight energy frameworks with only marginal directionality, energy frameworks of their brominated relatives display noticeable directionality along layers in the crystal. The lattice of 2-BG is mainly stabilised by a set of H-bonds involving all hydroxyl groups, as well as some aliphatic hydrogens and bromine atoms, which form a relatively dense network of interactions leading to the formation of a bilayered supramolecular entity. The major contribution to the bonding in the bilayer comes from electrostatic interactions. Bilayers are held together mainly by dispersive C⋯H interactions and weak H-bonds. The most important dimers are 1–3 (within the layer, energies are −56.7, −41.1, and −39.3 kJ mol−1, respectively) and 4 and 5 (inter-layer, energies −19.8, and −18.3 kJ mol−1, respectively). 2-BM possesses a more intricate energy framework composed of many relatively weak interactions within and between layers. Directionality of the energy framework shows that molecules in layer B interact stronger than molecules in layer A. Dimer 6 (energy −32.1 kJ mol−1) has the main contribution to intermolecular interactions in layer B whilst, dimer 7 (energy −11.5 kJ mol−1) provides additional stabilisation, mainly from the dispersive term. Interactions within layer A are considerably weaker, and the differences between the stabilization of these layers are caused primarily by the dense packing. Despite strong electrostatic interactions in layer A, molecular crowding increases the repulsive term, significantly reducing overall stability. Molecules within layer B have weaker electrostatic interactions, however, owing to relatively strong dispersive interactions and less intermolecular repulsion, the total energy is higher. Several different interactions connect molecules between layers, and some of them (dimers 8–11) are depicted in Fig. S5,† in which electrostatics plays a crucial role. More additional details concerning the energy framework for 2-BM are presented in Fig. S6 and S7.† The overall pattern of energy frameworks for 2-BG and 2-CG is very similar, which is also reflected in their supramolecular landscapes. Bilayers in 2-CG crystals are similarly stabilised by three distinctive molecular dimers and connected by mainly dispersive interactions. The introduction of bromine atom to sucrose has a less prominent effect on the directionality of the energy framework than in the case of 2-DG. Both sucrose and 4-BS form three-dimensional structures with almost no preferred orientation. Yet, their energy frameworks display some differences. The sucrose framework possesses some level of directionality since the strongest electrostatic interactions are observed along a axis while the most important contribution from dispersive interactions comes from b axis. On the other hand, the energy framework of 4-BS is devoid of any preferred orientation. The crystal is stabilized mainly by a few pivotal interactions among adjacent molecules. Dimers 12 and 13 provide the highest contribution (−88.5 and −74.3 kJ mol−1), and interestingly in the case of dimer 12, the dispersive term has significant value, which mainly results from Br engagement in H-bonding. The donation of dimers 14 and 15 is less noticeable but still important (−56.0 and −46.2 kJ mol−1, respectively) compared to the rest of the dimers.
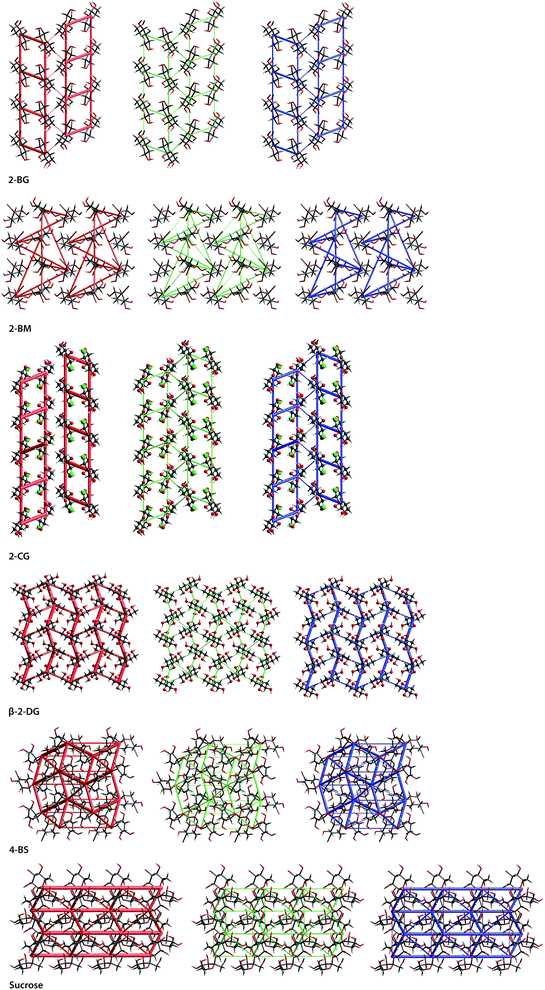 |
| | Fig. 7 Energy frameworks computed for investigating compounds and their relatives, viewed along a direction for electrostatic (red) and dispersion (green) components, and the total interaction energy (blue). The energy cut-off is 10 kJ mol−1. | |
2.7. Charge-density and bonding analysis
The electrostatic potential (ESP) and electron density for halogenated derivatives of 2-DG were calculated for an isolated molecule extracted from the crystal structure. Next, ESP and electron density for the procrystal were reconstructed using the required symmetry operations. For the calculations, we used previously computed wavefunction for the corresponding experimental HAR-refined structure (see Materials and methods for details). Computed values of the ESP were then mapped onto electron-density isosurface (contour 0.03 e A−3) to create the mapped electrostatic potential (MEP), which is discussed in the following text. The areas coloured red, white, and blue represent the highest to lowest ESP values (Fig. 8). To sort out and compare the substituent effect on the MEP we decided to use exactly the same colouring scheme for all compounds. In each case the overall potential was positive everywhere (what is expected for chosen level of electron density surface located nearby the positively charged nuclei). The rationale was to highlight the intermolecular interactions, which could be obscured if the isosurface near to van der Waals radii had been chosen. Despite this, the presence of both polarised bonds (O–H, H–X, where X is a halogen atom) and free electron pairs on O atoms still affect the MEP. The change of the substituent visibly affects MEP in its neighbourhood, shifting the potential towards more positive values. The main explanation of this effect is the total electron number and the sheer volume of the halogen atom since at this electron density isosurface, the participation of the core electron in screening nuclei charge plays a vital role in the ESP. Thus, the most pronounced effects are visible for I, whereas for Br and Cl these changes are more subtle. This observation is also in accordance with theoretical predictions for alkyl halogens.39,40 Importantly, these areas of more positive potential extend also to some hydrogen atoms indicating the presence of strong hydrogen bonds in the crystal. This is also evident for Br and Cl atoms, where a spot of increased positive potential indicates the location of the H-bond.
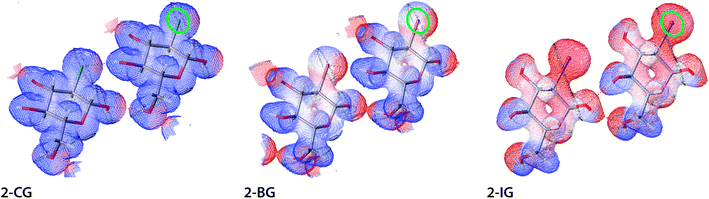 |
| | Fig. 8 Electrostatic potential for selected dimers of halogenated derivatives of 2-DG. All physical parameters are described in atomic units. ESP are mapped on isosurfaces of electron density (contour 0.03 e A−3) in atomic units using BWR colouring scheme, where low ESP values are blue and high are red (gradient from 18 to 35 Eh e−1). A green oval indicates the C2 position for each structure, and short noncovalent interactions are depicted as dotted lines. | |
For further bonding analysis, we choose two related indicators of bonding, ED Laplacian ∇2ρ(r) and electron localizability indicator (ELI-D), which are widespread in the literature and relatively easy to interpret. Both are real space methods in which the bonding is studied on the ground of electron density (ED). In contrast to bonding analysis based on the specific representation of the orbitals, such an approach can be directly applied to experimental data such as electron densities determined by X-ray crystallography. In this study, both indicators were derived from the wavefunction calculated for selected dimers extracted from the crystal to emulate the environment of the local crystal field. Laplacian is related to local charge accumulations or depletions, at a point where ∇2ρ < 0, the ED accumulates (in close vicinity of the point) whereas ED is depleted if the Laplacian is positive.41 In other words, ∇2ρ is the measure of ED curvature, facilitating analysis of the electronic structure of the molecules, including lone electron pairs and atomic shells. ELI-D is a quasi-continuous and dimensionless distribution of charges rescaled by a proportionality factor (eqn (1)), where g(ru) is the Fermi hole curvature. Since ELI-D indicates the average electron population required to form a fixed amount of electron pair, it also shows the correlation of electronic motion, thereby indicating bonding situation.42 ELI-D has also been applied to bonding analysis in the solid-state for several relatively simple systems such as epoxides or small inorganic compounds.43,44
| |
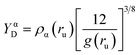 | (1) |
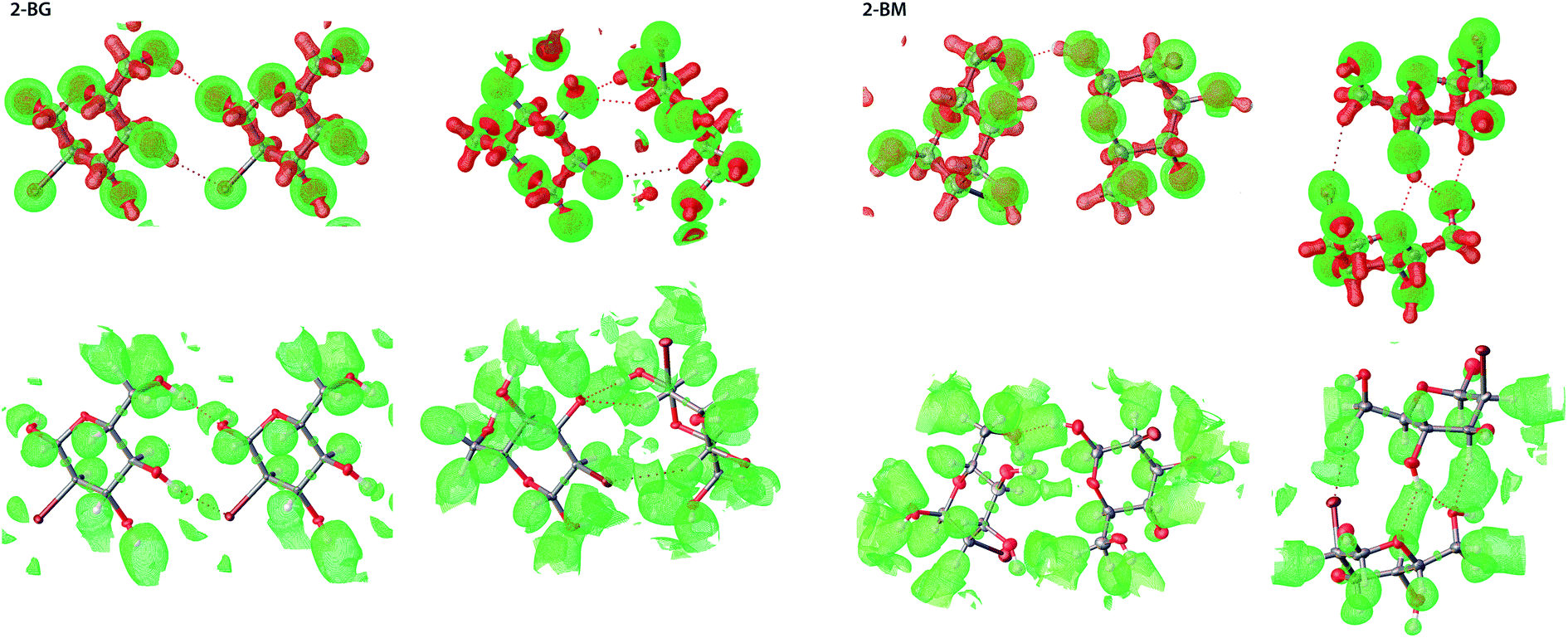
For both 2-BG and 2-BM, the shape of the Laplacian is very similar (Fig. 9), and at contour 0.8 e A−5 charge accumulation indicates covalent bonding within the molecules as well as charge depletion nearby oxygen atoms, which encapsulate hydrogen atoms in the OH groups with exceptions of “holes” indicating the directionality of H-bonds. In the case of ELI-D, the situation is somewhat similar, and the maps are alike for both structures. Since values of this functional are inversely related to overlap integrals, strong interactions and covalent bonds are represented as small and well-defined basins, whereas large and diffuse blobs indicate non-covalent interaction such as H-bonds and dispersive H⋯H interactions. Weakest intermolecular interactions, including dispersive H⋯H contacts not visible at the selected contour level. Even at lower ELI-D values, these interactions remain elusive due to partial overlap of ELI-D basins from different molecules in the crystal. We also computed Laplacian and ELI-D for molecules of several related compounds (Fig. S8†). All these maps are highly similar, and only regions in the vicinity of the C2 position display noticeable differences, which indicate that crystal packing has only a minor influence on the electronic structure of the molecules in the lattice. All charge concentration occurs within the covalent bonds in molecules, and the occurrence of intermolecular interactions is determined by geometric factors imposed by the crystal packing. Finally, we also compared the experimental and static deformation electron densities (ED) for 2-BG and 2-BM. To avoid overlap between ED of adjacent molecules, we chose a somewhat high contour level of 0.095 e A−3 (Fig. S8†). The experimental deformation ED is computed as a Fourier map of asymmetric structure factors convoluted with time-averaged thermal displacement factors. Hence it also contains information about the dynamic behavior of the lattice.45 On the other hand, the static deformation density was derived from molecular wavefunction calculated for selected dimers extracted from the crystal structure. In this approach firstly, the total ED is calculated, and then deformation density is obtained by extraction of spherical ED from the total ED. Thus, it does not include contribution from thermal motions in the crystal lattice. For both 2-BG and 2-BM, maps of experimental and static ED show a good agreement. Slightly diminished values of experimental deformation density are mainly due to limited completeness of the Fourier sum, which is caused by truncation of the collected data – information from very high and low resolution is not present. Nevertheless, most aspects of electronic structure, including charge accumulation on covalent bonds, lone electron pairs, and charge displacement caused by H-bondings, are visible on both maps. For the bromine atom, the ED pattern suggests the existence of σ-hole, which is not visible on MEP, but the geometry of this predicted σ-hole excludes the possibility of any type of halogen bonding in either 2-BG or 2-BM. On static deformation, ED maps are also visible diffused blobs of negative charge near the hydrogen involved in H-bonding, which are not present on experimental deformation ED maps. The most plausible explanation is the contribution of thermal motions, which “smears” ED in these places, making it unclear. To sum up, in our case of simple and qualitative charge-density analysis, we found that ELI-D is the most straightforward indicator of strong-to-moderate intermolecular interactions confirming the position of predicted H-bond and estimating their relative strength. However, it is difficult to compare these results to earlier published studies since, up to our knowledge, only small systems of organic molecules have been investigated. None of them forms an intricate network of H-bonds between various types of atoms.
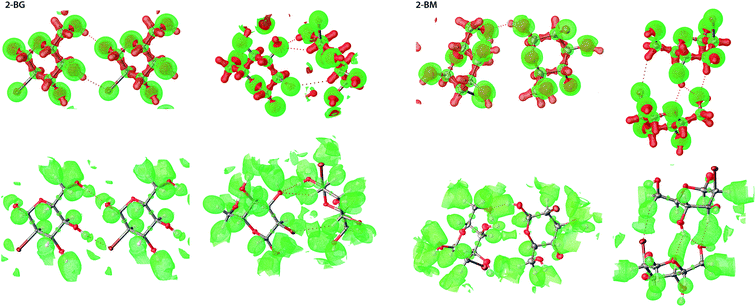 |
| | Fig. 9 3D representation of both ED Laplacian (top) and ELI-D (bottom) for selected dimers of 2-BM and 2-BG, in which Br is involved in intermolecular interactions. For the Laplacian, green represents charge depletion (CD) whilst red represents charge concentration (CC). Contour levels are 0.8 e A−5 and 2.2 YD for Laplacian and ELI-D, respectively. | |
2.8. Structure in aqueous solution
To investigate the conformation of pyranose ring and rotational equilibria of both 2-BG and 2-BM, we employed NMR spectroscopy and recorded a set of 13C HSQC and NOESY correlation spectra. On that base, we determined their anomeric equilibria, torsion angles within the pyranose ring, proton–proton distances, and distribution of C5–C6 rotamers. All these data are compiled in Table S19,† whereas NMR shifts and coupling constants from 1H–13C HSQC and 1H NMR spectra are available in Table S20.† For the pyranose form of glucose and mannose, the contribution of the energetically unfavourable axial (α) anomer is 37% and 69%, respectively.46 The contribution of α-anomer in mannopyranose is higher than for glucopyranose due to the endo-anomeric effect, caused mostly by electrostatic dipole–dipole interactions between the O1 and O5 oxygens.47,48 Another factor contributing to the prevalence of α-anomer is the Δ2 effect stemmed from additional dipolar interaction of 2′OH group bisecting C1–O1 and C1–O5 bonds, which augments the electronic destabilisation of the pyranose ring.49 It also has been established that the contribution of α-anomer increases along with higher electronegativity of adjacent substituents, especially at the C2 position. Nevertheless, in the case of halogenated mannose derivatives, such a tendency was not observed even for highly electronegative fluorine.25 The volume of substituent has major importance, which explains the diminishing contribution of α-anomer in the following order F > Cl ≈ Br > I. For the 2-BM, the population of α-anomeric form is 47%. Apart from the increased volume of a halogen atom, reduced electronegativity of Br in comparison to O reduces the Δ2 effect, hence the overall preference for axial anomer. In contrast to mannopyranose derivatives, halogen substitution at the C2 position in the glucopyranose ring has a negligible impact on the anomeric equilibrium. Thus, the contribution of α-anomer for 2-BG is 34%, virtually the same as for glucose. 3JH,H constants for 2-BG and 2-BM were derived from either HSQC or 1D 1H spectra and then used to calculate H–CX–CY–H torsion angles. For this purpose, we applied the expanded Altona equation, which considers the electronegativity of the adjacent substituents (see Materials and methods for the details).50 These results show that all studied compounds exist almost only in the energetically preferred 4C1 chair conformation. Additional data from NOESY spectra for β-anomers also support this claim since calculated proton–proton distances within the ring are roughly similar, which is expected for 4C1 conformation. Unfortunately, owing to the somewhat limited precision of the data obtained solely from NOESY and 3JH,H constants, we were unable to confirm existence of other high-energy conformers (i.e., 1C4 or twisted chair) or the acyclic form of studied compounds. The ring conformation of both 2-BG and 2-BM is very similar to the parent compound. Hence observed changes are mainly limited to H1–C1–C2–H2 and H2–C2–C3–H3 torsion angles directly affected by the bromine atom and do not alter the overall geometry of the ring. The molecular structure of these compounds in the solid state and aqueous solution are very alike. Observable differences are caused by intermolecular H-bonds and dispersive interactions imposing a given geometry in the crystal, contrary to the solution, where no such constrains apply. Moreover, the crystal of 2-BM is dense-packed which may lead to some unfavourable conformational changes imposed by molecular crowding. Differences in proton–proton distances in solution and solid-state are primarily caused by the internal dynamic of molecules in solution. In a fast-exchange regime, molecules may adopt a set of various conformations which are not detected by NOESY and COSY methods, hence only an average over the structural ensemble is available, where rare or short-lived conformational states contribute to a very limited extent. This is especially pronounced for distances between H4–H5 and H5–H6 since the CH2OH group is capable of rotating freely due to a lack of geometrical restrains.
3. Materials and methods
3.1. Inspection of crystal morphology
Obtained crystals of both 2-BG and 2-BM were studied using the optical microscope (Opta-Tech X2000) with a polarization filter. Images were taken using a build-in camera and further processed using Photoshop CS6 (Adobe Inc.) to adjust gamma and contrast levels.
3.2. X-ray data collection and initial structure refinement
Good quality single crystals of 2-BG and 2-BM were selected for the X-ray diffraction experiments at T = 100(2) K. Diffraction data were collected on the Agilent Technologies SuperNova Dual Source diffractometer with CuKα radiation (λ = 1.54184 Å) using CrysAlis RED software (CrysAlisPRO, Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England). For both compounds, the analytical numerical absorption correction using a multifaceted crystal model implemented in the SCALE3 ABSPACK scaling algorithm was applied.51 Determination of the structure was carried out using the SHELX software package.52 The structures were solved with direct methods followed by successive least-square refinement based on the full-matrix least-squares method on F2 using the SHELXL program. Hydrogen atoms linked to oxygen atoms were located from the Fourier difference electron density map and refined with Uiso(H) = 1.5Oeq(O).
3.3. Non-spherical structure refinement
The final aspherical structural refinement was carried out using olex.refine program with NoSpherA2 functionalities implemented in Olex2-1.3 software. ORCA 4.0 package.53,54 was applied to compute molecular wavefunction at the B3LYP/def2-TZVPP level of theory,55 and all SCF calculations were made for a single asymmetric unit extracted from the crystal lattice. Aspherical scattering factors were calculated at a high level of integration precision. In the case of 2-BG and 2-BM, all hydrogen atoms were refined isotropically, and for sucrose anisotropic refinement was applied. It was necessary to use DFIX restrains or AFIX 0/13 constrains for several H-atoms to avoid significant steric clashes within the crystal structure. In such situations, C–H bonds were fixed to 1.09 Å, and O–H bonds were fixed preferentially to 0.98 Å. Molecular interactions in crystals of all compounds as well as CP parameters were identified using PLATON program.56 The figures for this publication were prepared using Olex2, Mercury, and ChimeraX programs.57–59
3.4. Cohesive energy calculations, Hirshfeld surface analysis and energy frameworks
The optimizations of the molecular geometries at fixed lattice parameters of BG-2, BM-2, B4-sucrose, and sucrose have been performed using Crystal09 with the DFT method and B3LYP functional.60 We applied 6-31 G(d,p) basis set61 for H, C, N, and O and pob-TZVP basis set62,63 for Br atoms. The crystal lattice energies have been calculated with Grimme dispersion64,65 and BSSE corrections available in the used version of the software and according to the manual provided by the software developer (Crystal Solutions). Energy frameworks have been calculated with the B3LYP functional and 6-31 G(d,p) basis set using CrystalExplorer 21.66 Hirshfeld surfaces and two-dimensional fingerprint plots were also calculated using Crystal Explorer 21, and the atomic coordinates used in the calculations were taken from the structures optimised in Crystal09. The enrichment ratios of interatomic contacts on HS were calculated accordingly to the literature using the following equations:
where CXY is a contribution of a given type of atomic contact to the total HS and EXY is the enrichment ratio.67
3.5. Charge-density studies
For the structures of 2-BG and 2-BM after final structural refinement, molecular wavefunctions necessary for further calculations were computed in ORCA 4.0 package at the B3LYP/def2-TZVPP level of theory. All electron density maps and their derivatives, i.e., static and experimental deformation density, residual density, Laplacian of electron density, and ELI-D, were calculated in NoSpherA2, existing structures of sucrose, 2-CG, 2-FG, and 2-FM, previously refined using the IAM model, were corrected to non-spherical structures in NoSpherA2 using the same methodology as described in the previous paragraph. The molecular wavefunction for 2-DG was calculated for the previously optimized molecular geometry at fixed lattice parameters using Crystal09. Molecular wavefunctions for ESP calculations were computed in ORCA. 4.0 at the B3LYP/def2-SVP level of theory. ESP was also computed in NoSpherA2, all aforementioned maps were visualised and rendered in Olex2.
3.6. NMR spectroscopy
3.6.1. Data collection and processing. The NMR experiments were performed on Agilent DD2 spectrometer of 600 MHz proton resonance frequency, using a standard 5 mm 1H–13C–15N triple-resonance room temperature probe head. Measurements were conducted at 298 K. All 1D experiments were displayed and analysed in the MNova (Mestrelab Research) program, and 2D in either MNova or UCSF Sparky.68 The resonance assignment was performed using a set of three experiments for each sample: COSY, 13C-HSQC, and HMBC. Scalar coupling constants were measured using two types of experiments: 1H 1D spectrum (water suppression was included) for resonances that did not overlap with others and a modified version of 13C-HSQC, 13C-HSQC-AP, for resonances overlapping in a 1D spectrum.25 In 13C-HSQC-AP no carbon decoupling was performed during the acquisition period. It made long acquisition time (1.2 s) feasible, which resulted with narrow spectral linewidths, crucial for proper determination of J-coupling constants. For J-coupling constants determination with 1D data sets, line fitting was employed in order to determine small coupling constants (e.g., between C1 and C2 protons) accurately. Determination of J-coupling constants with 2D spectra, the fitting procedure was not employed. For each compound NOE factors were determined using a set of 2D NOESY experiments, consisting of spectra recorded for the following NOESY mixing time values: 100, 200, 350, 500, 600, and 750 ms. Cross-relaxation rate constants σ were determined as a slope of the dependence of peak intensity versus mixing time. These values were used for determination of distances between nuclei (r), assuming a reference distance rref (between the two protons attached to the C6 atom in the β anomer of each compound) to be equal to 1.77 Å. The following formula was employed:
where σref is a cross-relaxation rate of a reference signal. Relevant experimental parameters for all experiments were gathered in the Table 3.
Table 3 Experimental parameters for the data acquisition for the NMR experiments
| |
COSY |
13C-HSQC |
HMBC |
NOESY |
1H |
13C-HSQC-AP |
| Relaxation delay [s] |
1 |
1.5 |
1 |
6 |
1 |
1.5 |
| Number of transients |
4 |
2 |
4 |
4 |
8 |
4 |
| Acquisition time [s] |
0.15 |
0.15 |
0.15 |
0.15 |
1.7 |
1.2 |
| Number of increments |
200 |
300 |
200 |
200 |
n.a. |
400 |
3.6.2. Conformational analysis. H–CX–CY–H torsion angles in the pyranose ring were determined using the following equation:
| 3JH,H = C0 + C1cos(ϕ) + C2cos(2ϕ) + C3cos(3ϕ) + S1sin(ϕ) + S2sin(2ϕ) |
where: ϕ a torsion angle and Ci and Si are parameters taken from the literature.50,69,70 C5–C6 rotamers populations were calculated using following set equations:
| 3JH5,H6R = 9.9A + 0.8B + 4.5C |
| 3JH5,H6S = 1.5A + 1.3B + 10.8C |
where number coefficients are limiting values of 3JH5,H6R and 3JH5,H6S coupling constants, and parameters A, B, C molar fractions of gt, gg and tg rotamers respectively. C6–O6 rotamer populations were estimated using a following set of linear equations:
| 2JH6R,H6S(gauche) = 12.7(A + B) + 11.5C |
| 2JH6R,H6S(trans) = 9.7(A + B) − 8.5C |
| 2JH6R,H6S(app) = 3JH6R,H6S(gauche)D + 3JH6R,H6S(trans)E |
where number coefficients are limiting values of 2JH6R,H6S(gauche), 2JH6,H6S(trans) coupling constants for a given rotamer, A, B, C are molar fractions of gt, gg and tg rotamers respectively whilst D and E molar fractions of gauche and trans rotamers respectively. Values of all dihedral angles and H–H distances for the crystal structures were measured using UCSF Chimera.71
4. Conclusions
Our research showed that single bromine substitution has a significant effect on the crystal structure of mono- and disaccharides. These changes are more prominent in the case of 2-DG derivatives and are mainly caused by steric effect of the bulky substituent leading to the formation of a layered structure. In contrast, changes in sucrose are more subtle, and the main contribution comes from H⋯Br interactions changing the global pattern of H-bonds in the crystal. Because of the strong H-bonds, none of the studied crystals contain halogen bonds, which “lose” the competition between these two related intermolecular interactions. Geometry imposed by these H-bond excludes the possibility of any noticeable halogen bonding or Br⋯Br contacts. Despite differences in their van der Waals radii, crystals of Cl- and Br-substituted monosaccharides bear a striking resemblance. Our previous study showed that it is necessary to introduce an even bigger substituent to disrupt H-bonding between molecular layers in the crystal structure.25 On the other hand, the geometry of the pyranose ring is mostly unaffected by the introduction of bromine, which was quantitatively shown using CP parameters in the conformational analysis. Examination of HS indicated that the global pattern of interatomic contacts in the crystal is preserved upon introducing of the Br atom. However, some of H⋯H contacts are replaced by H⋯Br contacts reducing their contribution. H-bonds and short-range H⋯H(Br) contacts play a vital role in the interaction between molecules, while other interactions (i.e., O⋯Br and Br⋯Br) are marginal. These findings are further supported by the energy frameworks, which show that strong H-bonds between H and O atoms substantially contribute to the lattice energy. Interestingly, H⋯Br interactions are not marginalised and have an impact on the stabilization of the supramolecular layers, or the whole crystal in the case of 4-BS. It should be emphasised that the overall effect of Br substitution on the stability of the crystals is difficult to predict for compounds displaying a complicated network of intermolecular interactions. In our case, it could be beneficial (4-BS) or slightly detrimental (2-BG), and currently available structural data are insufficient to discuss it in more detail. Charge-density analysis showed that electronic structures of 2-BG and 2-BM and some other halogenated sugars are relatively similar, which indicates that crystal packing cannot significantly alter the electronic properties of the molecules. However, these studies are only preliminary, and we are currently working on a quantitative description of bonding in 2-DG derivatives and some related compounds. Finally, NMR studies in the aqueous solution confirmed that introducing a Br atom does not impose any significant conformational changes. Both 2-BG and 2-BM exist predominantly in the most stable 4C1 chair conformation. To achieve higher precision in calculating torsion angles, we applied an extended Karplus-like equation, also considering the contribution from the electronegativity of adjacent substituents. Minor alterations in pyranose conformation for structures in solid-state and solution are rooted in a different chemical environment. In the aqueous solution, sugar molecules rotate freely and are solvated by a cluster of water molecules, whereas solid-state imposes a fixed location in the lattice where stable molecular interactions play a crucial role, and thermal motion is of minor importance.
Another important aspect of this study is to investigate whether more advanced structure refinement methods, e.g., HAR, improve model quality for the data of average resolution, usually collected on the daily basis during X-ray experiments. In our case, we found that using HAR implemented in existing crystallographic software, it is possible to improve the final models not only using high-quality data but also in the situation when collected diffraction patterns are far from being perfect, as in the case of damaged or mosaic crystals. Even if the global quality of the model was only slightly improved, we were able to obtain better geometry of hydrogen atoms almost without the use of constrains, usually necessary in the refinement procedure in IAM. Moreover, wavefunction computed for the final geometry optimization is a valuable tool in further bonding analysis as it can be applied to compute static ED and its derivatives such as ELI-D and Laplacian for the molecule in the crystal. This approach is more computationally demanding (but at B3LYP/def2-TZVPP level of theory, all calculations were made on a personal computer in a reasonable time) yet results in a better quality of the final structures facilitating their subsequent analysis and processing.
Funding
This work was supported by National Science Centre in Poland (grants no. UMO-2017/25/B/NZ3/00251 and UMO-2017/24/C/NZ1/00366). Grant no. UMO-2017/25/B/NZ3/00251 is carried out with cooperation of Professor Waldemar Priebe, MD Anderson Cancer Center, where new 2-DG derivatives were synthesized. The research was carried out at the Biological and Chemical Research Centre, University of Warsaw, established within the project co-financed by European Union from the European Regional Development Fund under the Operational Pro-gramme Innovative Economy, 2007–2013. The X-ray diffraction data collection was accomplished at the Core Facility for Crystallographic and Biophysical research to support the development of medicinal products. The “Core Facility for Crystallographic and Biophysical research to support the development of medicinal products” project is carried out within the TEAM-TECH Core Facility programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund.
Author contributions
Conceptualization and study design, M. Z., W. P., K. W., and B. P.; methodology and software, M. Z., A. Z.-K., S. P., P. M. D., D. T., W. K., and K. W.; validation, M. Z., A. Z.-K., P. M. D., D. T., and B. P.; formal analysis, M. Z., K. W., and B. P.; investigation, M. Z., A. Z.-K., S. P., P. M. D., and D. T.; resources, M. Z., W. K., I. F., R. Z., S. S., and W. P.; data curation, A. Z.-K. and D. T.; writing—original draft preparation, M. Z. and B. P.; writing—review and editing, M. Z., K. W., W. P., and B. P.; visualization, M. Z., S. P. and D. T.; supervision, K. W. and B. P.; project administration, M. Z. and B. P.; and funding acquisition, M. Z. and B. P. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
W. Priebe is an inventor on patents covering new derivatives of 2-DG. He is Chair of SAB and a shareholder of Moleculin Biotech, Inc., CNS Pharmaceuticals, and WPD Pharmaceuticals. His research is in part supported by the sponsor research grant from Moleculin Biotech, Inc. and CNS Pharmaceuticals. I. Fokt and R. Zielinski are listed as inventors on patents covering new analogues of 2-DG, are consultants of Moleculin Biotech, Inc., and are shareholders of Moleculin Biotech, Inc. and CNS Pharmaceuticals. M. Ziemniak is an expert in WPD Pharamceuticals. K. Wozniak has been involved as a consultant in WPD Pharmaceuticals. B. Pająk is CSO for WPD Pharmaceuticals. The other authors declare no conflict of interest.
Acknowledgements
Maciej Kozubal is gratefully acknowledged for help with charge-density studies.
References
- M. De Castro and C. H. Marzabadi, Tetrahedron, 2010, 66, 3395–3404 CrossRef CAS.
- I. Fokt, S. Szymanski, S. Skora, M. Cybulski, T. Madden and W. Priebe, Carbohydr. Res., 2009, 344, 1464–1473 CrossRef CAS PubMed.
- J. St-Gelais, M. Bouchard, V. Denavit and D. Giguère, J. Org. Chem., 2019, 84, 8509–8522 CrossRef CAS PubMed.
- I. Cumpstey, ISRN Org. Chem., 2013, 2013, 1–27 CrossRef PubMed.
- J. E. Harvey, R. J. Hewitt, P. W. Moore and K. K. Somarathne, Pure Appl. Chem., 2014, 86, 1377–1399 CAS.
- C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed.
- Y. Li, M. G. B. Drew, E. V. Welchman, R. K. Shirvastava, S. Jiang, R. Valentine and G. Singh, Tetrahedron, 2004, 60, 6523–6531 CrossRef CAS.
- V. Denavit, D. Lainé, J. St-Gelais, P. A. Johnson and D. Giguère, Nat. Commun., 2018, 9, 4721 CrossRef PubMed.
- M. J. Adam, J. Labelled Compd. Radiopharm., 2002, 45, 167–180 CrossRef CAS.
- A. Ioannou, E. Cini, R. S. Timofte, S. L. Flitsch, N. J. Turner and B. Linclau, Chem. Commun., 2011, 47, 11228–11230 RSC.
- S. Bresciani, T. Lebl, A. M. Z. Slawin and D. O'Hagan, Chem. Commun., 2010, 46, 5434 RSC.
- A. Benz, S. Immel and F. W. Lichtenthaler, Tetrahedron: Asymmetry, 2007, 18, 1108–1114 CrossRef CAS.
- F. W. Lichtenthaler, Chem. Rev., 2011, 111, 5569–5609 CrossRef CAS PubMed.
- S. Libnow, S. Wille, A. Christiansen, M. Hein, H. Reinke, M. Köckerling and R. Miethchen, Synthesis, 2006, 496–508 CAS.
- A. Syrota, Y. Samson, C. Boullais, P. Wajnberg, C. Loc'h, C. Crouzel, B. Mazière, F. Soussaline and J. C. Baron, J. Cereb. Blood Flow Metab., 1985, 5, 358–368 CrossRef CAS PubMed.
- B. Zha, J. Li, J. Wu, X. Miao and M. Zhang, New J. Chem., 2019, 43, 17182–17187 RSC.
- H. Ding, Y. Lu, W. Wu and H. Liu, Chem. Phys., 2014, 441, 30–37 CrossRef CAS.
- R. H. Jones, K. S. Knight, W. G. Marshall, S. J. Coles, P. N. Horton and M. B. Pitak, CrystEngComm, 2013, 15, 8572 RSC.
- L. Koskinen, P. Hirva, A. Hasu, S. Jääskeläinen, J. Koivistoinen, M. Pettersson and M. Haukka, CrystEngComm, 2015, 17, 2718–2727 RSC.
- C. L. Christenholz, D. A. Obenchain, R. A. Peebles and S. A. Peebles, J. Phys. Chem. A, 2014, 118, 1610–1616 CrossRef CAS PubMed.
- A. R. Choudhury and T. N. Guru Row, Cryst. Growth Des., 2004, 4, 47–52 CrossRef CAS.
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277–290 CrossRef CAS.
- M. S. Pavan, R. Pal, K. Nagarajan and T. N. Guru Row, Cryst. Growth Des., 2014, 14, 5477–5485 CrossRef CAS.
- A. Dey and G. R. Desiraju, CrystEngComm, 2004, 6, 642 RSC.
- M. Ziemniak, A. Zawadzka-Kazimierczuk, S. Pawlędzio, M. Malinska, M. Sołtyka, D. Trzybiński, W. Koźmiński, S. Skora, R. Zieliński, I. Fokt, W. Priebe, K. Woźniak and B. Pająk, Int. J. Mol. Sci., 2021, 22, 3720 CrossRef CAS PubMed.
- B. Pajak, E. Siwiak, M. Sołtyka, A. Priebe, R. Zieliński, I. Fokt, M. Ziemniak, A. Jaśkiewicz, R. Borowski, T. Domoradzki and W. Priebe, Int. J. Mol. Sci., 2019, 21, 234 CrossRef PubMed.
- A. Genoni and P. Macchi, Crystals, 2020, 10, 473 CrossRef CAS.
- N. K. Hansen and P. Coppens, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1978, 34, 909–921 CrossRef.
- D. Jayatilaka and B. Dittrich, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 383–393 CrossRef CAS PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- S. C. Capelli, H.-B. Bürgi, B. Dittrich, S. Grabowsky and D. Jayatilaka, IUCrJ, 2014, 1, 361–379 CrossRef CAS PubMed.
- F. Kleemiss, O. V. Dolomanov, M. Bodensteiner, N. Peyerimhoff, L. Midgley, L. J. Bourhis, A. Genoni, L. A. Malaspina, D. Jayatilaka, J. L. Spencer, F. White, B. Grundkötter-Stock, S. Steinhauer, D. Lentz, H. Puschmann and S. Grabowsky, Chem. Sci., 2021, 12, 1675–1692 RSC.
- A. A. Hoser and A. Ø. Madsen, Acta Crystallogr., Sect. A: Found. Adv., 2017, 73, 102–114 CrossRef CAS PubMed.
- L. K. Saunders, A. R. Pallipurath, M. J. Gutmann, H. Nowell, N. Zhang and D. R. Allan, CrystEngComm, 2021, 23, 6180–6190 RSC.
- S. Pawlędzio, M. Malinska, M. Woińska, J. Wojciechowski, L. Andrade Malaspina, F. Kleemiss, S. Grabowsky and K. Woźniak, IUCrJ, 2021, 8, 608–620 CrossRef PubMed.
- M. Woińska, S. Grabowsky, P. M. Dominiak, K. Woźniak and D. Jayatilaka, Sci. Adv., 2016, 2, e1600192 CrossRef PubMed.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- J. S. Murray and P. Politzer, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 153–163 CAS.
- Molecular electrostatic potentials: concepts and applications, ed. J. S. Murray and K. D. Sen, Elsevier, Amsterdam, New York, 1996 Search PubMed.
- Complementary Bonding Analysis, ed. S. Grabowsky, De Gruyter, 2021 Search PubMed.
- M. Kohout, Int. J. Quantum Chem., 2004, 97, 651–658 CrossRef CAS.
- S. Grabowsky, D. Jayatilaka, S. Mebs and P. Luger, Chem.–Eur. J., 2010, 16, 12818–12821 CrossRef CAS PubMed.
- L. Chęcińska, S. Mebs, B. Ośmiałowski, A. Zakrzewska, K. Ejsmont and M. Kohout, ChemPhysChem, 2016, 17, 2395–2406 CrossRef PubMed.
- F. L. Hirshfeld, Crystallogr. Rev., 1991, 2, 169–200 CrossRef.
- M. Miljkovic, Carbohydrates, Springer, New York, NY, 2009 Search PubMed.
- G. Widmalm, Carbohydr. Res., 2013, 378, 123–132 CrossRef CAS PubMed.
- A. J. Kirby, The Anomeric Effect and Related Stereoelectronic Effects at Oxygen, Springer, Berlin, Heidelberg, 1983 Search PubMed.
- R. E. Reeves, J. Am. Chem. Soc., 1950, 72, 1499–1506 CrossRef CAS.
- L. A. Donders, F. A. A. M. De Leeuw and C. Altona, Magn. Reson. Chem., 1989, 27, 556–563 CrossRef CAS.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D'Arco, Y. Noël, M. Causà, M. Rérat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287–1317 CrossRef CAS.
- C. Gatti, F. Cargnoni and L. Bertini, J. Comput. Chem., 2003, 24, 422–436 CrossRef CAS PubMed.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451–459 CrossRef CAS PubMed.
- J. Laun, D. Vilela Oliveira and T. Bredow, J. Comput. Chem., 2018, 39, 1285–1290 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CrossRef CAS PubMed.
- D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco, 2000 Search PubMed.
- C. Altona, R. Francke, R. de Haan, J. H. Ippel, G. J. Daalmans, A. J. A. W. Hoekzema and J. van Wijk, Magn. Reson. Chem., 1994, 32, 670–678 CrossRef CAS.
- C. Altona, J. H. Ippel, A. J. A. W. Hoekzema, C. Erkelens, M. Groesbeek and L. A. Donders, Magn. Reson. Chem., 1989, 27, 564–576 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2121199–2121201. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra08312k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Paulina M. Dominiak
a,
Paulina M. Dominiak