
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfined-based catalyst investigation through the comparative functionalization and defunctionalization of Zr-MOF†
Meghdad Karimi *a,
Samira Sadeghi‡
a,
Haleh Mohebali‡a,
Hamzeh Bakhtib,
Alireza Mahjouba and
Akbar Heydari*a
*a,
Samira Sadeghi‡
a,
Haleh Mohebali‡a,
Hamzeh Bakhtib,
Alireza Mahjouba and
Akbar Heydari*a
aChemistry Department, Tarbiat Modares University, P.O. Box 14155-4838, Tehran, Iran. E-mail: heydar_a@modares.ac.ir; Tel: +98-21-82883444
bChemistry Department, Islamic Azad University Boroujerd Branch, Borujerd, Iran
First published on 6th June 2022
Abstract
In metal–organic frameworks, confined space as a chemical nanoreactor is as important as organocatalysis or coordinatively unsaturated metal site catalysis. In the present study, a set of mixed-ligand structures with UiO-66 architecture have been prepared. To the best of our knowledge, for the first time, structures derived by the solvothermal mixing ligand method and ultrasonic-assisted linker exchange approaches have been compared. Additionally, the relationship between the preparation method, structural properties, and catalytic efficiency of the prepared materials in the Knoevenagel condensation of aldehydes has been investigated. The prepared catalyst is very stable and can be recovered and reused for at least ten periods.
1. Introduction
Although functional groups within the pores of porous materials can significantly affect their catalytic activity, accessible intercrystalline void spaces to perform transformations are necessary from a classical catalytic point of view.1–3 The importance of relevant space makes research on the functionalization of these materials quite challenging, both qualitatively and quantitatively.4–6 Appropriate space is critical in condensation reactions because the manner in which the precursors approach and collide is essential to the reaction. Hence, merely having functional groups with an acidic or alkaline role without enough space to react cannot help advance the reaction.7–9Among the various porous compounds, metal–organic frameworks (MOFs) have attracted considerable attention in catalytic applications due to their various embedded functional groups and metal-containing units.10,11 Notably, MOFs can be a very appropriate example of functionalized materials for organocatalysis because of the significance of diversified organic units in their structures.12,13 These features have turned them into unparalleled materials that are used as porous and solid catalysts to study the interactions and factors affecting catalytic processes.14,15 In other words, these crystals have gathered much attention, particularly in two perspectives. Firstly, due to the presence of pores with shapes that depend on their topology, they can be a container for reactions regardless of the functional group.16–18 These structures act as nanoreactors to perform chemical reactions, which lead to more effective collisions and more likely reactions due to size and shape selectivity towards reactants, transition states, and products.19–26 Secondly, regarding the functional groups and the spatial effects, electronic effects are also involved, and the role of functional groups and open metal sites as Lewis acids and bases becomes very significant, especially in condensation reactions.25,27–29
Therefore, MOFs are distinguished candidates for the contemporaneous investigation of structurally and electronically driven catalysis, increasing our knowledge about catalytic reactions. As a result, structural features such as stability in different reaction media, the ability to create diversity while avoiding useless complications, the interactive effect of the catalytic centres and the pore's interior spaces are essential.30,31 It has been concluded that environmentally-friendly MOFs and eco-friendly strategies based on sustainable building blocks are ideal for the intended purpose.32–37 Frameworks based on terephthalic acid and 2-aminoterephthalic acid and iron, aluminium, chromium, cerium and zirconium clusters are essential in catalytic studies due to their unparalleled stability.38–46 Recently, structures based on zirconium and terephthalic acid-based ligands, namely the UiO-66 family, have captured much attention in various applications. Besides, UiO-66-(NH2) is used extensively in sensors, phototherapy, and photocatalytic applications due to its remarkable optical properties.47–52
Furthermore, the catalytic activity is strongly influenced by the balance between confined spaces and functional groups, or in other words, the presence or absence of functional groups. Undoubtedly, mixed-ligand MOFs are the best materials to study the aforementioned balance capability.53–55 A very important point in this regard, in addition to quantity, is the effect of the kind of functional group distribution on catalytic efficiency. This means that the method of functionalization and defunctionalization can also affect the catalytic efficiency. In fact, besides to be or not to be, how to be is also a vital issue. So, a major challenge in this area is the kind of relationship between the (de)functionalization strategies and the distribution of functional groups and ligands in the MOF texture.
Herein, to investigate the functional group and confined space-based catalysis simultaneously, a set of the mixed-ligand UiO-66 family with specific ratios of terephthalic acid and 2-aminoterephthalic acid ligands were prepared by mixing ligand and ultrasonic-assisted exchanged-ligand (de)functionalization strategies. Then, the prepared materials were used as versatile catalysts in the Knoevenagel condensation reaction under green conditions. Furthermore, the obtained materials were examined using different identification techniques such as thermogravimetric analysis (TGA), powder X-ray diffraction (PXRD), Fourier transform infrared (FTIR) spectroscopy, N2 sorption (BET surface area), ultraviolet-visible light (UV-vis) spectroscopy, UV-vis diffuse reflectance spectroscopy (UV-vis DRS), photoluminescence spectroscopy (PL spectroscopy) and zeta potential in order to determine the effect of (de)functionalization methods on structural and surface properties, porosity, and catalytic activity of the frameworks. The results confirmed the critical role of functional groups and confined spaces and, more importantly, the (de)functionalization methods on the catalytic activity. The results of catalytic process studies were analyzed according to the structure's spatial and electronic properties. In this study, UV-vis spectroscopy, zeta potential, and a comparative study of reaction kinetics were used to compare the functional group distribution in the obtained structures thoroughly.
2. Experimental
2.1. Materials and physical techniques
All required materials were bought from prestigious companies and utilized without any further purification. The as-prepared composites were also characterized by various identification techniques that are clarified: to perform FT-IR spectroscopy, NICOLET IR100 apparatus was used and carried out in the 400–4000 cm−1 region associated KBr disk method. Thermogravimetric analysis (TGA) of catalyst was obtained via the NETZSCH apparatus, which is run from room temperature to 700 °C with the heating speed of 10 °C min−1 under compressed nitrogen flow. Powder X-ray diffraction pattern was recorded by a Philips X'pert 1710 diffractometer at ambient temperature using Cu-Kα (α = 1.54056 Å) via charted Bragg–Brentano geometry over the 2θ of 10°–50°. In order to perform UV-visible DRS spectroscopy, an MPC-2200 spectrophotometer was utilized. SEM measurements were carried out by Philips XL 30 and S-4160 devices with a gold-aided coating laid out on nanoparticles within the EDX test. TEM images were taken at 120 kV using Philips apparatus with model CM120. The BET analysis was carried out by measuring N2 adsorption at 77 K using Quantachrome NOVA4000 apparatus, and the pore size distribution was obtained via the Barrett–Joyner–Halenda model. The non-local density functional theory, NLDFT, considering slit-shaped pores with energetically heterogeneous and rough walls, was used to estimate the pore size distributions. The yield of obtained products was achieved by gas chromatography (GC) (Yonglin 6100; BP-5; 30 m × 0.25 mm × 0.25 μm), and toluene was used as a solvent for injection and nitrogen as an inert carrier gas. 4802 UV/Vis spectrometer Unico attained the UV/Vis absorption graphs in the 200–800 nm wavelength range. In order to obtain the zeta potential of such samples, a Malvern Zetasizer nano particle analyzer was utilized.2.2. Synthesis of compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, 1:1, 2:1, and 1:0. All these compounds were identified using different characterization methods. These four kinds of proportions led to the production of four different frameworks, namely, UiO-66, UiO-66-NH2-33, UiO-66-NH2-50, UiO-66-NH2-66, and UiO-66-NH2-100, respectively (Scheme 1C).
:1, 1:2, 1:1, 2:1, and 1:0. All these compounds were identified using different characterization methods. These four kinds of proportions led to the production of four different frameworks, namely, UiO-66, UiO-66-NH2-33, UiO-66-NH2-50, UiO-66-NH2-66, and UiO-66-NH2-100, respectively (Scheme 1C).
 | ||
| Scheme 1 Synthesis of catalytic systems. | ||
:1), and the resulting mixture was stirred at ambient temperature for 30 min. Then, ethyl cyanoacetate (1.5 mmol) was added to the reaction mixture. The thin layered chromatography (TLC) was carried out to ensure that the reaction was complete. After that, the catalyst was centrifuged from the mixture and was eluted three times with ethanol. The remaining solution underwent organic-aqueous extraction using ethyl acetate and water, and column chromatography was utilized to separate the organic layer. In order to identify the obtained products, IR, Mass, 1H-NMR, and 13C-NMR techniques were employed, and the obtained spectral data were compared with the ones in the literature.3. Results and discussions
FT-IR spectra of UiO-66 structures were taken, and the results have been illustrated in Fig. 1A. An adsorption band around 1650 cm−1 in all graphs is ascribed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in DMF. The set of peaks at 650–900 cm−1, 1140 cm−1, and 1430–1625 cm−1 are related to the bending vibration of the C–H bond out of the plane, C–H in-plane, and C–C stretching vibrations, respectively.58–62 The absorption bands in the 600–800 cm−1 range are related to Zr–O as longitudinal and transverse modes, but the sharp doublet located at 1433 and 1389 cm−1 can be attributed to the stretching vibration of the carboxylic acid groups belonging to the NH2-BDC ligands. In addition, there are two bands at 1624 cm−1 and 1246 cm−1, in which the former corresponds to the N–H bending vibration, and the latter characterizes the C–N stretching of ligands containing the amine group. The double peaks at 3350–3500 cm−1 are obviously allocated to the amine functional group's asymmetrical and symmetrical stretching vibrations. More precisely, these double peaks are bold in the spectra of UiO-66 samples containing NH2 groups, proving the incorporation of 2-aminoterephthalic acid (NH2-BDC) molecules into the pores. There is a direct correlation between the peak intensities and the amount of NH2-BDC within the pores. Indeed, the rise in the peak intensities occurs upon increasing the amount of NH2-BDC according to this order: UiO-66, UiO-66-NH2-6h (Ex.), UiO-66-2h (Ex.), UiO-66-NH2-50, and UiO-66-NH2, respectively.
O in DMF. The set of peaks at 650–900 cm−1, 1140 cm−1, and 1430–1625 cm−1 are related to the bending vibration of the C–H bond out of the plane, C–H in-plane, and C–C stretching vibrations, respectively.58–62 The absorption bands in the 600–800 cm−1 range are related to Zr–O as longitudinal and transverse modes, but the sharp doublet located at 1433 and 1389 cm−1 can be attributed to the stretching vibration of the carboxylic acid groups belonging to the NH2-BDC ligands. In addition, there are two bands at 1624 cm−1 and 1246 cm−1, in which the former corresponds to the N–H bending vibration, and the latter characterizes the C–N stretching of ligands containing the amine group. The double peaks at 3350–3500 cm−1 are obviously allocated to the amine functional group's asymmetrical and symmetrical stretching vibrations. More precisely, these double peaks are bold in the spectra of UiO-66 samples containing NH2 groups, proving the incorporation of 2-aminoterephthalic acid (NH2-BDC) molecules into the pores. There is a direct correlation between the peak intensities and the amount of NH2-BDC within the pores. Indeed, the rise in the peak intensities occurs upon increasing the amount of NH2-BDC according to this order: UiO-66, UiO-66-NH2-6h (Ex.), UiO-66-2h (Ex.), UiO-66-NH2-50, and UiO-66-NH2, respectively.
 | ||
| Fig. 1 (A) FT-IR spectra, (B) PXRD results, (C) nitrogen adsorption–desorption isotherms and (D) TGA patterns of the UiO-66 family. | ||
Fig. 1B illustrates the PXRD patterns of UiO-66, UiO-66-NH2, and their derivatives. According to this analysis, the crystallinity of all compounds is in good consistency with the UiO-66 and UiO-66-NH2 patterns. All five compounds have similar X-ray diffraction patterns to UiO-66 materials reported in the literature and the simulated single crystal.63 Therefore, the preparation process of a dual-ligand UiO-66 does not lead to defects in the crystalline framework, revealing a specific topology. However, a more accurate look at the XRD pattern of the prepared compounds indicates the peaks related to these defects. Fortunately, they are not that noticeable, and their effects can convert them into desirable compounds. Furthermore, there are variable distances between the characteristic peaks of the crystalline planes of (200), (220), (311), and (222) in mixed-ligand UiO-66-(NH2), exchanged-ligand UiO-66, and exchanged-ligand UiO-66-NH2, demonstrating structural defects because of the absence of bridging ligands in the crystalline structure. In other words, some ligands are not replaced during the exchange process, and their positions remain empty in the structure. As a result, the distance between the planes changes, and the peaks in their XRD patterns shift slightly, but the results clearly show that the mixing-ligand method is more effective in creating structural defects.58,64,65
Nitrogen sorption isotherms of UiO-66, UiO-66-NH2, UiO-66-NH2-50, UiO-66-NH2-6h (Ex.), and UiO-66-2h (Ex.) were measured at 77 K (Fig. 1C). Before the nitrogen adsorption, all the prepared compounds were warmed at 120 °C for 24 h under vacuum. The type-I isotherms were obtained for all of the samples, demonstrating the microporous nature of the samples obtained from mixing and exchanged-ligand methods. UiO-66-NH2 functionalized by amino groups ultimately has less porosity than the parent UiO-66 due to the filling of confined spaces by NH2 groups.66 In other words, the surface area and pore volume of samples systematically decrease upon increasing the amount of NH2-BDC ligand within the framework. According to Table 1, the pore volume, pore size, and surface area (BET) of the samples have been obtained based on the data in the P/P0 range of 0–0.10. It is apparent that the more NH2 functional groups are embedded, the less internal volume would be obtained, whereas missing linkers function inversely. That means, by changing the linker proportions in the synthesis process, the number of missing linkers will be altered, banning the quantitative estimation of the exchanged ligand through absorption measurements solely.58,64 However, sometimes ligands are not replaced together, which functions as an obstacle to our calculations. By incorporating a considerable amount of ligands within the frameworks, in some specific ratios, pore size, pore volume, and BET surface area have been enhanced in the mixing-ligand method. In the case of the exchanged-ligand method, the insertion of –NH2 into UiO-66, the same results were achieved. In some cases, BDC has been removed, but NH2BDC has not been replaced. Therefore, the pore size, pore volume, and BET surface area were increased, especially after 2 h. In the second exchange method, the insertion of BDC into UiO-66-NH2, the more BDC has been inserted, the more BET surface area, pore size, and pore volume would be obtained, especially after 6 h.
| Name of sample | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (Å) |
|---|---|---|---|
| UiO-66 | 1007.03 | 0.77 | 39.59 |
| UiO-66-NH2-33 | 861.50 | 1.11 | 51.41 |
| UiO-66-NH2-50 | 1066.99 | 1.59 | 59.58 |
| UiO-66-NH2-66 | 935.39 | 0.99 | 33.27 |
| UiO-66-1h(Ex.) | 842.50 | 0.69 | 35.41 |
| UiO-66-2h(Ex.) | 907.04 | 0.73 | 37.50 |
| UiO-66-4h(Ex.) | 831.31 | 0.63 | 32.31 |
| UiO-66-6h(Ex.) | 810.22 | 0.61 | 31.36 |
| UiO-66-NH2-1h(Ex.) | 819.36 | 0.63 | 32.71 |
| UiO-66-NH2-2h(Ex.) | 873.09 | 0.66 | 35.39 |
| UiO-66-NH2-4h(Ex.) | 922.41 | 0.69 | 37.49 |
| UiO-66-NH2-6h(Ex.) | 964.35 | 0.73 | 37.56 |
| UiO-66-NH2 | 785.64 | 0.61 | 31.31 |
A comparison between the size and volume of the pores indicated that these two parameters would change upon an increase in the amount of 2-aminoterephthalic acid, confirming the presence of pores containing amine groups. The results have been presented in Table 1. The average pore size of all samples was determined using the Barett–Joyner–Halenda (BJH) model to make a rational comparison. As a general trend, and regardless of the occasional changes, in the case of materials prepared through the mixing-ligand method, by increasing the ratio of 2-aminoterephthalic acid, the volume and size of the pores increase, but in the exchanged-ligand method, they decrease. The results of the non-local density functional theory (NLDFT) model confirm this (Table S1†).
According to the XRD results, it can be realized that the occurrence of structural defects in the mixing-ligand method is more than those in the exchange-ligand method. The stability rate of UiO-66, UiO-66-NH2-50, and UiO-66-NH2 against temperature was investigated using TGA. According to the diagram (Fig. 1D), the prepared samples are decomposed in three weight-loss steps, showing the gradual decomposition of the as-synthesized samples upon increasing the temperature. 5–6% of the weight is lost by heating the samples to 80 °C, while in the 100–280 °C range, around 10 percent is decomposed. These weight losses can be due to the dehydroxylation of the zirconium oxo-clusters and the elimination of solvent. The third step at 450–550 °C shows the framework decomposition at almost different temperature ranges for each sample. About UiO-66-NH2-50, the third step is not separated from the second one entirely because the framework starts to decompose before the total removal of the solvent. The TGA curves also indicate an inverse correlation between the decomposition temperature of the materials and ABDC loading. As fewer ABDC ligands are embedded within the framework, the decomposition temperature of the framework increases.
According to UV-vis DRS of UiO-66 and UiO-66 compounds containing different amounts of NH2, we can observe a qualitative comparison of the fraction of ABDC in the samples (Fig. 2A). According to the sequence of UiO-66-NH2 > UiO-66-NH2-66 > UiO-66-NH2-50 > UiO-66-NH2-33 > UiO-66, the intensity of their peaks at around λmax = 350–370 nm, attributed to the electron transition in ABDC (n → π*), decreased.
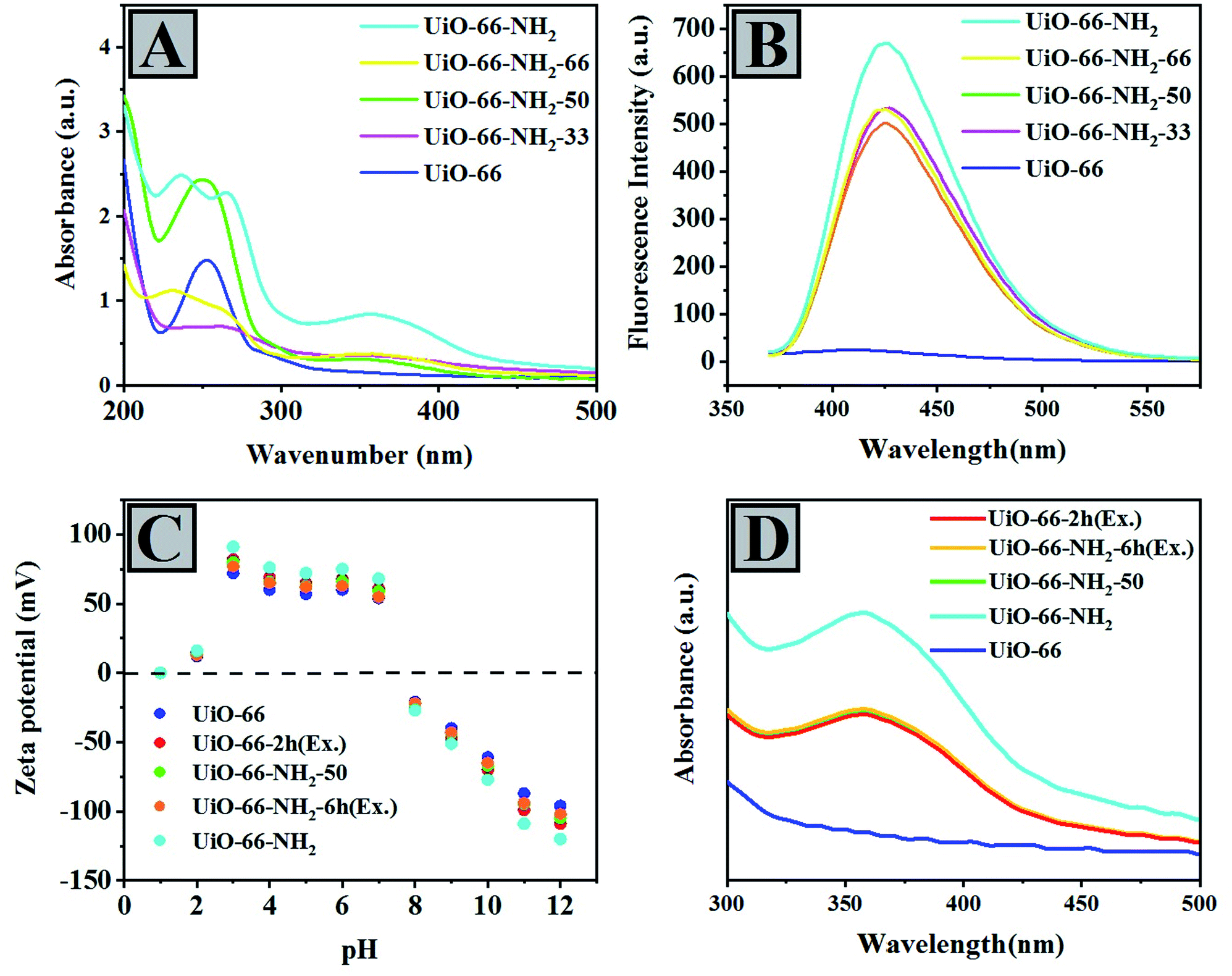 | ||
| Fig. 2 (A) UV-vis DRS and (B) PL spectroscopy of mixing-ligand prepared compounds. (C) Zeta potential and (D) UV-vis comparative analysis of 2-aminoterephthalic acid in 1 M NaOH solution. | ||
One of the most important electronic transfers in NH2BDC is n → π*. However, the peaks in the 200–270 nm range can be ascribed to the transfers from the ligand to the ZrO2 cluster. Moreover, the absorption bands of UiO-66-NH2-33, UiO-66-NH2-50, UiO-66-NH2-66, and UiO-66-NH2 move towards longer wavelengths named the red-shift phenomenon compared with UiO-66. Red-shift can be due to amino groups containing lone pair electrons, which increases visible light absorption (Fig. 2B).
The photoluminescence spectroscopy indicates the fluorescent and electronic properties of UiO-66, UiO-66-NH2-33, UiO-66-NH2-50, UiO-66-NH2-66, and UiO-66-NH2 qualitatively (Fig. 2B). As illustrated in Fig. 2B, UiO-66 is not sensitive to photons and shows no fluorescent properties. Hence, the presence of amino groups can lead to such properties in the UiO-66-NH2 systems. Such amino groups can have auxochromic properties, leading to charge–transfer interactions in the aromatic rings that existed in the framework.
In order to determine the charge state of UiO-66, UiO-66-2h (Ex.), UiO-66-NH2-50, UiO-66-NH2-6h (Ex.), and UiO-66-NH2 compounds, the zeta potential method was carried out in acidic and basic pH ranges (Fig. 2C). It is observed that the MOF samples showed positive zeta potential in the former pH range and negative zeta potential in the latter range. Moreover, these changes approved the production of protonated MOF particles and neutral or negatively charged particles in the acidic and basic pH ranges, respectively. Such obtained trends can result from hydroxyl ion adsorption or coordination on the surface of MOF samples and deprotonation of pendant ammonium groups. In a previous study, by adding more acid to the synthesis process of UiO-66-NH2, its zeta potential was increased.67,68 This behavior shows that the surface charge of UiO-66-(NH2) compounds can be due to the protonation of Zr clusters arising from amine functional groups at the surface of MOF samples. Regarding samples obtained from the exchanged-ligand method, the surface charge is more negative than a mixing-ligand method. Since the negative charge is related to the presence of amino groups, it can be found that compared to the samples obtained from the mixing-ligand method, the accumulation of 2-aminoterephthalic acid ligands on the surface is higher. This analysis shows that in the exchange method, the highest exchange rate is done at the surface of the nanoparticles, and as a result, the amount of amine functional groups is higher at the surface, which also affects the catalytic activity.
A comparison among some MOF samples obtained through different methods was carried out. Indeed, this survey aimed to compare the content of the ABDC linker qualitatively by analyzing the UV-vis measurements of the samples dissolved in 1 M NaOH solution.58 As shown in Fig. 2D, UV-vis spectroscopy analyses of mentioned samples indicated the qualitative fraction of ABDC in the following order UiO-66-NH2 > UiO-66-2h (Ex.) ∼ UiO-66-NH2-50 ∼ UiO-66-NH2-6h (Ex.) > UiO-66. To perform the UV-vis spectroscopy, initially, the sample is dissolved in a mixture of D2O and KOH to be ready for UV measurement. The results showed that UiO-66 and UiO-66-NH2 contain absolutely BDC and NH2-BDC, respectively, whereas the remaining samples contain BDC and NH2-BDC with no considerable difference between their amounts. As shown in Fig. 2D, UiO-66 compounds containing a mixture of both ligands and obtained through exchanged or mixing-ligand methods showed the same absorbance, and we concluded that the amount of both ligands in these compounds should be almost equal.
4. Catalytic study
4.1. Optimization of the condensation reaction
After observing the efficiency of UiO-66 compounds in the Knoevenagel condensation reaction of aldehydes as catalysts, we decided to optimize various parameters, including solvent, the kind of catalyst, catalyst loading, and temperature. The reaction between benzaldehyde and ethyl cyanoacetate was chosen as the model reaction. As shown in Fig. 3A, various UiO-66 compounds prepared through different procedures were used as catalysts, and UiO-66-NH2-50 led to the highest yield of product (ca. 45%). The UiO-66-NH2-6h (Ex.) and UiO-66-2h (Ex.) were at the second and third places, respectively (ca. 40% and 36%, respectively). These results showed that the presence of amine functional groups is a must to advance the reaction efficiently. The basicity of the coordinated oxygen's of the carboxylate groups and the performance of the interior pores of framework’ as nanoreactors can reflect the pure catalytic activity of UiO-66. From UiO-66 to UiO-66-NH2-50, there was an increase in the product yield as NH2BDC ligands increased (Scheme 2). | ||
| Fig. 3 Optimization of the catalysts, solvent, catalyst loading, time and temperature (A–E) for the Knoevenagel condensation of aldehydes. | ||
 | ||
| Scheme 2 Model reaction for the Knoevenagel condensation reaction. | ||
By contrast, when UiO-66-NH2-50 was replaced by UiO-66-NH2-66, although the number of amine sites rose, its catalytic performance decreased. According to the literature, this phenomenon may be attributed to the distributional limitations because of the narrower pores and less availability of the active amine sites.69,70 Therefore, UiO-66-NH2-50 was chosen as the most appropriate catalyst. During other repeated experiments, efforts have been made to choose the best solvent for the model reaction, so some solvents with various dielectric constants were investigated. As illustrated in Fig. 3B, the more polar the solvent used, the more conversion would occur. Faster release of the protons of protonated amine and facilitation of benzaldimine intermediate formation leads to increased conversion. In other words, better results were obtained using more polar solvents such as ethanol, H2O, methanol, and DMF. To survey other solvents with lower polarity, we observed that methanol led to a higher conversion than ethanol. Ethanol indicated higher efficiency when combined with water in different ratios (1:1, 1:2, and 2:1 for ethanol and H2O, respectively). The 1:1 ratio of ethanol/H2O was selected as the optimal solvent because it raised the solubility of precursors and led to the highest yield. Ethanol has an amphiprotic feature, which activates protonated benzaldehyde and leads to the electrophilic polarization of carbonyl group.71–73 Different masses of the catalyst were also examined, and 40 mg was selected as the optimal catalyst loading (Fig. 3C). As shown in Fig. 3D, using UiO-66-NH2-50 as a catalyst at room temperature increases the reaction efficiency until the reaction progresses very slowly after 800 min. Further studies on the effect of time on the reaction rate are presented in the Kinetic Studies section. Eventually, we investigated how the temperature changes affect the efficiency of the model reaction (Fig. 3E). In the early stages of the reaction, the temperature changes had a remarkable effect on the yield of reaction; however, the longer the reaction (8 h), the less dependent it was. Therefore, 40 °C was considered the best temperature.
4.2. Pore size effect
The amino-free UiO-66 and UiO-66-NH2 have pore sizes of 39.59 Å and 31.31 Å, respectively (Table 1). Given the theoretical dimensions of benzaldehyde (4.1 × 6.2 Å) and ethyl cyanoacetate (2.4 × 6.6 Å), the reaction can undoubtedly occur within the pores of the framework. Interestingly, more than 50% of the reaction yield could be achieved without relying on the pore size. However, in order to assess the effect of the pore size, the catalytic activity of UiO-66, UiO-66-NH2, UiO-66-NH2-33, UiO-66-NH2-50, UiO-66-NH2-66, UiO-66-1h(Ex.), UiO-66-2h(Ex.), UiO-66-4h(Ex.), UiO-66-6h(Ex.), UiO-66-NH2-1h(Ex.), UiO-66-NH2-2h(Ex.), UiO-66-NH2-4h(Ex.), and UiO-66-NH2-6h(Ex.) compounds was investigated on the condensation of benzaldehyde and n-butyraldehyde as a linear and less bulky aldehyde under these reaction conditions: 40 °C, 40 mg of catalyst, the equimolar ratio of aldehyde and methylene compound precursors, and EtOH/H2O (1:1) as a solvent for 20 min. As illustrated in (Fig. S1†), the conversion percentage was decreased upon increasing the abundance of active sites. Although UiO-66-NH2 had narrower pores than other catalysts, it led to relatively good conversion, indicating slight diffusion limitation, which could be due to the interaction of the hydrogens belonging to the amine functional group with zirconium clusters. However, using UiO-66-NH2-50, a higher conversion was attained than UiO-66-NH2, indicating the simultaneous effect of both diffusion and active amine sites in the reactions. As the results show, although the presence of the amine functional group is essential for the conversion of both aldehydes, the penetration restriction has a more negligible effect on the linear aldehyde than on the aromatic types. The more negligible effect of diffusion constraints on linear aldehyde conversion can be attributed to the lack of stacking interactions between the aldehyde and the framework ligands. However, the lower conversion of linear aldehyde can be attributed to the lower reactivity of this species.
4.3. Application of UiO-66-NH2-50 for the Knoevenagel condensation of aldehydes
Concerning benzaldehyde (Table 2, entries 1a and ,b), with a theoretical dimension of (4.1 × 6.2 Å), mass transfer limitations were investigated in some UiO-66 compounds. It was observed that the percentage of benzaldehyde conversion was 97% (CN) using UiO-66-NH2-50. Regarding the bulkier aldehydes, the effect of the diffusional restrictions appeared, causing a decrease in the conversion (Table 2, entries 6a, b, 7a and b) and the catalytic performance depends on the size rather than the abundance of the active catalytic sites. Other aldehyde derivatives were examined as well, and they did not indicate any considerable differences in their ability to be converted during the catalytic reaction. 4-Nitro benzaldehyde reacting with malononitrile showed almost complete conversion (98–96%) across 720 min during the reaction, which can be due to the powerful electron-withdrawing feature of the nitro substituent, assisting the nucleophilic attack to the carbonyl group (Table 2, entries 4a and b). In terms of aldehydes containing electron donor substituents, para-methoxy benzaldehyde indicated slower reaction kinetics (Table 2, entries 5a and b). In separate experiments, ethyl cyanoacetate was also used as the second precursor. The lower yields of ethyl cyanoacetate products can be explained by the methylene group acid resistance, pKa = 13 for ethyl cyanoacetate, more than that of malononitrile (pKa = 11).74| a Reaction conditions: substrates (1 mmol), EtOH/H2O (1:1, 2 mL), 40 mg catalyst, 720 min at 40 °C. |
|---|
 |
 |
4.4. Condensation transformation mechanism study
According to Gascon et al.,69 Cortese and Duca,75 and Wirz et al.,76 the mechanism of the reaction is in agreement with the formation of imine compound (Scheme 3) because IR spectroscopy and catalytic experiments results of NH2-IRMOF-3 and NH2–SiO2 confirmed the –CN– bond formation. Both the rate and the path of the Knoevenagel reaction rely on the polarity of the solvent.69,77,78 Hence, whenever the polarity of the solvent was decreased, the reaction rate was diminished as well.69 In another investigation, a similar trend was also observed in the same reaction catalyzed by amino-containing silica in the polar medium, which led to the production of the ion-pair species by stabilizing the carbanion intermediate.79 As mentioned, UiO-66-NH2-50 had the most significant activity in polar solvents with a high dielectric constant (Fig. 3B). In the Knoevenagel reaction of benzaldehyde, 96% product was obtained after 720 min at 40 °C in EtOH/H2O (1:1); whereas, at the same time, the conversion of benzaldehyde just reached ∼5% after 720 min at 40 °C in n-hexane as solvent (Fig. 3B). The main reason why such solvents showed different behavior goes back to the ion-pair mechanism facilitated by protic solvents.
 | ||
| Scheme 3 Proposed mechanism of the Knoevenagel condensation catalyzed by UiO-66-NH2. | ||
Moreover, protic solvents assist the electrophilic polarization of the carbonyl group belonging to aldehyde. Therefore, according to the computational study by Cortese and Duca75 and the IR investigation by Wirz et al.,76 we concluded that the reaction advances through the mechanism suggested by Gascon et al.,69 including the deactivation process and nucleophilic attack on benzaldehyde (Scheme 3).
The study of reaction kinetics (under optimized conditions) on UiO-66 derivatives prepared through various strategies confirmed UV-vis and zeta potential analyses. Using materials prepared by the exchanged-ligand method as the catalyst, the reaction completion quickly occurred, and as time passed, no change in the process was observed. In contrast, using the materials obtained from the mixing-ligand method, the reaction progressed slower (Fig. S2–S7†). Further kinetic studies indicated a more prolonged reaction half-life using a mixing-ligand catalyst. The catalytic reaction began to progress on the surface of solid particles and then continued as the precursors diffused into the pores. Concerning materials prepared by the exchanged-ligand method, due to the accumulation of amino groups on the surface of particles, saturation occurred, and diffusion into the pores was carried out gradually. In the materials prepared through the mixing-ligand method, because of the uniform distribution of the amine functional groups in their texture, the diffusion was more comfortable. However, the reaction completion was achieved slowly, but we observed a higher yield of the product. By comparing the minimum and maximum pore sizes in the compounds derived from UiO-66 with ethyl (E)-2-cyano-3-phenylbut-2-enoate, resulting from the condensation reaction of benzaldehyde and ethyl cyanoacetate, the possibility of this reaction within the pores was confirmed. Therefore, how precursors penetrate the pores plays a crucial role in these reactions. Since amino groups, as catalytically active sites, also occupy the confined space of the cavities, their proper distribution in the structure can provide a convenient conversion for the condensation reaction.
On the other hand, zeta potential analysis results confirmed the direct effect of the procedure used to manipulate the abundant amine functional groups on the surface of particles. Comparing UV-vis analyses of the three types of prepared materials showed an almost equal amount of 2-aminoterephthalic acid and the difference in their distribution. Therefore, the study of reaction kinetics on these compounds confirmed the results of UV-vis and zeta potential analyses.
5. Heterogeneity test and catalyst reusability
The heterogeneity rate and catalyst reusability of UiO-66-NH2-50 was attained through the hot filtration test and in the model reaction under the same conditions. During the reaction, no changes in product yield were obtained, which shows that no catalyst particles permeated the reaction solution (Fig. S8A†), and it showed high reusability up to the 10th run (Fig. S8B†). Moreover, the FT-IR and PXRD analyses of the recovered catalyst revealed structural stability against the reaction conditions, and almost all UiO-66-NH2-50 functional groups and structural properties have been preserved just like the intact catalyst (Fig. S8C and D†).6. Conclusion
A set of mixing and exchanged-linker UiO-66 compounds as metal–organic frameworks (MOFs) have been successfully prepared through mixing-ligand and exchanged-ligand methods. The latter method was carried out by ultrasonic irradiation, and 2-aminobenzene-1,4-dicarboxylic acid (ABDC) and benzene-1,4-dicarboxylic acid (BDC) were used as ligands in both methods. The ratio of ABDC to BDC in the framework and the kind of method used for amine content modulation are two critical factors that affect the surface charge, porosity, and the catalytic activity of such frameworks. The Knoevenagel condensation reaction of various aldehydes with ethyl cyanoacetate was used to survey the catalytic activity of UiO-66 derivatives. Catalyst performance relied on the amount of amine modulation arising from the proportion between BDC and NH2-BDC ligands used in the MOF preparation process and the kind of method used for 2-aminobenzene-1,4-dicarboxylic modulation. Although the pore volume and specific surface area of the UiO-66 compounds diminish with the insertion of the NH2-BDC ligand, these amino groups can promote the reaction. Among the UiO-66 materials, UiO-66-NH2-50 exhibited the highest catalytic performance because of an optimal balance among the number of active sites within the cavities and the lack of considerable diffusion restrictions. Since the distribution of amine functional groups depends on the method used to incorporate them inside of pores, the mixing-ligand method has been known as an optimal procedure for this purpose. In contrast, in exchange-based methods, we observed the agglomeration of ligands on the surface of the frameworks, leading to the reduction of surface saturation and lack of a proper direction through which the precursors can penetrate. Interestingly, the qualitative investigation of UV-vis spectroscopy showed approximately an equal amount of 2-aminoterephthalic acid in the structures prepared by different methods and unlike zeta potential results, confirmed the greater abundance of this ligand on the particle surface. The kinetic study of the Knoevenagel condensation of benzaldehyde also confirmed zeta potential and UV-vis analyses. In this work, we could successfully determine the ABDC in mixing-linker MOFs qualitatively using zeta potential.Author contributions
Meghdad Karimi and Haleh Mohebali: investigation. Meghdad Karimi and Samira Sadeghi: writing – original draft. Meghdad Karimi and Haleh Mohebali: formal analysis and resources. Hamzeh Bakhti: manuscript correction based on the reviewers' comments. Alireza Mahjoub: data curation. Meghdad Karimi: conceptualization. Akbar Heydari: writing – review & editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Tarbiat Modares University of Iran.References
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC.
- B. Lebeau, A. Galarneau and M. Linden, Chem. Soc. Rev., 2013, 42, 3661–3662 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, ChemCatChem, 2020, 12, 4732–4753 CrossRef CAS.
- I. I. Ivanova and E. E. Knyazeva, Chem. Soc. Rev., 2013, 42, 3671–3688 RSC.
- S. Canossa and S. Wuttke, Adv. Funct. Mater., 2020, 30, 2003875 CrossRef CAS.
- L. F. Dumée, L. He, B. Lin, F.-M. Ailloux, J.-B. Lemoine, L. Velleman, F. She, M. C. Duke, J. D. Orbell, G. Erskine, P. D. Hodgson, S. Gray and L. Kong, J. Mater. Chem. A, 2013, 1, 15185–15206 RSC.
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Adv. Synth. Catal., 2013, 355, 247–268 CAS.
- E. Verde-Sesto, E. Merino, E. Rangel-Rangel, A. Corma, M. Iglesias and F. Sánchez, ACS Sustainable Chem. Eng., 2016, 4, 1078–1084 CrossRef CAS.
- R. J. Hooley and J. Rebek, Chem. Biol., 2009, 16, 255–264 CrossRef CAS PubMed.
- V. R. Remya and M. Kurian, Int. Nano Lett., 2019, 9, 17–29 CrossRef CAS.
- M. S. Alhumaimess, J. Saudi Chem. Soc., 2020, 24, 461–473 CrossRef CAS.
- V. Pascanu, G. González Miera, A. K. Inge and B. Martín-Matute, J. Am. Chem. Soc., 2019, 141, 7223–7234 CrossRef CAS PubMed.
- J. V. Alegre-Requena, E. Marqués-López, R. P. Herrera and D. D. Díaz, CrystEngComm, 2016, 18, 3985–3995 RSC.
- J. Casaban, Y. Zhang, R. Pacheco, C. Coney, C. Holmes, E. Sutherland, C. Hamill, J. Breen, S. James, D. Tufano, D. Wong, E. Stavrakakis, H. Annath and A. Moore, Faraday Discuss., 2021, 231, 312–325 RSC.
- L. Wang, H. Xu, J. Gao, J. Yao and Q. Zhang, Coord. Chem. Rev., 2019, 398, 213016 CrossRef.
- T. A. Goetjen, J. Liu, Y. Wu, J. Sui, X. Zhang, J. T. Hupp and O. K. Farha, Chem. Commun., 2020, 56, 10409–10418 RSC.
- X.-J. Kong, T. He, Y.-Z. Zhang, X.-Q. Wu, S.-N. Wang, M.-M. Xu, G.-R. Si and J.-R. Li, Chem. Sci., 2019, 10, 3949–3955 RSC.
- Y. Wang and C. Wöll, Catal. Lett., 2018, 148, 2201–2222 CrossRef CAS.
- S. Dutta and I. S. Lee, Mater. Chem. Front., 2021, 5, 3986–4021 RSC.
- R. Martínez, C. Carrillo-Carrión, P. Destito, A. Alvarez, M. Tomás-Gamasa, B. Pelaz, F. Lopez, J. L. Mascareñas and P. del Pino, Cell Rep. Phys. Sci., 2020, 1, 100076 CrossRef PubMed.
- M. Viciano-Chumillas, M. Mon, J. Ferrando-Soria, A. Corma, A. Leyva-Pérez, D. Armentano and E. Pardo, Acc. Chem. Res., 2020, 53, 520–531 CrossRef CAS PubMed.
- G. Mirth, J. Cejka and J. Lercher, J. Catal., 1993, 139, 24–33 CrossRef CAS.
- J. Martens, J. Perez-Pariente, E. Sastre, A. Corma and P. Jacobs, Appl. Catal., 1988, 45, 85–101 CrossRef CAS.
- T. L. Maesen, M. Schenk, T. Vlugt, J. De Jonge and B. Smit, J. Catal., 1999, 188, 403–412 CrossRef CAS.
- M. Karimia, S. Sadeghia, R. G. Gavinehroudi, H. Mohebali, A. Mahjoub and A. Heydari, New J. Chem., 2021, 45, 6671–6681 RSC.
- Y. Li, M. Karimi, Y.-N. Gong, N. Dai, V. Safarifard and H.-L. Jiang, Matter, 2021, 4, 2230–2265 CrossRef CAS.
- Z. S. Rozveh, S. Kazemi, M. Karimi, G. A. M. Ali and V. Safarifard, Polyhedron, 2020, 183, 114514 CrossRef CAS.
- P. V. Dau, K. K. Tanabe and S. M. Cohen, Chem. Commun., 2012, 48, 9370–9372 RSC.
- J. Noh, Y. Kim, H. Park, J. Lee, M. Yoon, M. H. Park, Y. Kim and M. Kim, J. Ind. Eng. Chem., 2018, 64, 478–483 CrossRef CAS.
- Z. R. Herm, B. M. Wiers, J. A. Mason, J. M. van Baten, M. R. Hudson, P. Zajdel, C. M. Brown, N. Masciocchi, R. Krishna and J. R. Long, Science, 2013, 340, 960–964 CrossRef CAS PubMed.
- C. Martínez and A. Corma, Coord. Chem. Rev., 2011, 255, 1558–1580 CrossRef.
- E.-S. M. El-Sayed and D. Yuan, Green Chem., 2020, 22, 4082–4104 RSC.
- F. S. Al-Mubaddel, M. Karimi, S. Sadeghi, R. G. Gavinehroudi, H. Mohebali, A. Mahjoub, R. Marzouki, M. El Ouni and A. Heydari, Mol. Catal., 2021, 516, 111949 CrossRef CAS.
- T. Hajiashrafi, M. Karimi, A. Heydari and A. A. Tehrani, Catal. Lett., 2017, 147, 453–462 CrossRef CAS.
- M. Karimi, S. Sadeghi, H. Mohebali, Z. Azarkhosh, V. Safarifard, A. R. Mahjoub and A. Heydari, New J. Chem., 2021, 45(31), 14024–14035 RSC.
- M. Karimi, T. Hajiashrafi, A. Heydari and A. Azhdari Tehrani, Appl. Organomet. Chem., 2017, 31 CrossRef CAS.
- N. Khosroshahi, M. Karimi, T. Taghvaei and V. Safarifard, Mater. Today Chem., 2021, 22, 100582 CrossRef CAS.
- P. S. Bárcia, D. Guimarães, P. A. Mendes, J. A. Silva, V. Guillerm, H. Chevreau, C. Serre and A. E. Rodrigues, Microporous Mesoporous Mater., 2011, 139, 67–73 CrossRef.
- K. Yang, Q. Sun, F. Xue and D. Lin, J. Hazard. Mater., 2011, 195, 124–131 CrossRef CAS PubMed.
- J. Tang, M. Yang, M. Yang, J. Wang, W. Dong and G. Wang, New J. Chem., 2015, 39, 4919–4923 RSC.
- M. Karimi, H. Mohebali, S. Sadeghi, V. Safarifard, A. Mahjoub and A. Heydari, Microporous Mesoporous Mater., 2021, 322, 111054 CrossRef CAS.
- A. A. Taha, L. Huang, S. Ramakrishna and Y. Liu, J. Water Process. Eng., 2020, 33, 101004 CrossRef.
- P. Serra-Crespo, E. V. Ramos-Fernandez, J. Gascon and F. Kapteijn, Chem. Mater., 2011, 23, 2565–2572 CrossRef CAS.
- E. V. Ramos-Fernandez, C. Pieters, B. van der Linden, J. Juan-Alcañiz, P. Serra-Crespo, M. W. G. M. Verhoeven, H. Niemantsverdriet, J. Gascon and F. Kapteijn, J. Catal., 2012, 289, 42–52 CrossRef CAS.
- E. Niknam, F. Panahi, F. Daneshgar, F. Bahrami and A. Khalafi-Nezhad, ACS Omega, 2018, 3, 17135–17144 CrossRef CAS PubMed.
- S. Babaee, M. Zarei, H. Sepehrmansourie, M. A. Zolfigol and S. Rostamnia, ACS Omega, 2020, 5, 6240–6249 CrossRef CAS PubMed.
- F. G. Cirujano and F. X. Llabrés i Xamena, J. Phys. Chem. Lett., 2020, 11, 4879–4890 CrossRef CAS PubMed.
- D. Jiang, G. Fang, Y. Tong, X. Wu, Y. Wang, D. Hong, W. Leng, Z. Liang, P. Tu, L. Liu, K. Xu, J. Ni and X. Li, ACS Catal., 2018, 8, 11973–11978 CrossRef CAS.
- K. Leus, P. Concepcion, M. Vandichel, M. Meledina, A. Grirrane, D. Esquivel, S. Turner, D. Poelman, M. Waroquier, V. Van Speybroeck, G. Van Tendeloo, H. García and P. Van Der Voort, RSC Adv., 2015, 5, 22334–22342 RSC.
- I. Strauss, K. Chakarova, A. Mundstock, M. Mihaylov, K. Hadjiivanov, N. Guschanski and J. Caro, Microporous Mesoporous Mater., 2020, 302, 110227 CrossRef CAS.
- Q. Zheng, X. Liu, Y. Zheng, K. W. K. Yeung, Z. Cui, Y. Liang, Z. Li, S. Zhu, X. Wang and S. Wu, Chem. Soc. Rev., 2021, 50, 5086–5125 RSC.
- J. Wu, X. Fang, Y. Zhu, N. Ma and W. Dai, Energy Fuels, 2020, 34, 12911–12917 CrossRef CAS.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed.
- Z.-w. Huang, K.-q. Hu, L. Mei, X.-h. Kong, J.-p. Yu, K. Liu, L.-w. Zeng, Z.-F. Chai and W.-Q. Shi, Dalton Trans., 2020, 49, 983–987 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Catal. Sci. Technol., 2016, 6, 5238–5261 RSC.
- M. Pintado-Sierra, A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sánchez, J. Catal., 2013, 299, 137–145 CrossRef CAS.
- Y.-J. Lee, Y.-J. Chang, D.-J. Lee, Z.-W. Chang and J.-P. Hsu, J. Taiwan Inst. Chem. Eng., 2019, 96, 483–486 CrossRef CAS.
- S. M. Chavan, G. C. Shearer, S. Svelle, U. Olsbye, F. Bonino, J. Ethiraj, K. P. Lillerud and S. Bordiga, Inorg. Chem., 2014, 53, 9509–9515 CrossRef CAS PubMed.
- X. Feng, J. Hajek, H. S. Jena, G. Wang, S. Kaliya Perumal Veerapandian, R. Morent, N. De Geyter, K. Leyssens, A. E. Hoffman and V. Meynen, J. Am. Chem. Soc., 2020, 142(6), 3174–3183 CrossRef CAS PubMed.
- J. Hajek, M. Vandichel, B. Van de Voorde, B. Bueken, D. De Vos, M. Waroquier and V. Van Speybroeck, J. Catal., 2015, 331, 1–12 CrossRef CAS.
- J. Dong, K. Zhang, X. Li, Y. Qian, H. Zhu, D. Yuan, Q.-H. Xu, J. Jiang and D. Zhao, Nat. Commun., 2017, 8, 1–14 CrossRef CAS PubMed.
- N. Goel and N. Kumar, RSC Adv., 2018, 8, 10746–10755 RSC.
- S. Friebe, B. Geppert, F. Steinbach and J. r. Caro, ACS Appl. Mater. Interfaces, 2017, 9, 12878–12885 CrossRef CAS PubMed.
- K. Vellingiri, A. Deep, K.-H. Kim, D. W. Boukhvalov, P. Kumar and Q. Yao, Sens. Actuators, B, 2017, 241, 938–948 CrossRef CAS.
- Y. Tang, H. Wu, J. Chen, J. Jia, J. Yu, W. Xu, Y. Fu, Q. He, H. Cao and J. Cheng, Dyes Pigm., 2019, 167, 10–15 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- W. Morris, S. Wang, D. Cho, E. Auyeung, P. Li, O. K. Farha and C. A. Mirkin, ACS Appl. Mater. Interfaces, 2017, 9, 33413–33418 CrossRef CAS.
- J. Qiu, Y. Feng, X. Zhang, M. Jia and J. Yao, J. Colloid Interface Sci., 2017, 499, 151–158 CrossRef CAS PubMed.
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS.
- V. N. Panchenko, M. M. Matrosova, J. Jeon, J. W. Jun, M. N. Timofeeva and S. H. Jhung, J. Catal., 2014, 316, 251–259 CrossRef CAS.
- K. K. Tanabe, Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508–8517 CrossRef CAS PubMed.
- I. Rodriguez, G. Sastre, A. Corma and S. Iborra, J. Catal., 1999, 183, 14–23 CrossRef CAS.
- F. Jérôme, G. Kharchafi, I. Adam and J. Barrault, Green Chem., 2004, 6, 72–74 RSC.
- Y. Yang, H.-F. Yao, F.-G. Xi and E.-Q. Gao, J. Mol. Catal. A: Chem., 2014, 390, 198–205 CrossRef CAS.
- R. Cortese and D. Duca, Phys. Chem. Chem. Phys., 2011, 13, 15995–16004 RSC.
- R. Wirz, D. Ferri and A. Baiker, Langmuir, 2006, 22, 3698–3706 CrossRef CAS PubMed.
- F. X. Llabrés i Xamena, F. García Cirujano and A. Corma Canós, Microporous Mesoporous Mater., 2012, 157, 112–117 CrossRef.
- M. Hartmann and M. Fischer, Microporous Mesoporous Mater., 2012, 164, 38–43 CrossRef CAS.
- S. L. Hruby and B. H. Shanks, J. Catal., 2009, 263, 181–188 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1ra07767h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |