
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of dual-functional catalysis for hydrazine oxidation by an organic p–n bilayer through in situ formation of a silver co-catalyst†
Mamoru Sato and
Toshiyuki Abe*
and
Toshiyuki Abe*
Department of Frontier Materials Chemistry, Graduate School of Science and Technology, Hirosaki University, 3 Bunkyo-cho, Hirosaki 036-8561, Japan. E-mail: tabe@hirosaki-u.ac.jp
First published on 12th January 2022
Abstract
Dual-functional catalysis indicates that an organic p–n bilayer induces the catalytic oxidation involved in downhill reactions, not only under illumination but also in the dark. When the organo-bilayer is composed of a perylene derivative (3,4,9,10-perylenetetracarboxylic-bis-benzimidazole (PTCBI), n-type) and cobalt phthalocyanine (CoPc, p-type), only the photocatalytic oxidation of hydrazine (N2H4) occurs. However, the loading of Ag co-catalyst onto the CoPc surface in the PTCBI/CoPc bilayer successfully led to dual catalysis in terms of the oxidation of N2H4 to N2. To develop the present dual catalysis Ag loading was essential to achieve the catalysis performance particularly without irradiation.
Introduction
Studies on photocatalytic reactions have been widely reported. In addition to the application in uphill reactions with ΔG° > 0 (e.g., water splitting),1–5 photocatalysts are also effectively applied in downhill reactions with ΔG° < 0 (e.g., decomposition of pollutants), particularly when kinetically severe oxidation with large activation energy is involved.6–10 Among the photocatalysts, TiO2 is recognized to be practically used in the degradation of several types of pollutant.11–15 However, the degrading targets are limited to only substances of low concentrations because of the ultraviolet (UV) response of TiO2. Moreover, the catalytic degradation of pollutants by TiO2 is expected to occur only under UV irradiation; in other words, TiO2 can never exhibit a catalytic performance for degradation in the dark similar to that under irradiation.Various types of photocatalysts and electrocatalysts towards energy applications are extensively investigated in terms of hydrogen evolution, oxygen evolution, CO2 reduction, oxygen reduction, alcohol oxidation, etc., and some reviews have been published recently.16–20 We have been studying organic p–n bilayer, including p-type and n-type, semiconductors for application in photoelectrochemical and photocatalytic reactions in the water phase.21–32 Some unique reactions, which had not been induced so far by conventional photocatalysts, were also found to occur.27–32 In our studies, an organic p–n bilayer using 3,4,9,10-perylenetetracarboxylic-bis-benzimidazole (PTCBI) and cobalt phthalocyanine (CoPc) as n-type and p-type semiconductors, respectively, achieved the catalytic oxidation of thiol under illumination and in the dark.28 As depicted in Scheme S1,† the dark reaction occurs according to the pathway indicated with a dashed line, in which the lower edge of the conduction band of CoPc (i.e., CoIIPc) corresponds to the potential for oxidizing thiol. While, under irradiation, the thiol oxidation proceeds as demonstrated by the solid-lined pathway, in which the oxidizing power is generated at the CoPc surface (i.e., CoIIIPc) via a series of photophysical events (i.e., formation of excitons based on visible light absorption by the PTCBI/CoPc bilayer, excitation energy transfer of the excitons, generation of carriers by the dissociation of excitons into electrons and holes at the p–n interface, and conduction of hole carriers through the valence band of CoPc). We named “dual-functional catalysis” for the aforementioned catalysis. In each case, electrons can attain the conduction band of PTCBI, followed by its consumption through a reduction reaction. However, such dual-functional catalysis has been achieved only for thiol oxidation.27,28
Previously, we reported that the photocatalytic oxidation of hydrazine (N2H4) occurs at the CoPc surface in the PTCBI/CoPc bilayer,32 but there was no evidence of the corresponding oxidation in the dark. In this study, to develop the catalytic oxidation of N2H4 in the dark, silver species was combined with the organo-bilayer. Ag33 and Ag2O34 are recognized as a catalyst in the dark and photocatalyst, respectively, for N2H4 oxidation; however, Ag2O may be reduced to Ag in the presence of N2H4 (i.e., reductant). As a result, by preparing the organo-bilayer modified with Ag2O, the dual-functional catalysis for N2H4 oxidation was accomplished. The details are discussed from the perspective of photoelectrochemistry.
Experimental
PTCBI was synthesized according to a reported procedure and purified by sublimation prior to use.35 CoPc was purchased from Tokyo Chemical Industry and used as received. Indium tin oxide (ITO)-coated glass plate (sheet resistance = 8 Ω cm−2; transmittance ≥ 85%; and ITO thickness = 174 nm) was acquired from AGC Inc. N2H4 and Nafion (Nf) alcoholic solution were purchased from Kanto Chemical and Sigma-Aldrich, respectively. All other chemicals employed were of extra pure grade.The PTCBI/CoPc bilayer was fabricated by vapor deposition (pressure ≤ 1.0 × 10−3 Pa; deposition speed = 0.03 nm s−1),32 in which PTCBI was first coated on an ITO, followed by coating CoPc on top of the PTCBI layer. The thickness of the organo-bilayer was determined by measuring a UV-VIS absorption spectrum. Determination procedure of the thickness with each layer has so far been described elsewhere.26 Silver(I) oxide was synthesized according to a reported procedure.36 Silver(I) nitrate (0.58 g) was dissolved in water (50 mL), and the pH value of the aforementioned AgNO3 solution was adjusted to pH = 14 with 2 M NaOH solution. The resulting solution was maintained by stirring overnight, following which Ag2O was collected by filtration and dried at 70 °C. The resulting particles of Ag2O (13 mg) were suspended in 1 wt% Nafion (Nf) alcoholic solution (1 mL). The mixture solution (23 μL) was dropped on the CoPc surface in the PTCBI/CoPc bilayer and dried at 70 °C. The fabricated electrode is denoted as PTCBI/CoPc-Nf[Ag2O] (i.e., effective area = 1 cm2; thickness of Nf = 1 μm; loaded amount of Ag2O = ca. 1.3 μmol cm−2). Nf membrane was employed as the absorbent for N2H4 and support for Ag2O. In the present study, the controlled electrode free of Ag2O was also prepared and used (denoted as PTCBI/CoPc-Nf).
The structural analyses of Ag2O and Ag were performed using by an X-ray diffractometer (XRD: Rigaku, SmartLab 9 kW), a tunneling electron microscope (TEM: JEOL, JEM-2100) and a scanning electron microscope (SEM: JEOL, JSM-7000F).
When measuring voltammograms and photocurrents for acquiring an action spectrum, a single-compartment cell was operated using a potentiostat (Hokuto Denko, HA-301) equipped with a function generator (Hokuto Denko, HB-104), a coulomb meter (Hokuto Denko, HF-201), and an X–Y recorder (see Scheme S2†). Particularly for the action spectral measurements, the light source was used in the combination with a monochromator (Soma Optics, Ltd, S-10) for irradiating monochromatic light.
The electrolysis study was performed in a twin-compartment cell separated by a salt bridge (Scheme 1). PTCBI/CoPc-Nf[Ag2O] and Pt were placed as oxidation site in a N2H4 solution (5 mM, pH = 11) and as reduction site in a phosphoric acid solution (pH = 0), respectively. Ag/AgCl reference was set along with the Pt counter. For preparing the salt bridge, both agar (1.3 g) and KNO3 (4.74 g) were dissolved in hot water (10 mL). Subsequently, the mixture was allowed to flow into the bridging part of the cell, followed by its solidification at room temperature. The twin-compartment cell for electrolysis reaction was operated using the aforementioned electrochemical apparatus.
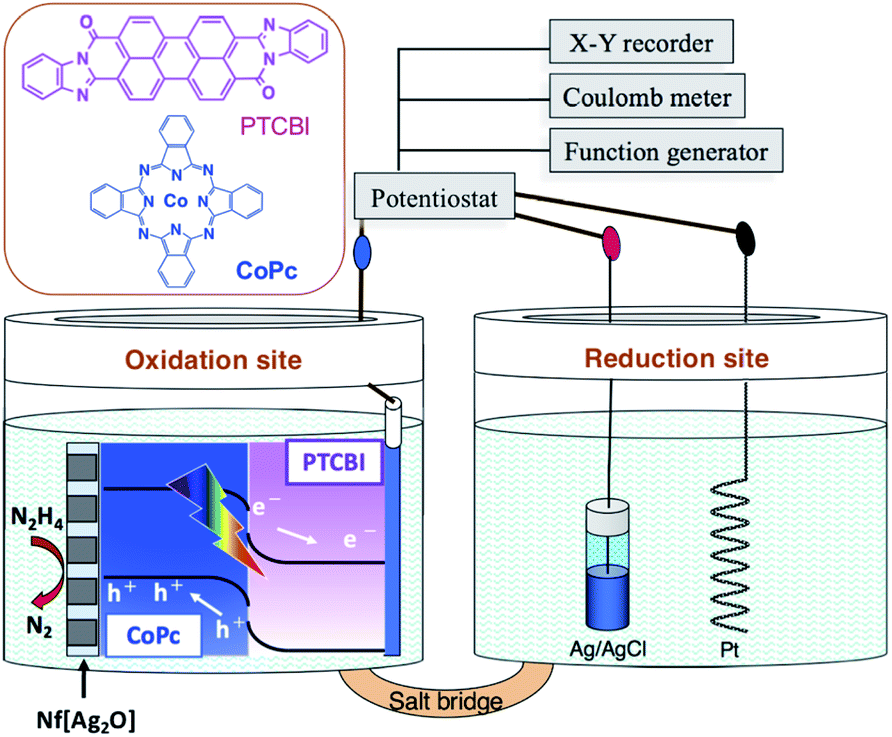 | ||
| Scheme 1 Illustration of the photoelectrolysis system employed for N2H4 oxidation and chemical structures of PTCBI and CoPc. | ||
A halogen lamp was used for irradiating the organo-bilayer. The light intensity was measured using a power meter (type 3A from Ophir Japan, Ltd), and the intensity was determined at approximately 100 mW cm−2, except for the action spectral measurement. The gaseous products of N2 and H2 were analyzed using a gas chromatograph (GL Sciences, GC-3200) equipped with a thermal conductivity detector (column, 5 Å molecular sieve; carrier gas, Ar). Additional experimental details are described in the ESI.†
Results and discussions
First, the voltammograms of PTCBI/CoPc-Nf[Ag2O] were measured in the dark and under irradiation (see Scheme S2†) and compared with those of PTCBI/CoPc-Nf. Similar to our previous study,32 PTCBI/CoPc-Nf induced the oxidation of N2H4 only under irradiation (Fig. 1(a)). However, when loading Ag2O on the PTCBI/CoPc bilayer, irrespective of irradiation, anodic currents occurred at PTCBI/CoPc-Nf[Ag2O] because of the N2H4 oxidation (Fig. 1(b)). Electrochemical oxidation of N2H4 was examined under potentiostatic conditions (see Scheme 1), and the electrolysis data are summarized in Table 1. The oxidative formation of N2 from N2H4 was confirmed in the dark along with the reduction of H+ to H2. Moreover, the N2 (oxidation product) and H2 (reduction product) amounts increased significantly under irradiation (note that in each case the faradaic efficiency of the N2 and H2 formation was estimated to be >85% and >90%, respectively), which are consistent with the aforementioned voltammetric characteristics of Fig. 1(b). As a supplementary explanation, the oxidation of N2H4 to N2H2 and subsequent spontaneous decomposition of N2H2 to N2 and H2 (ref. 37) are considered not to occur in the present system because no formation of H2 was confirmed in the oxidation site in Scheme 1.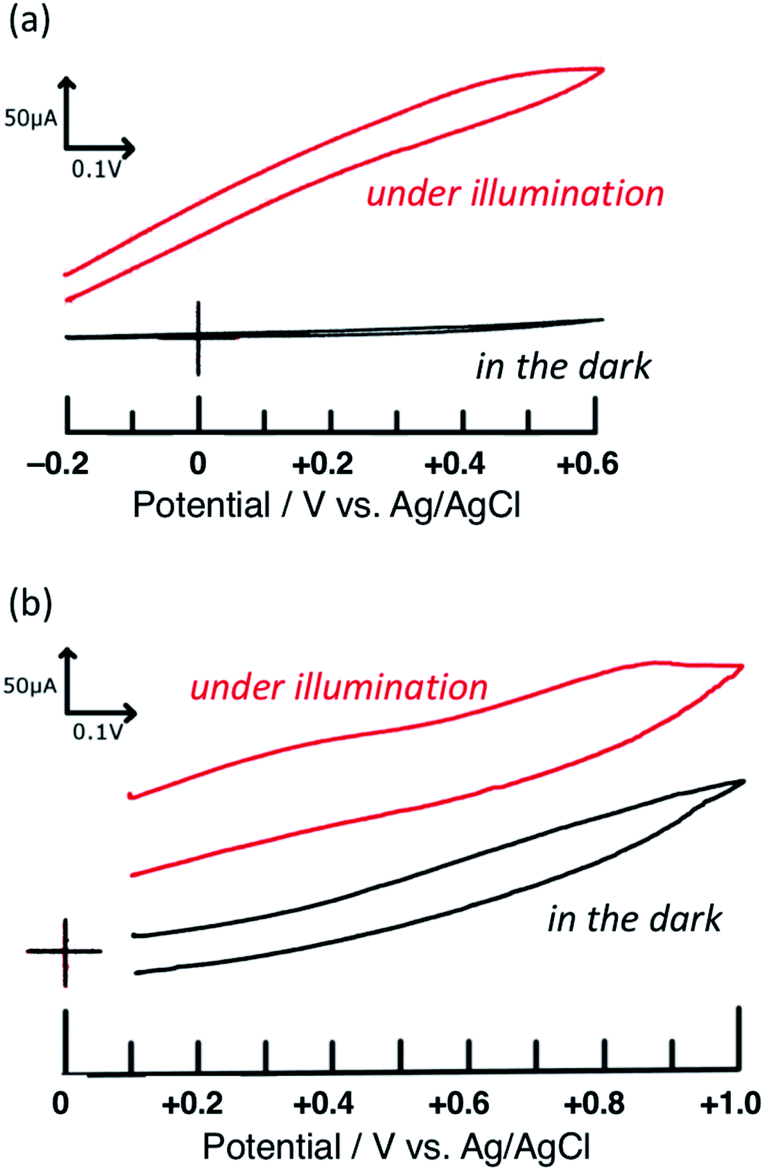 | ||
| Fig. 1 Voltammograms of (a) PTCBI/CoPc-Nf and (b) PTCBI/CoPc-Nf[Ag2O]. Film thickness: n-type PTCBI = 230 nm, p-type CoPc = 65 nm, and Nf = 1 μm; N2H4 solution, 5 mM (pH = 11); light intensity, 100 mW cm−2; scan rate = 20 mV s−1. | ||
| H2 evolved/μL | N2 evolved/μL | Note | |
|---|---|---|---|
| a Film thickness: PTCBI = 205 nm and CoPc = 60 nm; electrolyte solution (oxidation site), an aqueous N2H4 solution (5 mM, pH = 11); electrolyte solution (reduction site), an aqueous H3PO4 solution (pH = 0); applied potential, +0.3 V (vs. Ag/AgCl); reaction time, 3 h.b Irradiation was conducted from the backside of ITO-coated face (light intensity, 100 mW cm−2). | |||
| Entry 1 | 14.3 | 8.08 | In the dark |
| Entry 2b | 94.8 | 48.4 | Under irradiation |
It is important to verify how Ag2O participated in the N2H4 oxidation, particularly in the dark (vide supra). To clarify the catalytically active Ag species for the N2H4 oxidation, X-ray diffractometer (XRD) patterns were measured (Fig. 2). For reference, the XRD pattern of PTCBI/CoPc-Nf is shown in Fig. 2(a). In the unused PTCBI/CoPc-Nf[Ag2O] (Fig. 2(b)), the resulting XRD pattern was characterized by cubic Ag2O.38 From the XRD patterns after the electrolysis in the dark (Fig. 2(c)) and under irradiation (Fig. 2(d)), the formation of cubic Ag was confirmed,39 indicating a reductive transformation of Ag2O in the presence of N2H4. The XRD pattern depicted in Fig. S1† indicates that the Ag formation is probably occurring during the Ar purge of the electrolyte solution (30 min) prior to the electrochemical measurements. Comparing the potentials of +0.70 V vs. SHE (pH = 11) for E° (Ag2O/Ag)40 and −0.98 vs. SHE (pH = 11) for E° (N2H4/N2),41 the reduction of Ag2O to Ag can occur reasonably using N2H4 as reductant (i.e., N2H4 + 2Ag2O → N2 + 4Ag + 2H2O). Thus, the present dual catalysis for N2H4 oxidation originates from the in situ formation of Ag co-catalyst at the PTCBI/CoPc bilayer, revealing that Ag2O does not collaboratively show photocatalytic activity for the N2H4 oxidation along with the organo-bilayer (vide supra). Some TEM and SEM images of Ag2O (or Ag) dispersed in Nf membrane were observed. As for the samples prior to electrochemical study, those TEM images indicated the particle sizes were approximately <10 nm and 3–35 nm for Ag2O and Ag, respectively (Fig. S2(a) and (b)†). The SEM images were observed for PTCBI/CoPc-Nf[Ag2O] exposed to the N2H4 solution under Ar purge (Fig. S3(a)†) as well as that after photoelectrolysis (Fig. S3(b)†). Each image was almost the same as each other, indicating that Ag transformed from Ag2O remains unchanged even after the photoelectrolysis. That is, the aggregation and growth of Ag particles was not be recognized after its use.
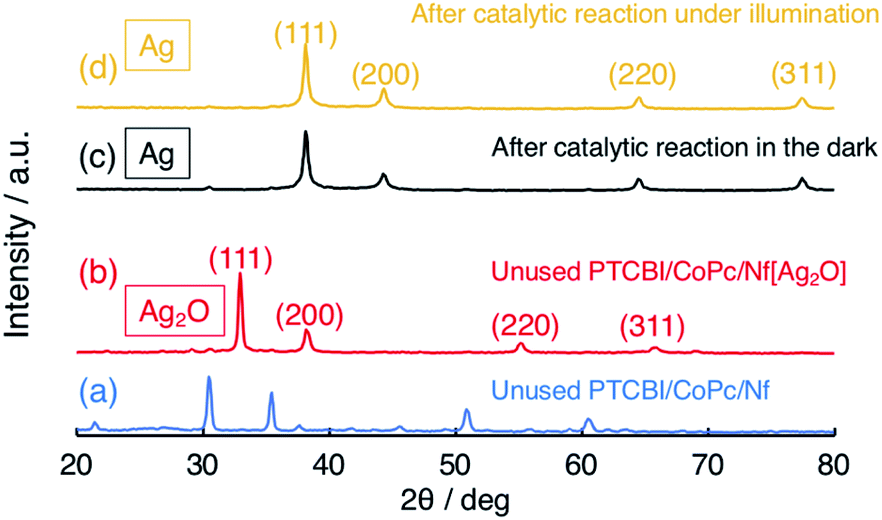 | ||
| Fig. 2 XRD patterns of (a) PTCBI/CoPc-Nf, (b) unused PTCBI/CoPc-Nf[Ag2O], and PTCBI/CoPc-Nf[Ag2O] after the N2H4 oxidation (c) in the dark and (d) under irradiation. | ||
The N2H4 oxidation occurring at PTCBI/CoPc-Nf[Ag2O] is represented in Scheme 2. In the dark, the potential of CoIIPc (corresponding to the lower edge of the conduction band: −0.32 V vs. SHE, pH = 11)42 is available for the N2H4 oxidation, and thus, N2H4 is catalytically oxidized at the Ag-loaded CoPc surface. When the photoinduced oxidation of N2H4 occurs, the oxidizing power is generated at the top edge of the valence band of CoPc (i.e., CoIIIPc, +0.93 V vs. SHE, pH = 11)42 through a series of the photophysical events within the organo-bilayer (vide supra). According to the resulting action spectrum for photocurrents (Fig. 3), the photoinduced N2H4 oxidation occurred originating in the absorption of PTCBI over the entire visible light region. This is a specific characteristic usually observed when using PTCBI as the n-type layer.25,27,28,32 The oxidizing power is larger under irradiation than in the dark; consequently, the photoinduced N2H4 oxidation is noticeably superior to the oxidation in the dark. In thermodynamic sense, the N2H4 oxidation can occur at the CoPc in the dark. However, to induce kinetically the forward oxidation of N2H4 to N2 particularly in the dark, the Ag co-cocatalyst needs to be loaded.
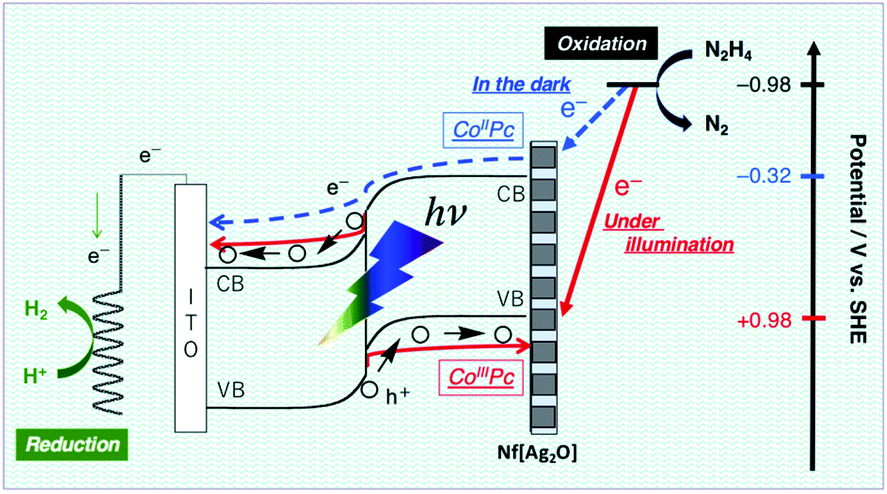 | ||
| Scheme 2 Mechanism of the dual-functional catalysis for N2H4 oxidation occurring at PTCBI/CoPc-Nf[Ag2O] under illumination and in the dark. | ||
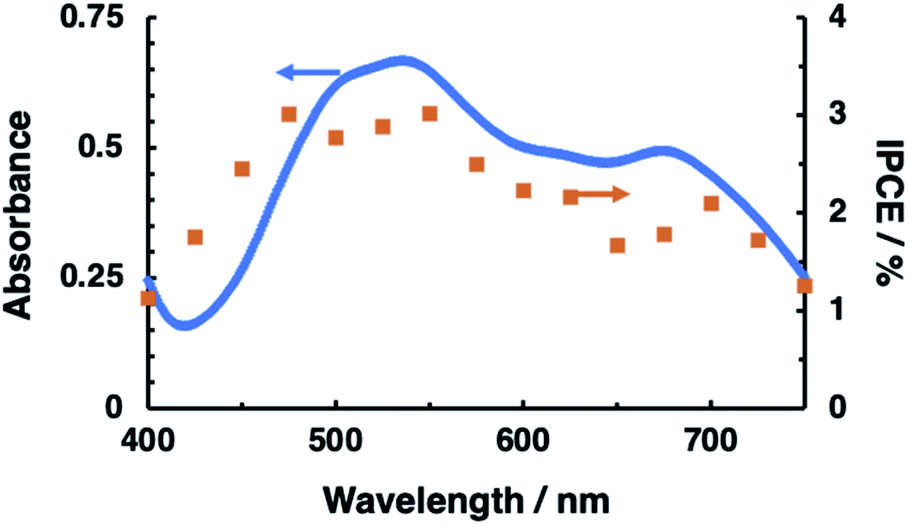 | ||
| Fig. 3 Action spectrum (closed squares) for photocurrents measured at PTCBI/CoPc-Nf (without loading Ag2O). The absorption spectrum of PTCBI (solid line) is also depicted. Irradiation was conducted from the backside of ITO-coated face. Film thickness: PTCBI = 210 nm and CoPc = 80 nm; applied potential, +0.3 V vs. Ag/AgCl; light intensity, 0.15 mW cm−2; electrolyte solution, an aqueous N2H4 solution (5 mM, pH = 11). | ||
Conclusion
In summary, the oxidation of N2H4 to N2 occurred successfully at the PTCBI/CoPc bilayer under irradiation and in the dark, particularly by loading Ag on the CoPc surface. The development of the present dual-functional catalysis was attributed to the in situ formation of Ag through the reductive transformation of Ag2O in the presence of N2H4 as reductant, whereby the catalytic oxidation of N2H4 effectively occurred even in the dark. Such catalysis for N2H4 oxidation did not occur at the Ag-free PTCBI/CoPc bilayer. Therefore, the so-called dual-functional catalysis is a novel catalytic process for oxidation reactions, irrespective of irradiation. The loading of a co-catalyst on an organo-bilayer is expected to expand the application for several types of downhill reactions, opening new opportunities in the field of pollutant degradation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partly supported by JSPS KAKENHI Grant Number JP18K05287 (T. A.).References
- X. Li, P. Cheng, X. Zhang, T. Shen, J. Liu, J.-C. Ren, H. Wang, S. Li and W. Liu, J. Mater. Chem. A, 2021, 9, 14515 RSC.
- C. Shi, S. Ye, X. Wang, F. Meng, J. Liu, T. Yang, W. Zhang, J. Wei, N. Ta, G. Q. Lu, M. Hu and J. Liu, Adv. Sci., 2021, 8, 2001987 CrossRef CAS PubMed.
- D. Zheng, L. Yang, W. Chen, Y. Fang and X. Wang, ChemSusChem, 2021, 14, 3821 CrossRef CAS PubMed.
- J. Cui, X. Yang, Z. Yang, Y. Sun, X. Chen, X. Liu, D. Wang, S. Jiang and J. Ye, Chem. Commun., 2021, 57, 10640 RSC.
- C. S. Gopinath and N. Nalajaja, J. Mater. Chem. A, 2021, 9, 1353 RSC.
- H. J. Biswal, A. Yadav, P. R. Vundavilli and A. Gupta, RSC Adv., 2021, 11, 1623 RSC.
- A. P. Kumar, D. Bilehal, A. Tadesse and D. Kumar, RSC Adv., 2021, 11, 6396 RSC.
- V. Balakumar, R. Manivannan, C. Chuaicham, S. Karthikeyan and K. Sasaki, Chem. Commun., 2021, 57, 6772 RSC.
- B. Zhu, D. Song, T. Jia, W. Sun, D. Wang, L. Wang, J. Guo, L. Jin, L. Zhang and H. Tao, ACS Omega, 2021, 6, 1647 CrossRef CAS PubMed.
- K. Akber, E. Moretti and A. Vomiero, Adv. Opt. Mater., 2021, 9, 2100532 CrossRef.
- J. He, P. Lyu, D. Li, C. Cheng, S. Chang, L. Qin, C. Zheng and J. Zhu, Chem. Commun., 2021, 57, 6883 RSC.
- Y. Wang, C. Zhu, G. Zuo, Y. Guo, W. Xiao, Y. Dai, J. Kong, X. Xu, Y. Zhou, A. Xie, C. Sun and Q. Xian, Appl. Catal., B, 2020, 278, 119298 CrossRef CAS.
- D. Benz, K. M. Felter, J. Köser, J. Thöming, G. Mul, F. C. Grozema, H. T. Hintzen, M. T. Kreutzer and J. R. van Ommen, J. Phys. Chem. C, 2020, 124, 8269 CrossRef CAS.
- H. Maleki and V. Bertola, ACS Appl. Nano Mater., 2019, 2, 7237 CrossRef CAS.
- A. Torres-Pinto, C. G. Silva, J. L. Faria and A. M. T. Silva, Adv. Sci., 2021, 8, 2003900 CrossRef CAS PubMed.
- C. S. Gopinath and N. Nalajala, J. Mater. Chem. A, 2021, 9, 1353 RSC.
- Z. P. Ifkovits, J. K. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2021, 14, 4740 RSC.
- B. Qiu, M. Du, Y. Ma, Q. Zhu, M. Xing and J. Zhang, Energy Environ. Sci., 2021, 14, 5260 RSC.
- L. Chai, Z. Hu, X. Wang, L. Zhang, T.-T. Li, Y. Hu, J. Pan, J. Qian and S. Huang, Carbon, 2021, 174, 531 CrossRef CAS.
- J. Wang, Z. Zhang, J. Ding, C. Zhong, Y. Deng, X. Han and W. Hu, Sci. China Mater., 2021, 64, 1 CrossRef CAS.
- T. Murakami, K. Ikezoi, K. Nagai, H. Kato and T. Abe, ChemElectroChem, 2020, 7, 5029 CrossRef CAS.
- K. Fujine, Y. Sato, K. Nagai and T. Abe, J. Electroanal. Chem., 2018, 823, 322 CrossRef CAS.
- T. Abe, Y. Tanno, T. Ebina, S. Miyakushi and K. Nagai, ACS Appl. Mater. Interfaces, 2013, 5, 1248 CrossRef CAS PubMed.
- T. Abe, S. Tobinai, N. Taira, J. Chiba, T. Itoh and K. Nagai, J. Phys. Chem. C, 2011, 115, 7701 CrossRef CAS.
- T. Abe, S. Miyakushi, K. Nagai and T. Norimatsu, Phys. Chem. Chem. Phys., 2008, 10, 1562 RSC.
- T. Abe, K. Nagai, S. Kabutomori, M. Kaneko, A. Tajiri and T. Norimatsu, Angew. Chem., Int. Ed., 2006, 45, 2778 CrossRef CAS PubMed.
- R. Watanabe and T. Abe, Int. J. Electrochem. Sci., 2019, 14, 3315 CrossRef CAS.
- T. Abe, M. Okumura, Y. Kikuchi, T. Itoh and K. Nagai, J. Mater. Chem. A, 2017, 5, 7445 RSC.
- D. Mendori, T. Hiroya, M. Ueda, M. Sanyoushi, K. Nagai and T. Abe, Appl. Catal., B, 2017, 205, 514 CrossRef CAS.
- T. Abe, K. Fukui, Y. Kawai, K. Nagai and H. Kato, Chem. Commun., 2016, 52, 7735 RSC.
- T. Abe, N. Taira, Y. Tanno, Y. Kikuchi and K. Nagai, Chem. Commun., 2014, 50, 1950 RSC.
- T. Abe, Y. Tanno, N. Taira and K. Nagai, RSC Adv., 2015, 5, 46325 RSC.
- V. Bansal, V. Li, A. P. O'Mullane and S. K. Bhargava, CrystEngComm, 2010, 12, 4280 RSC.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543 CrossRef CAS.
- T. Maki and H. Hashimoto, Bull. Chem. Soc. Jpn., 1952, 25, 411 CrossRef CAS.
- X. Xu, C. Hu and R. Wang, Appl. Catal., B, 2015, 176–177, 637 Search PubMed.
- H. Yuzawa, T. Mori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 4126 CrossRef CAS.
- JSPDS Card No. 01-075-1532.
- JSPDS Card No. 03-065-2871.
- H. Gil, C. P. Buitrago and A. Echavarria, J. Solid State Electrochem., 2015, 19, 1817 CrossRef CAS.
- J. Sanabria-Chinchilla, K. Asazawa, T. Sakamoto, K. Yamada, H. Tanaka and P. Strasser, J. Am. Chem. Soc., 2011, 133, 5425 CrossRef CAS PubMed.
- J. H. Zagal, M. A. Gulppi, C. Depretz and D. Leliévre, J. Porphyrins Phthalocyanines, 1999, 3, 355 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07960c |
| This journal is © The Royal Society of Chemistry 2022 |