
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBright red emission with high color purity from Eu(III) complexes with π-conjugated polycyclic aromatic ligands and their sensing applications†
Yuichi Kitagawa *ac,
Makoto Tsuruib and
Yasuchika Hasegawa*ac
*ac,
Makoto Tsuruib and
Yasuchika Hasegawa*ac
aFaculty of Engineering, Hokkaido University, N13W8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan. E-mail: y-kitagawa@eng.hokudai.ac.jp; hasegaway@eng.hokudai.ac.jp
bGraduate School of Chemical Sciences and Engineering, Hokkaido University, N13W8, Kita-ku, Sapporo, Hokkaido 060-8628, Japan
cInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, N21 W10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan
First published on 4th January 2022
Abstract
Eu(III) complexes emit red light with a high color purity and have consequently attracted attention for development toward display and physical sensing applications. The characteristic pure color emission originates from the intra-4f–4f transition, and the brightness strongly depends on the electronic and steric structures of organic ligands. A large π-conjugated ligand design with a large absorption coefficient has been actively studied for achieving bright emission. The π-conjugated Eu(III) luminophores also provide oxygen and temperature sensing properties by controlling their excited state dynamics based on π-electron systems. A comprehensive understanding of the design strategy of large π-conjugated ligands is crucial for the further development of luminescent Eu(III) complexes. In this review, we summarize the research progress on π-conjugated Eu(III) luminophores exhibiting bright emission and their physical sensing applications.
1 Introduction
Highly bright monochromatic luminescent compounds have become increasingly important for the development of display and sensing materials. A considerable number of studies have been conducted on the development of various luminescent compounds such as luminescent organic dyes,1–4 metal complexes,5–7 and inorganic compounds (nanoparticles and ceramics).8–15 Recently, Hatakeyama et al. successfully prepared a blue luminescent boron-based organic dye with high color purity (full-width at half-maximum (FWHM) of 28 nm) for the fabrication of efficient organic light emitting diodes (LEDs).16 Jang et al. prepared InP/ZnSe/ZnS quantum dots exhibiting strong red luminescence with high color purity (FWHM of 35 nm).17 Xia et al. synthesized the RbNa3(Li3SiO4)4:Eu2+ phosphor that exhibited a narrow red emission band (FWHM of 22.4 nm).18 Besides these luminescent systems, Eu(III) complexes have emerged as a promising candidate for achieving characteristic pure red luminescence (Fig. 1a, FWHM ≈ 10 nm).19–23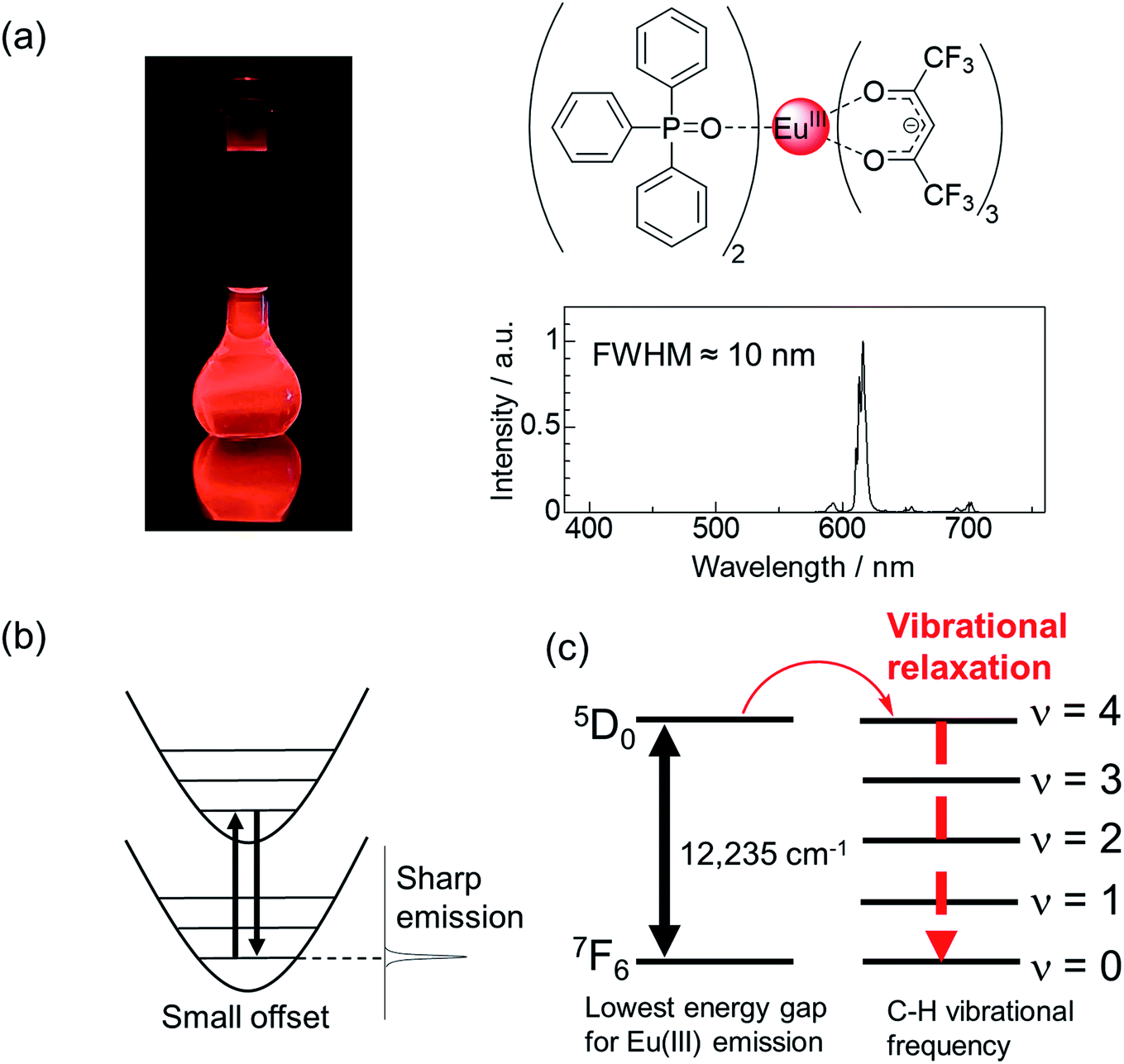 | ||
| Fig. 1 (a) Emission photograph, chemical structure of a highly luminescent Eu(III) complex, and its emission spectrum. (b) Emission due to 4f–4f transition in Eu(III) ions. (c) Matching of the vibrational overtone of the C–H bonds in the Eu(III) complex. | ||
Eu(III) complexes are organic–inorganic hybrid compounds in which organic ligands are attached to the Eu(III) center. The Eu(III) ion has an incompletely filled 4f orbital, which is shielded by the outer shells such as the filled 5s2 and 5p6 orbitals. In a configurational coordinate diagram, these levels appear as parallel parabolas (a small offset case: Fig. 1b) because the 4f electrons are well shielded from their surroundings.24 Therefore, sharp emission lines (FWHM ≈ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm) corresponding to the 5D0 → 7FJ (J = 0, 1, 2, 3, 4, 5, and 6) transitions are observed. Among these, the 5D0 → 7F2 transition is dramatically affected upon changing the ligand field, and pure red emission can be obtained using appropriate ligands. Eu(III) ions exhibit extremely weak absorption (molar absorption coefficient (ε) < 5 M−1 cm−1).25 This limitation can be overcome by using organic compounds with high light-harvesting ability (ε = 103 to 105 M−1 cm−1) as ligands in the Eu(III) complexes. Eu(III) complexes with such ligands can exhibit strong luminescence through energy transfer from the organic ligands to the metal center.
nm) corresponding to the 5D0 → 7FJ (J = 0, 1, 2, 3, 4, 5, and 6) transitions are observed. Among these, the 5D0 → 7F2 transition is dramatically affected upon changing the ligand field, and pure red emission can be obtained using appropriate ligands. Eu(III) ions exhibit extremely weak absorption (molar absorption coefficient (ε) < 5 M−1 cm−1).25 This limitation can be overcome by using organic compounds with high light-harvesting ability (ε = 103 to 105 M−1 cm−1) as ligands in the Eu(III) complexes. Eu(III) complexes with such ligands can exhibit strong luminescence through energy transfer from the organic ligands to the metal center.
The brightness of Eu(III) complexes is a key factor in the development of Eu(III)-based luminescent materials. Brightness is defined as the product of the light absorption coefficient and emission quantum yield. The brightness (IB) is expressed as follows:26
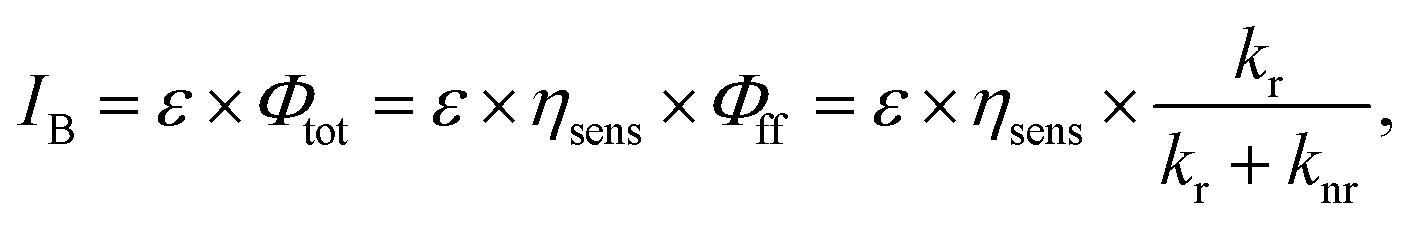
Eu(III) complexes containing an anionic β-diketonate ligand (e.g., thenoyltrifluoroacetonate (tta) or hexafluoroacetylacetonate (hfa) ligand) with large polarizability have a large kr value,27,28 and exhibit efficient energy transfer to the Eu(III) center.29,30 Asymmetric coordination geometries originating due to the presence of anionic ligands and neutral ligands are also a key factor in increasing kr, as they promote the mixing of the 4f–5d excited states with the 4f–4f excited states.31,32 The non-radiative deactivation of the Eu(III) emissive states is promoted by proximate energy-matched OH, NH, and CH oscillators (e.g., water, methanol, and amine; high vibrational frequency, >3000 cm−1, Fig. 1c).33,34 Thus, neutral ligands (e.g., phosphine oxide) with low vibrational frequencies provide small knr by suppressing vibrational quenching. From the viewpoint of kr and knr, it is evident that Eu(III) complexes containing β-diketonate ligands and low vibrational neutral ligands (e.g., Fig. 1a) are effective for achieving high emission quantum yields.35
Improving the light harvesting ability is the key to enhance the brightness of Eu(III) luminophores. Several large π-conjugated systems have extremely high absorption coefficients (εmax > 105 M−1 cm−1).36 A comprehensive understanding of the design strategies for large π-conjugated ligands is crucial for the development of efficient luminescent Eu(III) complexes. In this review, we have summarized the research progress and physical sensing properties of π-conjugated Eu(III) complexes that exhibit bright emission.
2 Historical interpretation of the efficient energy transfer from ligand-to-Eu(III)
First, we discuss the historical interpretation of the efficient energy transfer from the ligand to Eu(III) ions. The UV-light-sensitized luminescence of Eu(III) complexes with organic ligands was observed by Weissman in 1942.37 Based on the finding, organic ligands were designed for the preparation of strong luminescent Eu(III) complexes. The organic ligands undergo intersystem crossing (ISC) from the lowest singlet excited state (S1) to the lowest triplet excited state (T1) after excitation, thereby transferring their electronic energy to the Eu(III) ion (Fig. 2a). Eu(III) ions have several states (5D0: 17250 cm−1, 5D1: 19000 cm−1, and 5D2: 21500 cm−1) that can accept the energy. In 1970, Sato and coworkers demonstrated the importance of the T1 state for energy transfer to the 5D1 level in Eu(III) complexes with β-diketonate ligands.38 They showed that the emission quantum yield due to ligand excitation reached a maximum when the T1 level was ∼1200 cm−1 above the 5D1 level. Latva and coworkers performed a further detailed investigation of the energy transfer using amino-carboxylate-type ligands.39 They showed a clear relationship between the emission quantum yield (due to ligand excitation) and the T1 energy level. The results showed that the 5D2 level of the Eu(III) ion could also accept energy. These studies38,39 suggested that the T1 state should be higher in energy than the 5D1 state (19000 cm−1) for effective energy transfer.
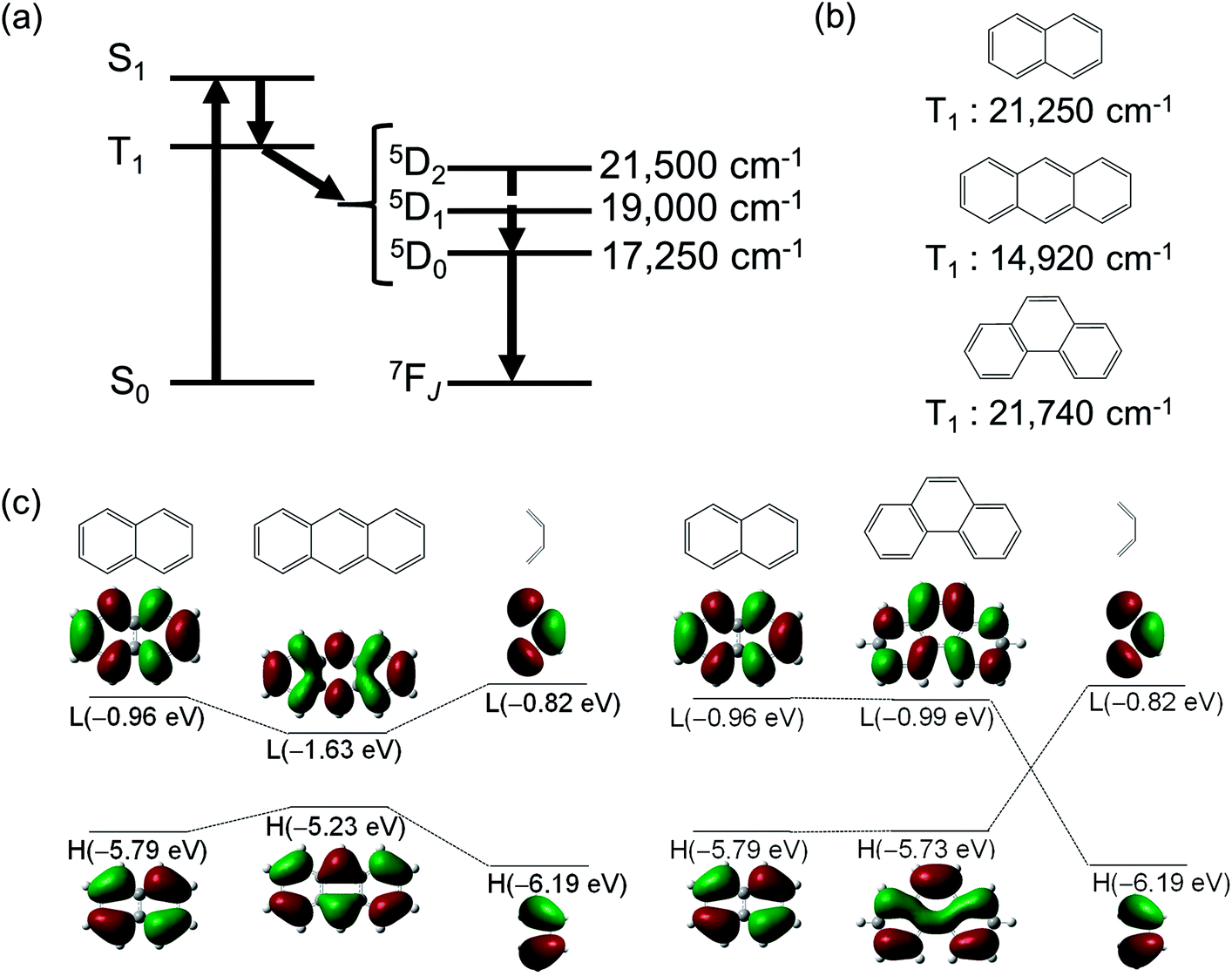 | ||
| Fig. 2 (a) Energy level diagrams of luminescent Eu(III) complex. (b) T1 energy levels of naphthalene, anthracene, and phenanthrene. (c) Fragment molecular orbitals for anthracene and phenanthrene. Redrawn from ref. 44. | ||
The T1 states of aromatic benzene, naphthalene, and anthracene lie at 29470 cm−1, 21250 cm−1, and 14920 cm−1, respectively (Fig. 2b).36 The T1 level of anthracene (14π-electron system) is lower than the emitting levels of Eu(III). In contrast, the T1 level of ligands such as phenanthrene, which has the same π-conjugation length (14π-electron), is higher (Fig. 2b, >19000 cm−1) and facilitates photosensitized energy transfer to Eu(III) ions.40,41 This suggests that the extension of π-conjugation to tailor the T1 levels of ligands can broaden the scope of ligand design. Using the fragment molecular orbital method and DFT calculations (B3LYP/6-31G(D)42,43),44 we have previously shown a simple method for manipulating the T1 energy level.
The energy of the T1 level (ΔE(T1)) is expressed as follows:
| ΔE(T1) = E(T1) − E(S0) = εf − εi − Jif | (1) |
As a standard for π-conjugated molecules, we used naphthalene, whose excited state energy (ΔE(T1) = 21250 cm−1) is higher than the emission levels of Eu(III) ions. Extending the π-conjugation by coupling naphthalene and butadiene affords anthracene or phenanthrene. The HOMOs of naphthalene and butadiene (−5.79 and −6.19 eV, respectively) and their LUMOs (−0.96 and −0.82 eV, respectively) are electronically coupled in-phase, resulting in a more destabilized HOMO (−5.23 eV) and more stabilized LUMO (−1.63 eV) for anthracene (Fig. 2c, left). This smaller HOMO–LUMO energy gap yields a low T1 level for anthracene (14920 cm−1). However, for phenanthrene (Fig. 2c, right), the HOMOs and LUMOs of naphthalene and butadiene are electronically coupled in such a manner that the HOMO (−5.73 eV) and LUMO (−0.99 eV) energies are almost unchanged relative to those of naphthalene. Consequently, the T1 level of phenanthrene was nearly unchanged (ΔE(T1) = 21740 cm−1).45 Thus, controlling the electronic structure can maintain the T1 level at an appropriate position in extended π-conjugation systems. Conjugated systems with more than 18 π electron, such as [4]-helicene (ΔE(T1) = 19870 cm−1),46 triphenylene (ΔE(T1) = 23580 cm−1),47 chrysene (ΔE(T1) = 20000 cm−1),47 [5]-helicene (ΔE(T1) = 19750 cm−1),46 picene (ΔE(T1) = 20010 cm−1),48 [6]-helicene (ΔE(T1) = 18990 cm−1),46 coronene (ΔE(T1) = 19400 cm−1),47 and phenacene (ΔE(T1) = 19380 cm−1),48 have relatively high T1 energies (Fig. 3). Among these, luminescent Eu(III) complexes with triphenylene,49–53 chrysene,54–56 [5]-helicene,44 picene,57 and coronene58,59 frameworks have already been reported (chemical structures and their photophysical properties are shown in Fig. S1 and S2 in the ESI†). Several hetero-conjugated systems,60–82 such as 1,4,8,9-tetraazatriphenylene (ΔE(T1) = 23500 cm−1), are well known to possess high T1 energy for photosensitized Eu(III) emission (chemical structures and their photophysical properties are shown in Fig. S3–S9 in the ESI†). These appropriate T1 levels are expected to facilitate photoinduced energy transfer to Eu(III) ions. The S1 energy of a molecule is expressed as follows:
| ΔE(S1) = E(S1) − E(S0) = εf − εi − Jif + 2Kif | (2) |
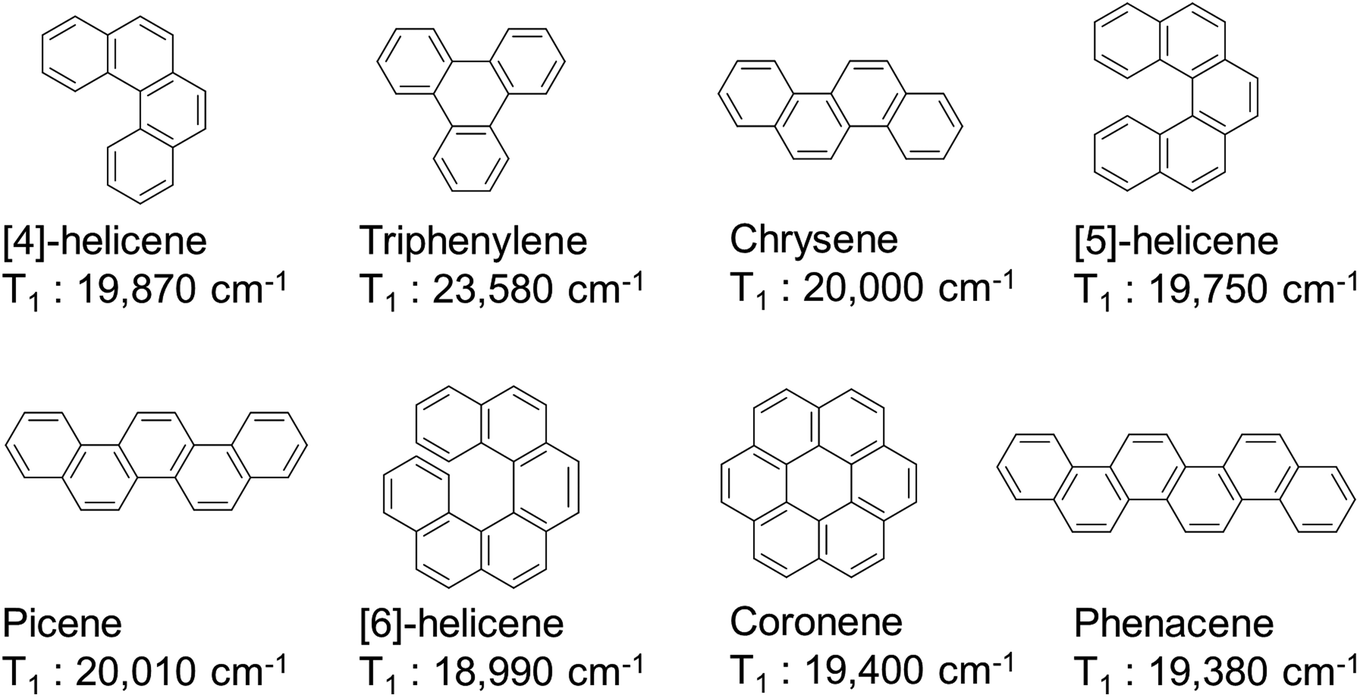 | ||
| Fig. 3 T1 energy levels of polycyclic aromatic hydrocarbon. | ||
3 Photophysics of Eu(III) complexes with large π-conjugated systems and their applications
Eu(III) complexes with β-diketonate ligands have a high radiative rate constant and high color purity emission due to the strong 5D0 → 7F2 electronic dipole transition. In this section, we mainly review the photophysics of extended π-conjugated Eu(III) complexes with β-diketonate ligands and their applications.3.1 Basic photophysical properties of Eu(III) complexes with large π-conjugated system
200 cm−1), 4,4,4-trifluoro-1-phenyl-1,3-butanedione (ΔE(T1) = 21400 cm−1), or 3-(2-naphthoyl)-1,1,1-trifluoroacetonate (ΔE(T1) = 19600 cm−1) ligands and neutral ligand with a low vibrational frequency (bis[2-(diphenylphosphino)phenyl]ether oxide),88 were investigated in detail (Fig. 4a–c, Eu-hfa, Eu-btfa, and Eu-ntfa). The thermal stability (thermal decomposition point: Td) of the Eu(III) complexes bearing the extended π-conjugated ligands (Td = 320 °C and 318 °C for Eu-btfa and Eu-ntfa, respectively) was much higher than that of Eu-hfa (Td = 228 °C). The longest absorption edge was observed for Eu-ntfa (394 nm), followed by Eu-btfa (380 nm) and Eu-hfa (361 nm); this order was consistent with the length of the π-conjugation. In contrast, the emission quantum yields of π-extended Eu(III) complexes Eu-btfa (Φtot = 38%) and Eu-ntfa (Φtot = 45%) were lower than that of Eu-hfa (Φtot = 57%). The lower emission quantum yield in a large π-conjugated system can be attributed to the increased non-radiative rate constants (knr = 280, 340, and 590 s−1 for Eu-hfa, Eu-btfa, and Eu-ntfa, respectively). knr is affected by secondary vibrational quenching due to the high C–H vibrational frequency of aromatic units in electronically delocalized β-diketonate ligands (Fig. 4d). The effect on knr owing to vibrational quenching originating from the β-diketonate ligand was also demonstrated by a deuterium replacement experiment in hfa ligands.35 Thus, a π-extended β-diketonate ligand containing aromatic moieties is not a reasonable ligand for Eu(III) complexes with high 4f–4f emission quantum yields.
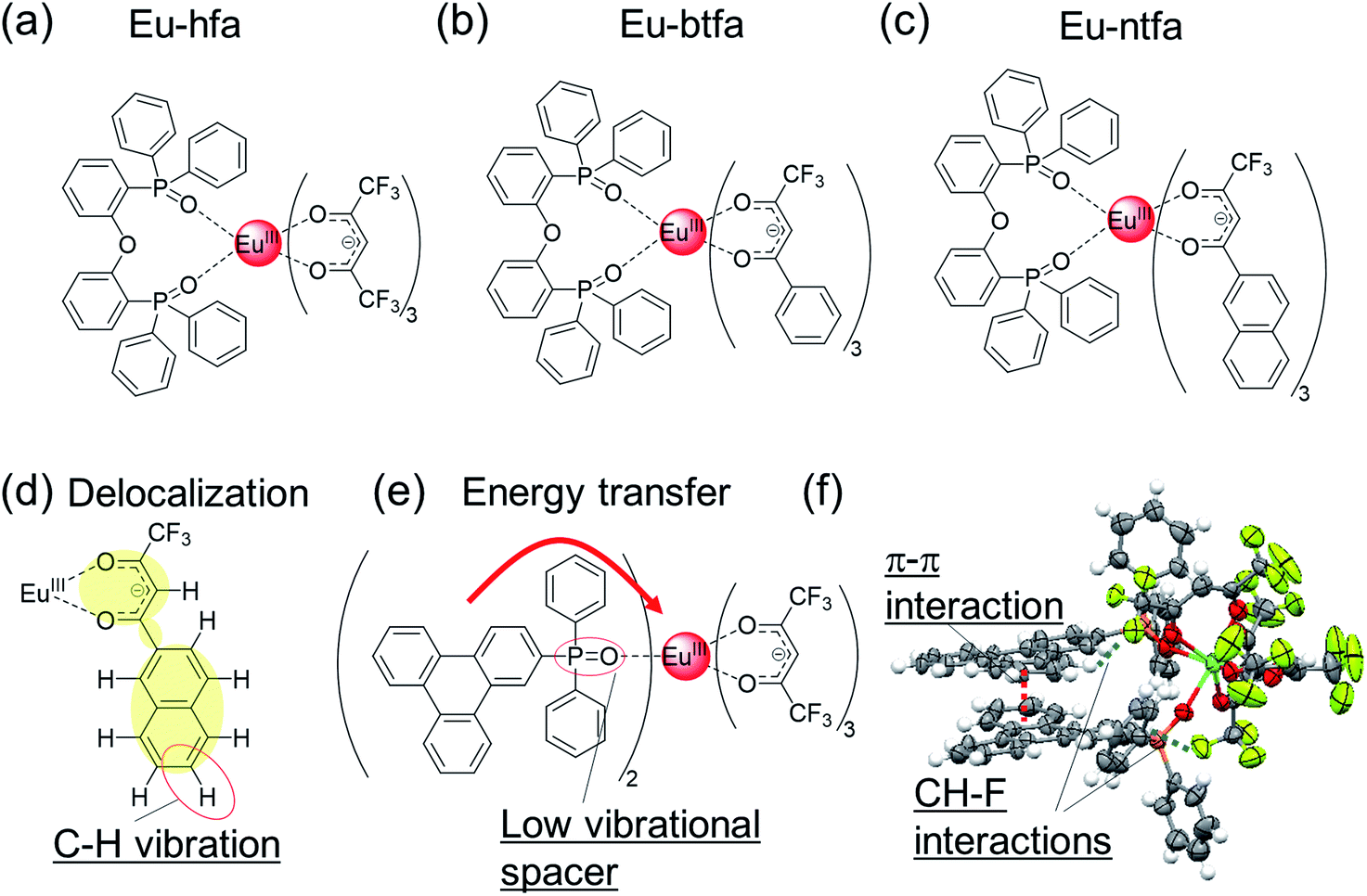 | ||
| Fig. 4 Chemical structures of (a) Eu-hfa, (b) Eu-btfa, and (c) Eu-ntfa. (d) Delocalization in Eu-ntfa. (e) Eu(III) complex with triphenylene frameworks. (f) X-ray crystal structure (ORTEP drawings) of Eu(III) complex with triphenylene frameworks. Redrawn from ref. 52. | ||
Based on the photophysical properties of Eu(III) complexes with the β-diketonate ligand, we focused on the electronic separation between the energy-donating aromatic orbital and the energy-accepting Eu(III) orbital via a phosphine spacer with a low vibrational frequency (Fig. 4e).52 This electronic separation is expected to suppress the vibrational relaxation of the Eu(III) ion and lower the rate of energy transfer. To construct an efficient energy transfer system based on the weak electronic interaction between the energy donor (aromatic ligand) and energy acceptor (Eu(III)), a triphenylene unit with high triplet state (T1) energy and long lifetime was employed. Thus, we designed an Eu(III) complex with low vibrational frequency hfa ligand and phosphine oxide ligand containing triphenylene frameworks. The crystal structure is shown in Fig. 4f. The intra- and intermolecular π–π (3.3 Å, triphenylene ligand/triphenylene ligand) and CH–F (2.6–2.9 Å, triphenylene ligand/hfa ligand) interactions result in the formation of a rigid structure, endowing high thermal stability (Td = 310 °C) by suppressing the dissociation of the hfa ligand. The UV light absorption ability of the triphenylene ligand (ε > 100000 cm−1 M−1) is much higher than that of the hfa ligand. The emission quantum yield owing to ligand excitation was estimated to be 63% (in CH2Cl2); thus, the Eu(III) complexes have remarkable thermal stability and exhibit high brightness. The detailed energy transfer mechanism in the triphenylene-based highly luminescent Eu(III) complex was revealed by time-resolved spectroscopy,89 which provides useful information on photosensitized energy transfer mechanism in luminescent Eu(III) complexes.
 | ||
| Fig. 5 (a) Energy diagram showing energy loss during photosensitized Eu(III) emission; (b) Eu(III) complex with carbazole framework (left), Eu(III) complex with N,N′-bis(salicylidene)-1,4-butanediamine and its luminescence image of LED package (right). Redrawn from ref. 95 Copyright (2018) American Chemical Society. (c) Schematic energy diagram for S–T transition (left) and Eu(III) complex with Ir(III) complex photosensitizer (right). (d) Schematic energy diagram for excited state equilibrium (left) and Eu(III) complex with stacked nanocarbon photosensitizer (right). | ||
Gong et al. successfully demonstrated blue light-sensitized emission from Eu(III) complexes bearing extended π-conjugated β-diketonate ligand containing carbazole frameworks (Fig. 5b, left).94 A characteristic pure red-emitting diode was fabricated by coating the complex phosphor onto a ∼460 nm-emitting InGaN chip. The emission quantum yield owing to ligand excitation was estimated to be 16%. The relatively low yield can be attributed to the low T1 level (18800 cm−1; below the 5D1 level). Koizuka et al. successfully prepared Eu(III) complexes with hfa and N,N′-bis(salicylidene)-1,4-butanediamine ligands, which could be excited by a blue LED chip (Fig. 5b, right, ε450 nm = 190 M−1 cm−1, Φtot = 47%).95 The blue light absorption was dependent on the effective electronic interactions between the hfa ligands and N,N′-bis(salicylidene)-1,4-butanediamine.
Chen et al. prepared a blue light-sensitized Eu(III) complex using an Ir(III) complex photosensitizer (Fig. 5c, ε450 nm = 600 M−1 cm−1, Φtot = 18%).96 The excitation window extended up to 530 nm, which can be related to the effective S–T transition, without any energy loss due to ISC (ΔES1–T1). The S–T transition is the key to photosensitization via the triplet states for enabling low-energy light excitation for photochemical processes.97–99 The T1 energy level (21200 cm−1) of the Ir(III)-based photosensitizer is appropriate for efficient energy transfer. Despite this, the emission quantum yield owing to ligand excitation was relatively low (Φtot = 18%). This can be attributed to the rapid deactivation of T1 due to the heavy-atom effect (T1 lifetime of photosensitizer, τ < 0.15 ms), leading to ineffective energy transfer from T1 to 5D1.
Recently, we reported the highest brightness in blue light sensitized Eu(III) complexes prepared using a stacked nanocarbon photosensitizer (Fig. 5d, ε450 nm = 1700 cm−1 M−1, Φtot = 36%).59 The two nanocarbon ligands are located between Eu(III) centers and form intramolecular π–π interactions (3.5Å). The stacked nanocarbon ligands are surrounded by hfa ligands, forming effective intramolecular CH–F interactions (3.0Å). The photosensitized T1 level (18800 cm−1) was lower than that of the 5D1 level, and the back energy transfer occurred from 5D0 to T1. Notably, the nanocarbon photosensitizers in rigid environments have a longer T1 lifetime (40 ms) compared to that of the Eu(III) ion (∼1 ms). The long T1 lifetime is expected to facilitate the efficient use of photons even in the case of low T1 levels, with an excited state equilibrium between 5D0 and T1. The excited state equilibrium was confirmed from the emission lifetime measurements based on the oxygen concentrations100,101 (Ar: 0.7 ms, Air: 0.5 ms). The large π-conjugated nanocarbon also induces a small ΔES1–T1 (=3700 cm−1, ΔE(S1) = 22600 cm−1). Thus, the stacked nanocarbon with long-lived photons and small ΔES1–T1 and ΔET1–5D0 energy gaps aids in low-energy light absorption and efficient energy transfer.102
3.2 Physical sensing applications of Eu(III) complexes with large π-conjugated system
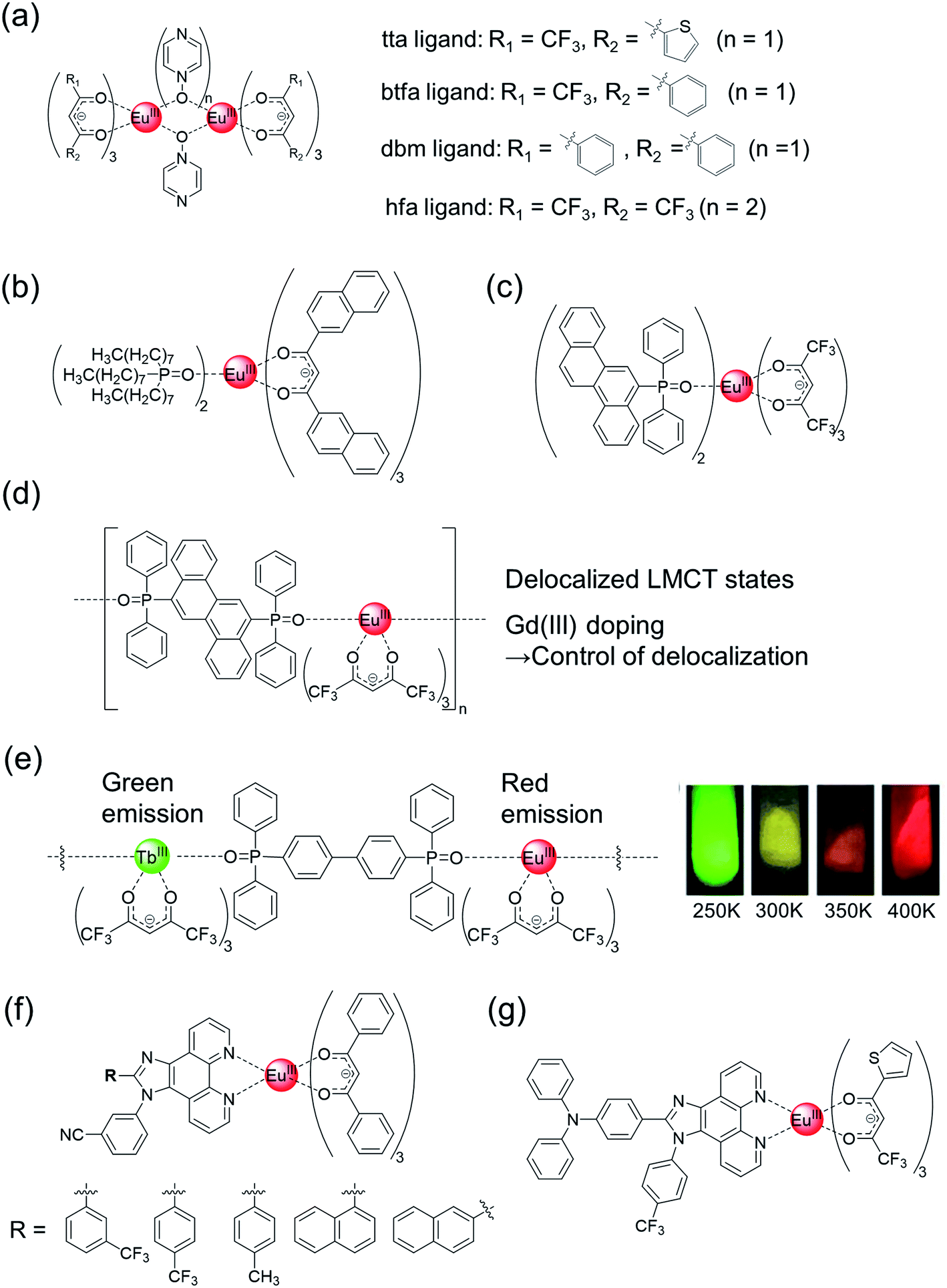 | ||
| Fig. 6 Chemical structures of Eu(III) complexes with (a) emission-intensity-based thermometer property and (b and c) emission-lifetime-based thermometer property. Chemical structures of Eu(III) coordination polymers with (d) emission-lifetime-based thermometer property and (e, left) ratiometric (Eu(III)/Tb(III))-emission-based thermometer property. (e, right) Emission photograph of Eu(III)/Tb(III) (1/99) coordination polymer under UV (365 nm) irradiation. Redrawn from ref. 111, Copyright (2013) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (f and g) Chemical structures of Eu(III) complexes with ratiometric (Eu(III)/ligand)-emission-based thermometer property. | ||
On the other hand, emission lifetime-based thermometers and ratiometric luminescence thermometers are not sensitive to variations in the luminophore concentration, its surrounding environment, or the sample viscosity. Wolfbeis et al. prepared luminescent Eu(III) nanoparticles with extended π-conjugated β-diketonate ligands for sensing and imaging temperature in the physiological range (Fig. 6b).108 We also demonstrated emission lifetime-based thermometers of the Eu(III) complexes with hfa and chrysene frameworks, which exhibited an extremely high molar absorption coefficient (490000 cm−1 M−1) in the UV region, a high intrinsic emission quantum yield (73%), and temperature-dependent energy transfer between ligands and Eu(III) ions (Fig. 6c).55 The characteristic energy transfer was explained by the LMCT based on π–f orbital interactions. The high thermostability (Td = 281 °C) was attributed to the multiple CH–F interactions, as evident from the crystal structure. This high thermostability is advantageous for developing molecular thermometers. The long range π–4f interactions in the chrysene frameworks were also investigated using an Eu(III) coordination polymer (Fig. 6d).56 The single polymer chains show the characteristic zig-zag orientation, inducing multiple CH–F interactions, and exhibit higher thermal stability (Td = 358 °C) than the mononuclear Eu(III) complex. The relative thermal sensitivity (Sm) of the Eu(III) coordination polymer with chrysene linkers was higher (Sm = 2.70% K−1 at 475 K) than those of mononuclear Eu(III) complexes (Sm = 0.89% K−1 at 475 K). The extent of LMCT delocalization was controlled by doping with Gd(III) ion, which resulted in an increased emission quantum yield and thermal sensitivity (Sm = 3.70% K−1 at 475 K). Thus, a luminescence-lifetime-based thermometer of Eu(III) complexes with high brightness, high thermostability, and high thermo-sensitivity was successfully demonstrated. The study on Eu(III) LMCT excited states of the ligand (π)- and 4f-orbitals is relatively unestablished; thus, the results also provide an useful information for future Eu(III) photo-physics study.109
Eu(III)-based ratiometric luminescence has also been utilized for the construction of effective thermometers. Historically, the most common case is the ratiomteric emission using coordination polymers composed of red-luminescent Eu(III) and green-luminescent Tb(III) centers (Fig. 6e, left).110,111 The mixed Eu(III)–Tb(III) coordination polymers exhibit strong green, yellow, orange, and red luminescence under UV irradiation (365 nm) at 250, 300, 350, and 400 K, respectively (Fig. 6e, right), which is mainly based on the temperature-dependent back energy transfer from Tb(III) to organic ligand.111 Both Eu(III) and Tb(III) exhibited long emission lifetimes (sub-millisecond), allowing for characteristic time-gated detection.112 There are several reports of ratiometric emission originating from Eu(III) phosphorescence and ligand fluorescence.113–115 Vaidyanathan et al. prepared five novel Eu(III) complexes with dibenzoylmethanate and phenantro-imidazole derivatives (Fig. 6f).114 The asymmetric hetero-conjugated system endowed additional fluorescence properties, and the five Eu(III) complexes could be utilized for thermometry owing to the ratiometric emission originating from Eu(III) phosphorescence and ligand fluorescence. In particular, the Eu(III) complex with a 4-(trifluoromethyl)phenyl substituent behaves as an effective ratiometric temperature sensor in the temperature range of 303–353 K, with a relative sensitivity of 1.97% K−1 at 313 K. Achieving high emission quantum yields in Eu(III) complexes with dual luminescence (Eu(III) phosphorescence and ligand fluorescence) is difficult (<20%, in ref. 114). Recently, it has been demonstrated that the emission quantum yield could be improved by changing the β-diketonate ligands (Fig. 6g, Φ ≈ 24%).115 These phenantro-imidazole based Eu(III) complexes are also shown to have versatile luminescent applications, such as vapour sensors and white LED, using the dual luminescent properties.113–124
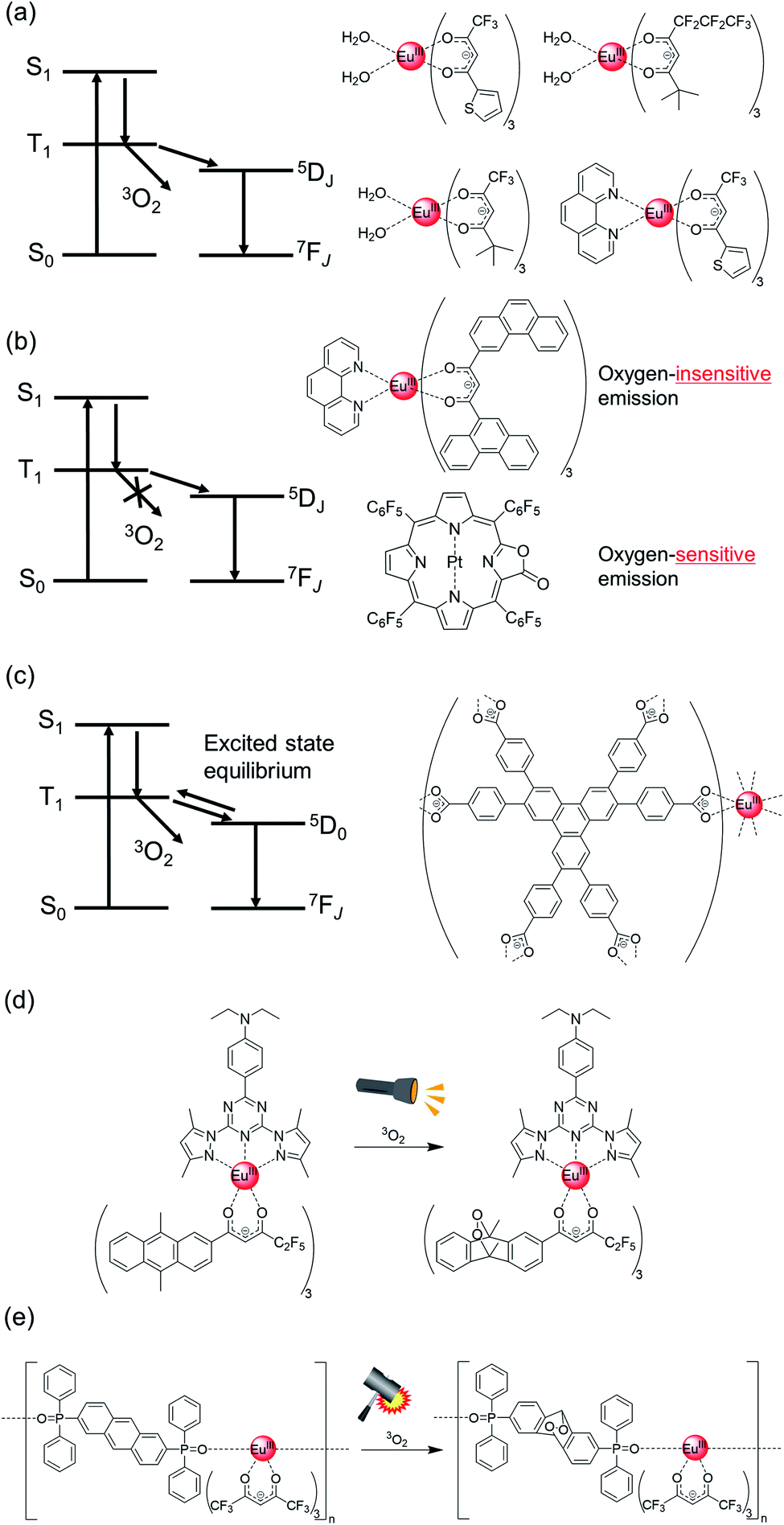 | ||
| Fig. 7 (a) Schematic energy diagram showing the quenching of T1 state by 3O2 (left) and chemical structure of Eu(III) complex with oxygen-sensitive emission (right). (b) Schematic energy diagram wherein the T1 state is not quenched by 3O2 (left) and chemical structures of Eu(III) complex with oxygen-insensitive emission and Pt(II) porphyrin with oxygen-sensitive emission (right). (c) Schematic energy diagram showing the excited state equilibrium between 5D0 and T1 (left), and chemical structure of Eu(III) complex with oxygen-sensitive emission (right). (d) Chemical structure of Eu(III) complex and chemical reaction induced by 3O2 and light irradiation. (e) Chemical structure of Eu(III) complex and chemical reaction induced by 3O2 and mechanical stress. | ||
The singlet oxygen generated by triplet quenching has also received attention because of its vital role in biological and environmental systems. Song et al. reported a visible light-excitable Eu(III) complex-based luminescent probe, which contains 2-(N,N-diethylanilin-4-yl)-4,6-bis(3,5-dimethylpyrazol-1-yl)-1,3,5-triazine as a photosensitized ligand and β-diketonate with anthracene as the reactive part (Fig. 7d).133 Anthracene reacts with the singlet oxygen generated by energy transfer from triplet anthracene to triplet oxygen. The T1 level of the anthracene unit is lower than the emitting level of Eu(III). The Eu(III) emission is strongly quenched by the β-diketonate-containing anthracene framework; however, Eu(III) shows strong emission due to the oxidization reaction because of the high T1 level in oxidized anthracene. This Eu(III) complex can also specifically localize in the mitochondria of live cells. This allows it to be used for tracing the generation of 1O2 in the mitochondria of living cells. The long emission lifetime of Eu(III) is also an important factor for distinguishing it from the strong auto-fluorescence of living cells.
In contrast to the light-excited oxygen reaction, we demonstrated a tribo-excited oxygen reaction using Eu(III) complexes. Eu(III) complexes show triboluminescence, which is a fascinating emission phenomenon that involves the transformation of mechanical energy to UV-visible light.134–139 We focused on the role of Eu(III) excitation energy in excited state chemical reactions to achieve tribo-excitation.140 Based on this concept, we prepared a Eu(III) coordination polymer with hfa and phosphine oxide ligands containing a reactive anthracene unit (bpa: 2,6-bis(diphenylphosphine oxide)anthracene). The stacked structure between single polymer chains was formed in the coordination polymer via hydrogen bonding. The anthracene unit in the phosphine oxide ligand was transformed to anthracene peroxide by mechanical stress, which was based on the singlet oxygen reaction initiated by energy transfer from the anthracene ligand to triplet oxygen. The oxidized anthracene ligands formed an emissive Eu(III) complex that allowed the monitoring of tribo-excited chemical reactions using luminescence spectroscopy. The tribo-chemical reaction in the “excited state” are fundamentally different from the general mechano-chemical reactions in the “ground states”.141–143 Such tribo-excited chemical reactions using lanthanide coordination polymers are expected to provide a new avenue for development in the fields of physical chemistry, material chemistry, and organic chemical reactions.
4 Conclusion
In this review, we have summarized the research progress on π-conjugated Eu(III) luminophores that exhibit high brightness, and we have also discussed their physical sensing applications. The electronic and steric control of large π-conjugated ligands provides high brightness and good thermostability to Eu(III) complexes. The key design points for prominent Eu(IIII) complexes are ligand–ligand interactions in rigid structures and poly-aromatic-type energy donors with a long-lived T1 state. The control of the excited state dynamics through the use of π-conjugated ligands also endows effective temperature and oxygen sensing properties. Studies on a design for luminescent Eu(III) complexes open up the frontier field of research in coordination chemistry, photochemistry, and materials science.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by grant-in-aid for grant numbers JP20H02748, JP20H04653, JP20H05197, JP20K21201, JP21K18969, and JP21J20980. This work was also supported by the Institute for Chemical Reaction Design and Discovery (ICReDD), established by the World Premier International Research Initiative (WPI) of MEXT, Japan.References
- M. Hu, H.-T. Feng, Y.-X. Yuan, Y.-S. Zheng and B. Z. Tang, Coord. Chem. Rev., 2020, 416, 213329 CrossRef CAS.
- Z. Xu, B. Z. Tang, Y. Wang and D. Ma, J. Mater. Chem. C, 2020, 8, 2614–2642 RSC.
- T. Chatterjee and K.-T. Wong, Adv. Opt. Mater., 2019, 7, 1800565 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Coord. Chem. Rev., 2018, 374, 55–92 CrossRef CAS.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef.
- M. Pan, W.-M. Liao, S.-Y. Yin, S.-S. Sun and C.-Y. Su, Chem. Rev., 2018, 118, 8889–8935 CrossRef CAS.
- M. Lu, Y. Zhang, S. Wang, J. Guo, W. W. Yu and A. L. Rogach, Adv. Funct. Mater., 2019, 29, 1902008 CrossRef.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- X. Qin, X. Liu, W. Huang, M. Bettinelli and X. Liu, Chem. Rev., 2017, 117, 4488–4527 CrossRef CAS.
- G. Li, Y. Tian, Y. Zhao and J. Lin, Chem. Soc. Rev., 2015, 44, 8688–8713 RSC.
- J. Dalal, M. Dalal, S. Devi, R. Devi, A. Hooda, A. Khatkar, V. B. Taxak and S. P. Khatkar, J. Lumin., 2019, 210, 293–302 CrossRef CAS.
- J. Dalal, M. Dalal, S. Devi, A. Hooda, A. Khatkar, V. B. Taxak and S. P. Khatkar, J. Lumin., 2019, 216, 116697 CrossRef CAS.
- J. Dalal, M. Dalal, S. Devi, P. Dhankhar, A. Hooda, A. Khatkar, V. B. Taxak and S. P. Khatkar, Mater. Chem. Phys., 2020, 243, 122631 CrossRef CAS.
- P. Phogat, S. P. Khatkar, R. K. Malik, J. Dalal, A. Hooda and V. B. Taxak, J. Lumin., 2021, 234, 117984 CrossRef CAS.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- Y.-H. Won, O. Cho, T. Kim, D.-Y. Chung, T. Kim, H. Chung, H. Jang, J. Lee, D. Kim and E. Jang, Nature, 2019, 575, 634–638 CrossRef CAS.
- H. Liao, M. Zhao, M. S. Molokeev, Q. Liu and Z. Xia, Angew. Chem., Int. Ed., 2018, 57, 11728–11731 CrossRef CAS.
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef.
- H. Xu, Q. Sun, Z. An, Y. Wei and X. Liu, Coord. Chem. Rev., 2015, 293–294, 228–249 CrossRef CAS.
- M. Gaft, R. Reisfeld and G. Panczer, Modern Luminescence Spectroscopy of Minerals and Materials, Springer, Heidelberg, 2nd edn, 2015 Search PubMed.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4450–4455 CrossRef CAS.
- K. A. Gschneidner, J.-C. G. Bünzli and V. K. Pecharsky, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, Amsterdam, 2007, vol. 37 Search PubMed.
- K. Binnemans, Handb. Phys. Chem. Rare Earths, 2005, 35, 107–272 Search PubMed.
- A. F. Kirby and F. S. Richardson, J. Phys. Chem., 1983, 87, 2544–2556 CrossRef CAS.
- M. Tsurui, Y. Kitagawa, K. Fushimi, M. Gon, K. Tanaka and Y. Hasegawa, Dalton Trans., 2020, 49, 5352–5361 RSC.
- E. E. S. Teotonio, G. M. Fett, H. F. Brito, W. M. Faustino, G. F. de Sá, M. C. F. C. Felinto and R. H. A. Santos, J. Lumin., 2008, 128, 190–198 CrossRef CAS.
- K. Yanagisawa, T. Nakanishi, Y. Kitagawa, T. Seki, T. Akama, M. Kobayashi, T. Taketsugu, H. Ito, K. Fushimi and Y. Hasegawa, Eur. J. Inorg. Chem., 2015, 2015, 4769–4774 CrossRef CAS.
- N. B. D. Lima, S. M. C. Gonçalves, S. A. Júnior and A. M. Simas, Sci. Rep., 2013, 3, 2395–2402 CrossRef.
- Y. Hasegawa, K. Murakoshi, Y. Wada, S. Yanagida, J.-H. Kim, N. Nakashima and T. Yamanaka, Chem. Phys. Lett., 1996, 248, 8–12 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- Y. Hasegawa, M. Yamamuro, Y. Wada, N. Kanehisa, Y. Kai and S. Yanagida, J. Phys. Chem. A, 2003, 107, 1697–1702 CrossRef CAS.
- N. Nijegorodov, V. Ramachandran and D. P. Winkoun, Spectrochim. Acta, Part A, 1997, 53, 1813 CrossRef.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- S. Sato and M. Wada, Bull. Chem. Soc. Jpn., 1970, 43, 1955–1962 CrossRef CAS.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- J. R. Darwent, W. Dong, C. D. Flint and N. W. Sharpe, J. Chem. Soc., Faraday Trans., 1993, 89, 873–880 RSC.
- M. L. P. Reddy, V. Divya and R. Pavithran, Dalton Trans., 2013, 42, 15249–15262 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Y. Kitagawa, R. Ohno, T. Nakanishi, K. Fushimi and Y. Hasegawa, Phys. Chem. Chem. Phys., 2016, 18, 31012–31016 RSC.
- M. Kasha, Chem. Rev., 1947, 41, 401–419 CrossRef CAS.
- K. Palewska and H. Chojnacki, Mol. Cryst. Liq. Cryst., 1993, 229, 31–36 CrossRef CAS.
- D. D. Morgan, D. Warshawsky and T. Atkinson, Photochem. Photobiol., 1977, 25, 31–38 CrossRef CAS.
- H. Okamoto, M. Yamaji, S. Gohda, K. Sato, H. Sugino and K. Satake, Res. Chem. Intermed., 2013, 39, 147–159 CrossRef CAS.
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. van der Tol and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 9408–9414 CrossRef CAS.
- O. Pietraszkiewicz, S. Mal, M. Pietraszkiewicz, M. Maciejczyk, I. Czerski, T. Borowiak, G. Dutkiewicz, O. Drobchak, L. Penninck, J. Beeckman and K. Neyts, J. Photochem. Photobiol., A, 2012, 250, 85–91 CrossRef CAS.
- S. I. Klink, L. Grave, D. N. Reinhoudt, F. C. van Veggel, M. H. V. Werts, F. A. J. Geurts and J. W. Hofstraat, J. Phys. Chem. A, 2000, 104, 5457–5468 CrossRef CAS.
- Y. Kitagawa, F. Suzue, T. Nakanishi, K. Fushimi and Y. Hasegawa, Dalton Trans., 2018, 47, 7327–7332 RSC.
- Y. Hasegawa, T. Sawanobori, Y. Kitagawa, S. Shoji, K. Fushimi, Y. Nakasaka, T. Masuda and I. Hisaki, ChemPlusChem, 2020, 85, 1989–1993 CrossRef CAS.
- F. J. Steemers, H. G. Meuris, W. Verboom, D. N. Reinhoudt, E. B. van Der Tol and J. W. Verhoeven, J. Org. Chem., 1997, 62, 4229–4235 CrossRef CAS.
- Y. Kitagawa, M. Kumagai, T. Nakanishi, K. Fushimi and Y. Hasegawa, Inorg. Chem., 2020, 59, 5865–5871 CrossRef CAS.
- Y. Kitagawa, M. Kumagai, P. P. Ferreira da Rosa, K. Fushimi and Y. Hasegawa, Chem. – Eur. J., 2021, 27, 264–269 CrossRef CAS.
- Y. Kitagawa, M. Kumagai, P. P. Ferreira da Rosa, K. Fushimi and Y. Hasegawa, Dalton Trans., 2020, 49, 3098–3101 RSC.
- M. Planells, E. Klampaftis, M. Congiu, R. Shivanna, K. V. Rao, O. Chepelin, A. C. Jones, B. S. Richards, S. J. George, K. S. Narayan and N. Robertson, Eur. J. Inorg. Chem., 2014, 2014, 3095–3100 CrossRef CAS.
- Y. Kitagawa, F. Suzue, T. Nakanishi, K. Fushimi, T. Seki, H. Ito and Y. Hasegawa, Commun. Chem., 2020, 3, 3 CrossRef CAS.
- G.-J. Chen, X. Qiao, J.-L. Tian, J.-Y. Xu, W. Gu, X. Liu and S.-P. Yan, Dalton Trans., 2010, 39, 10637–10643 RSC.
- H. Song, C. Fan, R. Wang, Z. Wang and S. Pu, J. Coord. Chem., 2020, 73, 2311–2327 CrossRef CAS.
- Z. Abbas, P. Singh, S. Dasari, S. Sivakumar and A. K. Patra, New J. Chem., 2020, 44, 15685–15697 RSC.
- Y. Kitagawa, M. Kumagai, K. Fushimi and Y. Hasegawa, Chem. Phys. Lett., 2020, 749, 137437 CrossRef CAS.
- S. Dasari, S. Singh, S. Sivakumar and A. K. Patra, Chem. – Eur. J., 2016, 22, 17387–17396 CrossRef CAS.
- D. Qiang, X. Yang, X. Deng and H.-L. Sun, CrystEngComm, 2016, 18, 8159–8163 RSC.
- T.-A. Uchida, K. Nozaki and M. Iwamura, Chem. –Asian J., 2016, 11, 2415–2422 CrossRef CAS.
- S. Dasari and A. K. Patra, Dalton Trans., 2015, 44, 19844–19855 RSC.
- L.-N. Zhang, A.-L. Liu, Y.-X. Liu, J.-X. Shen, C.-X. Du and H.-W. Hou, Inorg. Chem. Commun., 2015, 56, 137–140 CrossRef CAS.
- Z. Weng, D. Liu, Z. Chen, H. Zou, S. Qin and F. Liang, Cryst. Growth Des., 2009, 9, 4163–4170 CrossRef CAS.
- X. Li, D. Zhang and J. Li, Spectrochim. Acta, Part A, 2014, 127, 1–9 CrossRef CAS.
- L. Wang, B. Li, L. Zhang, P. Li and H. Jiang, Dyes Pigm., 2013, 97, 26–31 CrossRef CAS.
- K. Gislason and S. T. Sigurdsson, Bioorg. Med. Chem. Lett., 2013, 23, 264–267 CrossRef CAS.
- S. Deslandes, C. Galaup, R. Poole, B. Mestre-Voegtlé, S. Soldevila, N. Leygue, H. Bazin, L. Lamarque and C. Picard, Org. Biomol. Chem., 2012, 10, 8509–8523 RSC.
- N. Petkova, S. Gutzov, N. Lesev, S. Kaloyanova, S. Stoyanov and T. Deligeorgiev, Opt. Mater., 2011, 33, 1715–1720 CrossRef CAS.
- L. Zhang and B. Li, J. Lumin., 2009, 129, 1304–1308 CrossRef CAS.
- G.-L. Law, D. Parker, S. L. Richardson and K.-L. Wong, Dalton Trans., 2009, 8481–8484 RSC.
- R. Zong, G. Zhang, S. V. Eliseeva, J.-C. G. Bünzli and R. P. Thummel, Inorg. Chem., 2010, 49, 4657–4664 CrossRef CAS.
- L. Zhang, B. Li, L. Zhang and Z. Su, ACS Appl. Mater. Interfaces, 2009, 1, 1852–1855 CrossRef CAS.
- E. J. New, D. Parker and R. D. Peacock, Dalton Trans., 2009, 672–679 RSC.
- R. A. Poole, F. Kiela, S. L. Richardson, P. A. Stenson and D. Parker, Chem. Commun., 2006, 4084 RSC.
- R. A. Poole, G. Bobba, M. J. Cann, J.-C. Frias, D. Parker and R. D. Peacock, Org. Biomol. Chem., 2005, 3, 1013–1024 RSC.
- S. I. Klink, G. A. Hebbink, L. Grave, P. G. Oude Alink, F. C. van Veggel and M. H. V. Werts, J. Phys. Chem. A, 2002, 106, 3681–3689 CrossRef CAS.
- A. Köhler and D. Beljonne, Adv. Funct. Mater., 2004, 14, 11–18 CrossRef.
- Naphthalene (ΔE(S1) = 31170 cm−1),36 phenanthrene (ΔE(S1) = 28920 cm−1),47 [4]-helicene (ΔE(S1) = 26870 cm−1),46 triphenylene
(ΔE(S1) = 29420 cm−1),47 chrysene (ΔE(S1) = 27750 cm−1),47 [5]-helicene (ΔE(S1) = 25250 cm−1),46 picene (ΔE(S1) = 26620 cm−1),48 [6]-helicene (ΔE(S1) = 24390 cm−1),46 coronene (ΔE(S1) = 23320 cm−1),47 and phenacene (ΔE(S1) = 25710 cm−1).48.
- J. Yuan and K. Matsumoto, Anal. Sci., 1996, 12, 31–36 CrossRef CAS.
- H. Iwanaga, J. Lumin., 2018, 200, 233–239 CrossRef CAS.
- D. B. A. Raj, S. Biju and M. L. P. Reddy, Inorg. Chem., 2008, 47, 8091–8100 CrossRef CAS.
- T. Koizuka, M. Yamamoto, Y. Kitagawa, T. Nakanishi, K. Fushimi and Y. Hasegawa, Bull. Chem. Soc. Jpn., 2017, 90, 1287–1292 CrossRef CAS.
- S. Miyazaki, K. Miyata, H. Sakamoto, F. Suzue, Y. Kitagawa, Y. Hasegawa and K. Onda, J. Phys. Chem. A, 2020, 124, 6601–6606 CrossRef CAS.
- M. L. P. Reddy, V. Divya and R. Pavithran, Dalton Trans., 2013, 42, 15249–15262 RSC.
- T. Wu, C.-W. Sher, Y. Lin, C.-F. Lee, S. Liang, Y. Lu, S.-W. H. Chen, W. Guo, H.-C. Kuo and Z. Chen, Appl. Sci., 2018, 8, 1557 CrossRef.
- Z. Liu, C. H. Lin, B. R. Hyun, C. W. Sher, Z. Lv, B. Luo, F. Jiang, T. Wu, C. H. Ho, H. C. Kuo and J. H. He, Light: Sci. Appl., 2020, 9, 83 CrossRef CAS.
- K. Ding, V. Avrutin, N. Izyumskaya, Ü. Özgür and H. Morkoç, Appl. Sci., 2019, 9, 1206 CrossRef CAS.
- P. He, H. H. Wang, H. G. Yan, W. Hu, J. X. Shi and M. L. Gong, Dalton Trans., 2010, 39, 8919–8924 RSC.
- T. Koizuka, K. Yanagisawa, Y. Hirai, Y. Kitagawa, T. Nakanishi, K. Fushimi and Y. Hasegawa, Inorg. Chem., 2018, 57, 7097–7103 CrossRef CAS.
- F.-F. Chen, Z.-Q. Bian, Z.-W. Liu, D.-B. Nie, Z.-Q. Chen and C.-H. Huang, Inorg. Chem., 2008, 47, 2507–2513 CrossRef CAS.
- Y. Yamazaki and O. Ishitani, Chem. Sci., 2018, 9, 1031–1041 RSC.
- T. K. K. Nonomura, N. J. Jeon, F. Giordano, A. Abate, S. Uchida, T. Kubo, S. I. Seok, M. K. Nazeeruddin, A. Hagfeldt, M. Grätzel and H. Segawa, Nat. Commun., 2015, 6, 8834 CrossRef.
- Y. Sasaki, S. Amemori, H. Kouno, N. Yanai and N. Kimizuka, J. Mater. Chem. C, 2017, 5, 5063–5067 RSC.
- S. Quici, M. Cavazzini, G. Marzanni, G. Accorsi, N. Armaroli, B. Ventura and F. Barigelletti, Inorg. Chem., 2005, 44, 529–537 CrossRef CAS.
- T. J. Sørensen, A. M. Kenwright and S. Faulkner, Chem. Sci., 2015, 6, 2054–2059 RSC.
- At the present stage, the existence or non-existence of effective energy transfer process from S1 state to 5DJ (J = 0, 1, and 2) is unknown, but the energy transfer process via triplet states is confirmed by the emission lifetime depending on oxygen concentration.59.
- X.-D. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- Y. Cui, F. Zhu, B. Chen and G. Qian, Chem. Commun., 2015, 51, 7420–7431 RSC.
- J. Rocha, C. D. S. Brites and L. D. Carlos, Chem. - Eur. J., 2016, 22, 14782–14795 CrossRef CAS.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- L. Bellucci, G. Bottaro, L. Labella, V. Causin, F. Marchetti, S. Samaritani, D. Belli Dell'Amico and L. Armelao, Inorg. Chem., 2020, 59, 18156–18167 CrossRef CAS.
- H. Peng, M. I. J. Stich, J. Yu, L.-N. Sun, L. H. Fischer and O. S. Wolfbeis, Adv. Mater., 2010, 22, 716–719 CrossRef CAS.
- Y. Kitagawa, P. P. Ferreira da Rosa and Y. Hasegawa, Dalton Trans., 2021, 50, 14978–14984 RSC.
- Y. Cui, H. Xu, Y. Yue, Z. Guo, J. Yu, Z. Chen, J. Gao, Y. Yang, G. Qian and B. Chen, J. Am. Chem. Soc., 2012, 134, 3979–3982 CrossRef CAS.
- K. Miyata, Y. Konno, T. Nakanishi, A. Kobayashi, M. Kato, K. Fushimi and Y. Hasegawa, Angew. Chem., Int. Ed., 2013, 52, 6413–6416 CrossRef CAS.
- M. Tropiano and S. Faulkner, Chem. Commun., 2014, 50, 4696–4698 RSC.
- R. Boddula, K. Singh, S. Giri and S. Vaidyanathan, Inorg. Chem., 2017, 56, 10127–10130 CrossRef CAS.
- R. Devi, M. Rajendran, K. Singh, R. Pal and S. Vaidyanathan, J. Mater. Chem. C, 2021, 9, 6618–6633 RSC.
- R. Boddula, J. Tagare, K. Singh and S. Vaidyanathan, Mater. Chem. Front., 2021, 5, 3159–3175 RSC.
- M. Rajendran, R. Devi, S. Mund, K. Singh and S. Vaidyanathan, J. Mater. Chem. C, 2021, 9, 15034–15046 RSC.
- R. Devi, K. Singh and S. Vaidyanathan, J. Mater. Chem. C, 2020, 8, 8643–8653 RSC.
- R. Devi, R. Boddula, J. Tagare, A. B. Kajjam, K. Singh and S. Vaidyanathan, J. Mater. Chem. C, 2020, 8, 11715–11726 RSC.
- R. Devi and S. Vaidyanthan, Dalton Trans., 2020, 49, 6205–6219 RSC.
- B. Rajamouli, R. Devi, A. Mohanty, V. Krishnan and S. Vaidyanathan, New J. Chem., 2017, 41, 9826–9839 RSC.
- B. Rajamouli and V. Sivakumar, New J. Chem., 2017, 41, 1017–1027 RSC.
- K. Singh, R. Boddula and S. Vaidyanathan, Inorg. Chem., 2017, 56, 9376–9390 CrossRef CAS.
- B. Rajamouli, P. Sood, S. Giri, V. Krishnan and S. Vaidyanathan, Eur. J. Inorg. Chem., 2016, 2016, 3900–3911 CrossRef CAS.
- B. Rajamouli and V. Sivakumar, J. Photochem. Photobiol., A, 2017, 347, 26–40 CrossRef.
- X.-D. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- B. M. Luby, C. D. Walsh and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 2558–2569 CrossRef CAS.
- J. W. Gregory, H. Sakaue, T. Liu and J. P. Sullivan, Annu. Rev. Fluid. Mech., 2014, 46, 303–330 CrossRef.
- T. Yoshihara, Y. Hirakawa, M. Hosaka, M. Nangaku and S. Tobita, J. Photochem. Photobiol., C, 2017, 30, 71–95 CrossRef CAS.
- Y. Amao, Microchim. Acta, 2003, 143, 1–12 CrossRef CAS.
- A. Ruggi, F. W. B. van Leeuwen and A. H. Velders, Coord. Chem. Rev., 2011, 255, 2542–2554 CrossRef CAS.
- Y. Amao, I. Okura and T. Miyashita, Bull. Chem. Soc. Jpn., 2000, 73, 2663–2668 CrossRef CAS.
- B. Zelelow, G. E. Khalil, G. Phelan, B. Carlson, M. Gouterman, J. B. Callis and L. R. Dalton, Sens. Actuators, B, 2003, 96, 304–314 CrossRef CAS.
- H. Ma, X. Wang, B. Song, L. Wang, Z. Tang, T. Luo and J. Yuan, Dalton Trans., 2018, 47, 12852–12857 RSC.
- X.-F. Chen, X.-H. Zhu, Y.-H. Xu, S. S. S. Raj, S. Öztürk, H.-K. Fun, J. Ma and X.-Z. You, J. Mater. Chem., 1999, 9, 2919–2922 RSC.
- S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J.-C. G. Bünzli and N. P. Kuzmina, Inorg. Chem., 2010, 49, 9300–9311 CrossRef CAS.
- Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito and Y. Hasegawa, Angew. Chem., Int. Ed., 2017, 56, 7171–7175 CrossRef CAS.
- C. R. Hurt, N. Mcavoy, S. Bjorklund and N. Filipescu, Nature, 1966, 212, 179–180 CrossRef CAS.
- L. M. Sweeting and A. L. Rheingold, J. Am. Chem. Soc., 1987, 109, 2652–2658 CrossRef CAS.
- Y. Hasegawa, R. Hieda, K. Miyata, T. Nakagawa and T. Kawai, Eur. J. Inorg. Chem., 2011, 2011, 4978–4984 CrossRef CAS.
- Y. Kitagawa, A. Naito, K. Fushimi and Y. Hasegawa, Chem. - Eur. J., 2021, 27, 2279–2283 CrossRef CAS.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- D. Tan and T. Friščić, Eur. J. Org. Chem., 2018, 2018, 18–33 CrossRef CAS.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical structures and photophysical parameters of Eu(III) complexes (photophysical parameters of several Eu(III) complexes are not available). See DOI: 10.1039/d1ra08233g |
| This journal is © The Royal Society of Chemistry 2022 |