
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic resonance imaging of tumor-associated-macrophages (TAMs) with a nanoparticle contrast agent†
Junhan Zhou a,
Vijaykumar S. Meli‡
b,
Esther Yu-Tin Chen‡b,
Rohan Kaprecd,
Raji Nagallab,
Wenwu Xiaoef,
Alexander D. Borowskyfgh,
Kit S. Lamaefi,
Wendy F. Liub and
Angelique Y. Louie*ac
a,
Vijaykumar S. Meli‡
b,
Esther Yu-Tin Chen‡b,
Rohan Kaprecd,
Raji Nagallab,
Wenwu Xiaoef,
Alexander D. Borowskyfgh,
Kit S. Lamaefi,
Wendy F. Liub and
Angelique Y. Louie*ac
aChemistry Graduate Group, University of California, Davis, CA 95616, USA. E-mail: aylouie@ucdavis.edu
bDepartment of Biomedical Engineering, University of California, Irvine, CA 92697, USA
cDepartment of Biomedical Engineering, University of California, Davis, CA 95616, USA
dBiostatistics Graduate Group, University of California, Davis, CA 95616, USA
eDepartment of Biochemistry and Molecular Medicine, University of California, Davis, CA 95616, USA
fComprehensive Cancer Center, University of California, Davis, CA 95616, USA
gDepartment of Pathology and Laboratory Medicine, University of California, Davis, CA 95616, USA
hCenter for Immunology and Infectious Diseases, University of California, Davis, CA 95616, USA
iDivision of Hematology &Oncology, Department of Internal Medicine, University of California, Davis, CA 95616, USA
First published on 8th March 2022
Abstract
In the tumor micro-environment, tumor associated macrophages (TAMs) represent a predominant component of the total tumor mass, and TAMs play a complex and diverse role in cancer pathogenesis with potential for either tumor suppressive, or tumor promoting biology. Thus, understanding macrophage localization and function are essential for cancer diagnosis and treatment. Typically, tissue biopsy is used to evaluate the density and polarization of TAMs, but provides a limited “snapshot” in time of a dynamic and potentially heterogeneous tumor immune microenvironment. Imaging has the potential for three-dimensional mapping; however, there is a paucity of macrophage-targeted contrast agents to specifically detect TAM subtypes. We have previously found that sulfated-dextran coated iron oxide nanoparticles (SDIO) can target macrophage scavenger receptor A (SR-A, also known as CD204). Since CD204 (SR-A) is considered a biomarker for the M2 macrophage polarization, these SDIO might provide M2-specific imaging probes for MRI. In this work, we investigate whether SDIO can label M2-polarized cells in vitro. We evaluate the effect of degree of sulfation on uptake by primary cultured bone marrow derived macrophages (BMDM) and found that a higher degree of sulfation led to higher uptake, but there were no differences across the subtypes. Further analysis of the BMDM showed similar SR-A expression across stimulation conditions, suggesting that this classic model for macrophage subtypes may not be ideal for definitive M2 subtype marker expression, especially SR-A. We further examine the localization of SDIO in TAMs in vivo, in the mammary fat pad mouse model of breast cancer. We demonstrate that uptake by TAMs expressing SR-A scales with degree of sulfation, consistent with the in vitro studies. The TAMs demonstrate M2-like function and secrete Arg-1 but not iNOS. Uptake by these M2-like TAMs is validated by immunohistochemistry. SDIO show promise as a valuable addition to the toolkit of imaging probes targeted to different biomarkers for TAMs.
Introduction
Macrophages are active members of the immune system and play versatile roles in phagocytosis, presentation of foreign antigens, and producing biochemical signals in inflammation. The complexity of the macrophage response to diseases and crucial metabolic events has only recently been appreciated, and it is now understood that macrophages polarize between functions that inhibit or promote disease.1–4 At one extreme, the M1 phenotype expresses markers such as CD40 and CD86, secretes inflammatory cytokines, and is associated with a host-defense, pro-inflammatory function. M1 macrophages can be produced in culture by exposing cells to interferon-γ (IFN-γ) and lipopolysaccharide (LPS). At the opposite polarity, M2 macrophages are elevated in markers such as CD206, CD204 (SR-A) and CD163, and are associated with tissue remodeling and immune suppression.5–8 M2 macrophages can be produced in culture by stimulation with IL-4 and IL-13.9,10Increasingly, distinct macrophage subtypes have been associated with disease progression or resolution. Macrophages can sometimes represent 50% of tumor mass and the presence of tumor-infiltrating macrophages has been connected to poor prognosis in the majority of human malignant tumors.11 Because of this correlation, it has been assumed that the TAMs are primarily M2-like and there is an interest in co-therapies to reduce macrophage infiltration.12 However, there also have been reports of cases in which the opposite was observed: in studies of prostate cancer13 and osteosarcoma patients14 TAM infiltration was associated with better survival and thus, macrophage activating agents were proposed.15 The function of TAMs seems to have a complex relationship with cancer type, stage, and microenvironment. These previous studies identified TAMs with generic macrophage markers such as CD68, which do not inform on subtype, only total macrophage burden. The differences in prognostic ability may be due to differences in polarization state of the TAMs. When and how macrophages are recruited to tumors and polarized to each subtype is not well understood. Elimination at the wrong time or in the wrong type of cancer may exacerbate the disease. It is clear that the ability to assess polarization in vivo is crucial to understanding macrophage impact, and ultimately, will aid in the design of appropriate therapies to modulate macrophages. However, TAMs are challenging to identify as they exist in a range of polarization and thus, express overlapping markers; one cannot identify specific TAM phenotypes with a single marker.16
Most clinical data on macrophage polarization markers have been obtained from histology on extracted patient tumor tissues. However, biopsy is non-ideal for assessment for TAMs, which have heterogeneous localization in tumors.17 TAMs can be found throughout the tumor, such as in perivascular, vascular and hypoxic spaces; moreover, their polarization state can differ in each microenvironment.18 Given the lack of uniformity in tumors and other diseases, noninvasive imaging offers a far superior method to identify subtypes of macrophages over biopsy. Imaging macrophage subtypes is still a nascent effort and there is no method, currently, to reliably identify polarization subtypes in vivo. There is a strong need for better in vivo tools to characterize macrophage polarization.
Noninvasive imaging modalities have great potential for detecting macrophages, but are limited in capabilities for detailed subtype imaging.19 For example, CT (computed tomography) is the most commonly used imaging modality in clinic. It provides high spatial resolution but low soft tissue contrast. PET (positron emission tomography) and SPECT (single-photon emission computed tomography) have high contrast sensitivity but poor spatial resolution. Comparatively, MRI (magnetic resonance imaging) offers high soft tissue contrast and high spatial resolution.20
Macrophage subtype-targeted probes for PET and/or MRI have primarily utilized antibody conjugated probes.21–24 There have been efforts to image macrophage subtypes in vivo, primarily through optical imaging with antibody conjugated fluorescent probes.25–29 Antibodies have high specificity and affinity to their targets, but still have limitations. For example, high-cost, short half-life, low efficacy, and potential for immunogenicity.30–32
We have previously developed nanoparticulate contrast agents for magnetic resonance imaging (MRI) that are targeted to macrophage scavenger receptors. We showed that these nanoparticles label macrophages in vivo in cardiovascular inflammation models.33 Since CD204 (SR-A) is a biomarker for M2 like macrophages,5–8 these sulfated dextran coated iron oxide nanoparticles could have promise as novel M2-specific imaging probes for MRI. In this work we sought to characterize the uptake of these agents by different macrophage subtypes, and to evaluate if uptake of the agent alters viability or phenotype. We further examine uptake by tumor-associated macrophages in vivo in a mouse breast tumor model.
Results and discussion
Synthesis and physical properties of SDIO/DIO
The synthesis routes were as previously reported34 and shown in Scheme 1. In brief, reduced dextran and FeCl3⋅6H2O were dissolved in degassed deionized ultra-filtered water. FeCl2⋅4H2O solution and concentrated ammonia were added dropwise at 0–5 °C under argon. After 3 hours at 85 °C, DIO was formed and purified by dialysis against deionized water (50 kDa cutoff) for 72 hours. DIO was obtained as a brown powder after lyophilization.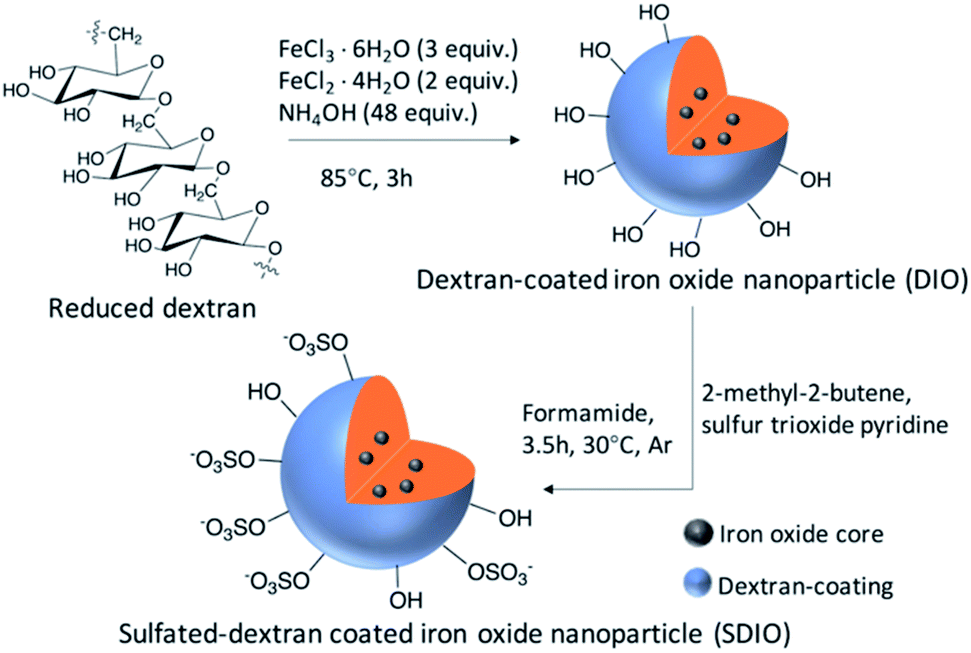 | ||
| Scheme 1 Synthetic route of SDIO/DIO. | ||
DIO was dissolved in dry formamide, and 2-methyl-2-butene was added to the DIO solution under argon. Sulfur trioxide pyridine was added into the mixture. Then, the mixture was stirred at 30 °C under argon for 3.5 hours with refluxing. Saturated NaHCO3 aqueous solution was used to quench the reaction. The product was purified by dialysis against deionized water (50 kDa cutoff) for 72 h. SDIO was obtained as a brown powder after lyophilization. Stoichiometries of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 7:1, 5:1 and 1:1 of S:OH SDIO were synthesized.
:1, 7:1, 5:1 and 1:1 of S:OH SDIO were synthesized.
The average hydrodynamic diameters were 37.06 nm, 66.06 nm, 74.84 nm, 78.00 nm and 70.94 nm for DIO, 1:1 SDIO, 5:1 SDIO, 7:1 SDIO, and 10:1 SDIO, respectively (Table 1). The mean size of SDIO was larger than DIO, which we hypothesize is due to the much more negative surface charge, shown by zeta-potential, which increases the solvent layer thickness around the nanoparticles. The core sizes were similar both in SDIO and DIO, which were measured by transmission electron microscopy (TEM). The elemental analysis results showed that the sulfur content (wt%) of SDIO scaled up with higher S:OH. The wt% of iron decreases with higher S:OH due to the increase of sulfur in SDIO. The r1 and r2 relaxivities at 1.4 T magnetic field were calculated and show that DIO and SDIO are good T2 contrast agents, with high r2/r1 ratios (Table 1). The r1 and r2 relaxivities at 7 T magnetic field were determined for the particles used in subsequent in vitro and in vivo studies. The physical properties are summarized and shown in Table 1. The longitudinal and transverse relaxivity plots from data collected at 1.4 T and 7 T are shown in ESI as Fig. S1 and S2.†
| Type | DIO | 1:1 SDIO |
5:1 SDIO |
7:1 SDIO |
10:1SDIO |
|---|---|---|---|---|---|
| Hydrodynamic diameter (nm) | 37.06 ± 1.64 | 66.06 ± 2.37 | 74.84 ± 1.34 | 78.00 ± 1.17 | 70.94 ± 2.25 |
| Core size (nm) | 4–6 | 4–6 | — | — | 4–6 |
| Zeta-potential (mV) | 5.4 ± 0.8 | −39.4 ± 4.6 | −31.6 ± 5.13 | −39.2 ± 3.4 | −58.2 ± 5.3 |
| Fe% (wt%) | 16.14 | 12.50 | 8.63 | 8.61 | 8.38 |
| S% (wt%) | n/a | 5.41 | 13.50 | 13.90 | 13.95 |
| r1 (mM−1 s−1) at 1.4 T | 16.60 | 14.76 | 17.89 | 16.14 | 14.90 |
| r2 (mM−1 s−1) at 1.4 T | 119.40 | 95.08 | 108.57 | 104.78 | 90.80 |
| r1 (mM−1 s−1) at 7 T | 1.675 | 1.649 | — | — | 1.343 |
| r2 (mM−1 s−1) at 7 T | 145.06 | 114.13 | — | — | 97.62 |
Bone marrow derived macrophages (BMDMs) uptake studies
M0 (no stimulation), low dose M1 stimulation (M1L, 0.3 ng mL−1 LPS, 1 ng mL−1 IFN-γ), high dose M1 stimulation (M1H, 10 ng mL−1 LPS, 10 ng mL−1 IFN-γ) and M2 (20 ng mL−1 IL-4, 20 ng mL−1 IL-13) macrophage subtypes all showed much greater uptake of 1:1 SDIO and/or 10:1 SDIO compared to DIO and media only conditions, as measured by relaxometry (lower T2 values = higher [Fe]) and ICP-OES on cell lysates. By both measures, BMDMs took up more SDIO than DIO.
As shown in Fig. 1, BMDMs took up very little DIO as there were no significant T2 and/or [Fe] differences between cells incubated with DIO (red) and untreated controls (grey). BMDMs incubated with 1:1 SDIO (light blue) and 10:1 SDIO (dark blue) showed significantly shorter T2 (Fig. 1a) and greater uptake of iron (Fig. 1b) compared to DIO or untreated controls. 10:1 SDIO showed significantly shorter T2 than 1:1 SDIO in M0, M1 low and M1 high stimulated macrophages (Fig. 1a). And, a significantly higher Fe concentration was observed in all types of stimulated BMDMs for 10:1 SDIO compared to 1:1 SDIO (Fig. 1b). The iron concentration for M0, M1L, M1H and M2 stimulated macrophages incubated with 10:1 SDIO was 2.38, 1.59, 2.70 and 2.70 greater than for the same subtypes incubated with DIO, respectively. The iron content and 1/T2 value data showed good agreement with a Pearson correlation of 0.74 (95% CI: 0.67–0.80).
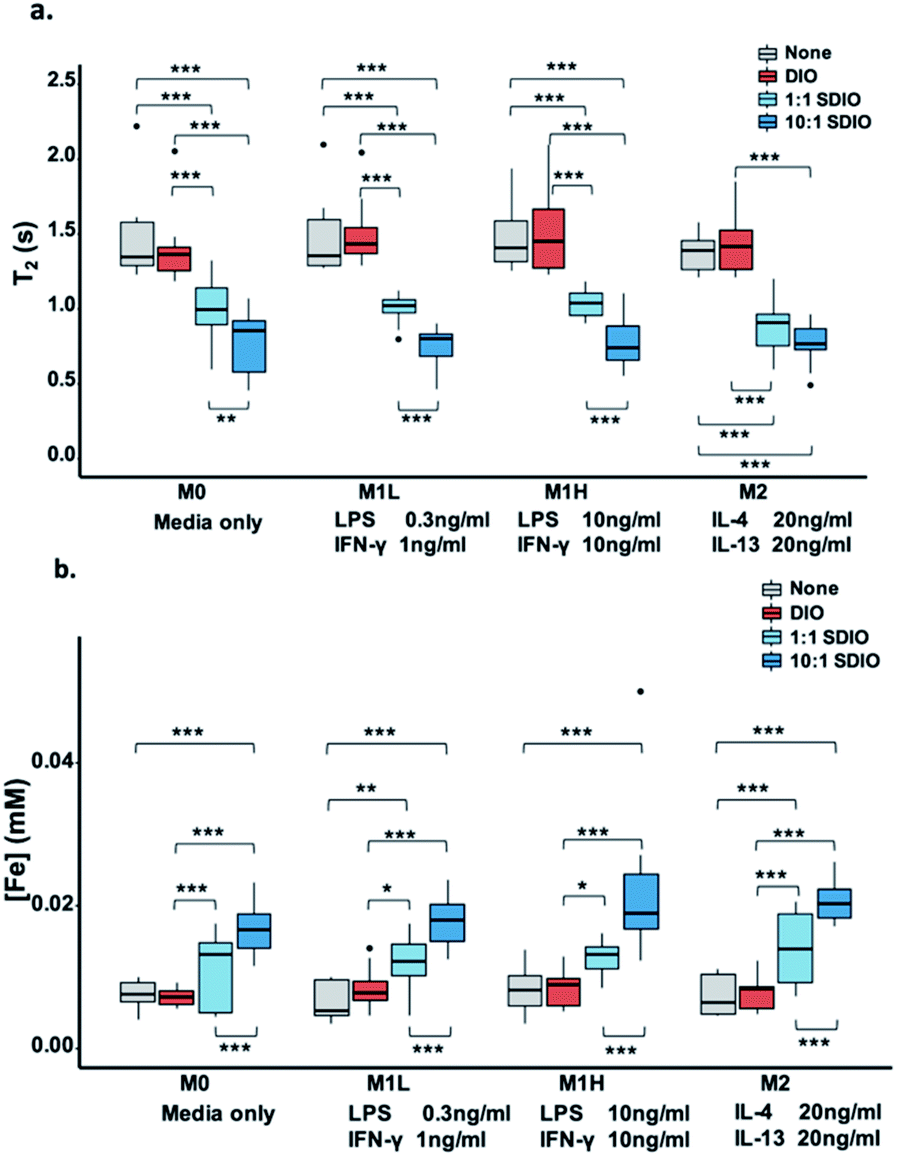 | ||
| Fig. 1 BMDMs cell uptake study. (a) T2 values of BMDMs lysates. 10:1 SDIO and 1:1 SDIO have significant higher uptake compared to untargeted DIO and untreated control. (b) Iron concentration of BMDMs lysates. 10:1 SDIO has significant higher uptake compared to 1:1 SDIO, untargeted DIO and untreated control. The differences were considered significant with P values * < 0.05, ** < 0.01, and *** < 0.001 as shown. P values are corrected for multiple comparisons within a given outcome with the Bonferroni Holm–correction for 42 tests. | ||
These data confirm that SDIO are taken up more avidly by BMDMs than the untargeted DIO nanoparticles, and that DIO nanoparticles are not significantly taken up by BMDMs. This is in agreement with our earlier work on cultured macrophage cell lines,33 which also showed significantly greater uptake of SDIO than DIO. However, it was unexpected that no differences were noted between M0, M1L, M1H, and M2 stimulation conditions.
SR-A expression in polarized BMDMs
To examine the contribution of the scavenger receptor A, SR-A (CD204), on the uptake of the SDIO, we performed immunoblot analysis with the cell lysates from the polarized BMDMs (Fig. 2). The housekeeping gene, GADPH (glyceraldehyde-3-phosphate dehydrogenase), was used to normalize for the variation in total protein loading. The relative protein expression of SR-A is shown in Fig. 2b. No significant differences in SR-A expression were found between M0 and the other polarized subtypes. For the M1 low stimulation condition (0.3 ng mL−1 LPS, 1 ng mL−1 IFN-γ), SR-A protein expression level was significantly lower compared to M1 high and/or M2 stimulation. However, the cell uptake study (Fig. 1.) for SDIO did not show significant differences for SDIO uptake between M1 low and the other conditions.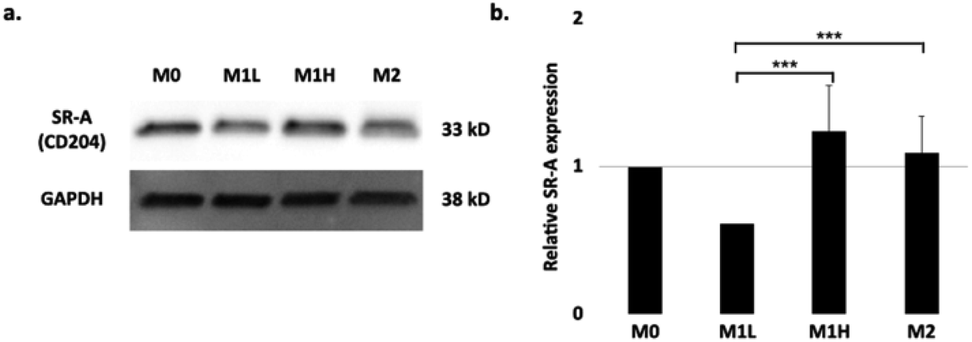 | ||
| Fig. 2 Expression of SR-A receptor in different polarization states of BMDMs. (a) Representative immunoblot of SR-A expression in polarized macrophages after 24 h of stimulation. GAPDH was used as a housekeeping gene. (b) Quantification of SR-A in macrophages polarized as in (a). Quantification is an average of three blots. Relative SR-A expression on stimulated BMDMs. The raw blot is shown in Fig. S3 in ESI.† The differences were considered significant with P values * < 0.05, ** < 0.01, and *** < 0.001 as shown. P-values were corrected for 3 tests via Bonferroni–Holm. | ||
This discrepancy may due to the natural variation of the primary macrophages between mice that might have resulted in differential activation and expression of SR-A protein. Also, the usage of M-CSF in the differentiation conditions for BMDMs, which are widely used in the field, has been shown to skew the macrophages towards M2 like phenotype.35 Thus, the SDIO uptake differences between cell types may have been masked during the duration of the experiments. Also, since phagocytosis is complex process, it is possible that particles themselves might alter the cell behaviors, which might be another reason for discrepancy. We examine this further below.
Magnetic resonance imaging studies
Magnetic resonance imaging (MRI) was performed on the cell lysate solutions used for the T2 measurements. Fig. 3a shows a representative MR image from (n = 3) containing reconstituted cell lysate samples from different macrophages subtype incubated with 1:1 SDIO (top row), DIO (2nd row), media only (3rd row), and a reference of pure water (bottom row). Lysate from cells incubated with 1:1 SDIO showed signal suppression characteristic of T2 agents (darker images), while DIO and untreated cells were similar to water. This trend was consistent across all three subtypes.
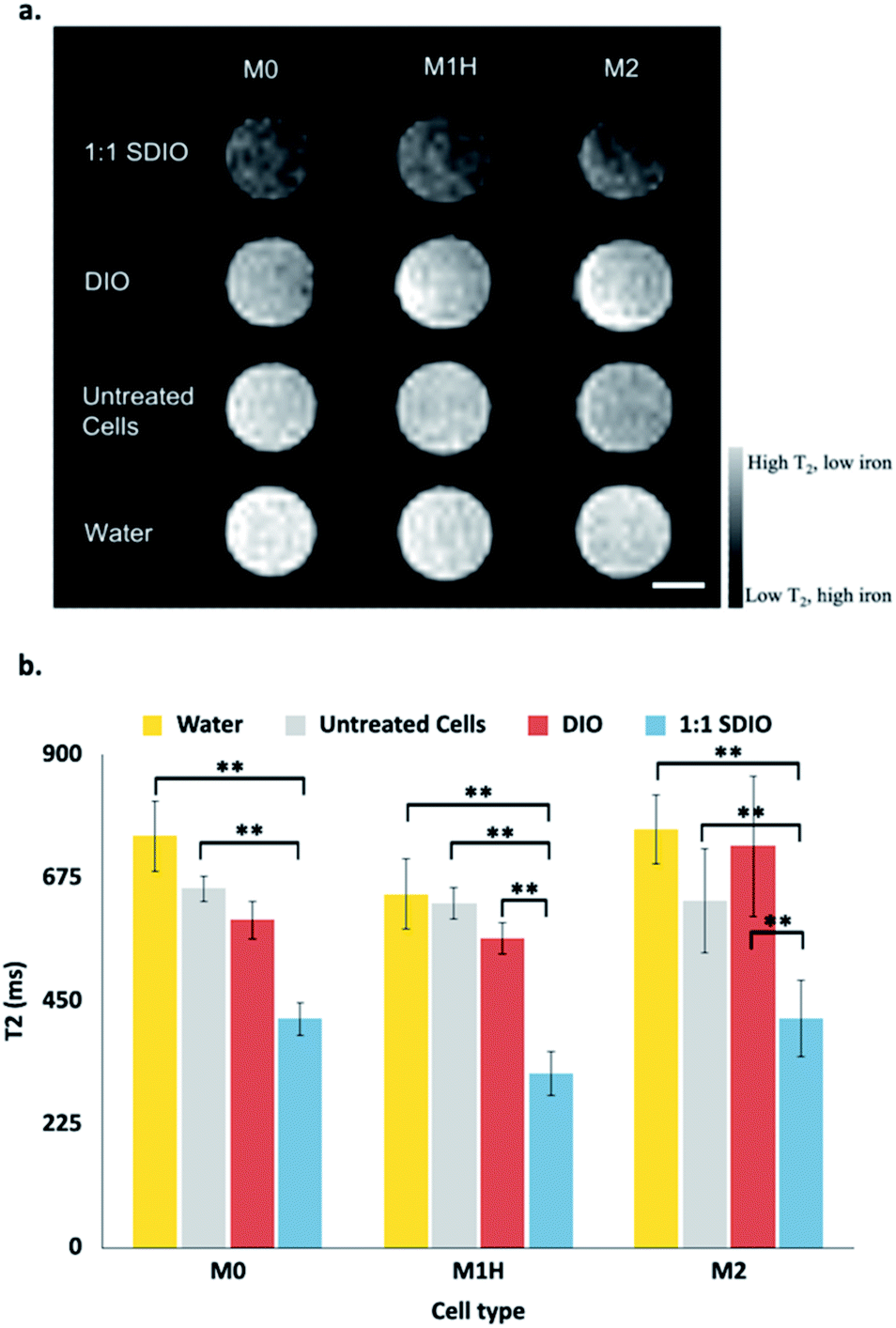 | ||
| Fig. 3 Representative reconstituted MR images of BMDMs lysates (a) and mean T2 values (b). Samples containing reconstituted cell lysate samples from different macrophages subtype incubated with 1:1 SDIO (top row), DIO (2nd row), media only (3rd row), and a reference of pure water (bottom row). (n = 3) All three subtypes of macrophage incubated with 1:1 SDIO showed the highest signal suppression characteristic of T2 agents (darker images), while DIO incubated cells and untreated cells were similar to pure water. Scale bar = 4 mm. The average T2 values (n = 3) were reported in panel b with standard deviation as the error bar. The differences were considered significant with P values * < 0.05, ** < 0.01, and *** < 0.001 as shown. P values are corrected for multiple comparisons within a given outcome with the post hoc Tukey HSD correction. | ||
The mean T2 values at 7 T are shown in Fig. 3b. The T2 values were significantly different between untreated cells and SDIO-treated cells in all subtypes (p < 0.01). In the polarized M1H and M2 condition, the T2 values were also significantly lower in SDIO-treated cells compared to DIO-treated cells. These results support that SDIO uptake by macrophages was significantly higher compared to DIO in M1H and M2 polarized cells. No differences were found between tubes of water, untreated cells and DIO-treated cells.
Cytokine studies
To evaluate whether the uptake of SDIO influenced cell physiology and inflammatory state of the macrophages, we assessed for markers specific to the M1 phenotype. The secretion of the inflammatory cytokines, TNF-α, IL-6, and MCP-1, were analyzed to assess the inflammatory state of the polarized macrophages.Fig. 4a illustrates that BMDMs incubated with 1:1 SDIO (light blue) had significantly higher secretion of TNF-α compared to cells incubated with 10:1 SDIO (dark blue) under M1L and M1H stimulated conditions. No significant differences in TNF-α secretion was found between cells incubated with DIO (red), dextran sulfate (yellow) and untreated controls (grey). These results indicate that 1:1 SDIO treatment synergistically enhance the TNF-α secretion by the macrophages stimulated with M1 polarizing cytokines.
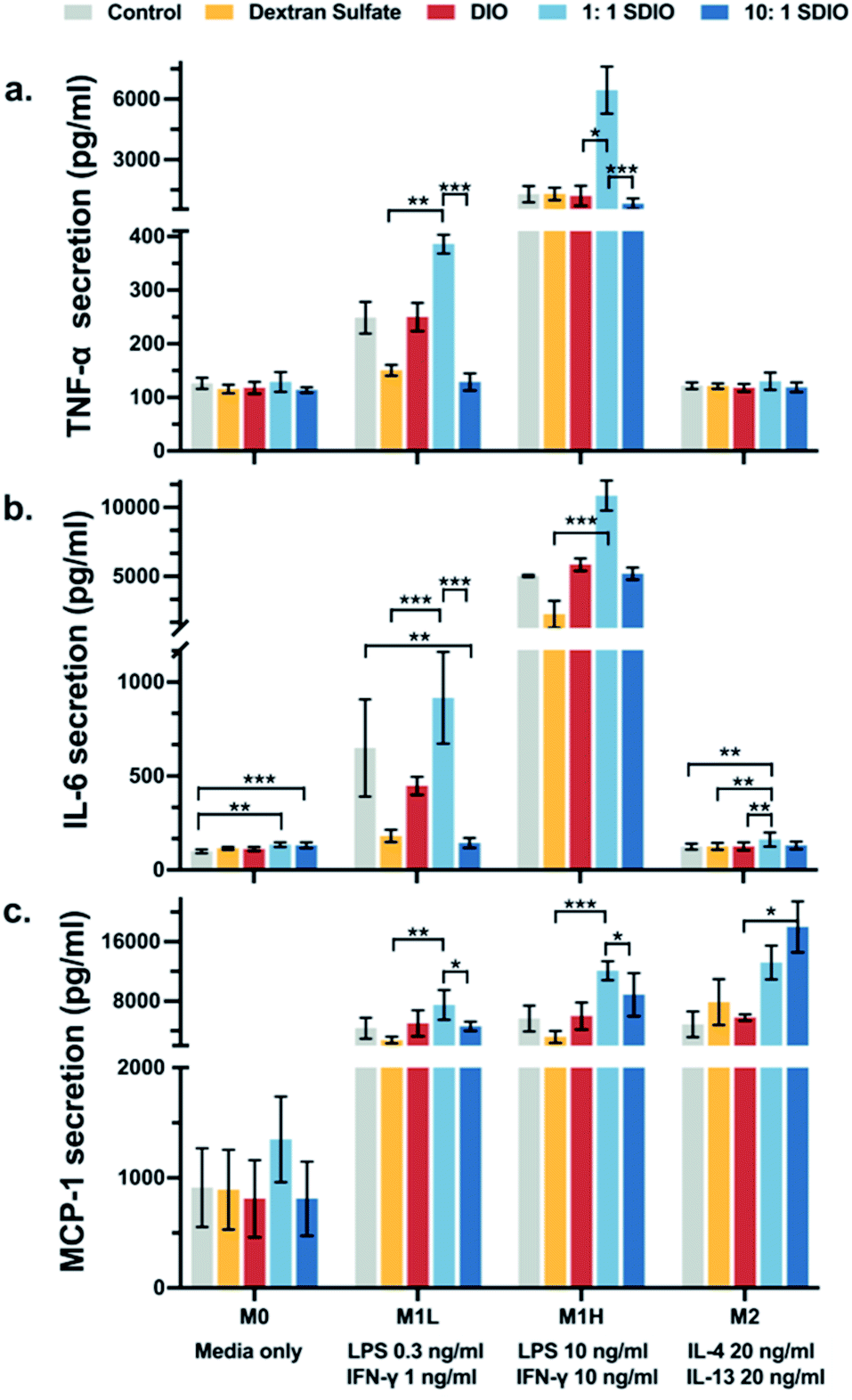 | ||
| Fig. 4 Relative TNF-α (a), IL-6 (b) and MCP-1 (c) secretion by bone marrow derived macrophages (BMDMs) treated with dextran sulfate, DIO, 1:1 SDIO, 10:1 SDIO and media. BMDMs incubated with 1:1 SDIO show more production of TNF-α, IL-6 and MCP-1 in M1 stimulated cells. 10:1 SDIO incubated M2 stimulated BMDMs showed significantly more production of MCP-1. The differences were considered significant with P values * < 0.05, ** < 0.01, and *** < 0.001 as shown. P values are corrected for multiple comparisons within a given outcome with the Bonferroni Holm–correction for 46 tests. | ||
Cells incubated with 1:1 SDIO showed significantly higher IL-6 secretion compared to cells incubated with dextran sulfate in M1L, M1H and M2 stimulated conditions (Fig. 4b), suggesting that 1:1 SDIO was able to suppress M2 macrophage phenotype and enhance the secretion of pro-inflammatory cytokines. We expected that the 10:1 SDIO treatment would have enhanced cytokine secretion more than 1:1 SDIO due to their higher uptake, but the cytokine secretion profiles showed that the dextran sulfate suppressed the IL-6 secretion in M1 polarized cells.
Similar trends were found in M1L and M1H stimulated conditions for MCP-1 secretion profiles (Fig. 4c). M2 stimulated BMDMs incubated with 10:1 SDIO had significantly higher MCP-1 secretion compared to cells incubated with DIO, which indicates that 10:1 SDIO may have the ability to alter cytokine section under M2 polarized conditions.
Overall, the pro-inflammatory cytokine secretion profiles indicate that different sulfation of SDIO has different effects on the inflammatory responses of polarized BMDMs.
MR imaging for breast cancer model
Given the validation of uptake of SDIO by BMDMs expressing SR-A (CD204) in primary culture, but the drawbacks of BMDMs as a model for expression of M2 surface markers, we sought to confirm that SDIO could target M2-like macrophages in a more realistic environment. The 4T1 breast cancer model is known for highly aggressive tumors accompanied by macrophage infiltration.36–38 In breast cancers, high macrophage content has been associated with poor prognosis,39,40 so we expected to find higher density of TAMs of tumor-permissive, M2-like function. Since breast cancer development activates multiple pathways that correlate to macrophage polarization and behavior,41,42 it is likely that a mixed effect will occur during the process, which meets our model needs. Fig. 5 shows the representative MRI images in axial plane of mouse body around the tumor site with 10:1 SDIO (15 mg Fe kg−1) injected. The tumor site was then defined, as outlined in orange, the tumor images were shown in Fig. 6 with all conditions.
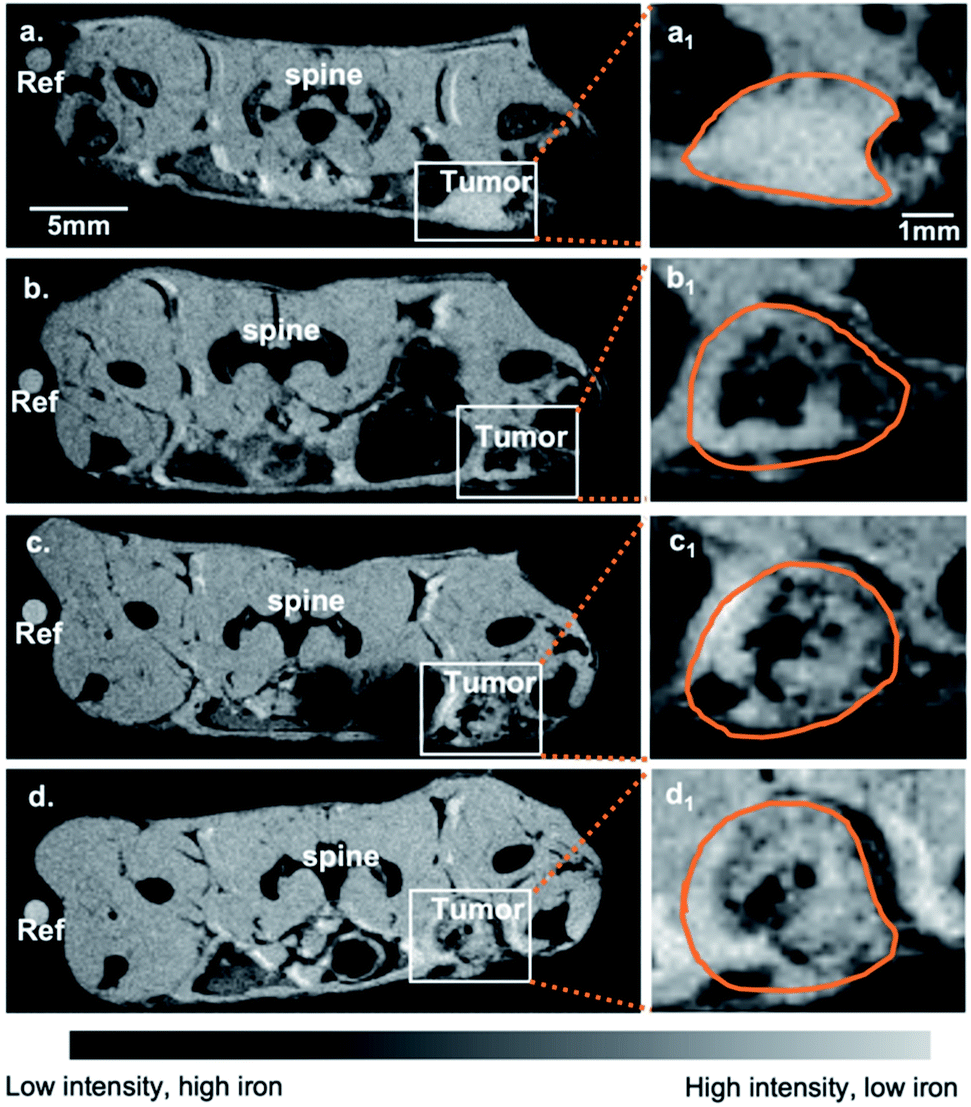 | ||
| Fig. 5 Representative MR images in axial plane of breast cancer models. MR images for a representative adult Balb/c mouse with 10:1 SDIO 15 mg Fe per kg I.V. injection (a) pre-injection scan, (b) 4 h-post injection scan, (c) 24 h-post injection scan, and (d) 48 h-post injection scan. The left tumor is indicated by a white box and the zoomed in images are shown in (a1–d1). Tumor margins are circled in orange. | ||
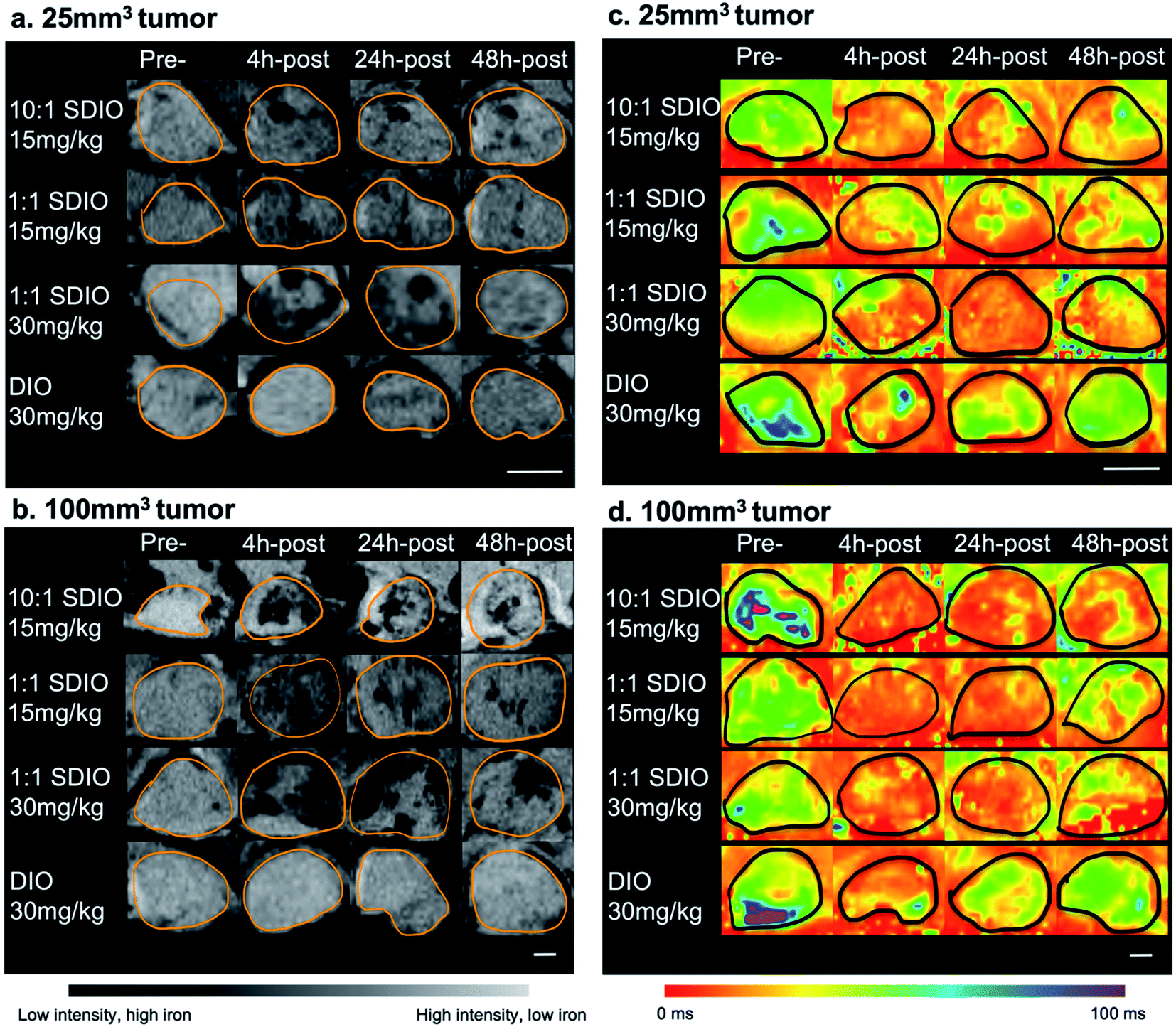 | ||
| Fig. 6 Representative zoomed in MR images and parametric T2* maps for 25 mm3 tumor (a and c) and 100 mm3 tumor models (b and d) injected with either 15 mg Fe kg−1 10:1 SDIO, 15 mg Fe kg−1 1:1 SDIO, 30 mg Fe kg−1 1:1 SDIO, or 30 mg Fe kg−1 DIO at Pre-, 4 h-post, 24 h-post, or 48 h-post injection time point (tumor margins are outlined in orange). SDIO-injected group in both sizes of tumors have stronger negative contrast and lower T2 * after injection compared to untargeted DIO-injected control. Scale bar = 2 mm. | ||
As SDIO and DIO were T2-based contrast agents, the regions with SDIO or DIO accumulation should appear darker than normal tissues. After IV injection, the DIO-injected group showed slightly enhanced T2 contrast within the tumor site (Fig. 6a last row and Fig. 6b last row), while SDIO-injected group displayed much stronger contrast at tumor site (Fig. 6a top three rows, Fig. 6b top three rows), which indicated that with the same dose of iron injected, SDIO accumulated preferentially compared to DIO. The negative contrast was strongest for the 1:1 SDIO 30 mg Fe per kg dose injection at 24 h-post injection imaging with 100 mm3 tumor and reduced by the 48 h-post injection imaging due to nanoparticle degradation in acidic tumor environment,43 and/or possibly the clearance of the nanoparticles. Comparing 1:1 SDIO and 10:1 SDIO with the same injection dosages, 10:1 SDIO showed more negative contrast after injection and stayed longer within the tumor region.
Images collected with the MGE sequence demonstrate similar trends. Fig. 6c and d show the T2* parametric maps at pre-, 4 h-post, 24 h-post and 48 h-post injection in 25 mm3 and 100 mm3 tumors. 10:1 SDIO and 1:1 SDIO treated animals show lower T2* values in the tumor region at 4 h-post injection and T2* values remain low until 48 h-post injection in both sizes of tumors. In contrast, in the DIO treated animals T2* values in the tumor region decrease at 4 h-post injection but recover to near pre-injection values by 48 h-post injection.
In mice with 25 mm3 tumors injected with SDIO the changes in mean T2* in the tumors obtained by 48-post injection MR images, compared to pre-scan, were 4.7 (15 mg Fe kg−1 10:1 SDIO), 3.4 (15 mg Fe kg−1 1:1 SDIO) and 5.4 (30 mg Fe kg−1 1:1 SDIO) fold greater than for DIO-injected controls, respectively (Fig. 7). In mice with 100 mm3 tumor injected with SDIO the changes in mean T2* in the tumors were 18.3 (15 mg Fe kg−1 10:1 SDIO), 11.6 (15 mg Fe kg−1 1:1 SDIO) and 20.8 (30 mg Fe kg−1 1:1 SDIO) fold greater than DIO-injected groups (Fig. 7).
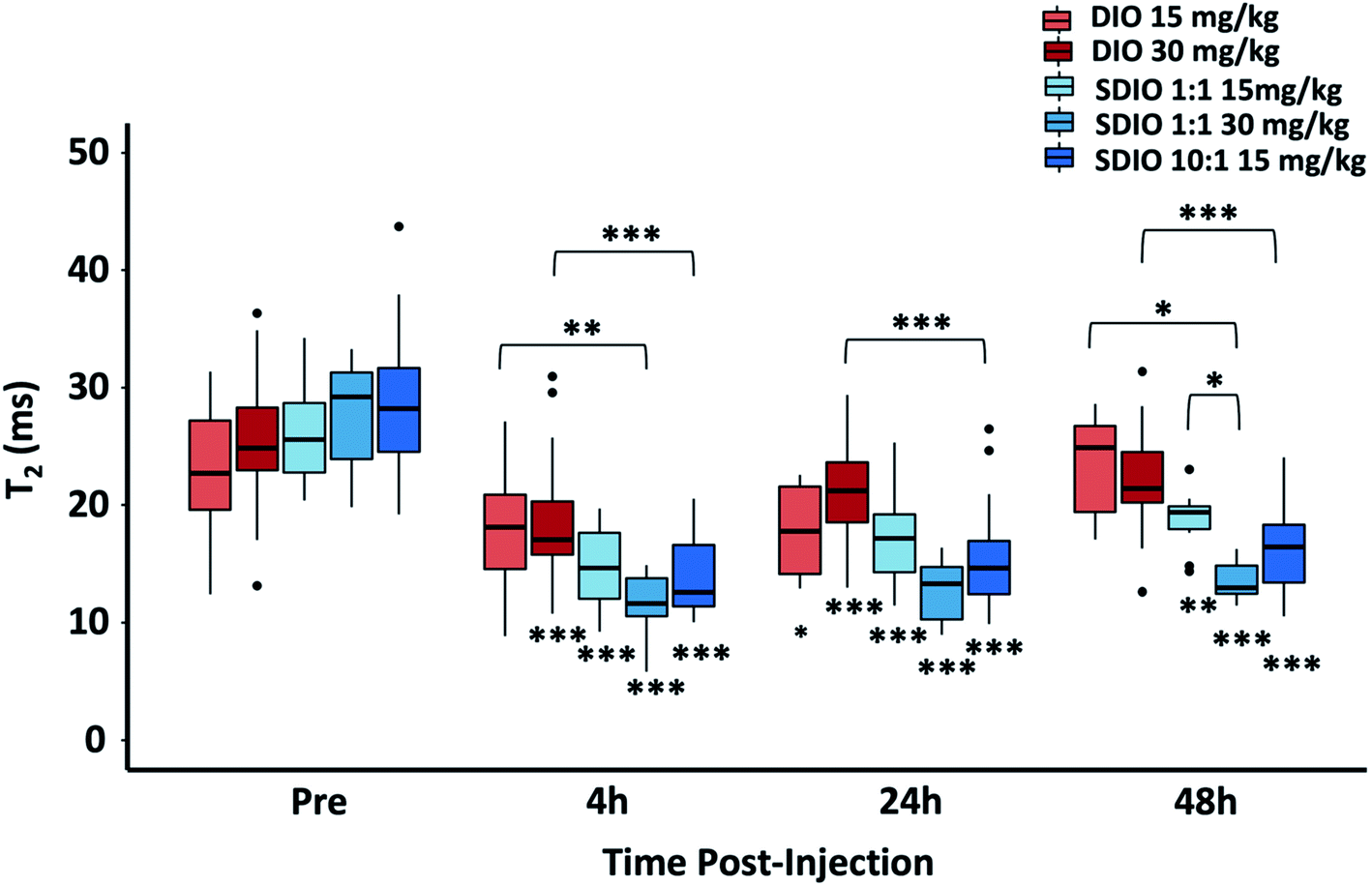 | ||
| Fig. 7 T2* of 25 mm3 tumor and 100 mm3 tumor at pre-, 4 h-post, 24 h-post, or 48 h-post injection imaging with SDIO/DIO injection. 10:1 SDIO with 15 mg Fe kg−1 dose (dark blue) have significantly higher accumulation within tumors compared to untargeted DIO-injected controls (orange and red) at all MR imaging time points. The 1:1 SDIO with 30 mg Fe kg−1 dose (medium blue) injected group has significant shorter T2 than untargeted DIO injection (orange) at 24 h-post and 48 h-post injection imaging, indicating higher uptake. The differences were considered significant with P values * < 0.05, ** < 0.01, and *** < 0.001 as shown. The *'s below the boxplot showed the P values when comparing to pre-injection. P-values were corrected for 33 tests via Bonferroni–Holm. | ||
These results illustrated that SDIO had higher accumulation within larger tumors (p = 0.043), probably because of the more extensive neovasculature in the larger tumors facilitating more efficient delivery. For both sizes of tumors, T2* showed similar results with 15 mg Fe kg−1 10:1 SDIO and 30 mg Fe kg−1 1:1 SDIO, which supports that SDIO with higher sulfation level has higher uptake efficiency.
Immunohistochemistry and histology
Tumors were collected and analyzed for morphology, CD204 (SR-A) expression and expression of cytokines associated with M1 and M2 phenotypes. H&E staining clearly showed, by morphology, aggressive tumor cells which were highly disorganized and distorted due to the rapid division of tumor cells, as well as a necrotic tumor core (Fig. 8a). CD204 (SR-A) was highly expressed within tumor region which contributes to the high accumulation of SDIO in the tumor. In order to confirm that the T2 signal decreases were due to the accumulation of SDIO or DIO contrast agents, Prussian blue staining for iron content was performed. Accumulation of iron containing particles in cells was evidenced as blue spots and the nuclei were stained with nucleus fast red (pink), as shown in Fig. 8a. The 10:1 SDIO injected group contained the highest amount of iron even after 48 h-post injection in tumor microenvironment, the same dose 1:1 SDIO injected group retained some, but less iron, while DIO injected mice were cleared out by 48 h-post injection, which correlates with the MR images and T2 quantitative analysis.
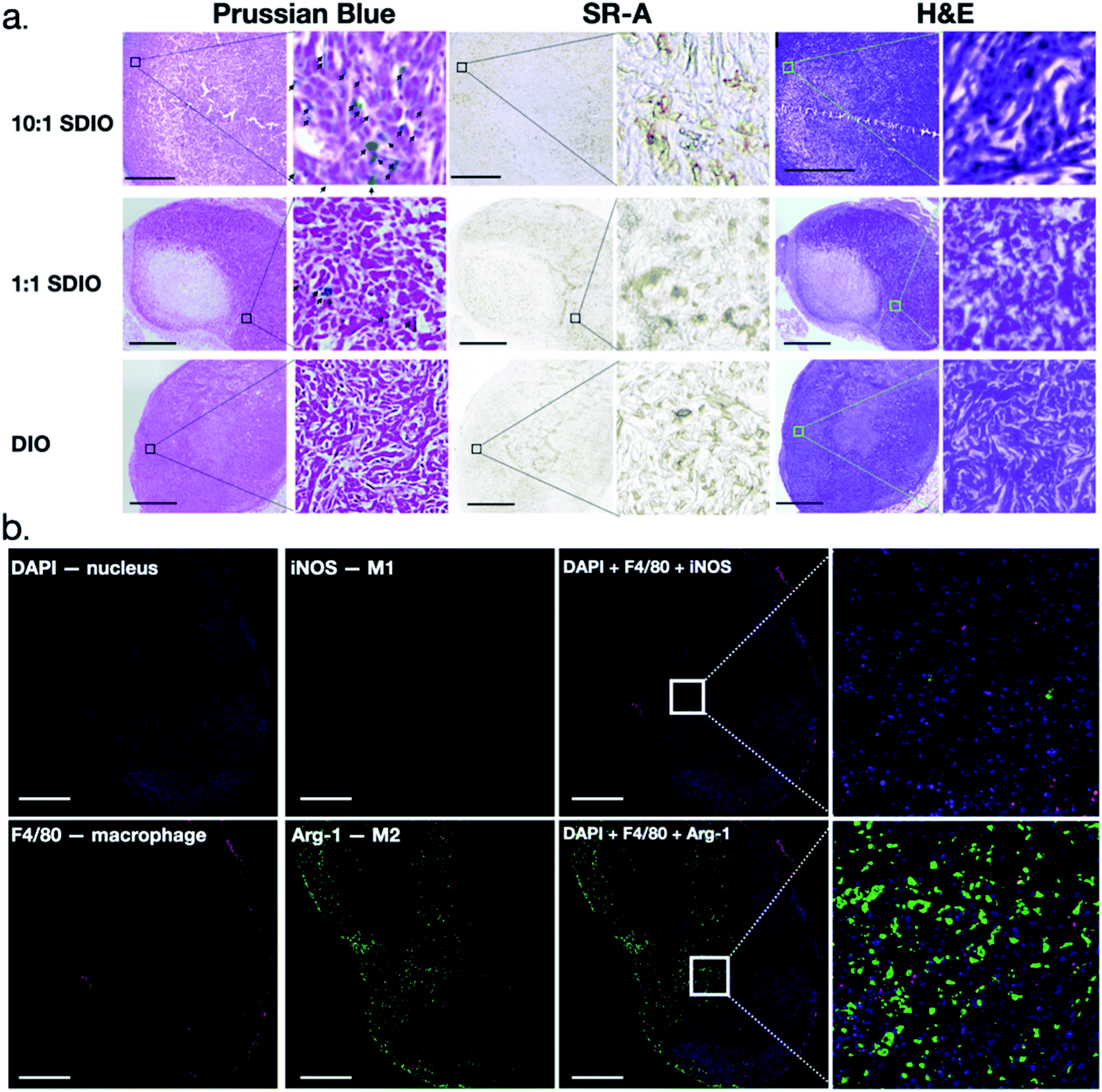 | ||
| Fig. 8 Representative histology and immunohistochemistry images of mouse breast cancer model. (a) Bright-field H&E, CD204, and Prussian Blue staining for iron after 48 hours post-injection MR imaging. Black arrowhead indicate iron containing cells. Scale bar = 1 mm. (b) Representative immunohistochemistry for Arginase-1, iNOS and F4/80 staining images after 48 h post-injection MR imaging. The cell nuclei were counterstained with Hoechst 33342. Scale bar = 500 μm. TAMs do not express iNOS, an M1 marker, but do express Arg-1, an M2 marker. | ||
CD204 (SR-A) was stained in an adjacent slide to validate the high expression within the tumor region in a similar pattern as Prussian blue iron staining. This is consistent with high accumulation of SR-A- targeted uptake of SDIO (iron) in the tumor microenvironment.
Fig. 9a illustrates through morphology that iron staining is found in macrophages, and does not overlap with 4T1 cells. The 4T1 tumor cells can be easily distinguished by their large, elongated nuclei (arrows), whereas the macrophages are smaller cells (arrowheads) between the tumor cells. We also confirmed through in vitro uptake studies that SDIO are not taken up by 4T1 cells in culture (Fig. S4†).
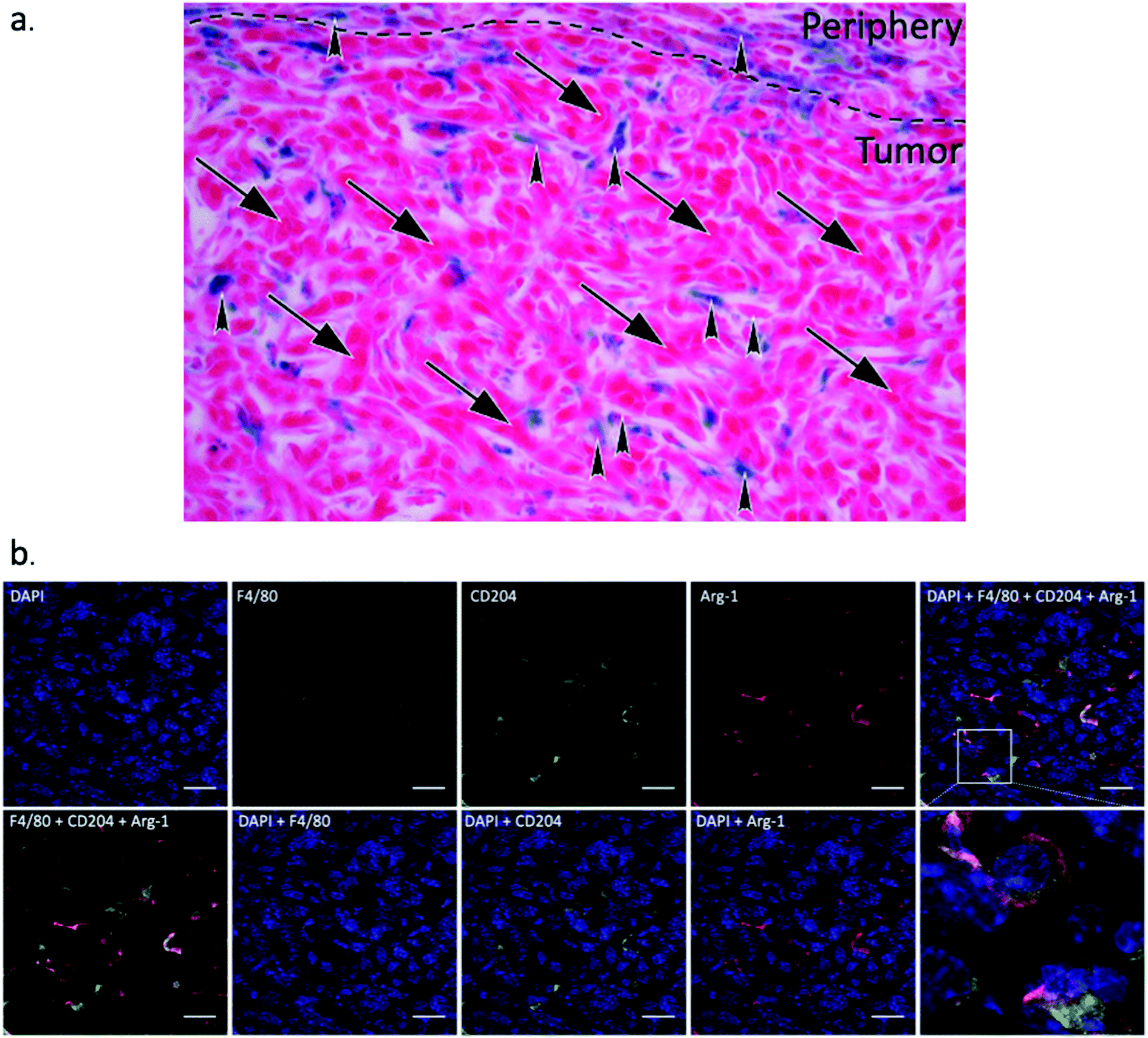 | ||
| Fig. 9 Representative zoomed in Prussian blue stained image (a) and immunohistochemistry for Arginase-1, CD204 and F4/80 staining images (b). (a) Prussian blue staining image for 10:1 SDIO injected animals after 48 h post-injection MRI, which clearly shows the morphology of the tumor cells (arrows) and macrophages (arrowheads) within the tumor microenvironment. Iron is found in macrophages, but not in tumor cells. (b) F4/80, CD204, and Arg-1 co-stained images after 48 h-post injection MRI. The cell nuclei were counterstained with Hoechst 33342. The tumor associated macrophages are CD204 and Arg-1 double positive. Scale bar = 20 μm. | ||
Liver and kidney were also harvested and stained with Prussian Blue, CD204, and H&E. No iron was found in kidney while high accumulation of iron was found in liver (Fig. S5 and S6†), which are as expected that both DIO and SDIO are cleared out by liver. The H&E staining shows a normal kidney and liver tissue morphology.
Macrophage infiltration in the tumor region was clear from the IHC as shown in Fig. 8b. The iNOs+ F4/80+ (classically activated macrophages/M1) cells are mostly in the peritumoral area while arginase-1+F4/80+ (alternatively activated macrophages/M2) cells were observed inside the tumor region, which is consistent with previous reports.44 We also performed co-staining with fluorescent antibodies against CD204, Arg-1 and F4/80 to demonstrate that nearly all tumor associated macrophages express Arg-1 and CD204 (Fig. 9b). These results also correlate with SR-A (CD204) and Prussian Blue staining (Fig. 8a), which confirmed that iron and SR-A receptors appear in the tumor microenvironment.
Conclusion
The results of these studies illustrated that an established primary culture model for macrophage subtypes (BMDM)45–48 exhibited inconsistent expression of the M2 surface marker, SR-A.6–8,46 There was no significant difference in uptake of SDIO, which is targeted to SR-A, between polarized phenotypes in BMDMs. CD204 (SR-A) protein expression was not distinguishable between high dose M1 stimulation and M2. Cytokine expression levels, however, support that M1L and M1H cells secrete higher amounts of TNF-α, Il-6 and MCP-1 than M0 or M2 cells, which is functionally characteristic of the M1-like subtype. The vagaries of the BMDM model highlighted that a single biomarker was not enough to identify macrophage subtypes in this primary culture model; and, that one should not expect faithful expression of all classical subtype markers in this model. The use of the BMDM model must be carefully considered for macrophage subtype studies, and utility will be dependent on the particular subtype markers of interest.Nonetheless, BMDMs did show preferential uptake of 10:1 SDIO compared to untargeted DIO or untreated cells for M0, M1L, M1H, and M2 polarized BMDMs. Furthermore, more highly sulfated SDIO showed greater labelling of M2-like macrophages in vivo in a mouse breast cancer model, illustrating the potential utility of SDIO as a tool for mapping macrophage subtype markers.
In summary, sulfated dextran coated iron oxide nanoparticles (SDIO) preferentially target and localize to M2-like TAMs in vivo. More highly sulfated iron oxide nanoparticles (10:1 SDIO), demonstrated higher uptake in primary cultured bone marrow derived macrophage as well as longer retention time in tumors in a mouse breast cancer model compared to untargeted controls (DIO) and 1:1 SDIO. Furthermore, from cytokine secretion profiles in primary culture BMDMs, 1:1 SDIO drives BMDMs to be more M1-like in cytokine secretion; thus, SDIO may have some therapeutic benefit by further polarizing M1 macrophages phenotype and represent a potential theranostic for subtypes expressing the M2 marker SR-A.
In the recent decades, immunotherapy combined with traditional method to treat cancer have showed great interests in both clinic and research, not only the T-cell immune-modulating agents, e.g., monoclonal antibodies (mAbs), immune checkpoint inhibitors, tumor vaccines, but medicines/strategies for macrophage reprogramming are merging in the field.49–55 It is important to know and evaluate the tumor microenvironment immunity for individuals as well as monitor the immunotherapy longitudinally. Thus, SDIO could also be promising to validate these macrophage modulating drugs in cancer immunotherapy and assist with customized treatment planning in the future.
Materials and methods
General materials
Materials were purchased from commercial suppliers and used directly, unless specifically noted. Dextran (from Leuconostoc, average mol. wt 9000–11000 kDa) and ferric chloride hexahydrate (FeCl3·6H2O, MW 270.29 g mol−1) were obtained from Sigma-Aldrich. Ferrous chloride tetrahydrate (FeCl2·4H2O, MW 198.81 g mol−1). Ammonium hydroxide (28–30%), sodium bicarbonate and sodium hydroxide were from by Fisher Scientific. Sulfur trioxide (SO3) pyridine complex and 2-methyl-2-butene (2M2B) were purchased from Acros. Anhydrous formamide was purchased from MP, Biomedicals, LLC. Spectra/por® dialysis membrane (mol. wt cut-off 50 kDa) was purchased from Spectrum Laboratories, Inc. Water was purified using a Millipore Milli-Q Synthesis purifier (18.0 MU cm, Barnstead).
Synthesis of DIO and SDIO
Dextran coated iron oxide (DIO) nanoparticles were synthesized by co-precipitation of Fe(II) and Fe(III) chlorides with ammonium hydroxide, as previously reported.33,34,56 In brief, reduced dextran and FeCl3⋅6H2O were dissolved in degassed deionized ultra-filtered water. FeCl2⋅4H2O solution and concentrated ammonia were added dropwise at 0–5 °C under argon. After 3 hours at 85 °C, DIO was formed and purified by dialysis against deionized water (50 kDa) for 72 hours. DIO was obtained as a brown powder after lyophilization. DIO (60 mg) was dissolved in 3 mL dry formamide, and 2-methyl-2-butene (0.48 mL) was added to the DIO solution under argon. 2-Methyl-2-butene was introduced as an acid scavenger before the addition of the sulfation agent, in order to clear the free acid produced in the system. Sulfur trioxide pyridine complex (96 mg) was added in the mixture. Then, the mixture was stirred at 30 °C under argon for 3.5 hours with refluxing. Saturated NaHCO3 aqueous solution was used to quench the reaction. The product was purified by dialysis against deionized water (50 kDa) for 72 hours. SDIO was obtained as a brown powder after lyophilization. Stoichiometries of 10:1, 7:1 and 1:1 of S:OH SDIO were synthesized.
Characterization of SDIO and DIO
Successful sulfation was verified by infrared spectroscopy with a Shimadzu IR Prestige 21 spectrophotometer. The samples were also sent to ALS (Environmental Analytical Service, Tucson, Arizona) for sulfur content analysis by combustion-infrared spectroscopy. The amount of iron in SDIO was measured with MP-AES or ICP-OES. The core sizes of DIO and SDIO were measured by transmission electron microscopy (TEM), using a Philips CM-12 operating at 80 kV. Sample solutions (5 μL) were loaded on a thin carbon film, 400 copper mesh grids and dried naturally. The average core diameter of particles was calculated based on 500 particles from different regions on the grids. The average hydrodynamic diameters and size distributions were measured by dynamic light scattering (DLS) using a Malvern Zetasizer. The zeta potential of SDIO was obtained by determining the electrophoretic mobility of the particles using a Malvern Zetasizer in deionized water and at room temperature.Longitudinal (T1) and transverse (T2) relaxation times of SDIO and DIO in deionized water with different iron concentrations were measured at 60 MHz (1.4 T) and 37 °C on a Bruker Minispec mq60 (Bruker, Billerica, MA, USA), as reported previously.33 The stock solutions of SDIO were prepared at 0.03125, 0.0625, 0.125, 0.25, and 0.5 mM [Fe]. Each solution was incubated at 37 °C for 8–10 min before measurement to reach the thermal equilibrium. T1 values were determined with an inversion recovery sequence with 10 to 15 data points, while T2 values were measured using a Carr–Purcell–Meiboom–Gill sequence, with τ = 1 ms and 200 data points. The longitudinal (r1) and transverse (r2) relaxivities were determined as the slope of the linear plots of 1/T1 or 1/T2 vs. iron concentration, with a correlation coefficient greater than 0.99.
Longitudinal (T1) and transverse (T2) relaxation times of SDIO and DIO in deionized water with different iron concentrations were measured in a 7 T small animal MRI (Bruker Biospec). The stock solutions of SDIO were prepared at 0.1, 0.2, 0.3, 0.4, and 0.5 mM [Fe]. Each solution was incubated at 25 °C for 15 min before measurement to reach the thermal equilibrium. The RAREVTR (RARE with variable TR, TR = 128–7500 ms, TE = 60 ms, matrix size = 128 × 128, FOV = 45 × 45 mm2, slice thickness = 1 mm) sequence was used to measure T1. The MSME (Multi-Slice Multi-Echo, TR = 1000 ms, TE = 10–160 ms, matrix size = 128 × 128, FOV = 45 × 45 mm2, slice thickness = 1 mm) sequence was used to measure T2. The longitudinal (r1) and transverse (r2) relaxivities were determined as the slope of the linear plots of 1/T1 or 1/T2 vs. iron concentration.
Cell isolation and culture
All protocols involving animals were approved by University of California's Institutional Animal Care and Use Committee, both UC Davis and UC Irvine are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Primary bone marrow derived macrophages (BMDM) were obtained by harvesting marrow from femurs of 6–12 week old female C57BL/6 mice, lysing red blood cells with ammonium-chloride-potassium (ACK) lysing buffer, and then culturing cells for seven days on bacteriological polystyrene plates in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2% penicillin/streptomycin, 2 mM L-glutamine, and 10% conditioned media from CMG 12–14 cells expressing recombinant mouse macrophage colony stimulating factor (M-CSF). One day 7 BMDM were plated and treated with the indicated concentrations of LPS (Sigma), IFN-γ, IL-4, or IL-13 (all from Biolegend) for the indicated time, then incubated with nanoparticles or control media, and assayed for cell uptake or cytokines as described below.BMDM uptake studies
To study the preferential cellular uptake of SDIO nanoparticles by BMDM, cells were plated into 6-well culture plates at a density of 800000 cell per well in DMEM supplemented with 10% FBS, 2% penicillin/streptomycin, 2 mM L-glutamine, and 10% conditioned media from CMG 12–14 cells expressing recombinant mouse M-CSF. 2 hours after seeding, cells were stimulated with cytokines under either low dose M1 phenotype conditions 0.3 ng mL−1 LPS (Sigma), 1 ng mL−1 IFN-γ, or high dose for M1 phenotype conditions 10 ng mL−1 LPS and IFN-γ. The M2 phenotype was induced using 20 ng mL−1 IL-4 and IL-13. Stimulated cells were incubated at 37 °C (5% CO2) for 24 h. Solutions of SDIO, DIO or no particles at 200 μM [Fe] were prepared by dissolving nanoparticles in D10 media without serum and incubated with cells for 1 hour at 37 °C. Cells were then washed 3 times with phosphate buffered saline (PBS), then lysed using deionized water (0.5 mL freeze, thaw) and the lysed cells lyophilized for analysis by ICP-OES and T2.
T2 relaxation time measurements of cell lysates
Lyophilized cell lysates were reconstituted in 400 μl deionized water and took 200 μl aliquot for the transverse (T2) relaxation measurements at 60 MHz (1.4 T) on a Bruker Minispec mq60 (Bruker, Billerica, MA, USA). Each solution was incubated at 37 °C for 8–10 min before measurement to reach the thermal equilibrium. T2 values were measured using a Carr–Purcell–Meiboom–Gill sequence, with τ = 1 ms and 200 data points.ICP-OES for cell lysates
Another 200 μl aliquot of reconstituted cell lysate samples were used for inductive coupled plasma optical emission spectrometry (ICP-OES) and were further diluted to 10 mL using aqueous nitric acid (2% v/v). Standard solutions were prepared by serial dilution of a Fe standard (Fisher Scientific). ICP-OES measurements were conducted with a PerkinElmer 5300 DV optical emission ICP with auto sampler.Western blotting
Cells were cultured for 24 h on tissue culture polystyrene before stimulating with as described above, and cultured for another 24 h before collecting the protein. Then the cells were lysed using RIPA lysis buffer (VWR) supplemented with 1× of Halt protease and phosphatase inhibitor cocktail (Thermo Scientific). 10 μg of the isolated total protein was resolved on 4–15% Mini-PROTEAN TGX precast gels (Bio-Rad). The protein was blotted onto nitrocellulose membrane using iBlot2 transfer system (Invitrogen). Then the blots were incubated with SRA-1 polyclonal primary antibody (Proteintech, cat no: 24655-1-AP) for 1 h at RT, after three washes it was further incubated with anti-rabbit HRP conjugated secondary antibody (Cell Signaling Technology) for 1 h. The blots were washed and incubated in Supersignal West Femto Maximum Sensitivity Substrate (Thermo scientific) for 5 min before imaging the blot using Biorad ChemiDoc XRS+ with Image Lab software.Cytokine studies
Cells were plated and stimulated as described above and treated with 10:1 SDIO, 1:1SDIO, DIO, dextran sulfate or fresh media (controls). BMDM were seeded at 300000 cells per mL in D-10 media (described above in cellular uptake study) and allowed 24 hours adherence period. We further stimulated BMDM with different combinations of phenotypic cytokines and particles. The stimulation cytokine used in this study includes no stimulus (M0), [0.3 ng mL−1 LPS, 1 ng mL−1 IFN-γ] (M1L), [10 ng mL−1 LPS, 10 ng mL−1 IFN-γ] (M1H) and [20 ng mL−1 IL-4, 20 ng mL−1 IL-13] (M2). Together with phenotype stimulation, we introduced BMDM with different particles including no particle, dextran sulfate, 200 μM [Fe] DIO, 200 μM [Fe] 1:1 SDIO, and 200 μM [Fe] 10:1 SDIO. The culture supernatant was collected after 24 h incubation and analyzed for TNF-α, IL-6, MCP-1 and IL-10 secretion by ELISA (BioLegend).
MRI of cell lysate solutions
Triplicated lysate samples containing M0, M1H, or M2 subtype incubated with 1:1 SDIO or DIO were suspended in deionized water in 0.2 mL tubes. The MR images and T2 values of the macrophage subtype lysate sample were measured by 7 T (Bruker Biospec) small animal magnet, RAREVTR (RARE with variable TR, TR = 3000 ms, TE = 30–450 ms, echo space = 60 ms, matrix size = 128 × 128, FOV = 47.843 × 40 mm2, slice thickness = 2 mm) sequence was used.
Images were imported in Fiji (Image J) and processed in Slicer 3D. Cell lysate samples were segmented to quantify T2 values within each ROI.
4T1 cell uptake studies
To study if SDIO nanoparticles are taken up by 4T1 tumor cells, cells were plated into 6-well culture plates at a density of 800000 cell per well in RPMI 1640 supplemented with 10% FBS, 2% penicillin/streptomycin, 2 mM L-glutamine. Cells were incubated at 37 °C (5% CO2) overnight for adherence. Solutions of 10:1 SDIO, 1:1 SDIO, DIO or no particles at 200 μM [Fe] were prepared by dissolving nanoparticles in RPMI 1640 medium without serum and incubated with cells for 1 hour at 37 °C. Cells were then washed 3 times with phosphate buffered saline (PBS), then lysed using deionized water (0.5 mL freeze, thaw) and the lysed cells lyophilized for analysis by T2 relaxation time.
Lyophilized cell lysates were reconstituted in 400 μl deionized water and took 200 μl aliquot for the transverse (T2) relaxation measurements at 60 MHz (1.4 T) on a Bruker Minispec mq60 (Bruker, Billerica, MA, USA). Each solution was incubated at 37 °C for 8–10 min before measurement to reach the thermal equilibrium. T2 values were measured using a Carr–Purcell–Meiboom–Gill sequence, with τ = 1 ms and 200 data points.
Breast cancer model MR imaging
To generate the breast cancer xenografts, 1 × 105 4T1 mouse breast cancer cells in 50 μL PBS, were implanted into the lower 4th mammary fat pad on both right and left side of Balb/C mice via subcutaneous injection. Tumors were allowed to develop from 5–10 days to achieve different size with a range of 25 mm3 to 100 mm3. All MR images were obtained on a 7 T (Bruker Biospec) small animal magnet. Animals were anesthetized with an 2.0% isoflurane:air mixture and maintained at 1.5% for the duration of imaging. Animals were monitored for physiological temperature and respiration (SAII), and sedation adjusted as needed. The indicated SDIO and their corresponding DIO controls were dissolved in sterile saline and injected via tail vein.
In order to have good anatomical images as well as obtain T2* values within the tissue, RARE (Rapid Acquisition with Refocused Echoes), FLASH (Fast Low Angle Shot), and MGE (Multi- Gradient Echo) sequences were chosen. After localizing the tumor region using the RARE (TR = 2000 ms, TE = 81 ms, matrix size = 160 × 160, FOV = 30 × 30 mm2, slice thickness = 1 mm), FLASH (TR = 540 ms, TE = 5 ms, F.A. = 30, matrix size = 260 × 120, FOV = 32.5 × 15 mm2, slice thickness = 0.3 mm) image were obtained. In addition, a T2* map was generated by MGE (TR = 900 ms, TE = 2.11–26.11 ms, echo spacing = 1.6 ms, F.A. = 45°, matrix size = 240 × 176, FOV = 32.5 × 15 mm2, slice thickness = 0.768 mm). Images were obtained before and 4 h, 24 h, 48 h after injection.
Images were imported in Fiji (Image J) and processed in Slicer 3D. Tumors, femur bones and phantom (0.02 mg mL−1 of [Fe] DIO-saline solution) were segmented to quantify mean signal intensity using FLASH images and mean T2* value using MGE-T2*-map within each ROI at each time point.
Immunohistochemistry and histology
Tissue samples were extracted after 48 hours post injection imaging scans and incubated in 4% paraformaldehyde overnight at 4 °C then transferred to 100% ethanol for storage at 4 °C until embedded. Samples were paraffin embedded and sectioned to 10 μm slices for histological staining. Sections were co-stained in the following combinations hematoxylin and eosin (H&E); CD204; Prussian blue; F4/80, iNOS and Arginase-1; F4/80, Arginase-1 and CD204.Tissue sections were deparaffinized and rehydrated by standard immunohistochemistry protocol. The antigen retrieval was achieved by placing slides in pre-warmed target retrieval solution (Dako) and steaming for 30 min. Endogenous peroxidase was blocked using peroxidase block solution (Dako) for 5 min. Protein blocking was performed using TBS-B buffer (TBS + 0.1% Triton X-100 + 2% BSA-MP biomedical + 1.5% donkey serum-Jackson Immunology) for 30 min. The slides were incubated with primary antibody CD204 (Thermo MA5-29733 1:100), F4/80 (Thermo BM8 1:200), Arginase-1 (ab60176 1:50), iNOS (Abcam 15323 1:100) at 4 °C overnight in the humid pan. Slides were then washed with TBS + 0.1% Triton X-100 for 5 min, and TBS-B buffer for 15 min. Then sections were stained for 1 hour with fluorescent conjugated secondary antibodies (Thermo A21209, A21447, A21206, respectively) and Hoechst 33342 dye (Fisher Scientific) at 1:1000 dilution or HRP conjugated secondary antibody (1:50). The slides were washed with TBS + 0.1% Triton X-100 for 5 min, and then TBS for three times, 5 min each, then rinsed with deionized water. Slides were mounted with Fluoromount (Southern Biotech) and imaged at 20× using the Olympus FV3000 laser scanning microscope, Nikon Ti-E widefield microscope and Keyence microscope. The F4/80, CD204 and Arginase-1 co-stained slides were imaged under 63× using Leica TCS SP8 confocal microscope.
Statistical analysis
Statistical analyses were performed in R version 4.0.2 using the base, tidyverse, lme4, lmerTest, emmeans, and doParallel packages. p < 0.05 was considered statistically significant. Multiple comparisons within each outcome were corrected separately. In the macrophage study, we used a Gamma GLM with the default inverse link function on the T2 data. The Gamma GLM accounts for a constant coefficient of variation in MRI relaxometry data as previously shown by our group.57 Cell (M0/M1L/M1H/M2), NP (none, DIO/SDIO 1:1/SDIO 10:1), and 3 trials were each included up to all 2-factor interactions. A total of 42 Bonferroni–Holm corrected pairwise comparisons were performed within each cell between NPs and between cells given SDIO 1:1 or SDIO 10:1. To account for possible within-trial batch variability, we permuted the NPs within each Cell:Trial and Cells within each NP:Trial combinations. The original p-values for each pairwise comparison were compared to the p-values under the null permutation distribution to obtain final p-values. The permutation test on the GLM was done using 100k runs and parallel computing. For the ICP-OES [Fe] data, we used the same approach except found that a Quasipoisson GLM (with default log link) was needed to stabilize the variance of the deviance residuals.
In the ELISA study with 3 outcomes (TNF-alpha, MCP-1, and IL-6), a Tweedie generalized linear mixed effects model (GLMM) (package: mgcv) with log link was used with the Mouse as a random effect, and with Cell, NP, and a Cell:NP interaction as fixed effects. The random effect accounts for within-Mouse correlations. A permutation test approach (100k runs) was also used to avoid parametric assumptions, and to further account for any baseline differences in the overall outcomes between mice. Within each Mouse:NP, the Cell was permuted to obtain the null distributions for the between-Cells comparisons overall and within each Mouse:Cell the NP was permuted to obtain the null distributions for the within Cell between NP comparisons. A total of 46 pairwise comparisons corrected via Bonferroni–Holm (within each outcome) were performed.
For the mouse breast cancer study, a linear mixed effects model (LMM) (package: lme4) was used incorporating the Mouse as a random effect. The model was fitted on the log transformed R2 data to stabilize the variance. The main factors of interest were NP (DIO/SDIO 1:1/SDIO 10:1) and dosing (15 mg kg−1 and 30 mg kg−1), and time post injection (Pre/4 h/24 h/48 h) all treated categorically. We also included the tumor size (25 mm3 and 100 mm3) as a categorical covariate. The model included all terms up to NP:Dose:Time and Size:Time to assess the difference in log(R2). 33 Bonferroni–Holm pairwise comparisons were performed between the NPs at the same dose combinations at each time point post-injection. A likelihood ratio test was performed relative to a model without the size term to assess the effect of tumor size. The pairwise comparisons were done in emmeans using Kenward–Roger T-tests (within the logLMM) on the back-transformed and bias-corrected R2 scale, since R2 (and not logR2) is linearly related to concentration of uptake.57
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank UC Davis Comprehensive Cancer Center for technical support and NIH/NIBIB R01 137887-01-A1 for funding support. The UC Davis Comprehensive Cancer Center In Vivo Translational Imaging Shared Resource is supported by the Cancer Center Support Grant P30 CA093373 from the National Cancer Institute, NIH. Dr Douglas J Rowland assisted with MR image sequence development and is supported by the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation [2019-198156 to D. J. R.]. We would like to thank UC Davis Advanced imaging facility for the technical support.References
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv and T. Lawrence, et al., Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines, Immunity, 2014, 41(1), 14–20, DOI:10.1016/j.immuni.2014.06.008.
- A. Sica, P. Invernizzi and A. Mantovani, Macrophage Plasticity and Polarization in Liver Homeostasis and Pathology, Hepatology, 2014, 2035–2043, DOI:10.1002/hep.26754.
- J. L. Stöger, M. J. J. Gijbels, S. V. D. Velden, M. Manca, C. M. V. D. Loos, E. A. L. Biessen, M. J. A. P. Daemen, E. Lutgens and M. P. J. D. Winther, Distribution of Macrophage Polarization Markers in Human Atherosclerosis, Atherosclerosis, 2012, 225(2), 461–468, DOI:10.1016/j.atherosclerosis.2012.09.013.
- H. P. Corporation, Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms, Mediat. Inflamm., 2015, 2015, 16–18 Search PubMed.
- M. Lewis and A. J. Merched, Tumor-Associated Macrophages, Inflammation and Pathogenesis of Hepatocellular Carcinoma, J. Mol. Genet. Med., 2014, 8(3), 132, DOI:10.4172/1747-0862.1000132.
- A. Kawachi, H. Yoshida, S. Kitano, Y. Ino, T. Kato and N. Hiraoka, Tumor-Associated CD204+ M2 Macrophages are Unfavorable Prognostic Indicators in Uterine Cervical Adenocarcinoma, Cancer Sci., 2018, 109(3), 863–870, DOI:10.1111/cas.13476.
- A. Kouketsu, I. Sato, M. Oikawa, Y. Shimizu, H. Saito, K. Tashiro, Y. Yamashita, T. Takahashi and H. Kumamoto, Regulatory T Cells and M2-Polarized Tumour-Associated Macrophages Are Associated with the Oncogenesis and Progression of Oral Squamous Cell Carcinoma, Int. J. Oral Maxillofac. Surg., 2019, 48(10), 1279–1288, DOI:10.1016/j.ijom.2019.04.004.
- J. Sato, S. Kitano, N. Motoi, Y. Ino, N. Yamamoto, S. Watanabe, Y. Ohe and N. Hiraoka, CD20+ Tumor-Infiltrating Immune Cells and CD204+ M2 Macrophages Are Associated with Prognosis in Thymic Carcinoma, Cancer Sci., 2020, 111(6), 1921–1932, DOI:10.1111/cas.14409.
- W. Ying, P. S. Cheruku, F. W. Bazer, S. H. Safe and B. Zhou, Investigation of Macrophage Polarization Using Bone Marrow Derived Macrophages, J. Vis. Exp., 2013, 76, 1–8, DOI:10.3791/50323.
- W. Ying, H. Gao, F. C. G. Dos Reis, G. Bandyopadhyay, J. M. Ofrecio, Z. Luo, Y. Ji, Z. Jin, C. Ly and J. M. Olefsky, MiR-690, an Exosomal-Derived MiRNA from M2-Polarized Macrophages, Improves Insulin Sensitivity in Obese Mice, Cell Metab., 2021, 33(4), 781–790, DOI:10.1016/j.cmet.2020.12.019.
- C. D. Mills, L. L. Lenz and K. Ley, Macrophages at the Fork in the Road to Health or Disease, Front. Immunol., 2015, 6, 1–6, DOI:10.3389/fimmu.2015.00059.
- W. M. Gallagher, N. Wadhwani, S. D. Keil, S. A. Junaid, H. S. Rugo and E. Shelley, Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy, Cancer Discov., 2011, 1(1), 54–67, DOI:10.1158/2159-8274.cd-10-0028.
- S. Shimura, G. Yang, S. Ebara, T. M. Wheeler, A. Frolov and T. C. Thompson, Reduced Infiltration of Tumor-Associated Macrophages in Human Prostate Cancer: Association with Cancer Progression 1, Cancer Res., 2000, 5857–5861 CAS.
- K. Agelopoulos, O. Myklebost, M. Serra, F. Mertens, P. C. W. Hogendoorn and A. C. Lankester, Tumor-Infiltrating Macrophages Are Associated with Metastasis Suppression in High-Grade Osteosarcoma: A Rationale for Treatment with Macrophage Activating Agents, Hum. Cancer Biol., 2011, 17(7), 2110–2120, DOI:10.1158/1078-0432.ccr-10-2047.
- Y. Komohara, M. Jinushi and M. Takeya, Clinical Significance of Macrophage Heterogeneity in Human Malignant Tumors, Cancer Sci., 2014, 105(1), 1–8, DOI:10.1111/cas.12314.
- A. J. Petty and Y. Yang, Tumor-Associated Macrophages: Implications in Cancer Immunotherapy, Immunotherapy, 2017, 9, 289–302 CrossRef CAS PubMed.
- M. F. Cuccarese, J. M. Dubach, C. Pfirschke, C. Engblom, C. Garris, M. A. Miller, M. J. Pittet and R. Weissleder, Heterogeneity of Macrophage Infiltration and Therapeutic Response in Lung Carcinoma Revealed by 3D Organ Imaging, Nat. Commun., 2017, 8(1), 1–10, DOI:10.1038/ncomms14293.
- M. De Palma and C. E. Lewis, Macrophage Regulation of Tumor Responses to Anticancer Therapies, Cancer Cell, 2013, 23(3), 277–286, DOI:10.1016/j.ccr.2013.02.013.
- R. Toy, L. Bauer, C. Hoimes, K. B. Ghaghada and E. Karathanasis, Targeted Nanotechnology for Cancer Imaging, Adv. Drug Deliv. Rev., 2014, 76(1), 79–97, DOI:10.1016/j.addr.2014.08.002.
- J. V. Frangioni, New Technologies for Human Cancer Imaging, J. Clin. Oncol., 2008, 26(24), 4012–4021, DOI:10.1200/jco.2007.14.3065.
- C. Zhang, X. Yu, L. Gao, Y. Zhao, J. Lai, D. Lu, R. Bao and B. Jia, Noninvasive Imaging of CD206-Positive M2 Macrophages as an Early Biomarker for Post-Chemotherapy Tumor Relapse and Lymph Node Metastasis, Theranostics, 2017, 7(17), 4276, DOI:10.7150/thno.20999.
- A. Faraj and D. Press, MR Imaging and Targeting of a Specific Alveolar Macrophage Subpopulation in LPS-Induced COPD Animal Model Using Antibody-Conjugated Magnetic Nanoparticles, Int. J. Nanomed., 2020, 1491–1503 Search PubMed.
- Y. Shimizu, H. Hanzawa, Y. Zhao, S. Fukura, K. Nishijima, T. Sakamoto, S. Zhao, N. Tamaki, M. Ogawa and Y. Kuge, Immunoglobulin G (IgG)-Based Imaging Probe Accumulates in M1 Macrophage-Infiltrated Atherosclerotic Plaques Independent of IgG Target Molecule Expression, Mol. Imaging Biol., 2017, 531–539, DOI:10.1007/s11307-016-1036-8.
- S. Mukherjee, D. Sonanini, A. Maurer and H. E. Daldrup-Link, The Yin and Yang of Imaging Tumor Associated Macrophages with PET and MRI, Theranostics, 2019, 9(25), 7730–7748, DOI:10.7150/thno.37306.
- J. Klohs, M. Gra, K. Graf, J. Steinbrink, T. Dietrich, D. Stibenz, P. Bahmani, G. Kronenberg, C. Harms and M. Endres, et al., In Vivo Imaging of the Inflammatory Receptor CD40 After Cerebral Ischemia Using a Fluorescent Antibody, Stroke, 2008, 2845–2852, DOI:10.1161/strokeaha.107.509844.
- C. Jiang, H. Cai, X. Peng and P. Zhang, Targeted Imaging of Tumor-Associated Macrophages by Cyanine 7-Labeled Mannose in Xenograft Tumors, Mol. Imag., 2017, 16, 1–10, DOI:10.1177/1536012116689499.
- S. A. Fernández, M. Vendrell, A. Ferna and M. Vendrell, Smart Fluorescent Probes for Imaging Macrophage Activity, Chem. Soc. Rev., 2016, 45(5), 1182–1196, 10.1039/c5cs00567a.
- X. Sun, D. Gao, L. Gao, C. Zhang, X. Yu, B. Jia, F. Wang and Z. Liu, Molecular Imaging of Tumor-Infiltrating Macrophages in a Preclinical Mouse Model of Breast Cancer, Theranostics, 2015, 5(6), 597–608, DOI:10.7150/thno.11546.
- S. Park, B. Kim, S. Choi, S. Balasubramaniam, S. Lee, J. Y. Lee, H. S. Kim, J. Kim, J. Kim and Y. Lee, et al., Imaging Inflammation Using an Activated Macrophage Probe with Slc18b1 as the Activation-Selective Gating Target, Nat. Commun., 2019, 1–7, DOI:10.1038/s41467-019-08990-9.
- S. D. Steichen, M. Caldorera-moore and N. A. Peppas, A Review of Current Nanoparticle and Targeting Moieties for the Delivery of Cancer Therapeutics, Eur. J. Pharm. Sci., 2013, 48(3), 416–427, DOI:10.1016/j.ejps.2012.12.006.
- C. M. Dawidczyk, L. M. Russell and P. C. Searson, Nanomedicines for cancer therapy: state-of-the-art and limitations to pre-clinical studies that hinder future developments, Front. Chem., 2014, 2(69), 1–13, DOI:10.3389/fchem.2014.00069.
- P. Chames, M. V. Regenmortel, E. Weiss and D. Baty, Therapeutic Antibodies: Successes, Limitations and Hopes for the Future, Br. J. Pharmacol., 2009, 157(2), 220–233, DOI:10.1111/j.1476-5381.2009.00190.x.
- C. Tu, T. S. C. Ng, H. K. Sohi, H. A. Palko, A. House, R. E. Jacobs and A. Y. Louie, Receptor-Targeted Iron Oxide Nanoparticles for Molecular MR Imaging of Inflamed Atherosclerotic Plaques, Biomaterials, 2011, 32(29), 7209–7216, DOI:10.1016/j.biomaterials.2011.06.026.
- B. R. Jarrett, M. Frendo, J. Vogan and A. Y. Louie, Size-Controlled Synthesis of Dextran Sulfate Coated Iron Oxide Nanoparticles for Magnetic Resonance Imaging, Nanotechnology, 2007, 18(3), 035603, DOI:10.1088/0957-4484/18/3/035603.
- T. A. Hamilton, C. Zhao, P. G. Pavicic and S. Datta, Myeloid Colony-Stimulating Factors as Regulators of Macrophage Polarization, Front. Immunol., 2014, 5, 1–6, DOI:10.3389/fimmu.2014.00554.
- J. Y. Jang, J. K. Lee, Y. K. Jeon and C. W. Kim, Exosome Derived from Epigallocatechin Gallate Treated Breast Cancer Cells Suppresses Tumor Growth by Inhibiting Tumor-Associated Macrophage Infiltration and M2 Polarization, BMC Cancer, 2013, 13, 421, DOI:10.1186/1471-2407-13-421.
- R. Deng, S. M. Wang, T. Yin, T. H. Ye, G. B. Shen, L. Li, J. Y. Zhao, Y. X. Sang, X. G. Duan and Y. Q. Wei, Dimethyl Sulfoxide Suppresses Mouse 4T1 Breast Cancer Growth by Modulating Tumor-Associated Macrophage Differentiation, J. Breast Cancer, 2014, 17(1), 25–32, DOI:10.4048/jbc.2014.17.1.25.
- I. S. Kim, Y. Gao, T. Welte, H. Wang, J. Liu, M. Janghorban, K. Sheng, Y. Niu, A. Goldstein and N. Zhao, et al., Immuno-Subtyping of Breast Cancer Reveals Distinct Myeloid Cell Profiles and Immunotherapy Resistance Mechanisms, Nat. Cell Biol., 2019, 21(9), 1113–1126, DOI:10.1038/s41556-019-0373-7.
- J. W. Pollard, Macrophages Define the Invasive Microenvironment in Breast Cancer, J. Leukoc. Biol., 2008, 84(3), 623–630, DOI:10.1189/jlb.1107762.
- R. D. Leek and A. L. Harris, Tumor-Associated Macrophages in Breast Cancer, J. Mammary Gland Biol. Neoplasia, 2002, 7(2), 177–189, DOI:10.1023/A:1020304003704.
- A. Sica, T. Schioppa, A. Mantovani and P. Allavena, Tumour-Associated Macrophages Are a Distinct M2 Polarised Population Promoting Tumour Progression: Potential Targets of Anti-Cancer Therapy, Eur. J. Cancer, 2006, 42(6), 717–727, DOI:10.1016/j.ejca.2006.01.003.
- S. Parveen, S. Siddharth, L. S. Cheung, A. Kumar, J. Shen, J. R. Murphy, D. Sharma and W. R. Bishai, Therapeutic Targeting with DABIL-4 Depletes Myeloid Suppressor Cells in 4T1 Triple-Negative Breast Cancer Model, Mol. Oncol., 2021, 15(5), 1330–1344, DOI:10.1002/1878-0261.12938.
- R. Jin, B. Lin, D. Li and H. Ai, Superparamagnetic Iron Oxide Nanoparticles for MR Imaging and Therapy: Design Considerations and Clinical Applications, Curr. Opin. Pharmacol., 2014, 18, 18–27, DOI:10.1016/j.coph.2014.08.002.
- M. P. Melancon, W. Lu, Q. Huang, P. Thapa, D. Zhou, C. Ng and C. Li, Targeted Imaging of Tumor-Associated M2 Macrophages Using a Macromolecular Contrast Agent PG-Gd-NIR813, Biomaterials, 2010, 31(25), 6567–6573, DOI:10.1016/j.biomaterials.2010.05.001.
- J. Müller, W. Von Bernstorff, C. D. Heidecke and T. Schulze, Differential S1P Receptor Profiles on M1- and M2-Polarized Macrophages Affect Macrophage Cytokine Production and Migration, Biomed Res. Int., 2017, 2017, 9–11, DOI:10.1155/2017/7584621.
- Y. C. Koh, G. Yang, C. S. Lai, M. Weerawatanakorn and M. H. Pan, Chemopreventive Effects of Phytochemicals and Medicines on M1/M2 Polarized Macrophage Role in Inflammation-Related Diseases, Int. J. Mol. Sci., 2018, 19(8), 1–30, DOI:10.3390/ijms19082208.
- S. Wang; S. Xu; J. Zhou; L. Zhang; X. Mao; X. Yao and C. Liu Luteolin Transforms the Polarity of Bone Marrow-Derived Macrophages to Regulate the Cytokine Storm, 2021, vol. 1–12 Search PubMed.
- H. Sun, S. Sun, G. Chen, H. Xie, S. Yu, X. Lin, J. Qian, C. Mao, H. Peng and H. Chen, Ceramides and Sphingosine-1-Phosphate Mediate the Distinct Effects of M1/M2-Macrophage Infusion on Liver Recovery after Hepatectomy, Cell Death Dis., 2021, 12(4), 1–16, DOI:10.1038/s41419-021-03616-9.
- A. M. Georgoudaki, K. E. Prokopec, V. F. Boura, E. Hellqvist, S. Sohn, J. Östling, R. Dahan, R. A. Harris, M. Rantalainen and D. Klevebring, et al., Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis, Cell Rep., 2016, 15(9), 2000–2011, DOI:10.1016/j.celrep.2016.04.084.
- L. Cassetta and J. W. Pollard, Repolarizing Macrophages Improves Breast Cancer Therapy, Cell Res., 2017, 27(8), 963–964, DOI:10.1038/cr.2017.63.
- Y. Huang, C. Mei, Y. Tian, T. Nie, Z. Liu and T. Chen, Bioinspired Tumor-Homing Nanosystem for Precise Cancer Therapy via Reprogramming of Tumor-Associated Macrophages, NPG Asia Mater., 2018, 10(10), 1002–1015, DOI:10.1038/s41427-018-0091-9.
- M. J. Su, H. Aldawsari and M. Amiji, Pancreatic Cancer Cell Exosome-Mediated Macrophage Reprogramming and the Role of MicroRNAs 155 and 125b2 Transfection Using Nanoparticle Delivery Systems, Sci. Rep., 2016, 6, 1–15, DOI:10.1038/srep30110.
- T. Cooks, I. S. Pateras, L. M. Jenkins, K. M. Patel, A. I. Robles, J. Morris, T. Forshew, E. Appella, V. G. Gorgoulis and C. C. Harris, Mutant P53 Cancers Reprogram Macrophages to Tumor Supporting Macrophages via Exosomal MiR-1246, Nat. Commun., 2018, 9(1) DOI:10.1038/s41467-018-03224-w.
- Z. Duan and Y. Luo, Targeting Macrophages in Cancer Immunotherapy, Signal Transduct. Target. Ther., 2021, 6(1), 1–21, DOI:10.1038/s41392-021-00506-6.
- A. S. Poltavets, P. A. Vishnyakova, A. V. Elchaninov, G.T. Sukhikh and T. K. Fatkhudinov, Macrophage Modification Strategies for Efficient Cell Therapy, Cells, 2020, 9(6), 1–19 CrossRef PubMed.
- B. R. Jarrett, D. L. Kukis and A. Y. Louie, Synthesis of Cu-64-Labeled Magnetic Nanoparticles for Multimodal Imaging, Bioconjugate Chem., 2008, 1496–1504 CrossRef CAS PubMed.
- R. Kapre, J. Zhou, X. Li, L. Beckett and A. Y. Louie, A Novel Gamma GLM Approach to MRI Relaxometry Comparisons, Magn. Reson. Med., 2020, 84(3), 1592–1604, DOI:10.1002/mrm.28192.
Footnotes |
| † Electronic supplementary information (ESI) available: Relaxivity fits at 1.4 T and 7 T, raw data of western blot, 4T1 cells uptake study, kidney and liver histology and immunohistochemistry images would be found. See DOI: 10.1039/d1ra08061j |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |