
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStyrene-based polymerised high internal phase emulsions using monomers in the internal phase as co-surfactants for improved liquid chromatography†
Christopher T. Desire ab,
R. Dario Arrua*b,
Fotouh R. Mansourc,
Stefan A. F. Bond and
Emily F. Hilder*b
ab,
R. Dario Arrua*b,
Fotouh R. Mansourc,
Stefan A. F. Bond and
Emily F. Hilder*b
aAustralian Centre for Research on Separation Science (ACROSS), School of Physical Sciences, University of Tasmania, Hobart, Australia
bUniversity of South Australia, STEM, Future Industries Institute, SA 5000, Australia. E-mail: Dario.Arrua@unisa.edu.au; Emily.Hilder@unisa.edu.au
cDepartment of Pharmaceutical Analytical Chemistry, Tanta University, Tanta, Egypt
dDepartment of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK
First published on 29th March 2022
Abstract
Poly(styrene-co-divinylbenzene)-based monoliths were prepared from the polymerisation of water-in-monomer high internal phase emulsions, where the water-soluble monomers acrylamide (AAm) or poly(ethylene glycol) diacrylate (PEGDA) (Mw 258) were also included in the 90 vol% internal phase. Both AAm and PEGDA were found to act as co-surfactants, resulting in the obtainment of monoliths with greater homogeneity in some cases. As a result these materials demonstrated significantly improved chromatographic performance for the separation of a standard mixture of proteins using reversed-phase liquid chromatography, in comparison to monoliths prepared with no internal phase monomer. In particular, the columns grafted with PEGDA were capable of separating a more complex mixture consisting of seven components. The inclusion of monomers in the internal phase also allowed for the functionalisation of the monolith's surface where the degree of polymerisation that occurred in the internal phase, which was governed by the monomer content in the internal phase and initiation location, determined whether polymeric chains or a hydrogel were grafted to the surface. A monolith grafted with AAm was also found to be capable of retaining polar analytes as a result of the increase in surface hydrophilicity.
Introduction
Since their development in the 1990s1,2 polymer monoliths have attracted significant interest for use as stationary phases in liquid chromatography (LC). These materials consist of a permanently rigid continuous piece of porous material2 and typically possess a crosslinked inter-connected macroporous structure.3 This structure provides these materials with significantly higher permeabilities and flow that is convective in nature, in comparison to traditional formats such as packed columns.4,5 This flow profile enhances the mass transport of analytes, which is particularly important for large molecules such as proteins, whose low diffusion coefficients can cause issues for traditional formats, which rely on diffusion.5 As such polymer monoliths have been demonstrated to be excellent stationary phases for the separation of large molecules, including: proteins,6,7 antibodies,8,9 DNA10,11 and polymers.12 Traditionally polymer monoliths are prepared by free radical polymerisation involving phase separation from a solvent mixture,13,14 which results in a random globular morphology.3 This therefore results in a degree of column bed heterogeneity and is considered to be one of the limiting factors for their application as stationary phases for LC.15,16 As an example the pore size distribution can vary over several orders of magnitude.15 Recent work has therefore focused on the use of templated techniques in an effort to improve their open-cellular homogeneity. For example, polymer-based cryogels have been prepared by directional freezing and applied for the separation of proteins by high performance liquid chromatography (HPLC).17Another technique that has been employed is the polymerisation of high internal phase emulsions (HIPEs) to form so called poly(HIPE)s.18–23 HIPEs are simply emulsions with an internal phase that exceeds 74 vol%,24,25 which is the characteristic packing density of face centred cubic (fcc) or hexagonally closed packed (hcp) arranged uniform spheres. If the continuous phase contains one or more monomeric species and non-ionic surfactants are employed as stabilisers, poly(HIPE)s with open cellular morphology are typically obtained. Here individual voids, which are directly templated from the emulsion droplets, are interconnected by the presence of pores that are called windows. These windows allow for transport throughout the material. An alteration in the emulsions structure therefore directly influences the morphology of the resulting material, affording a degree of control over their structure. Their application in LC has also been demonstrated for a variety of analytes including alkylbenzenes,20,22,26 proteins,18,19,27 cytokinins,28 plant extracts23 and nanoparticles.22 However their chromatographic performance has so far been inferior to that of conventional polymer monoliths, potentially from the presence of column heterogeneity.29
In most cases these poly(HIPE)s were prepared using low shear emulsification,18–20,22,27 however our group recently demonstrated that the use of a high energy mixer resulted in columns with significantly improved column bed homogeneity for poly(styrene-co-divinylbenzene) [poly(Sty-co-DVB)] poly(HIPE)s when prepared in capillary format.30 In accordance, these columns were demonstrated to possess improved chromatographic performance for the separation of a standard protein mixture. However the degree of column bed homogeneity was still a limiting factor in their performance as a result of broad void size distributions. The preparation of poly(HIPE)s with narrower void size distributions is therefore expected to result in further improvements in chromatographic performance, and this can potentially be achieved by increasing the emulsions stability.
Increases in the stability of water-in-oil HIPEs can be achieved through increases in the emulsification energy,30–32 by the addition of salt,24,33,34 or by the inclusion of co-surfactants.35 For example, Gitli and Silverstein35 previously observed an increase in the homogeneity of poly(styrene)-based poly(HIPE)s upon the inclusion of acrylamide (AAm) to the internal aqueous phase. While an improvement in column homogeneity can potentially be achieved, this approach also offers an alternative route for the functionalisation of the monolithic surface through the grafting of AAm, which may allow for the use of these materials for other applications and/or other chromatographic modes.
In this work we explored the possibility of preparing poly(Sty-co-DVB) poly(HIPE)s in capillary format with improved column homogeneity by including monomers as co-surfactants in the internal phase and emulsifying with high shear. Initial experiments focused on the inclusion of the hydrophilic monomer AAm, where both the monomer content and initiator were varied to establish the influence this had on the morphology and resulting chromatographic performance for a standard protein mixture using RPLC. The inclusion of PEGDA (Mw 258) in the internal phase was also investigated as this monomer is expected to be a more efficient co-surfactant due to its more amphiphilic nature. Finally the surface hydrophilicity of a poly(HIPE)s grafted with AAm was evaluated by accessing its applicability to retain analytes through polar interactions.
Experimental
Materials
Acetic acid (≥99.7%), albumin from chicken egg white (ovalbumin) (≥98%), basic alumina (Brockman activity I, 60–325 mesh), α-chymotrypsinogen A from bovine pancreas, cytochrome c from equine heart (≥95%), divinylbenzene (DVB, 80%), lysozyme from chicken egg white (≥90%), myoglobin from horse heart (≥90%), PEG diacrylate (PEGDA, Mw 258), potassium persulfate (KPS, ≥99.0%), ribonuclease A, type I-A, from bovine pancreas (≥60%), sodium hydroxide (≥98.0%), sodium sulfate (anhydrous) (≥99.0%), styrene (Sty, 99%) and 3-(trimethoxysilyl)propyl methacrylate (≥98%) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Acrylamide (AAm, ≥98.0%), Span® 80 (≥60%), formic acid (≥98.0%) and guanosine (≥99%) were obtained from Fluka (Seelze, Hannover, Germany). Acetone (>98%) and ethanol (>99%) were obtained from Chem-Supply (Gillman, South Australia, Australia). Hydrochloric acid (37%) and sodium carbonate (anhydrous) (≥99.9%) were obtained from Merck (Kilsyth, VIC, Australia). Acetonitrile (ACN, ≥99.8%) was obtained from VWR (Radnor, PA, USA). 2,2′-Azobis(2-methylpropionitrile) (AIBN) was obtained from MP Biomedicals (Santa Ana, CA, USA). Calcium chloride dihydrate (≥98.0%) was obtained from Ajax Chemicals (Sydney, NSW, Australia). Dichloromethane (>99%) was obtained from Unilab (Mandaluyong City, Philippines). Methanol (MeOH, 99.9%) was obtained from Fisher Scientific (Pittsburgh, Pennsylvania, United States). The monomers (Sty and DVB) were passed through a column of basic alumina to remove inhibitors. KPS was re-crystalized from H2O, while AIBN was re-crystalized from MeOH. PEGDA was purified in accordance with a previous procedure.8 All other chemicals were used as received. H2O used in all experiments was first purified using a Milli-Q system (Millipore). Polyimide-coated capillaries of 150 μm i.d. (360 μm o.d.) were obtained from Polymicro Technologies.Modification of fused silica capillaries
The polyimide-coated fused silica capillaries were surface modified based on a previous procedure by Rohr et al.36 Here, capillaries were first rinsed with acetone and then H2O, before being activated by pumping a solution of 0.2 M NaOH through the capillaries at a rate of 30 μL h−1 for 30 min using a syringe pump. These were then rinsed with H2O, before 0.2 M HCl was pumped through at the same rate for 30 min. The capillaries were then rinsed with H2O, and ethanol at pH 5 (adjusted using acetic acid). A 20 wt% solution of 3-(trimethoxysilyl)propyl methacrylate in ethanol at pH 5 was then pumped through the capillaries at 30 μL h−1 for 1 h, after which these were rinsed with acetone and purged with nitrogen for 2 min. Finally, these were left at room temperature for 24 h to allow for the completion of the condensation reaction.Preparation of poly(HIPE)s
The poly(Sty-co-DVB)-based poly(HIPE)s were prepared based on the conditions established in a previous publication.30 The internal phase was prepared by dissolving 0.006 g of calcium chloride dihydrate and internal phase monomer in 9 mL of H2O. This was then added dropwise at a rate of 1 drop per second to a continuous phase consisting of 0.2970 g of Span® 80, 0.8 mL of Sty and 0.2 mL of DVB with constant stirring at 300 rpm. 0.02 g (0.074 mmol) of KPS or 0.012 g (0.074 mmol) of AIBN were also dissolved in the internal or continuous phase, respectively, before addition of the internal phase. The emulsion was then blended using an IKA Ultra Turrax T 25 homogeniser equipped with an S 25 N 10 G dispersing element (7.5 mm rotor) at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 2 min. This was then passed through 20 cm of 150 μm i.d. surface-modified fused silica capillaries by hand using a 250 μL Hamilton® syringe. The emulsion emerging from the outlet was collected in 4 mL glass vials. At least three capillaries were filled for each emulsion prepared and they were each filled multiple times to limit the number of air bubbles or voids present before the ends were sealed with rubber. They were then placed horizontally in the water bath at 60 °C, as vertical placement can result in column heterogeneity due to the influence of gravity for conventional polymer monoliths.13 These were cured for 48 h, while the remaining emulsion was transferred to a 25 mL glass vials as a bulk sample and, along with the 4 mL vials, also immersed in the water bath. Once cured the bulk material from the vials was removed, cut into smaller pieces and washed using MeOH with a Soxhlet apparatus for 24 h to remove the internal phase and any impurities. These were then left to dry at 25 °C in a vacuum oven for 1 week. The capillaries were flushed with MeOH for 2 h and then H2O for 2 h using the capillary LC system with a flow rate of 2 μL min−1. These samples were referred to as follows: [wt% of monomer (w.r.t. internal phase), monomer, (initiator)].
000 rpm for 2 min. This was then passed through 20 cm of 150 μm i.d. surface-modified fused silica capillaries by hand using a 250 μL Hamilton® syringe. The emulsion emerging from the outlet was collected in 4 mL glass vials. At least three capillaries were filled for each emulsion prepared and they were each filled multiple times to limit the number of air bubbles or voids present before the ends were sealed with rubber. They were then placed horizontally in the water bath at 60 °C, as vertical placement can result in column heterogeneity due to the influence of gravity for conventional polymer monoliths.13 These were cured for 48 h, while the remaining emulsion was transferred to a 25 mL glass vials as a bulk sample and, along with the 4 mL vials, also immersed in the water bath. Once cured the bulk material from the vials was removed, cut into smaller pieces and washed using MeOH with a Soxhlet apparatus for 24 h to remove the internal phase and any impurities. These were then left to dry at 25 °C in a vacuum oven for 1 week. The capillaries were flushed with MeOH for 2 h and then H2O for 2 h using the capillary LC system with a flow rate of 2 μL min−1. These samples were referred to as follows: [wt% of monomer (w.r.t. internal phase), monomer, (initiator)].
Polymer disks for porosity measurements were prepared as described above, except the emulsion was transferred to 10 mL disposable syringes (∼1.5 cm in diameter). These were then sealed and placed in the water bath at 60 °C at an angle of ∼45° from the horizontal to ensure any air bubbles migrated to the top of the syringe and then cured for 48 h. Once cured they were removed and cut into 0.5 cm thick pieces and then washed with MeOH using the Soxhlet apparatus for 24 h. They were dried in a vacuum at 25 °C for 1 week before use.
Characterisation
Optical microscopy images of the emulsions were taken immediately after preparation using a Nikon Eclipse E200 microscope equipped with a 10× objective and a 30.5 mm 0.5× C-mount adapter connected to a 5.0 MP Tucsen IS500 Camera (Fuzhou Xintu Photonics). A few drops of emulsion were placed on a glass slide, which had a piece of 500 μm thick Teflon covering the perimeter. Another glass slide was placed on top to limit evaporation and allow stable images to be obtained. This was also performed for the emulsion that was collected from the capillary outlets during filling.Scanning electron micrographs of the poly(HIPE)s were obtained using a Hitachi-SU-70 field emission scanning electron microscope operated in high vacuum mode with an acceleration voltage of 1.5 kV. Secondary electrons were detected using a Hitachi scintillator-type detector. Samples were platinum coated (2–3 nm thick coating) using a Bal-Tec SCD 050 Sputter Coater. The average void and window diameters were obtained by measuring the diameter of at least 300 voids and windows using ImageJ (NIH Image). The values obtained are lower than the true values so it is necessary to introduce a statistical correction,37,38 where the values are essentially multiplied by a factor of 2/(31/2). The derivation of this correction factor can be found in Barbetta and Cameron.38 The radial distribution of voids across the capillary cross-sections were also investigated by calculating the average void diameter for voids present within the annulus formed from concentric circles, which differed in diameter by 15 μm originating from the capillary wall. Additionally, the average number of windows per void was estimated for some samples by counting the number of observed windows for 300 voids, as a rough estimate.
The specific surface area of the poly(HIPE)s was obtained using the Brunauer–Emmett–Teller method39 with a Micromeritics Tristar II 2020 automated nitrogen sorption-desorption instrument. All samples were dried using a Micromeritics SmartPrep at 70 °C for 24 h prior to analysis. Measurements were conducted in triplicate using ∼100 mg of sample.
The porosity of the bulk samples were estimated by immersing dry polymer disks in MeOH following a modified method from Greig and Sherrington.40 Here the mass and dimensions of the dry disk (diameter and height) was recorded prior to immersion. The disks were then placed in centrifuge tubes, which contained MeOH for 1 h before being centrifuged at 2600 rpm for 15 min, after which their mass was re-measured. They were then re-immersed in MeOH for 5 min before being centrifuged again. This process was repeated until a constant mass was achieved, after which the dimensions of the polymer disk were re-measured. The porosity in the wet state (φw) can then be calculated from the following equation:41
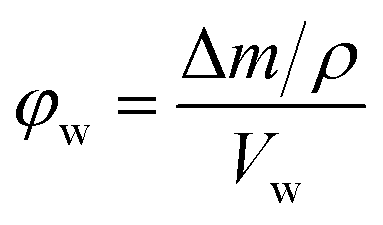 | (1) |
The nitrogen and sulfur content of the bulk poly(HIPE)s was determined with a Thermo Finnigan EA 1112 Series Flash Elemental Analyser, while FTIR spectra were recorded using a Bruker Vertex 70 infrared spectrometer equipped with an ATR probe and redrawn using Origin® 8.5 (OriginLab).
Chromatography
Chromatographic separations were performed using a Dionex UltiMate™ 3000 RSLCnano system equipped with a NCS-3500RS capillary LC gradient pump including a membrane degasser unit and integrated column compartment, a VWD-3400RS UV detector equipped with a 45 nL flow cell and a WPS-3000TPLC RS autosampler fitted with a 1 μL sample loop. Chromeleon® software (Ver. 6.80) was used for system control and data processing (data collection rate was 2.5 Hz). Chromatograms were converted to ASCII files and redrawn using Origin® 8.5. The LC experiments were conducted under gradient conditions and 1 μL injections were performed with the aid of an autosampler. UV detection was employed at both 214 and 280 nm. For all chromatograms the baseline drift caused by the gradient was subtracted. Comparisons were made to the original chromatograms to ensure that structures observed were not artefacts of the subtraction process. For the RPLC separations eluent A consisted of 0.1 vol% formic acid in H2O and eluent B consisted of 0.1 vol% formic acid in ACN and these were degassed prior to use. Samples were dissolved in and diluted with H2O to the appropriate concentrations. For the HILIC separations eluent A was ACN and eluent B was Milli-Q H2O and these were also degassed prior to use. Here, samples were dissolved in and diluted with ACN to the appropriate concentrations.Permeability measurements were performed for columns of various lengths by recording the column back pressure at various flow rates between 0.5 and 2.5 μL min−1 in both MeOH and H2O at 25 °C. Before being recorded the pressure was allowed to stabilise for 5 to 10 min. The permeability was then calculated using Darcy's law,42,43 from the slope of a plot of column back pressure against flow rate. The values measured actually contain contributions from the back pressure of the system,44 so this was corrected for by subtracting the slope obtained from a plot of back pressure against flow rate, over the same range of flow rates, in the absence of the column from the slope obtained with the column. The resulting value was then used to calculate the permeability. Viscosities of 0.544 mPa.s and 0.890 mPa.s for MeOH and H2O at 25 °C, respectively, were used in the calculations,43 and this was performed for at least three columns prepared from the same batch. In the case of 0.4 wt% PEGDA (AIBN) and 4 wt% PEGDA (AIBN) the flow rate was varied between 2 and 9 μL min−1 due to their significantly higher permeabilities.
Results and discussion
Preparation of poly(HIPE)s grafted with AAm
Poly(Sty-co-DVB)-based poly(HIPE)s were first prepared by including AAm in the internal phase at two different concentrations, 0.1 and 1 wt% (w.r.t. internal phase), in order to investigate the influence of its inclusion on the morphology of the resulting poly(HIPE)s. In addition the initiation location was also varied, by employing KPS as a water-soluble initiator or AIBN as an oil-soluble initiator. Two control samples without monomer in the internal phase, but using the different initiators, were also prepared for comparison.SEM analysis (Fig. 1 and Table 1) of the resulting poly(HIPE)s revealed an apparent reduction in the average void diameter upon the inclusion of AAm for both initiators. For example, this was reduced from 3.4 ± 0.7 to 2.3 ± 0.7 μm for KPS and from 7 ± 2 to 4 ± 1 μm for AIBN with the inclusion of 0.1 wt% AAm to the internal phase. This suggests that AAm is acting as a co-surfactant where its inclusion is resulting in a reduction in the average droplet diameter and resulting void size, as previously observed by Gitli and Silverstein.35 Increasing the AAm content further to 1 wt% resulted in an additional reduction in the void size to 2.9 ± 0.6 μm for AIBN, however the value obtained for KPS of 2.5 ± 0.9 μm was not statistically different to that of 0.1 wt% AAm. The same trend was also observed for the inter-connecting window size (Table 1).
 | ||
| Fig. 1 SEM images of poly(HIPE)s prepared with different amounts of AAm or PEGDA (w.r.t. internal phase) in the internal phase and using different initiators. Scale bar is 3 μm. | ||
| Sample | VBb/μm | WBc/μm | DBd/μm | Porositye/% | Surface areaf/m2 g−1 | VCb/μm | WCc/μm |
|---|---|---|---|---|---|---|---|
| a B, indicates the bulk material. C, indicates the material in capillary.b Average void diameter for the poly(HIPE)s as determined from SEM (n = 300).c Average window diameter for the poly(HIPE)s as determined from SEM (n = 300).d Average droplet diameter immediately after preparation for the emulsions (n = 300).e Porosity of bulk poly(HIPE)s determined by immersion in MeOH.f Specific surface area of bulk poly(HIPE) determined from BET. | |||||||
| 0 wt% (KPS) | 3.4 ± 0.7 | 0.8 ± 0.3 | 3 ± 1 | 97 ± 4 | 19.7 ± 0.8 | 4 ± 2 | 1.1 ± 0.4 |
| 0.1 wt% AAm (KPS) | 2.3 ± 0.7 | 0.6 ± 0.2 | 2 ± 1 | 86 ± 5 | 43.7 ± 0.4 | 5 ± 2 | 1.2 ± 0.5 |
| 1 wt% AAm (KPS) | 2.5 ± 0.9 | 0.5 ± 0.2 | 3 ± 1 | 83 ± 7 | 30.2 ± 0.3 | 3 ± 2 | 0.6 ± 0.2 |
| 0.4 wt% PEGDA (KPS) | 1.9 ± 0.5 | 0.5 ± 0.2 | 2 ± 1 | 98 ± 7 | 37.7 ± 0.5 | 2 ± 1 | 0.6 ± 0.2 |
| 4 wt% PEGDA (KPS) | 4 ± 3 | 0.7 ± 0.3 | 5 ± 2 | 97.5 ± 0.3 | 29 ± 1 | 4 ± 4 | 0.8 ± 0.3 |
| 0 wt% (AIBN) | 7 ± 2 | 1.6 ± 0.7 | 7 ± 3 | 96 ± 8 | 14.1 ± 0.4 | 7 ± 2 | 1.8 ± 0.6 |
| 0.1 wt% AAm (AIBN) | 4 ± 1 | 1.2 ± 0.4 | 3 ± 2 | 108 ± 8 | 13.4 ± 0.3 | 7 ± 4 | 0.9 ± 0.4 |
| 1 wt% AAm (AIBN) | 2.9 ± 0.6 | 0.7 ± 0.2 | 3 ± 1 | 89 ± 7 | 17.5 ± 0.8 | 6 ± 4 | 1.9 ± 0.8 |
| 0.4 wt% PEGDA (AIBN) | 4 ± 2 | 1.2 ± 0.5 | 4 ± 2 | 98 ± 5 | 15 ± 1 | 5 ± 2 | 1.5 ± 0.5 |
| 4 wt% PEGDA (AIBN) | 4 ± 1 | 1.3 ± 0.4 | 4 ± 2 | 105 ± 5 | 13.1 ± 0.8 | 3 ± 3 | 1.7 ± 0.5 |
In all cases the poly(HIPE)s prepared using AIBN as initiator possessed larger void sizes and therefore lower specific surface areas (Table 1) than the equivalent materials prepared using KPS as initiator. This is well documented for poly(styrene)-based systems33,34 and is due to the reduced salt concentration when AIBN is used as initiator. As discussed the addition of salt, such as KPS, can have a stabilising effect on the oil–water interface and therefore can reduce the droplet size and aid in emulsion stability.24,33,34 As such KPS is commonly used as the initiator in the preparation of poly(HIPE)s from water-in-oil emulsions.
This enhanced stability could also explain why the influence of increasing the AAm content on the resulting void size was less pronounced for KPS in comparison to AIBN. Regardless, the droplet sizes observed immediately after preparation (Table 1 and Fig. S19†) for all emulsions were consistent with the void sizes obtained, suggesting that no significant coalescence occurred during curing, even when AIBN was used as the initiator. In addition, the porosity (Table 1) for these materials were also consistent with the 90 vol% internal phase utilised, which suggests that no or limited creaming of these emulsions occurred.24 Even though the increase in AAm content from 0.1 to 1 wt% did not result in a shift in the void size in the case of KPS, a significantly different void surface was obtained (Fig. 1). In the case of 0.1 wt% AAm a smooth void surface was observed, however at 1 wt% AAm the resulting void surface appeared wrinkled and crumpled regions were present on the void walls. This texturing is indicative of the presence of a collapsed hydrogel,45 which has also previously been observed when monomers are included in the internal phase.35,46–48 A comparison of these two contrasting materials at a higher magnification can be found in Fig. S1.† The presence of a hydrogel that filled the voids of this poly(HIPE) was also supported by the decrease in porosity observed from 97 ± 4% for 0.1 wt% AAm to 83 ± 7% for 1 wt% AAm.
This hydrogel appeared to be AAm-based with elemental analysis (Table S1†) indicating a significant increase in nitrogen content from 0.03 wt% for 0 wt% AAm in the internal phase to 1.05 wt% for 1 wt% AAm in the internal phase, with AAm the only plausible source. This corresponded to an estimated incorporation of 58% of the internal phase monomer into the resulting poly(HIPE). In addition, the characteristic amide bands at 3402, 3192 and 1672 cm−1 were clearly observed by FTIR (Fig. S2†), indicating the presence of polymerised AAm at the surface.35 In addition to the presence of a hydrogel, particles of ∼500 nm in diameter were also observed for 1 wt% AAm (KPS). These were also present for 0.1 wt% AAm (KPS), but in smaller number. This suggests that some degree of phase inversion occurred,49 which was also associated with the increase in AAm content.
This was in contrast to that of AIBN where the increase in AAm content simply resulted in a decrease in the void size, with no alteration in the texture of the void surface, suggesting no hydrogel was formed in this case. However, elemental analysis (Table S1†) also revealed the presence of a significant level of nitrogen with a value of 1.63 wt for 1 wt% AAm (AIBN), which corresponded to an estimated incorporation of 90% of the internal phase monomer. In addition, the characteristic amide bands were also observed by FTIR (Fig. S2†). This clearly indicates that although a hydrogel was not observed in the case of AIBN, AAm was still incorporated into the resulting structure.
To understand the reason for the presence or absence of an AAm-based hydrogel in the case of KPS and AIBN, respectively, it is important to consider the mechanism responsible for the incorporation of AAm into the material. Given the partition co-efficient for AAm between toluene and H2O is relatively low,50 the amount of AAm located in the continuous phase is not considered to be significant.35 The incorporation of AAm into the resulting poly(HIPE) therefore occurs through the co-polymerisation of AAm from the internal phase and external phase monomers at the oil–water interface.35,45 Given AAm is acting as a co-surfactant a proportion of AAm is expected to be located at the interface, where it is available to undergo co-polymerisation. Consideration of the relative reactivity ratios indicates that AAm is expected to undergo a higher degree of co-polymerisation than homopolymerisation,51 however this will also be influenced by both the radical and AAm concentration in the internal phase.
When AIBN is used as initiator, the radicals are generated in the external phase, and the polymerisation of this phase is predominant. This results in the grafting of AAm-chains rather than a gel to the surface.35,47 This was also previously observed by Gitli and Silverstein,35 who incorporated AAm and MBAm into the internal phase, when using the oil-soluble benzoyl peroxide initiator.
In contrast when KPS is utilised as initiator the radicals are generated in the internal phase and the polymerisation of the external phase is initiated at the interface.35 Given the higher concentration of radicals present in the internal phase the AAm present can undergo a higher degree of homopolymerisation. If AAm is present at a sufficient concentration a hydrogel can form which is grafted to the surface without the presence of an additional crosslinker. Hayward et al.47 observed a similar result upon increasing the concentration of acrylic acid (AA) in the internal phase when using KPS as initiator.
A higher degree of homopolymerisation could also explain the lower estimated incorporation of 58%, in comparison to 90% when AIBN was used (Table S1†), as a larger proportion of AAm was preferentially bound to other AAm chains and not the surface, resulting in its removal during purification, in contrast to AIBN where the amount of AAm radicals generated in the internal phase is significantly lower and thus homopolymerisation of AAm is limited. Alternatively, the formation of AAm-based particles, through phase-inversion, that were not bound to the surface may explain the lower incorporation.
In terms of the lower concentration of AAm, both 0.1 wt% AAm (KPS) and 0.1 wt% AAm (AIBN) showed a negligible increase in nitrogen content (Table S1†) and no obvious amide signals were observed (Fig. S2†). It is likely that the low AAm content resulted in significantly less co-polymerisation at the oil–water interface, as the likelihood for a growing AAm chain to be captured by the surface is significantly lower when the concentration is low, regardless of the initiator utilised. As a result a significant proportion of the AAm in the internal phase was unbound and simply removed during the purification process, resulting in an estimated incorporation of internal phase monomer of only 17 and 11%, respectively. In this case the incorporation of AAm was slightly higher when KPS was utilised, in comparison to AIBN, presumably due to the generation of a higher number of AAm radicals in the internal phase which tended to undergo co-polymerisation with styrene at the interface.
Preparation of poly(HIPE)s grafted with AAm in capillary format
In order to evaluate the chromatographic performance of these materials, they were first prepared in capillary format using 150 μm i.d. fused silica capillaries. In most cases the average void and window size obtained within the capillaries were consistent with that obtained with the bulk materials (Fig. 2 and Table 1). In addition, the average droplet and resulting void and window sizes for the emulsions that were passed through these capillaries and then cured were also consistent, in most cases, with that of the bulk materials (Table S2 and Fig. S4, S20†). This suggested that passing these particular emulsions through the capillary inlet and/or confining them within the capillary itself did not compromise their structure or stability. However, the void size distributions within the capillaries were broader (Table 1), and this was potentially related to the presence of a small number of larger voids, for example one can clearly be seen in the cross-section for 0.1 wt% AAm (KPS) in Fig. 2.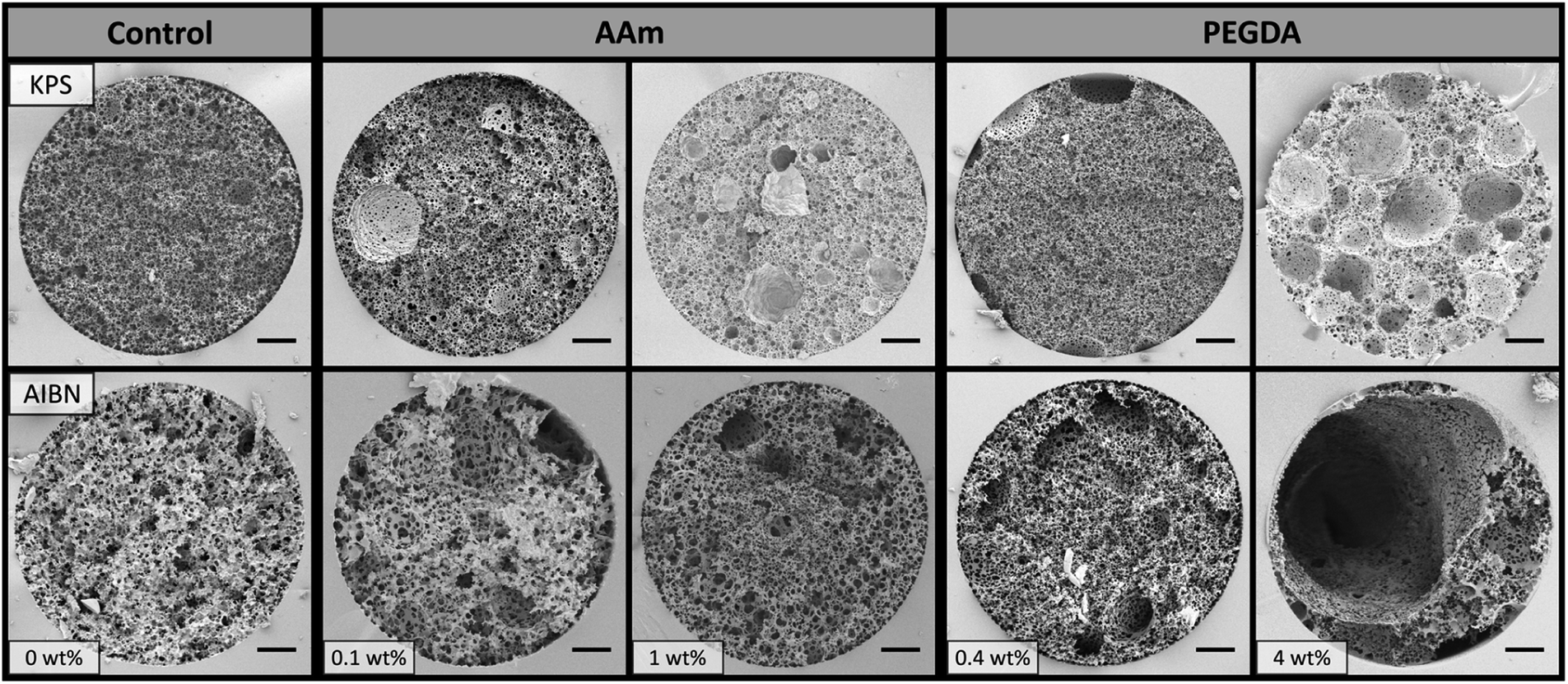 | ||
| Fig. 2 SEM images of poly(HIPE)s prepared with different amounts of AAm or PEGDA (w.r.t. internal phase) in the internal phase in 150 μm i.d. capillaries using different initiators. Scale bar is 20 μm. | ||
As discussed in a previous publication,30 the origin of these larger voids is most likely from the introduction of air bubbles associated with the use of a high energy mixer and/or the capillary filling process, and their presence will contribute towards band broadening. While the utilisation of lower shear emulsification or another emulsification technique may reduce their presence, the use of high energy mixers was shown to result in poly(HIPE)s with increased homogeneity, in particular minimal radial heterogeneity, and structures that resembled the bulk material when prepared in capillary format.
While all the columns prepared in this work exhibited no significant radial heterogeneity (Fig. S5†), not all the materials prepared in capillary format reflected that of their bulk counterparts. Both 0.1 wt% AAm (KPS) and 1 wt% AAm (AIBN) possessed larger void and window sizes when confined within the capillaries in comparison to when cured in glass vials (Table 1). For example, the void and window size increased from 2.3 ± 0.7 and 0.6 ± 0.2 μm, respectively, to 5 ± 2 and 1.2 ± 0.5 μm for 0.1 wt% AAm (KPS), while the void and window size increased from 2.9 ± 0.6 and 0.7 ± 0.2 μm, respectively, to 6 ± 4 and 1.9 ± 0.8 μm for 1 wt% AAm (AIBN), when confined within the capillary. The emulsions that were passed through these capillaries actually had similar droplet sizes to that of the original emulsion (Table S2†), suggesting no initial alteration in the emulsions structure occurred. However after curing these emulsions, larger void and window sizes were also obtained. This suggests that a degree of coalescence occurred during the curing process, and suggests that passing the emulsion through the narrow capillary inlet has resulted in a reduction in the stability of these particular emulsions. It should also be pointed out that the void size for 0.1 wt% AAm (AIBN) also appeared to increase when confined within the capillary (though this was not statistically significant), however this did not appear to be related to the stability, as this was not observed for the emulsion passed through the capillary and polymerised (Table S2†). It is more likely in this case that the larger voids corresponding to introduced air bubbles resulted in the shift in the observed distribution. The stability for all other emulsions also did not appear to be compromised (Table S2†).
SEM analysis (Fig. 2) revealed good attachment of the monoliths to the capillary wall and the presence of a wrinkled surface in the case of 1 wt% AAm (KPS) was again observed, indicating the presence of a hydrogel that filled the voids of this poly(HIPE), even when prepared within the capillary format. The back pressure for these columns was found to vary linearly with flow rate when both MeOH and H2O were pumped through these columns using flow rates between 0.5 and 2.5 μL min−1 (Fig. S10–S14†), which indicated no mechanical failure or compression of these monoliths occurred.52 The column permeabilities (Table 2) also reflected that of the porous properties observed. For example a reduction in permeability from 1.3 ± 0.1 × 10−13 m2 to 0.6 ± 0.3 × 10−13 m2 in MeOH was observed with an increase in AAm content from 0 to 0.1 wt% when KPS was used as initiator, consistent with the apparent reduction in void and window size observed (Table 1).
| Column i.d./μm | kavga (MeOH)/×10−13 m2 | kavga (H2O)/×10−13 m2 |
|---|---|---|
| a Average permeability calculated from at least three columns from the same batch of emulsion.b NP indicates the column was non-permeable. | ||
| 0 wt% AAm (KPS) | 1.3 ± 0.1 | 1.4 ± 0.2 |
| 0.1 wt% AAm (KPS) | 0.6 ± 0.3 | 0.7 ± 0.3 |
| 1 wt% AAm (KPS) | 0.08 ± 0.01 | 0.08 ± 0.01 |
| 0.4 wt% PEGDA (KPS) | 2.6 ± 0.5 | 4 ± 1 |
| 4 wt% PEGDA (KPS) | 3.4 ± 0.2 | 3.9 ± 0.2 |
| 0 wt% AAm (AIBN) | NPb | NPb |
| 0.1 wt% AAm (AIBN) | 0.3 ± 0.1 | 0.3 ± 0.1 |
| 1 wt% AAm (AIBN) | 2.3 ± 0.3 | 2.3 ± 0.3 |
| 0.4 wt% PEGDA (AIBN) | 21 ± 3 | 23 ± 3 |
| 4 wt% PEGDA (AIBN) | 200 ± 200 | 300 ± 300 |
Unfortunately the columns obtained using AIBN without AAm were not permeable, even though windows with an average size of 1.8 ± 0.6 μm (Table 1) were present (Fig. 2). This is in contrast to the bulk material, which had a porosity reflective of the internal phase volume utilised (Table 1), suggesting that it was permeable (as liquid had to fill the voids for the porosity measurement). It is likely that there exist a number of non-permeable voids located along the columns length restricting the flow (as cutting the column at different lengths did not allow for liquid to flow).
The use of different initiators is known to influence window formation,53 and it is possible that the use of AIBN (without monomer in the internal phase) resulted in reduced window formation. This could simply have been a result of the larger droplets this emulsion possessed,31,54 and this only appeared to be significant when confined within the narrower dimensions of the capillary. This is consistent with a closer inspection of the capillary cross-sections (Fig. 2), which revealed a larger proportion of voids with no windows in the case of AIBN, in contrast to the control column prepared with KPS. In fact, the average number of windows per void was estimated to be 1 ± 1 and 6 ± 4, respectively. The inclusion of a co-surfactant such as AAm, which lowers the interfacial tension and reduces the droplet size, is expected to promote window formation.55 In accordance, the inclusion of 0.1 wt% AAm resulted in an increase in the average number of windows per void to 3 ± 3. This was consistent with the columns prepared with AAm present in the internal phase being permeable when AIBN was used as the initiator, in contrast to without AAm.
The column permeability for 1 wt% AAm (KPS) was significantly lower than that of 0.1 wt% AAm (KPS) with a value of 0.08 ± 0.01 × 10−13 m2 in MeOH, even though their average void and window sizes were not statistically different. It is important to consider that these values for the void and window size are obtained in the dry state, and may not be reflective of the solvated morphology. In particular in the case of a hydrogel, with the reduction in permeability consistent with the presence of a hydrogel that filled the voids of this poly(HIPE) and potentially reduced the window size in the solvated state. This could also have been the result of a higher frequency of voids containing a lower number of windows, particularly in the regions observed to exhibit the collapsed hydrogel morphology, which may have ultimately impacted the flow through the material. The permeability when H2O was used as the mobile phase was identical, suggesting that any difference in the swelling of this gel between MeOH and H2O was not significant enough to further restrict the window size or the flow path. In contrast, when the AAm content was increased from 0.1 to 1 wt% using AIBN as initiator, the permeability in MeOH also increased from 0.3 ± 0.1 × 10−13 m2 to 2.3 ± 0.3 × 10−13 m2, consistent with the larger than expected voids and windows in the case of 1 wt% AAm (AIBN).
In addition to 1 wt% AAm (KPS), all other columns exhibited similar permeabilities in H2O, suggesting that negligible shrinkage or swelling occurred in these solvents. This is particularly important for their suitability to be utilised as stationary phases for liquid chromatography using a solvent gradient. It is important to note that the permeability values obtained for these poly(HIPE)s, except for 1 wt% AAm (KPS), are significantly larger than that of conventional polymer monoliths prepared using a porogen, which typically have permeabilities in the range of (1–10) × 10−14.42,56 This potentially allows these materials to be used for rapid separations with minimal increase in back pressure.
Chromatographic performance of poly(HIPE)s grafted with AAm
The separation performance of poly(Sty-co-DVB) monoliths for the separation of proteins is well documented.57 In particular, in a previous publication we demonstrated the applicability of the 0 wt% AAm (KPS) poly(HIPE) for the separation of a standard protein mixture consisting of ribonuclease A, lysozyme and α-chymotrypsinogen A by RPLC.30 The chromatographic performance of these poly(HIPE)s was therefore first evaluated for the separation of this mixture using this chromatographic mode and the chromatograms obtained are shown in Fig. 3.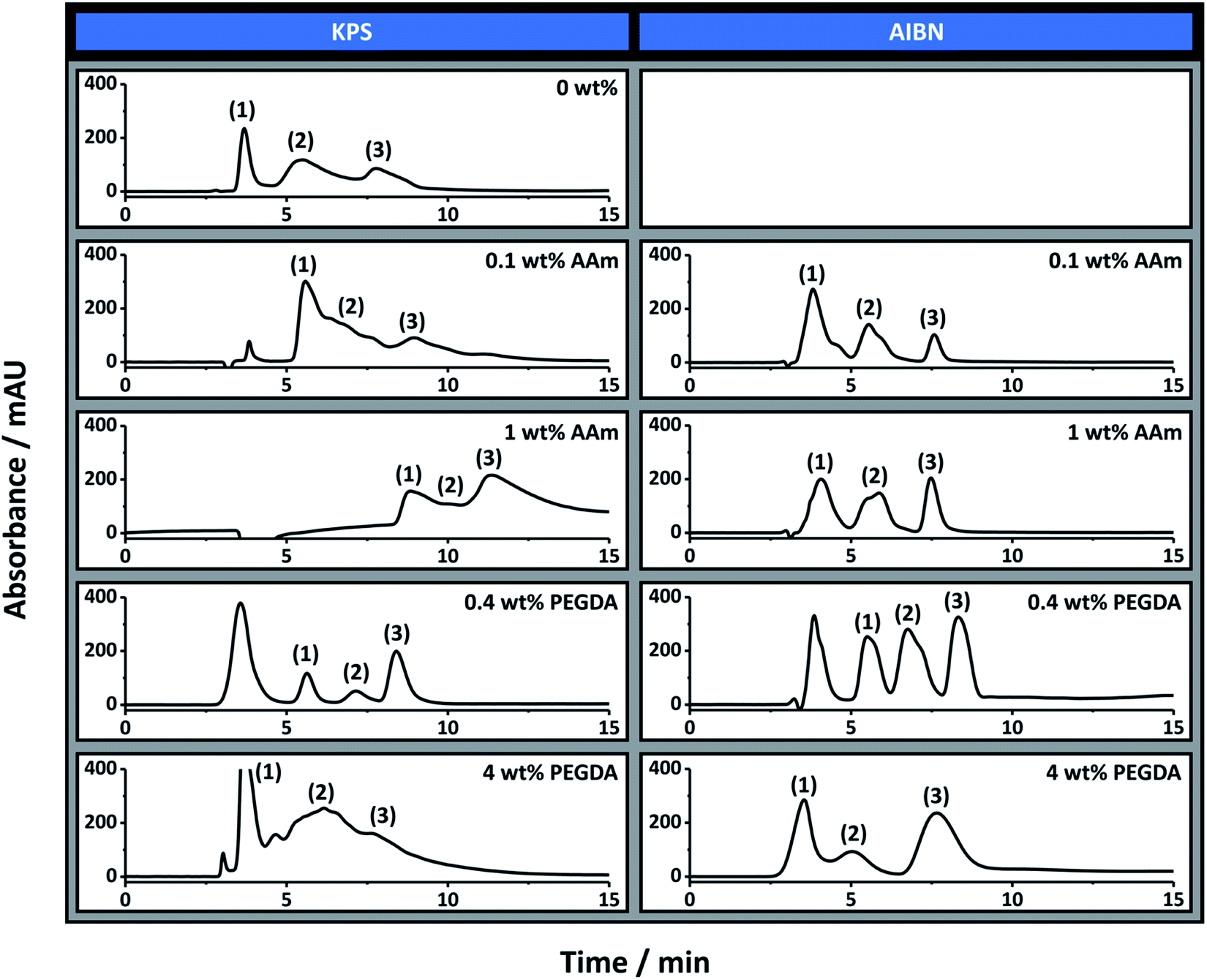 | ||
| Fig. 3 The separation of ribonuclease A (1), lysozyme (2) and α-chymotrypsinogen A (3) under reversed-phase conditions using columns prepared with different amounts of monomer in the internal phase using different initiators. Conditions: 18 cm × 150 μm i.d. columns; eluent A was 0.1 vol% formic acid in Milli-Q H2O, and eluent B was 0.1 vol% formic acid in ACN; linear gradient 15 to 90% B in 15 min and then isocratic elution at 90% B for 5 min before returning to 15% B in 5 min; flow rate, 2.0 μL min−1; injection volume, 1 μL; protein concentration, 0.05 mg mL−1 except for 0.1 wt% AAm (KPS) and 4 wt% PEGDA (KPS) which was 0.1 mg mL−1; UV detection at 214 nm. The 0 wt% column prepared with AIBN was not permeable so was not included. | ||
As previously demonstrated, almost baseline resolution was achieved for the 0 wt% AAm (KPS) column. However, as the AAm content was increased the co-elution between these proteins also increased. For example, significant co-elution was observed between ribonuclease A and lysozyme for 0.1 wt% AAm (KPS), while significant co-elution between all three proteins was observed for 1 wt% AAm (KPS). In all cases the peaks corresponding to lysozyme and α-chymotrypsinogen A exhibited significant rear tailing, while both the retention time and peak width increased for all proteins as the AAm content was increased. While a reduction in resolution between these proteins could simply be the result of an increase in surface hydrophilicity due to the increase in AAm content, given these analytes are separated based on hydrophobic interactions, this is inconsistent with the longer retention time observed.
The increase in retention time could be related to a decrease in void and window size observed for these poly(HIPE)s when the AAm content was increased from 0 wt% to 0.1 wt%. Smaller voids and windows often correlate to increased surface areas and therefore a potentially greater interaction of the analytes with the column surface. This could also explain the broader nature of these peaks, as analytes that spend more time within the column naturally have broader peaks. However, when prepared in capillary format the broader nature of the voids and windows resulted in the difference between them not being as significant as when prepared in bulk (Table 1). Another more plausible explanation is column bed heterogeneity.
As mentioned the 0 wt% AAm (AIBN) poly(HIPE) was not permeable under the pressures investigated and so its chromatographic performance could not be evaluated. However, both 0.1 wt% AAm (AIBN) and 1 wt% AAm (AIBN) exhibited a significantly improved chromatographic separation in contrast to that achieved with 0 wt% AAm (KPS). For these columns baseline resolution of these proteins was readily achieved, in particular when using a shallower solvent gradient (Fig. S7†). The peaks corresponding to lysozyme and α-chymotrypsinogen A also appeared narrower and more Gaussian in nature, consistent with columns that possessed greater homogeneity. Of these, 1 wt% AAm (AIBN) appeared to have the most symmetrical peaks.
The reduction in rear tailing of these peaks could also have been related to the incorporation of AAm onto the surface of these poly(HIPE)s, which reduced non-specific interactions as a result of a potential increase in surface hydrophilicity. However the retention times were identical to that of the 0 wt% AAm (KPS) column, under these conditions, suggesting they possessed similar surface hydrophobicities. This could also have been a result of the different porous morphologies observed for these columns, but if this were the case different retention times would also have been expected. It therefore appears that the inclusion of AAm, which is acting as a co-surfactant, has resulted in poly(HIPE)s with increased column bed homogeneity, in the case of the columns prepared with AIBN, which has resulted in a significantly improved separation of these proteins.
While the inclusion of AAm has resulted in improved RPLC performance, in some cases, its incorporation may also result in an increase in the surface hydrophilicity of these materials. Hydrophilic monoliths are important for extending the range of analytes that can be analysed and are often applied for life science applications such as metabolomics.58 For example, AAm-based monoliths have previously been utilised for the separation of polar compounds, using hydrophilic interactions between the analytes and the monolithic surface.59,60 Here, polar analytes are retained more strongly in high percentages of organic solvent and are eluted more easily when the H2O content is increased.58
Interestingly, the 1 wt% AAm (KPS) column was found to exhibit hydrophilic character when high acetonitrile content was employed (Fig. S8†), in contrast to the 0 wt% AAm (KPS) material, which exhibited no retention for the test analyte (data not shown), suggesting its potential suitability for these applications. This increase in hydrophilic character may also allow for applications elsewhere, for example hydrophilic poly(2-ethylhexyl acrylate-co-DVB) poly(HIPE)s have recently been announced for use in sanitary napkins by P&G.45
Preparation of poly(HIPE)s grafted with PEGDA
Given the improvement in chromatographic performance observed for the separation of proteins by RPLC upon the inclusion of AAm, the inclusion of the weakly hydrophilic PEGDA (Mw 258) into the emulsion formulation was also investigated. This divinyl monomer is expected to partition more strongly between the internal and external phase and therefore may be a more efficient co-surfactant than AAm, which could further improve the homogeneity of the resulting columns. This was included at the same mol% as AAm, to allow representative comparisons to be achieved, and Fig. 1 shows the resulting bulk materials.Similar trends were observed to that of the inclusion of AAm, with an initial reduction in void size upon the inclusion of 0.4 wt% PEGDA (Table 1) for both initiators. This decrease in void size was consistent with PEGDA also acting as a co-surfactant. Again the material prepared with AIBN possessed a larger void size with a value of 4 ± 2 μm compared to 1.9 ± 0.5 μm for KPS. No significant alteration in the void size was observed upon increasing the PEGDA content to 4 wt% in the case of AIBN, with an average void size of 4 ± 1 μm. However the void size increased from 1.9 ± 0.5 to 4 ± 3 μm for KPS. In all cases the void size obtained was consistent with the average droplet size measured immediately after preparation (Table 1 and Fig. S19†), suggesting that minimal coalescence occurred. This also indicated that coalescence was unlikely to have been the origin of the increase in void size observed for 4 wt% PEGDA (KPS).
While the inclusion of PEGDA initially appears to reduce the droplet size, it is possible that an increase in its concentration in the internal phase has resulted in migration of the Span® 80 emulsifier into the external phase37 as it competes with PEGDA at the interface. Alternatively, the inclusion of water-soluble organics, such as PEGDA and AAm, is known to enhance the solubility of Span® 80 in the aqueous phase.61 Both explanations would result in a reduction in the amount of Span® 80 at the interface and result in droplets of larger size with a broader distribution, which was reflected in the void structure of the resulting poly(HIPE) for 4 wt% PEGDA (KPS). This was also observed by Gitli and Silverstein35 for their poly(HIPE) system with an initial reduction in void size upon the addition of small amounts of AAm, however as the AAm content was increased the void size also increased.
The same effect was not observed for AAm in this work but the percentage of AAm was not increased above 1 wt% (w.r.t. internal phase). The reason why this was observed for PEGDA, even though it was present at the same mol% as AAm, is potentially related to its weakly hydrophilic nature. For example, a higher proportion of PEGDA is expected to be located at the interface, in comparison to the highly hydrophilic AAm, which is primarily located in the internal phase. It is not clear though why an increase in the void size was not also observed for 4 wt% PEGDA (AIBN). In light of this discussion, it is also important to consider that the weakly hydrophilic nature of PEGDA would also result in a greater proportion partitioning into the external phase. This would result in a poly(Sty-co-DVB-co-PEGDA) poly(HIPE) as the base material. As such a proportion of the PEGDA initially incorporated into the internal phase may be buried within the backbone of the monolith and not grafted to the surface.62
The window sizes observed (Table 1) also followed the same trends as the void size, and the porosities were again consistent with the internal phase volume utilised for most samples. In the case of 4 wt% PEGDA (KPS) and 4 wt% PEGDA (AIBN) these were slightly higher than expected (Table 1) suggesting some degree of creaming may have occurred. The specific surface areas were again higher when KPS was utilised, in comparison to AIBN (Table 1).
In terms of the surface morphology, particles were observed for both 4 wt% PEGDA (KPS) and 4 wt% PEGDA (AIBN) suggesting a degree of phase inversion occurred for these samples. This could be related to the possible migration of Span® 80 to the internal phase, which potentially stabilises (in conjunction with PEGDA) droplets of external phase monomer within the internal phase, resulting in particle formation. Apart from the presence of these particles, both 4 wt% PEGDA (KPS) and 4 wt% PEGDA (AIBN) possessed a smooth void surface (Fig. 1), suggesting the absence of a hydrogel even when KPS was used as initiator. This may suggest that the incorporation of PEGDA occurred primarily in the external phase, however FTIR (Fig. S3†) clearly showed the characteristic carbonyl stretch at 1732 cm−1 and ether stretch at 1161 cm−1, suggesting the incorporation of PEGDA did occur to some degree at the surface. It is important to note that these bonds are also present in the emulsifier Span® 80, however these signals were noticeably absent in the case of 0 wt% PEGDA (KPS) and 0 wt% PEGDA (AIBN), suggesting their origin was indeed from PEGDA. Similarly to AAm, these signals were also absent for the lower concentration of PEGDA (0.4 wt%) for both initiators, consistent with a lower incorporation of PEGDA into the resulting poly(HIPE). The extent of homopolymerisation and the degree of co-polymerisation between the internal phase and external phase monomers also depends on their relative reactivity ratios and these values could also potentially account for the different behaviour observed, in comparison to when AAm was included in the internal phase.
When these emulsions were confined within (Fig. 2 and Table 1) or passed through the 150 μm i.d. capillaries (Fig. S4, S20 and Table S2†) no significant alteration in the average droplet and/or resulting average void and window size was observed. Additionally, no radial heterogeneity in terms of void size was observed within the capillaries (Fig. S5†). In terms of homogeneity, 0.4 wt% PEGDA (KPS) appeared to have the narrowest void size distribution of these materials with an average void size of 2 ± 1 μm. However, some larger voids were again observed in its structure, in particular near the capillary wall (Fig. 2). Similar voids were also observed for 0.4 wt% PEGDA (AIBN) and as discussed are most likely from the introduction of air bubbles. Large voids were also observed for 4 wt% PEGDA (KPS), but unlike the other materials, voids of intermediate size were observed in this case. This suggests that a degree of coalescence occurred for this material, resulting in the void size distribution observed.
While most of the poly(HIPE)s prepared in capillary format, when PEGDA was included, resembled that of their bulk counterparts, a large crater was observed for 4 wt% PEGDA (AIBN), which persisted throughout a large proportion of the column (Fig. 2). This capillary actually resembled that of a wall coated open-tubular column.26 Interestingly, the poly(HIPE) present within the capillary still had voids and windows consistent with those present in the bulk material, suggesting that coalescence and phase separation was not responsible for this craters presence. Upon inspection of the emulsion that was passed through the capillary and cured, a significantly large proportion of particles (∼500 nm in diameter) were observed in some regions (Fig. S6A†). These particles were also observed to be attached to the poly(HIPE) that was present within the capillary (Fig. S6B†). This indicates that a degree of phase inversion has occurred, and the shear number of particles present for the material obtained after being passed through the capillary suggests this was quite significant. It is likely that this also occurred within the capillary, resulting in the generation of a large number of particles which were unbound to the poly(HIPE)s surface. As a result a significant proportion of these particles were simply removed during the purification process, resulting in the large crater observed.
As discussed, this phase inversion could have been promoted by the migration of Span® 80 to the internal phase as a result of the increased PEGDA content. However, this did not occur in the case of the emulsion that was prepared and cured without being passed through the capillary. This suggests that this phase inversion was promoted by passing this emulsion through the narrow capillary inlet. The reason for this and why this did not occur in the case of 4 wt% PEGDA (KPS) is not clear.
Apart from 4 wt% PEGDA (AIBN) all other poly(HIPE)s had good attachment to the capillary wall (Fig. 2). In addition the back pressure was observed to vary linearly with flow rate (Fig. S15–S18†) again suggesting that no mechanical failure or compression occurred. The permeabilities (Table 2) were also consistent with the trends observed with the void and window size (Table 1), however the back pressure for 4 wt% PEGDA (AIBN) was very similar to the back pressure of the system. This was a result of its open-tubular nature, which made it difficult to accurately determine its permeability.
Chromatographic performance of poly(HIPE)s grafted with PEGDA
These columns were also evaluated for the RPLC separation of the same protein mixture and the chromatograms obtained are also shown in Fig. 3. Both 0.4 wt% PEGDA (KPS) and 0.4 wt% PEGDA (AIBN) offered a significantly improved chromatographic separation for these proteins in comparison to the control column prepared with no PEGDA. These separations had significantly improved peak shape, suggesting the inclusion of PEGDA had also improved the column bed homogeneity. The separation achieved with these columns was similar to that achieved with 1 wt% AAm (AIBN), except both 0.4 wt% PEGDA (KPS) and 0.4 wt% PEGDA (AIBN) were capable of separating these proteins from the peak corresponding to impurities from ribonuclease A and lysozyme (the first peak in the corresponding chromatograms in Fig. 3). Interestingly both columns had very similar chromatographic separations, even though their porous properties were significantly different (Table 1). This clearly highlights the importance of column homogeneity on the separation performance.Significant co-elution of these proteins was observed in the case of 4 wt% PEGDA (KPS), which was consistent with the broader void size distribution observed (Fig. 2 and Table 1). Surprisingly the separation obtained with 4 wt% PEGDA (AIBN) was good, with only slight co-elution between ribonuclease A and lysozyme, which suggests the applicability of these materials for open-tubular liquid chromatography.26 The use of a shallower gradient again resulted in improvements in these separations, which is demonstrated for some of these columns in Fig. S9.†
The improvement in chromatographic performance under RPLC conditions for the separation of proteins upon the inclusion of PEGDA in the emulsion formulation was further demonstrated with the separation of a more complex protein mixture using the 0.4 wt% PEGDA (KPS) column (Fig. 4). Here a reasonable separation was obtained where seven components of the mixture were clearly identifiable. This was in contrast to that of the column without PEGDA where significant co-elution between these components was observed.
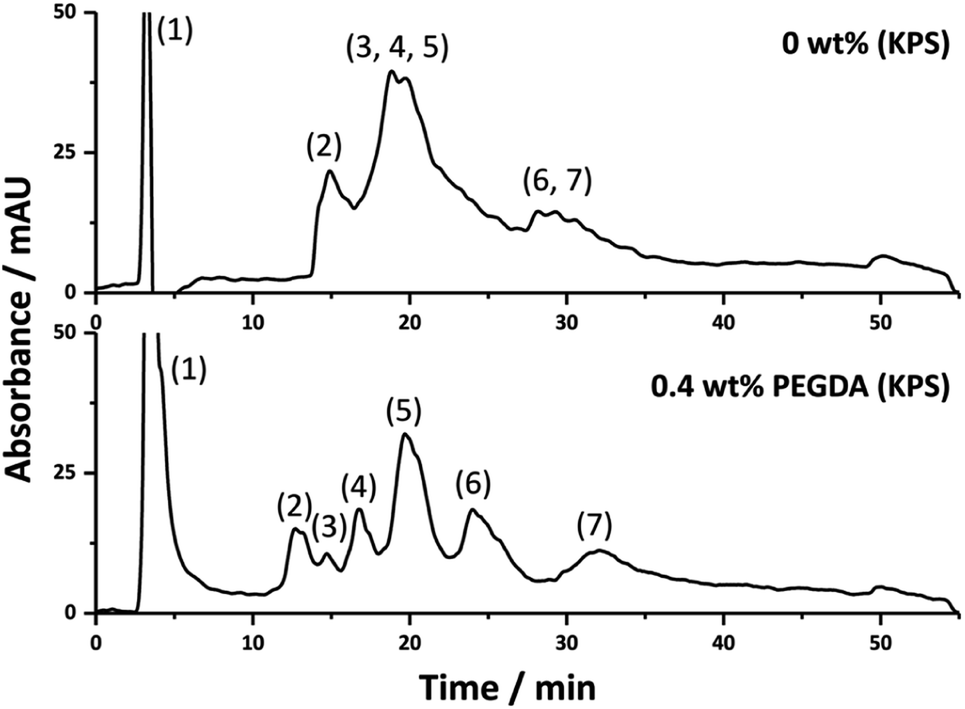 | ||
| Fig. 4 The separation of impurity from ribonuclease A and lysozyme (1), ribonuclease A (2) impurity from ovalbumin (3), cytochrome c (4), lysozyme (5), myoglobin (6) and ovalbumin (7) using columns prepared with different amounts of PEGDA. Conditions: 18 cm × 150 μm i.d. columns; eluent A was 0.1 vol% formic acid in Milli-Q H2O, and eluent B was 0.1 vol% formic acid in ACN; linear gradient 5 to 50% B in 40 min and then isocratic elution at 50% B for 5 min before returning to 5% B in 5 min; flow rate, 2.0 μL min−1; injection volume, 1 μL; protein concentration, 0.025 mg mL−1 except for ovalbumin which was 0.05 mg mL−1; UV detection at 214 nm. | ||
In addition to the improvement in RPLC, the inclusion of PEG chains onto the surface of these materials may also allow for applications as biocompatible stationary phases, capable of resisting the non-specific adsorption of proteins,63 or for stationary phases for hydrophobic interaction chromatography,43,64 which is a chromatographic mode that better preserves the proteins native conformation in contrast to RPLC and relies on the presence of both hydrophobic and hydrophilic patches on the surface.63
While the chromatographic performance has been significantly improved in this work, it is still inferior to that of conventional polymer monoliths where baseline separations of these and similar proteins can be readily achieved.65 However these materials possess significantly higher permeabilities, and as demonstrated in previous work,30 can allow for very rapid separations to be obtained. This is particularly important for applications requiring high-throughput, or when the pressure of the LC pump is limited, such as is the case with miniaturised platforms. In addition, some of these materials may also be beneficial for applications such as, and similar to, open-tubular-liquid chromatography, where they may offer improved capacities, or even for sample preparation.
Conclusions
In summary a series of poly(Sty-co-DVB)-based poly(HIPE)s were prepared by including the monomers AAm or PEGDA into the internal phase and emulsifying under high shear. It was found that both AAm and PEGDA acted as co-surfactants resulting in significantly improved column bed homogeneity when these poly(HIPE)s were prepared in capillary format. This resulted in significantly improved chromatographic performance for the separation of proteins by RPLC, where a poly(HIPE) grafted with PEGDA was capable of separating a more complex protein mixture, consisting of seven components. This highlights the benefit of including co-stabilisers in the emulsion formulation for obtaining columns with enhanced homogeneity.In addition, a poly(HIPE) grafted with AAm was found to be suitable for the retention of analytes through hydrophilic interactions. This demonstrated that the inclusion of monomers in the internal phase was also an appropriate method for the surface functionalisation of these materials. This route potentially allows for the preparation of poly(HIPE)s with improved homogeneity and tailored surface chemistry for various applications, by simply including monomers in the internal phase and optimising the monomer content and initiation location.
Author contributions
Christopher T. Desire: conceptualization, data curation, formal analysis, methodology, writing – original draft. R. Dario Arrua: conceptualization, methodology, supervision, writing – review and editing. Fotouh R. Mansour: conceptualization, methodology, writing – review and editing. Stefan A. F. Bon: conceptualization, writing – review and editing. Emily F. Hilder: conceptualization, supervision, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council's Discovery funding scheme (DP130101471). C. T. D. was the recipient of both an Endeavour Fellowship provided by the Australian Government and an Australian Government Research Training Program Scholarship. We gratefully acknowledge Dr Karsten Gömann and Dr Sandrin Feig, for their assistance with scanning electron microscopy, and Dr Thomas Rodemann for assistance with FTIR and elemental analysis, from the Central Science Laboratory (CSL), University of Tasmania.References
- S. Hjertén, J.-L. Liao and R. Zhang, J. Chromatogr. A, 1989, 473, 273–275 CrossRef.
- F. Svec and J. M. J. Fréchet, Anal. Chem., 1992, 64, 820–822 CrossRef CAS.
- C. Viklund, E. Pontén, B. Glad and K. Irgum, Chem. Mater., 1997, 9, 463–471 CrossRef CAS.
- G. Guiochon, J. Chromatogr. A, 2007, 1168, 101–168 CrossRef CAS PubMed.
- F. Svec and J. M. J. Fréchet, Ind. Eng. Chem. Res., 1999, 38, 34–48 CrossRef CAS.
- F. Svec and J. M. J. Fréchet, J. Chromatogr. A, 1995, 702, 89–95 CrossRef CAS PubMed.
- B. Gu, Y. Li and M. L. Lee, Anal. Chem., 2007, 79, 5848–5855 CrossRef CAS PubMed.
- C. T. Desire, R. D. Arrua, M. Talebi, N. A. Lacher and E. F. Hilder, J. Sep. Sci., 2013, 36, 2782–2792 CrossRef CAS PubMed.
- J. Krenkova, N. A. Lacher and F. Svec, Anal. Chem., 2010, 82, 8335–8341 CrossRef CAS PubMed.
- A. Premstaller, H. Oberacher and C. G. Huber, Anal. Chem., 2000, 72, 4386–4393 CrossRef CAS PubMed.
- H. Oberacher and C. G. Huber, Trends Anal. Chem., 2002, 21, 166–174 CrossRef CAS.
- M. Petro, F. Svec, I. Gitsov and J. M. J. Fréchet, Anal. Chem., 1996, 68, 315–321 CrossRef CAS PubMed.
- F. Svec and J. M. J. Fréchet, Chem. Mater., 1995, 7, 707–715 CrossRef CAS.
- C. Viklund, F. Svec and J. M. J. Fréchet, Chem. Mater., 1996, 8, 744–750 CrossRef CAS.
- H. Koku, R. S. Maier, K. J. Czymmek, M. R. Schure and A. M. Lenhoff, J. Chromatogr. A, 2011, 1218, 3466–3475 CrossRef CAS PubMed.
- T. Mullner, A. Zankel, C. Mayrhofer, H. Reingruber, A. Holtzel, Y. Lv, F. Svec and U. Tallarek, Langmuir, 2012, 28, 16733–16737 CrossRef PubMed.
- R. Dario Arrua and E. F. Hilder, RSC Adv., 2015, 5, 71131–71138 RSC.
- P. Krajnc, N. Leber, D. Štefanec, S. Kontrec and A. Podgornik, J. Chromatogr. A, 2005, 1065, 69–73 CrossRef CAS PubMed.
- C. Yao, L. Qi, H. Jia, P. Xin, G. Yang and Y. Chen, J. Mater. Chem., 2009, 19, 767–772 RSC.
- Y. Tunç, Ç. Gölgelioğlu, N. Hasirci, K. Ulubayram and A. Tuncel, J. Chromatogr. A, 2010, 1217, 1654–1659 CrossRef PubMed.
- S. Jerenec, M. Šimić, A. Savnik, A. Podgornik, M. Kolar, M. Turnšek and P. Krajnc, React. Funct. Polym., 2014, 78, 32–37 CrossRef CAS.
- S. Choudhury, L. Fitzhenry, B. White and D. Connolly, Materials, 2016, 9, 212–225 CrossRef PubMed.
- D. Yin, Y. Guan, H. Gu, Y. Jia and Q. zhang, RSC Adv., 2017, 7, 7303–7309 RSC.
- N. R. Cameron and D. C. Sherrington, Adv. Polym. Sci., 1996, 126, 162–214 CrossRef.
- S. D. Kimmins and N. R. Cameron, Adv. Funct. Mater., 2011, 21, 211–225 CrossRef CAS.
- S. Choudhury, D. Connolly and B. White, J. Appl. Polym. Sci., 2016, 133, 44237 CrossRef.
- C. Yao, L. Qi, G. Yang and F. Wang, J. Sep. Sci., 2010, 33, 475–483 CrossRef CAS PubMed.
- F. Du, L. Sun, X. Zhen, H. Nie, Y. Zheng, G. Ruan and J. Li, Anal. Bioanal. Chem., 2015, 407, 6071–6079 CrossRef CAS PubMed.
- J. Luo, Z. Huang, L. Liu, H. Wang, G. Ruan, C. Zhao and F. Du, J. Sep. Sci., 2021, 44, 169–187 CrossRef CAS PubMed.
- C. T. Desire, R. D. Arrua, F. R. Mansour, S. A. F. Bon and E. F. Hilder, RSC Adv., 2019, 9, 7301–7313 RSC.
- Y. Luo, A.-N. Wang and X. Gao, Soft Matter, 2012, 8, 7547–7551 RSC.
- M. Tebboth, A. Kogelbauer and A. Bismarck, Chem. Eng. Sci., 2015, 137, 786–795 CrossRef CAS.
- P. Viswanathan, D. W. Johnson, C. Hurley, N. R. Cameron and G. Battaglia, Macromolecules, 2014, 47, 7091–7098 CrossRef CAS.
- J. M. Williams, A. J. Gray and M. H. Wilkerson, Langmuir, 1990, 6, 437–444 CrossRef CAS.
- T. Gitli and M. S. Silverstein, Soft Matter, 2008, 4, 2475–2485 RSC.
- T. Rohr, E. F. Hilder, J. J. Donovan, F. Svec and J. M. J. Fréchet, Macromolecules, 2003, 36, 1677–1684 CrossRef CAS.
- R. J. Carnachan, M. Bokhari, S. A. Przyborski and N. R. Cameron, Soft Matter, 2006, 2, 608–616 RSC.
- A. Barbetta and N. R. Cameron, Macromolecules, 2004, 37, 3188–3201 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- J. A. Greig and D. C. Sherrington, Polymer, 1978, 19, 163–172 CrossRef CAS.
- C. T. Desire, A. Khodabandeh, T. L. Schiller, R. Wilson, R. D. Arrua, S. A. F. Bon and E. F. Hilder, Eur. Polym. J., 2018, 102, 56–67 CrossRef CAS.
- I. Nischang, F. Svec and J. M. Fréchet, J. Chromatogr. A, 2009, 1216, 2355–2361 CrossRef CAS PubMed.
- Y. Li, H. D. Tolley and M. L. Lee, J. Chromatogr. A, 2010, 1217, 4934–4945 CrossRef CAS PubMed.
- L. Geiser, S. Eeltink, F. Svec and J. M. Fréchet, J. Chromatogr. A, 2007, 1140, 140–146 CrossRef CAS PubMed.
- Q. Jiang, A. Menner and A. Bismarck, React. Funct. Polym., 2017, 114, 104–109 CrossRef CAS.
- S. Kovačič, K. Jeřabek and P. Krajnc, Macromol. Chem. Phys., 2011, 212, 2151–2158 CrossRef.
- A. S. Hayward, N. Sano, S. A. Przyborski and N. R. Cameron, Macromol. Rapid Commun., 2013, 34, 1844–1849 CrossRef CAS PubMed.
- T. Gitli and M. S. Silverstein, Polymer, 2011, 52, 107–115 CrossRef CAS.
- N. Cohen, D. C. Samoocha, D. David and M. S. Silverstein, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4369–4377 CrossRef CAS.
- C. Graillat, C. Pichot, A. Guyot and M. S. E. Aasser, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 427–449 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley & Sons, Inc, Hoboken, NJ, 4th edn, 1999 Search PubMed.
- I. Nischang, F. Svec and J. M. J. Fréchet, Anal. Chem., 2009, 81, 7390–7396 CrossRef CAS PubMed.
- A. Quell, B. d. Bergolis, W. Drenckhan and C. Stubenrauch, Macromolecules, 2016, 49, 5059–5067 CrossRef CAS.
- K. Mathieu, C. Jérôme and A. Debuigne, Polymer, 2016, 99, 157–165 CrossRef CAS.
- J. M. Williams and D. A. Wrobleski, Langmuir, 1988, 4, 656–662 CrossRef CAS.
- L. Trojer, S. H. Lubbad, C. P. Bisjak, W. Wieder and G. K. Bonn, J. Chromatogr. A, 2007, 1146, 216–224 CrossRef CAS PubMed.
- F. Svec, J. Sep. Sci., 2004, 27, 1419–1430 CrossRef CAS PubMed.
- Z. Jiang, N. W. Smith and Z. Liu, J. Chromatogr. A, 2011, 1218, 2350–2361 CrossRef CAS PubMed.
- R. Freitag, J. Chromatogr. A, 2004, 1033, 267–273 CrossRef CAS PubMed.
- Z. Jiang, N. W. Smith, P. D. Ferguson and M. R. Taylor, J. Sep. Sci., 2009, 32, 2544–2555 CrossRef CAS PubMed.
- J.-H. Zhu, F. Shao, Y.-H. Zhan, X.-L. Yan and B. Zhang, Colloids Surf., A, 2006, 290, 19–24 CrossRef CAS.
- F. Svec, J. Chromatogr. A, 2010, 1217, 902–924 CrossRef CAS PubMed.
- Y. Li and M. L. Lee, J. Sep. Sci., 2009, 32, 3369–3378 CrossRef CAS PubMed.
- Y. Li, H. D. Tolley and M. L. Lee, Anal. Chem., 2009, 81, 9416–9424 CrossRef CAS PubMed.
- Q. C. Wang, F. Svec and J. M. J. Fréchet, Anal. Chem., 1993, 65, 2243–2248 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07705h |
| This journal is © The Royal Society of Chemistry 2022 |