
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu(OAc)2 catalysed aerobic oxidation of aldehydes to nitriles under ligand-free conditions†
Asit Kumar Dasa,
Sneha Nandyb and
Sanjay Bhar *b
*b
aDepartment of Chemistry, Krishnath College, Berhampore, Murshidabad-742101, India
bDepartment of Chemistry, Organic Chemistry Section, Jadavpur University, Kolkata-700032, India. E-mail: sanjaybharin@yahoo.com; sanjay.bhar@jadavpuruniversity.in; Tel: +918697179547
First published on 4th February 2022
Abstract
An economically efficient and environmentally benign approach for the direct oxidative transformation of aldehydes to nitriles has been developed using commercially available non-toxic copper acetate as an inexpensive catalyst and ammonium acetate as the source of nitrogen in the presence of aerial oxygen as an eco-friendly oxidant under ligand-free conditions. The reactions were associated with high yield and various sensitive moieties like allyloxy, benzyloxy, t-butyldimethylsilyloxy, hetero-aryl, formyl, keto, chloro, bromo, methylenedioxy and cyano were well tolerated in the aforesaid method. The kinetic studies showed first order dependency on the aldehyde substrate in the reaction rate. The reaction was faster with the electron deficient aldehydes as confirmed by Hammett analysis. Moreover, the present oxidative method was effective on larger scales showing potential for industrial application.
Introduction
Nitrile is an important functional group which has been widely used for different organic transformations towards the synthesis of dyes, pigments, materials, polymers, natural products, agrochemical, and pharmaceuticals.1 Moreover, nitriles also serve as a recurrent pharmacophore in many commercially available drugs, such as Bicalutamide® (prostate cancer and breast cancer therapies), Citalopram® (antidepressant drug), Etravirine® (anti-HIV), Fadrozole® (oncolytic drug), Letrozole® (breast cancer therapy), and Periciazine® (anti-psychotic drug) and 5-lipoxygenase inhibitors have been recognized (Fig. 1).2 | ||
| Fig. 1 Some potent biologically active organonitrile drugs. | ||
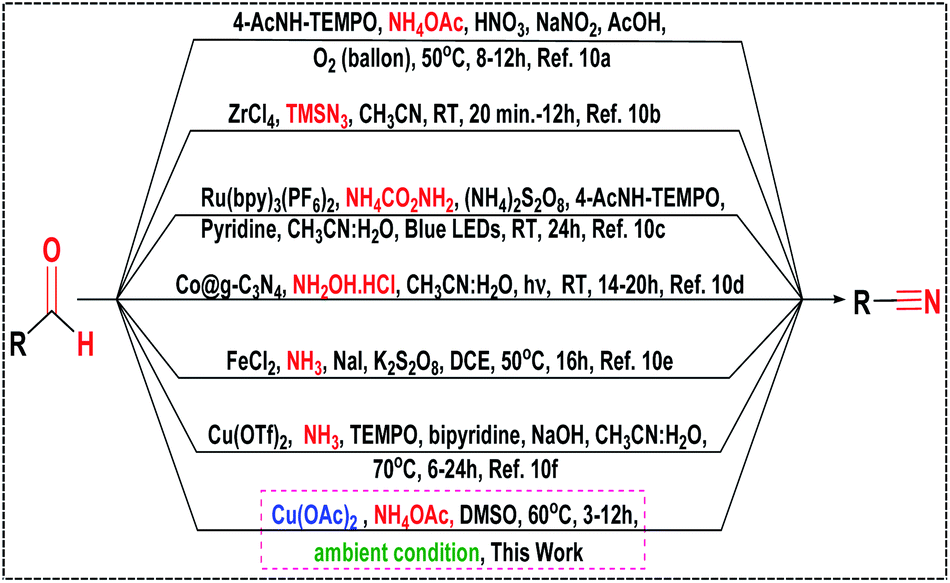
The classical methods for preparing aryl nitriles involve the Sandmeyer reaction3a–d of aromatic diazonium salts and the Rosenmund-von Braun reaction3e of aryl halides, which require stoichiometric amounts of highly toxic CuCN and harsh reaction conditions. Other alternative approaches for nitrile synthesis such as hydrocyanation of alkenes,4 Kolbe nitrile synthesis,5 methyl arenes,6 oxidative rearrangement of alkene,7a benzyl or allyl halides,7b ammoxidation of alcohols7c and cyanation of aryl halides8 were reported in the last few years, but most of these methods suffer limitations such as high temperature (>100 °C), use of harmful and expensive metal catalysts as well as toxic and corrosive reagents, requirement of capricious ligands, inert atmosphere, and poor functional group tolerance. Moreover, transition-metal free protocols such as trichloroisocyanuric acid (TCCA),9a tetrabutylammonium tribromide (TBATB),9b ceric ammonium nitrate (CAN),9c chloramine-T (CAT),9d TEMPO/HMDS/pyridine,9e TEMPO/KPF6/NaNO2/HMDS9f and SO2F2/Et3N9g have been documented. But still the requirements of highly sensitive and perilous reagents were an inevitable issue. Thus, the development of an alternative protocol for direct oxidative transformation of aldehydes to nitriles associated with some attributes like operational simplicity, ready accessibility of the substrates, cost-effectiveness as well as obviating the isolation of intermediates has received substantial interest in recent times. In this direction, several synthetic strategies using different nitrogen sources viz., NH4OAc,10a TMSN3,10b NH4CO2NH2,10c NH2OH·HCl,10d NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10e,f in the presence of various catalysts have been developed in the last few years (Scheme 1). However, despite the potential utility of the reported protocols listed in Scheme 1, applications of most of them remained limited due to the use of hazardous reagents, expensive ligands, requirement of additives (such as acids, bases and salts), laborious catalyst preparation and tedious work-up procedures which are less eco-compatible from the sustainable perspective.
10e,f in the presence of various catalysts have been developed in the last few years (Scheme 1). However, despite the potential utility of the reported protocols listed in Scheme 1, applications of most of them remained limited due to the use of hazardous reagents, expensive ligands, requirement of additives (such as acids, bases and salts), laborious catalyst preparation and tedious work-up procedures which are less eco-compatible from the sustainable perspective.
 | ||
| Scheme 1 Different strategies for the direct oxidative transformation of aldehydes to nitriles. | ||
Therefore, development of highly efficient strategy11 avoiding the use of toxic and expensive metal catalysts and utilizing the less hazardous and inexpensive reagents for the synthesis of nitriles is of great demand in the perspective of present environmental scenario. Of late, copper-catalyzed12 organic transformations has drawn tremendous interest because copper and its compounds are considerably more abundant, less toxic, cheaper in price and environmentally benign compared to other existing precious metal-based catalysts. In this pursuit, we report herein a mild, efficient and eco-compatible protocol13 for the direct oxidative transformation of aldehydes to nitriles using commercially available non-toxic copper acetate as an inexpensive catalyst and ammonium acetate as the nitrogen source under ambient conditions with a broad substrate scope and tolerance of various sensitive moieties during the reaction (Scheme 1).
Results and discussion
Firstly, we conducted a series of experiments to optimize the reaction conditions for the oxidative transformation of 4-methoxybenzaldehyde 1a to the corresponding nitrile 2a in the presence of different copper salts as well as various nitrogen sources and solvents. The results are presented in Table 1. The reaction did neither occur at all in the absence of any copper salt (entry 1) nor with CuSO4 in combination with (NH4)2SO4 in EtOH solvent (entry 5), the unreacted substrates were isolated intact. The conversion was little improved when the reaction was performed in the presence of CuSO4 with aqueous NH3 in DMSO medium (entry 2), CuSO4 with (NH4)2SO4 in DMSO and CH3CN medium (entries 3 and 4), CuCl2 with NH4Cl in DMSO and DMF solvent (entries 6 and 7) and Cu(NO3)2 with NH4NO3 in DMSO and CH3CN medium (entries 8 and 9). Interestingly the extent of conversion was increased to 73% when the reaction was studied in the presence of Cu(OAc)2 along with NH4OAc at 60 °C in CH3CN medium (entry 10). Surprisingly when the reaction was performed using Cu(OAc)2 along with NH4OAc in DMSO medium under ambient atmosphere, the extent of conversion was increased to 90% within a shorter reaction time (entry 11).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cu salt (mol%) | Nitrogen source (mmol) | Solvent (mL) | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (1.0 mmol), Cu salt (10 mol%), nitrogen source (1.5 mmol), solvent (3 mL), temperature (as indicated), under ambient condition.b Yield of isolated product.c The reaction was carried out under inert (argon) atmosphere. | ||||||
| 1 | — | NH4OAc | DMSO | 60 °C | 12 h | — |
| 2 | CuSO4 | Aq. NH3 | DMSO | 60 °C | 12 h | 28 |
| 3 | CuSO4 | (NH4)2SO4 | DMSO | 60 °C | 12 h | 47 |
| 4 | CuSO4 | (NH4)2SO4 | CH3CN | 60 °C | 12 h | 35 |
| 5 | CuSO4 | (NH4)2SO4 | EtOH | 60 °C | 12 h | — |
| 6 | CuCl2 | NH4Cl | DMSO | 60 °C | 12 h | 45 |
| 7 | CuCl2 | NH4Cl | DMF | 60 °C | 12 h | 49 |
| 8 | Cu(NO3)2 | NH4NO3 | DMSO | 60 °C | 12 h | 53 |
| 9 | Cu(NO3)2 | NH4NO3 | CH3CN | 60 °C | 12 h | 42 |
| 10 | Cu(OAc)2 | NH4OAc | CH3CN | 60 °C | 12 h | 73 |
| 11 | Cu(OAc)2 | NH4OAc | DMSO | 60 °C | 10 h/12 h | 90/91 |
| 12c | Cu(OAc)2 | NH4OAc | DMSO | 60 °C | 10 h/12 h | — |
| 13 | Cu(OAc)2 | NH4OAc | H2O | 60 °C | 10 h/12 h | 36/38 |
| 14 | Cu(OAc)2 | NH4OAc | EtOH | Reflux | 12 h/14 h | 51/54 |
| 15 | Cu(OAc)2 | NH4OAc | DCM | Reflux | 12 h/14 h | 48/49 |
| 16 | Cu(OAc)2 | HCOONH4 | DMSO | 60 °C | 8 h/12 h | — |
| 17 | Cu(OAc)2 | Aq. NH3 | DMSO | 60 °C | 10 h/12 h | 23/24 |
| 18 | CuO | NH4OAc | DMSO | 60 °C | 8 h/12 h | — |
| 19 | CuCl | NH4OAc | DMSO | 60 °C | 8 h/12 h | — |
| 20 | Cu(OAc)2·H2O | NH4OAc | DMSO | 60 °C | 12 h | 78 |

But, when the reaction was performed under an inert (Ar) atmosphere in the absence of aerial oxygen, no trace of nitrile 2a was detected in the reaction mixture, the substrate 1a remained intact (entry 12). This observation indicated the importance of atmospheric oxygen as the eco-friendly oxidant during the aforementioned transformation. Moreover, this observation also indicated that DMSO does not have any role as an oxidant towards this oxidative transformation. It is simply used as the solvent in the aforementioned protocol. The effective role of DMSO towards this reaction might be speculated to originate from the better solubilization of the organic substrate as well as ionic reagent and catalyst along with rendering some stabilization towards the polar intermediates through solvation. The yield was much less in the presence of Cu(OAc)2 with NH4OAc in H2O medium (entry 13). This reaction was less responsive in ethanol (EtOH) and dichloromethane (DCM) as the solvent even under reflux and a longer period of reaction time (entries 14 and 15). Interestingly, no reaction took place when ammonium formate (HCOONH4) was used as the nitrogen source instead of ammonium acetate (CH3COONH4) in the presence of copper acetate (entry 16). Yield was quite low in the presence of aqueous NH3 (entry 17). The inferior performance was observed in the case of CuO and CuCl (entries 18 and 19). The reaction with Cu(OAc)2 H2O in the presence of NH4OAc produces the desired nitrile with 78% yield (entry 20) which is lower than the yield obtained with Cu(OAc)2 (91%, entry 11). Among the screened copper salts and nitrogen sources, Cu(OAc)2 was the most effective catalyst and NH4OAc was the best option as the nitrogen source. Less toxicity, good stability and cheaper price of ammonium acetate (NH4OAc) were found to be good attributes to be a better alternative nitrogen source of NH3. Therefore, the conditions, as delineated in entry 11, have been chosen as the optimized reaction condition for further studies.
To explore the substrate scope and limitation of this oxidative protocol, a systematic investigation on all kinds of aromatic, heterocyclic, naphthyl, and aliphatic aldehydes 1 was carried out under the optimized reaction condition to obtain the corresponding nitriles 2 (Scheme 2). The results are furnished in Table 2.
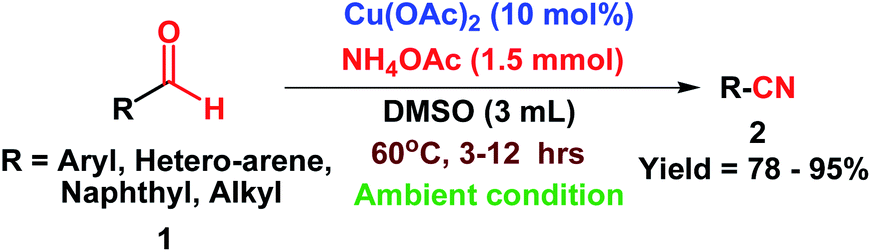 | ||
| Scheme 2 Cu(OAc)2 catalyzed direct oxidative transformations of aldehydes to nitriles. | ||
| Entry | Substrate | Product | Time (h) | Yieldd (%) |
|---|---|---|---|---|
| a Reaction conditions: 1a (1.0 mmol), Cu(OAc)2 (10 mol%), NH4OAc (1.5 mmol), DMSO (3 mL), 60 °C, under ambient atmosphere.b NH4OAc (1.5 mmol).c NH4OAc (3.0 mmol) were used for 1 mmol of substrate.d Yield of isolated and purified product. | ||||
| Synthesis of benzonitriles bearing electron-donating substituents | ||||
| 1 |  |
 |
10 | 90 |
| 2 |  |
 |
10 | 91 |
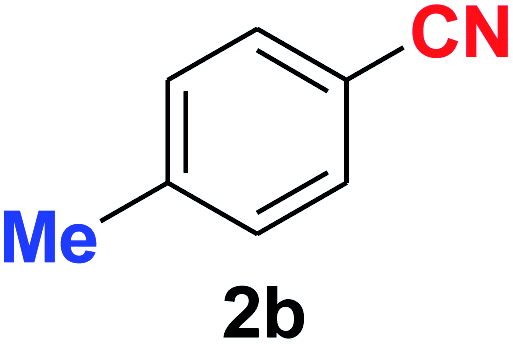
| 3 | 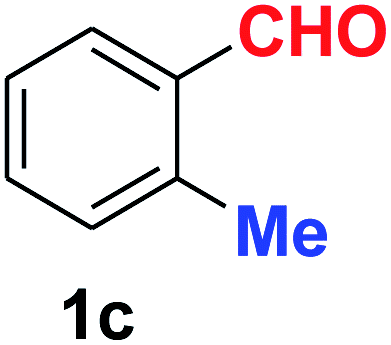 |
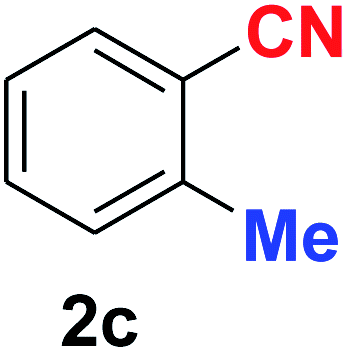 |
10 | 89 |
| 4 |  |
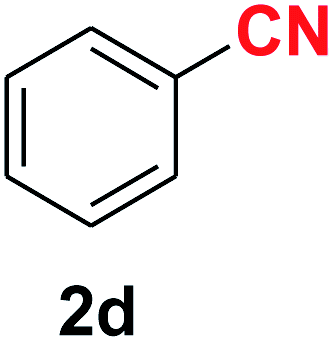 |
7.5 | 90 |
| 5 | 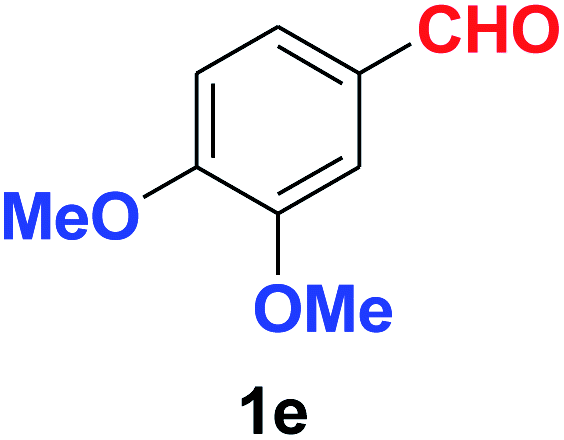 |
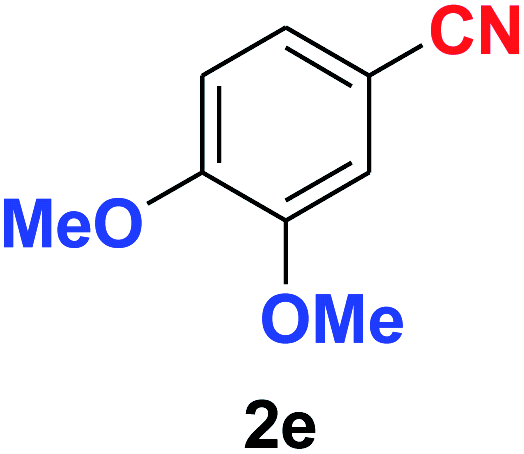 |
10 | 90 |
|
||||
| Synthesis of hydroxy functionalized benzonitriles | ||||
| 6 | 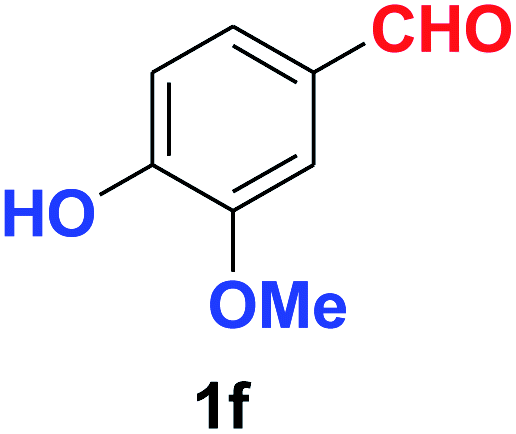 |
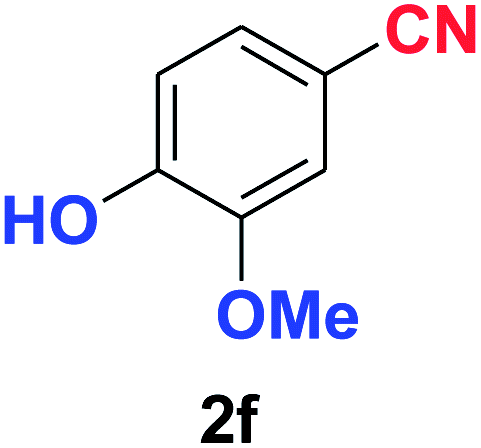 |
10 | 89 |
| 7 | 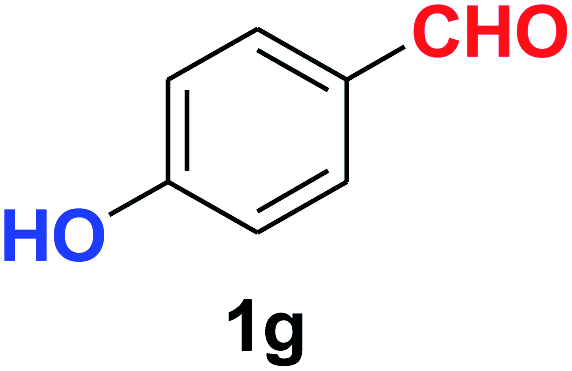 |
 |
10 | 87 |
| 8 | 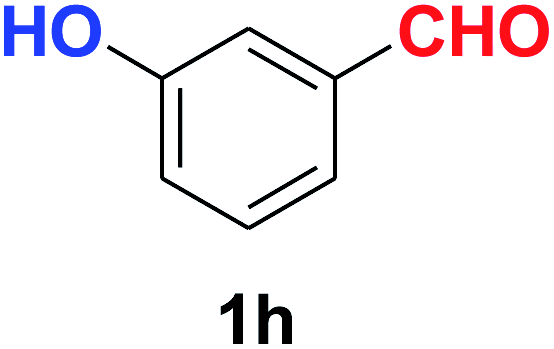 |
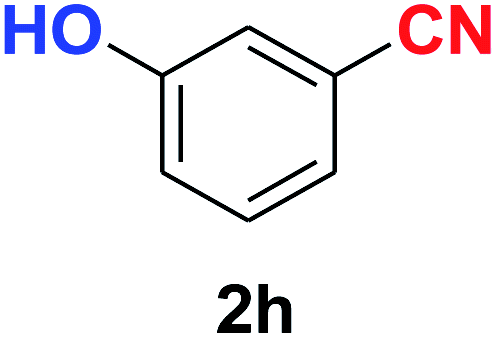 |
10 | 89 |
| 9 | 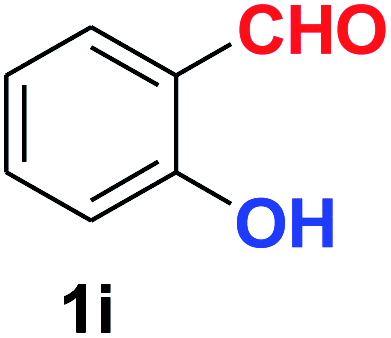 |
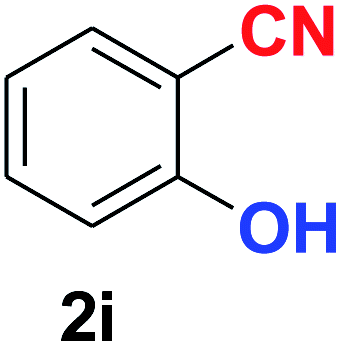 |
10 | 88 |
|
||||
| Synthesis of amino functionalized benzonitriles | ||||
| 10 | 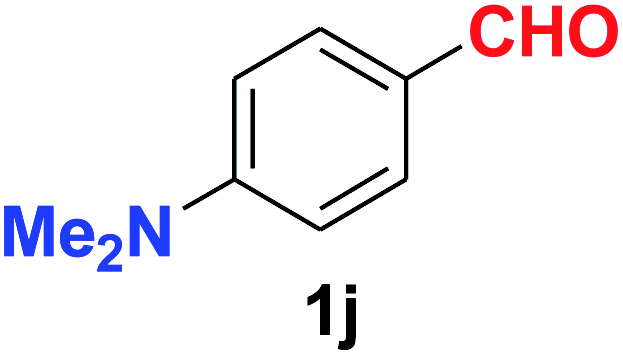 |
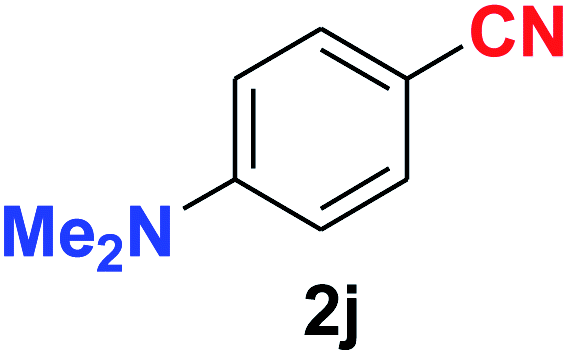 |
9.0 | 88 |
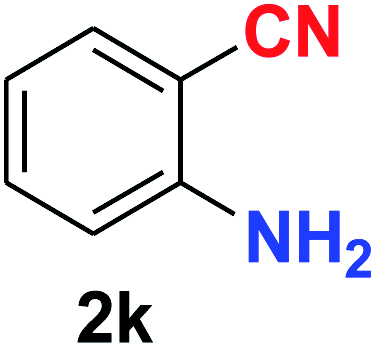
| 11 |  |
 |
9.0 | 89 |
|
||||
| Synthesis of halogenated benzonitriles | ||||
| 12 | 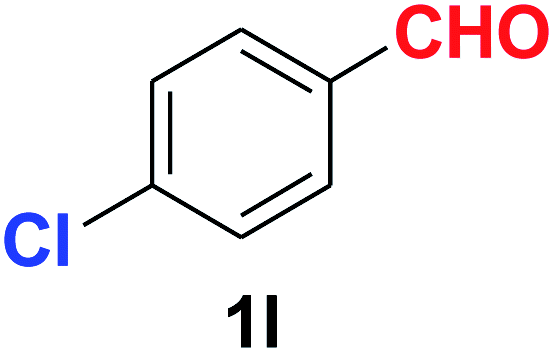 |
 |
6.0 | 93 |
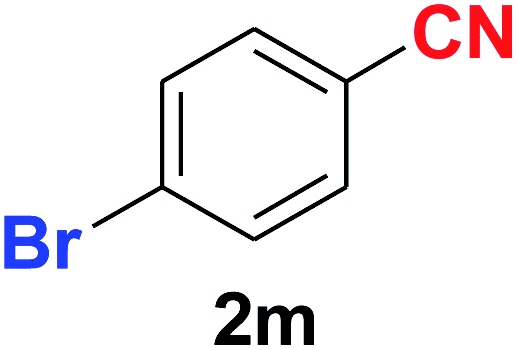
| 13 | 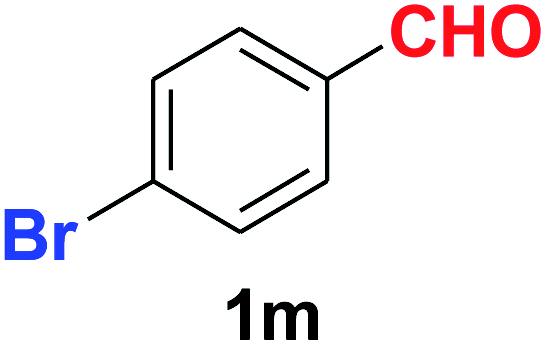 |
 |
6.0 | 91 |
|
||||
| Synthesis of nitro functionalized benzonitriles | ||||
| 14 | 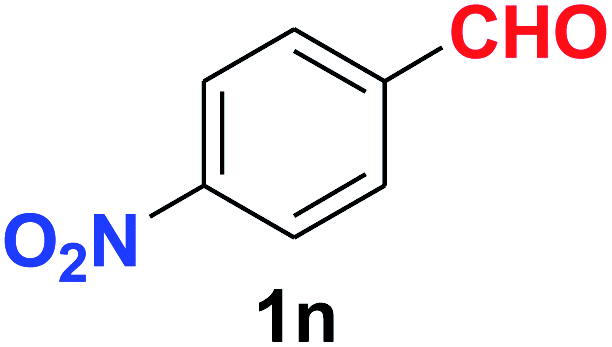 |
 |
3.0 | 95 |
| 15 | 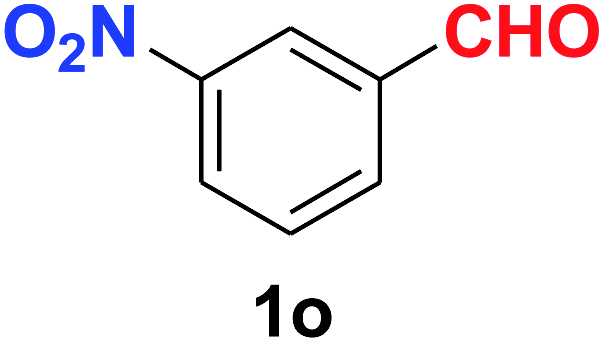 |
 |
3.0 | 94 |
|
||||
| Selectivity towards the synthesis of nitriles | ||||
| 16b | 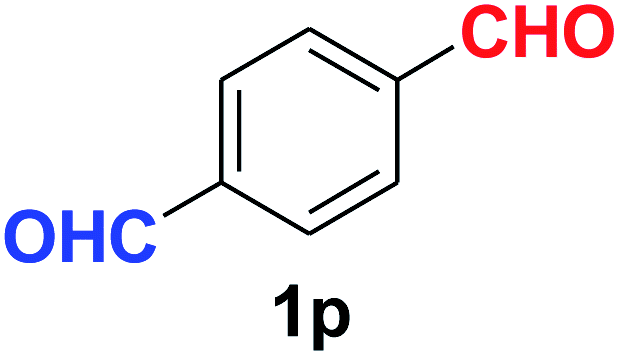 |
 |
6.0 | 88 |
| 17c | 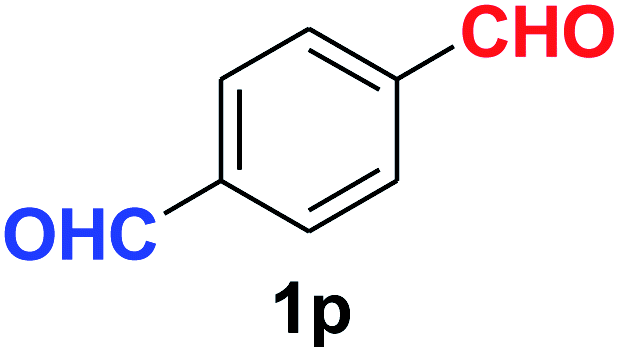 |
 |
6.0 | 87 |
| 18 | 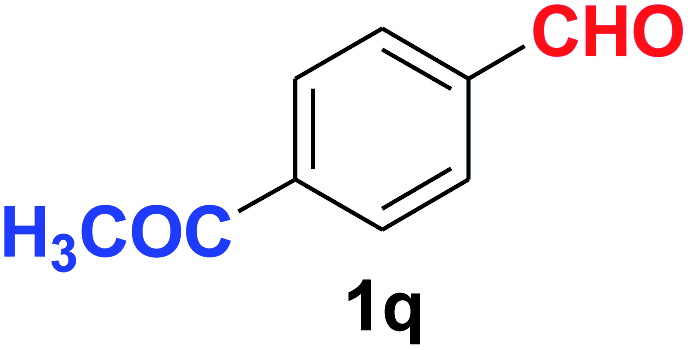 |
 |
6.0 | 88 |
|
||||
| Synthesis of nitriles bearing naphthyl, methylenedioxy and alkenyl moiety | ||||
| 19 | 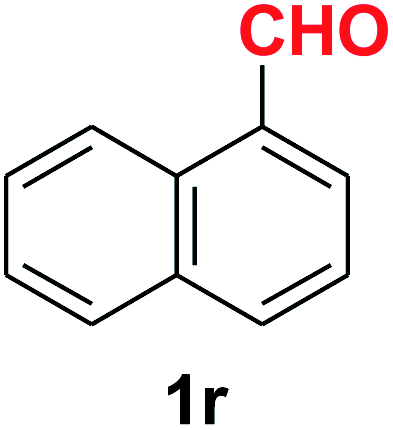 |
 |
11 | 86 |
| 20 | 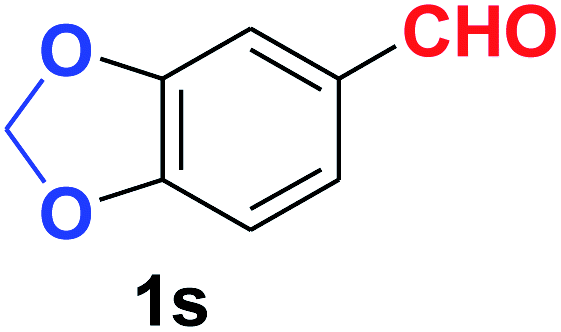 |
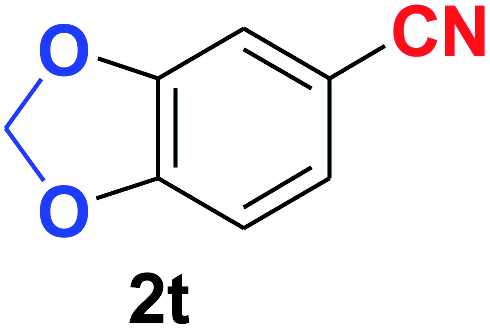 |
10 | 88 |
| 21 | 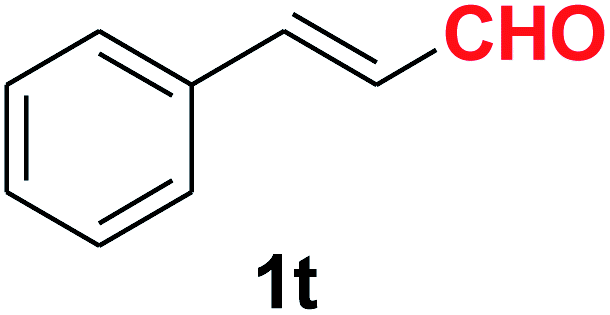 |
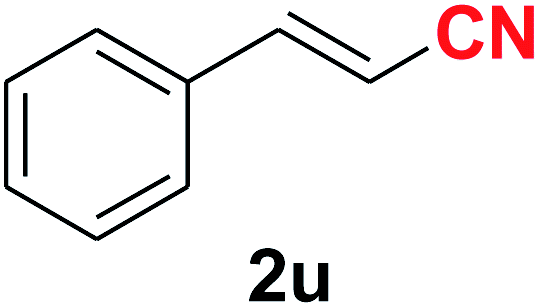 |
10 | 83 |
|
||||
| Synthesis of acid sensitive heterocyclic nitriles | ||||
| 22 | 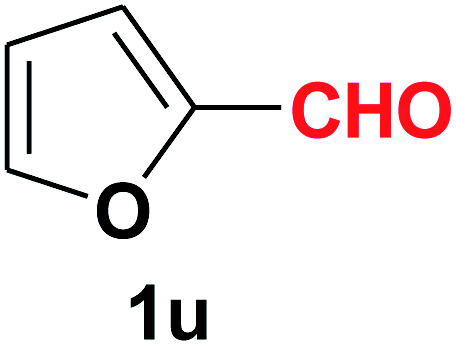 |
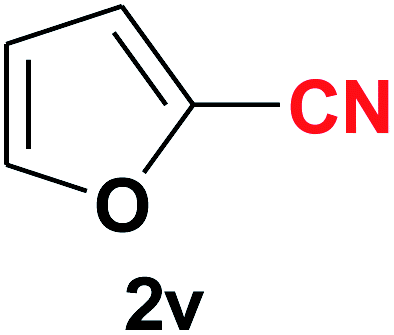 |
10 | 79 |
| 23 | 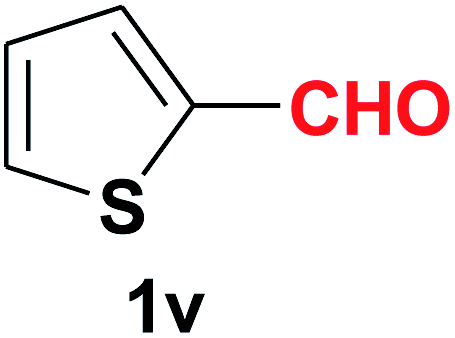 |
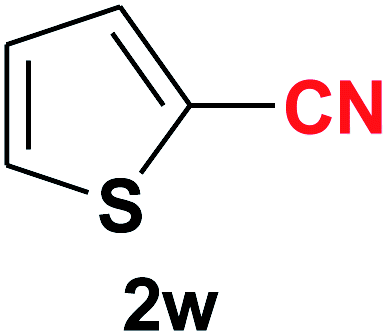 |
10 | 81 |
| 24 | 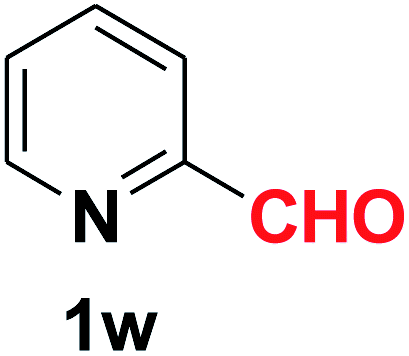 |
 |
10 | 83 |
| 25 |  |
 |
10 | 80 |
|
||||
| Synthesis of highly vulnerable nitriles | ||||
| 26 | 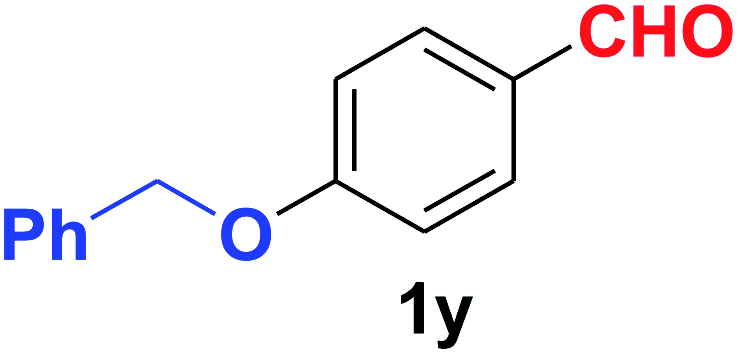 |
 |
10 | 86 |
| 27 | 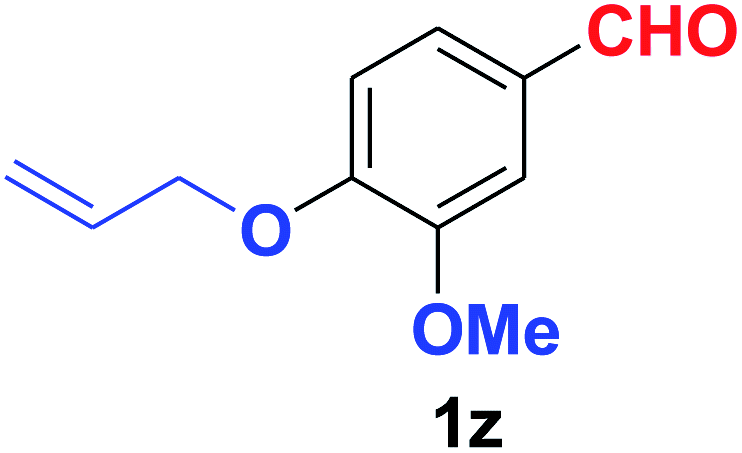 |
 |
10 | 84 |
| 28 | 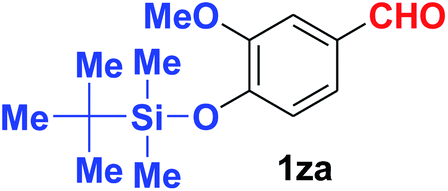 |
 |
10 | 85 |
|
||||
| Synthesis of aliphatic nitriles | ||||
| 29 |  |
 |
12 | 78 |
| 30 |  |
 |
12 | 79 |
| 31 | 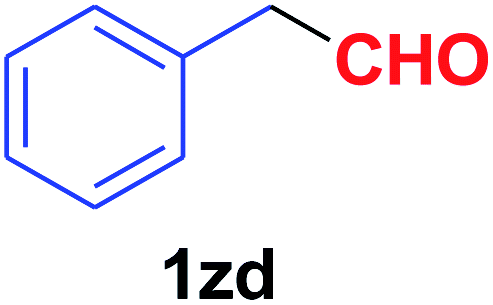 |
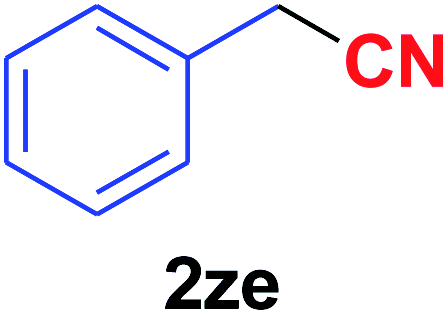 |
12 | 84 |
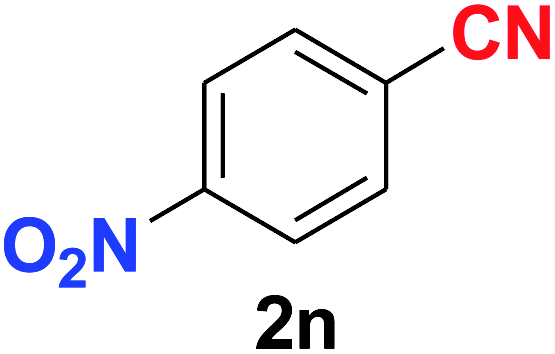
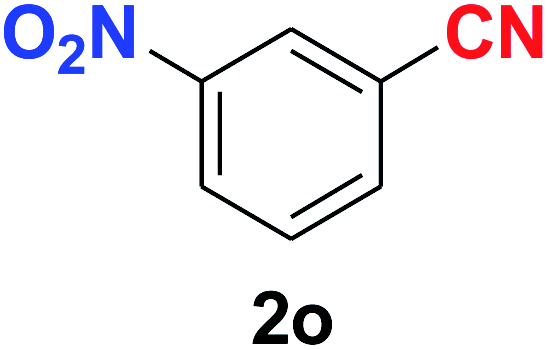
As evident from Table 2, benzaldehyde as well as other aryl aldehydes bearing electron-donating substituents (1a–e) showed excellent reactivity and furnished the products (2a–e) in high yields (entries 1–5). Aryl aldehydes bearing phenolic –OH group were equally efficient under the optimized reaction condition to produce the products 2f–i in good yield (entries 6–9). Deactivated aromatic aldehydes such as 4-N,N-dimethylaminobenzaldehyde and 2-aminobenzaldehyde afforded the corresponding nitriles 2j and 2k under the present reaction condition with 88% and 89% yield respectively (entries 10 and 11). This protocol was also effective for the substrates bearing halogen which produced the nitriles 2l and 2m in good yield within a shorter reaction time without any dehalogenated product (entries 12 and 13). Aldehydes with electron-withdrawing groups (–NO2) at m- and p-positions underwent efficient transformation to the corresponding nitriles 2n and 2o with 95% and 94% yields within 3 hours (entries 14 and 15).
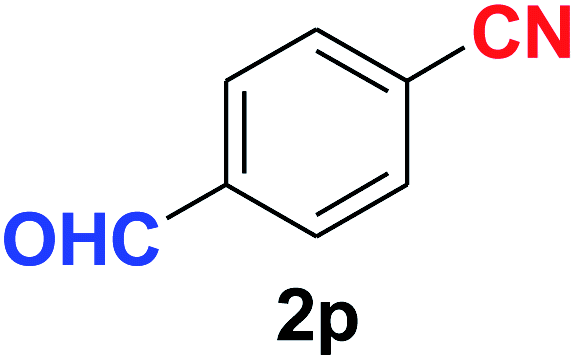
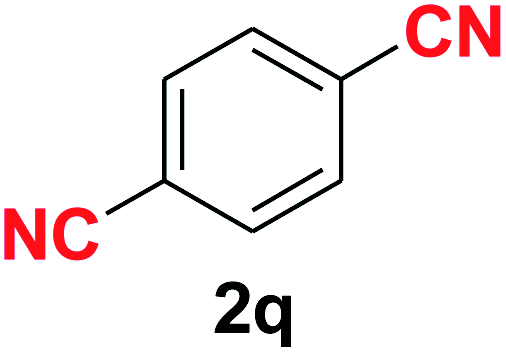
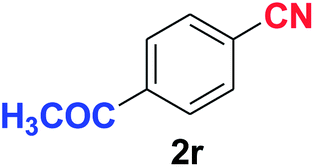
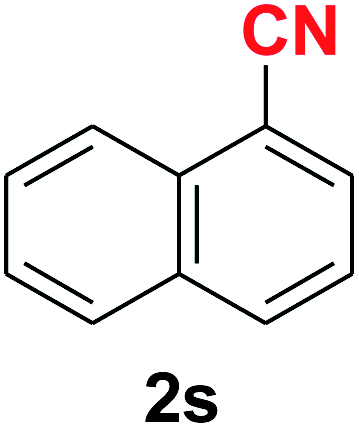
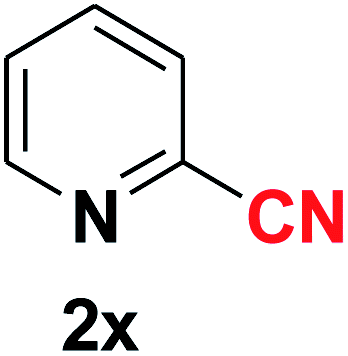
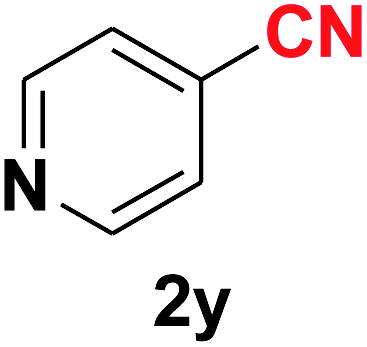
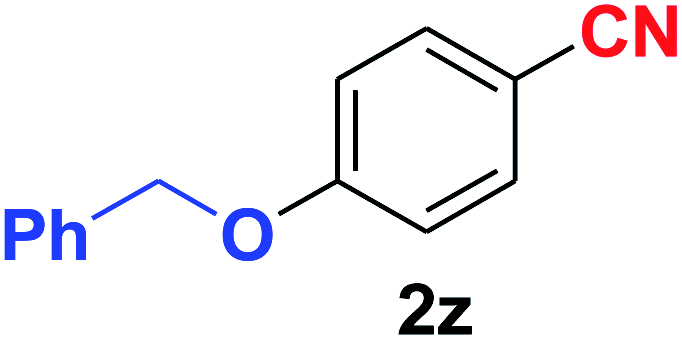
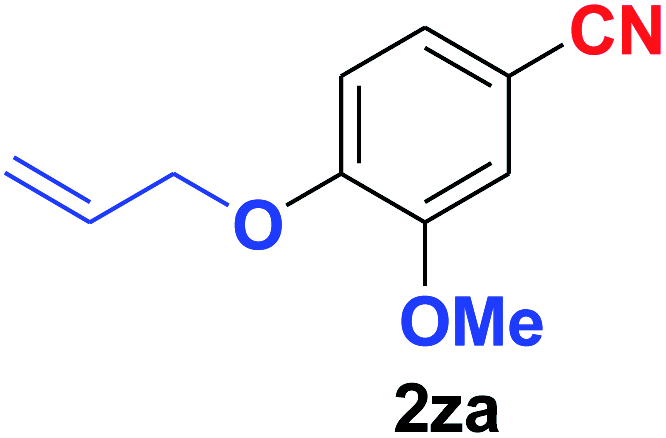
Quite interestingly, terephthaldehyde (1p) was converted to the 4-formylbenzonitrile (2p) and terephthalonitrile (2q) in 88% and 87% yield using 1.5 mmol and 3.0 mmol of ammonium acetate respectively (entries 16 and 17) as the source of nitrogen with respect to 1 mmol of substrate. The structure of 2p was substantiated by the singlet at δ 10.10 (due to –CHO) along with two doublets at δ 7.98 (due to aromatic protons ortho to –CHO) and at δ 7.84 (due to aromatic protons ortho to –CN). In the 13C NMR spectrum of 2p, simultaneous occurrence of two signals at δ 190.6 and δ 117.6 proves the co-existence of –CHO and –CN groups respectively. 4-Formylbenzonitrile (2p) furnished terephthalonitrile (2q) in 85% yield using 1.5 mmol of ammonium acetate. The formation of 2q was confirmed by the presence of only one singlet due to chemically equivalent aromatic hydrogens at δ 7.52 in its 1H NMR spectrum as well as from the signal (at δ 118.2) specific for –CN in the 13C NMR spectrum. This regioselectivity is an extremely important attribute of the present protocol in contrast to many reported methods where no such selectivity was observed.9d,f,10b,c,f We have also investigated the reaction using terephthaldehyde (1p) (1 mmol) and ammonium acetate (1.5 mmol) under oxygen atmosphere, the product 2p was obtained exclusively with 86% yield within 4 h without formation of any terephthalonitrile (2q) product. This observation further suggested the importance of oxygen as the eco-friendly oxidant as well as the selective formation of nitrile product during the aforementioned transformation. However, we restricted ourselves to the use of aerial oxygen for the entire study due to procedural simplicity involving the ambient atmosphere albeit longer reaction time for comparable conversion. 4-Acetylbenzaldehyde reacted smoothly to furnished 4-acetylbenzonitrile 2r with 88% yield within 6 hours (entry 18). Therefore, it can be concluded that the reaction was highly selective for aldehyde. The method was also successful for 2s containing naphthyl moiety with satisfactory yield within 11 hours (entry 19). Hydrolyzable groups like methylenedioxy in 2t also survived under the aforesaid protocol (entry 20). This is not commonly observed in some literature reports.9d,g,10b–f The present method was extended towards the efficient synthesis of α,β-unsaturated nitrile 2u (entry 21). Acid-sensitive electron-rich as well as electron-deficient heteroaromatic moieties also survived during this reaction (2v–y) which paved the way towards the construction of important molecular skeletons densely loaded with heterocycles in satisfactory yields (entries 22–25). It is extremely important to note the fact that highly vulnerable groups like O-benzyl, O-allyl, and O-t-butylsilyl were also tolerated under the optimized reaction condition to furnish to 2z, 2za, and 2zb respectively with good yields (entries 26–28). This is not commonly observed in some literature reports.9d–g,10a–f Furthermore, aliphatic nitriles 2zc, 2zd, and 2ze were also produced quite efficiently during a longer period under the aforesaid protocol (entries 29–31) from the corresponding aliphatic aldehydes (1zb–zd).
We next performed the kinetic experiments with the aforesaid protocol in order to determine the order of the reaction. Therefore, two identical experiments were carried out following the general procedure varying only the concentration of 4-methoxybenzaldehyde 1a (Table 3).
| Run | 1a | NH4OAc | Cu(OAc)2 | DMSO |
|---|---|---|---|---|
| Run 1 | 1 mmol | 1.5 mmol | 10 mol% | 3 mL |
| Run 2 | 2 mmol | 1.5 mmol | 10 mol% | 3 mL |
The initial rate of the reaction for different run was calculated to determine the order with respect to aldehyde 1a. The kinetic studies showed that the reaction rate depends on the concentration of 4-methoxybenzaldehyde 1a only (Fig. 2). Therefore, the aforesaid oxidative protocol follows first order kinetics (see ESI†).
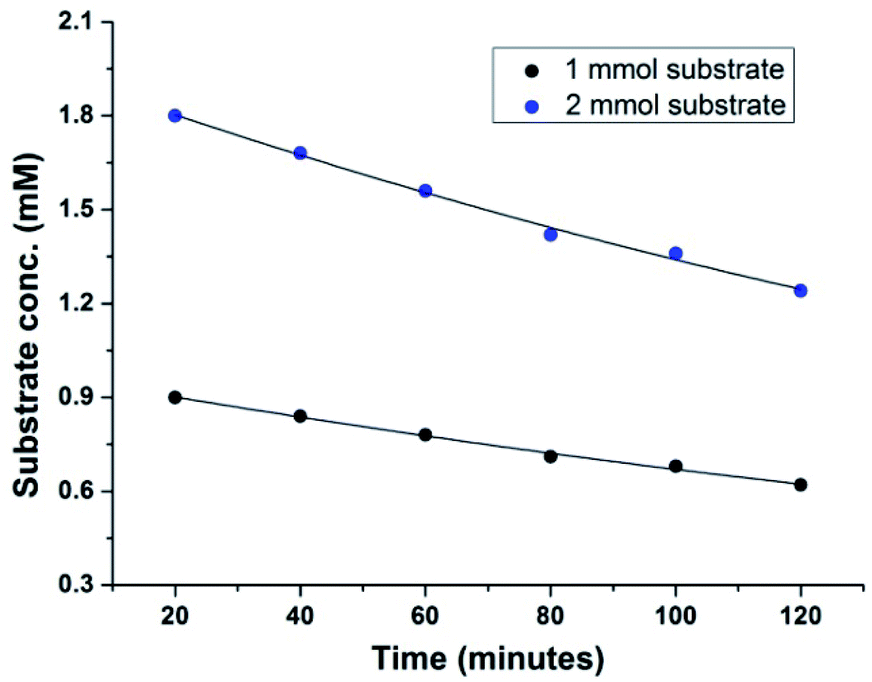 | ||
| Fig. 2 Dependence of the initial rate of the reaction on [4-methoxybenzaldehyde] using Cu(OAc)2 (10 mol%), NH4OAc (1.5 mmol), DMSO (3 mL), 60 °C, under ambient atmosphere. | ||
Table 2 demonstrated that both electron donating as well as electron withdrawing substituents showed an excellent reactivity and produced the desired products in excellent yield with different time intervals. Electronic effect was noted in this direct oxidative transformation of aldehydes to nitriles.
Therefore, kinetic experiments were carried out using several electronically disparate benzaldehydes following the general procedure (Fig. 3). It was evident from Fig. 3 that the reactions with electron withdrawing substituent were faster than with electron donating substituent and better conversion was achieved within shorter reaction time in the former case. It was also evident that the rate of the reaction with 4-nitrobenzaldehyde was nine times faster than with 4-methoxybenzaldehyde. Therefore, Hammett analysis (Table 4) was carried out using various substituted benzaldehydes under the optimized reaction conditions. A very good linear relationship was observed when relative rates [log(kX/kH)] with these substituted benzaldehydes were plotted against the substituent constant (σ) (Fig. 4). It was also observed that a positive ρ value of +0.95 and the reactivity sequence: p-NO2 > p-Cl > p-H > p-Me > p-OMe for this oxidative protocol. This observation further suggested that the electron-withdrawing substituent should enhance the reaction and the results were consistent with the reactivity of the substrates reported in Table 2.
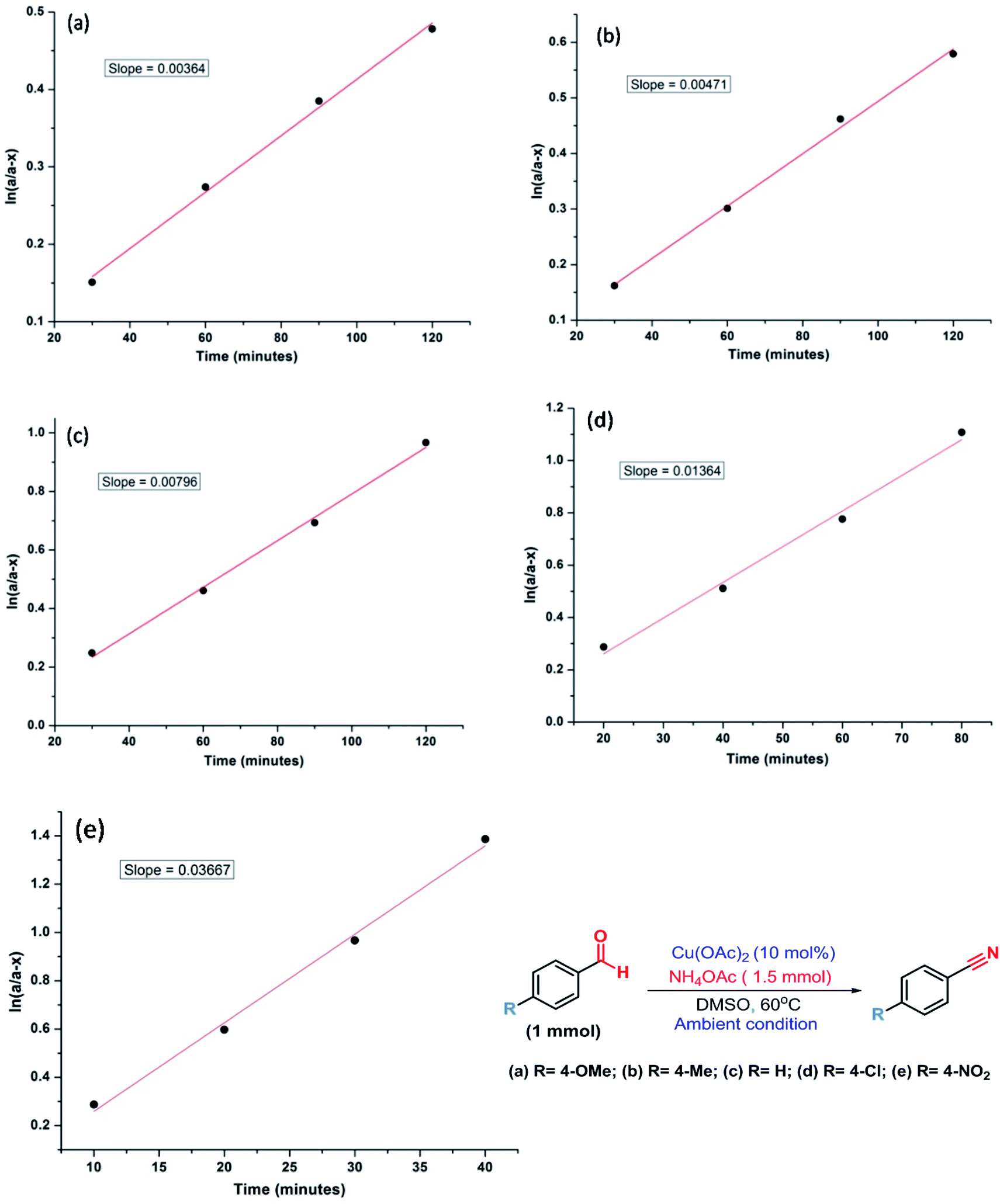 | ||
| Fig. 3 Determination of rate constant for the electronically disparate aldehydes during the synthesis of nitriles (a–e). | ||
| Substrate | k × 10−4 (min−1) | kX/kH | log(kX/kH) |
σP | ρ |
|---|---|---|---|---|---|
| 4-Methoxybenzaldehyde | 36.4 | 0.457 | −0.340 | −0.268 | +0.95 |
| 4-Methylbenzaldehyde | 47.1 | 0.591 | −0.228 | −0.170 | |
| Benzaldehyde | 79.6 | 1 | 0 | 0 | |
| 4-Chlorobenzaldehyde | 136.4 | 1.713 | 0.234 | 0.230 | |
| 4-Nitrobenzaldehyde | 366.7 | 4.606 | 0.663 | 0.780 |
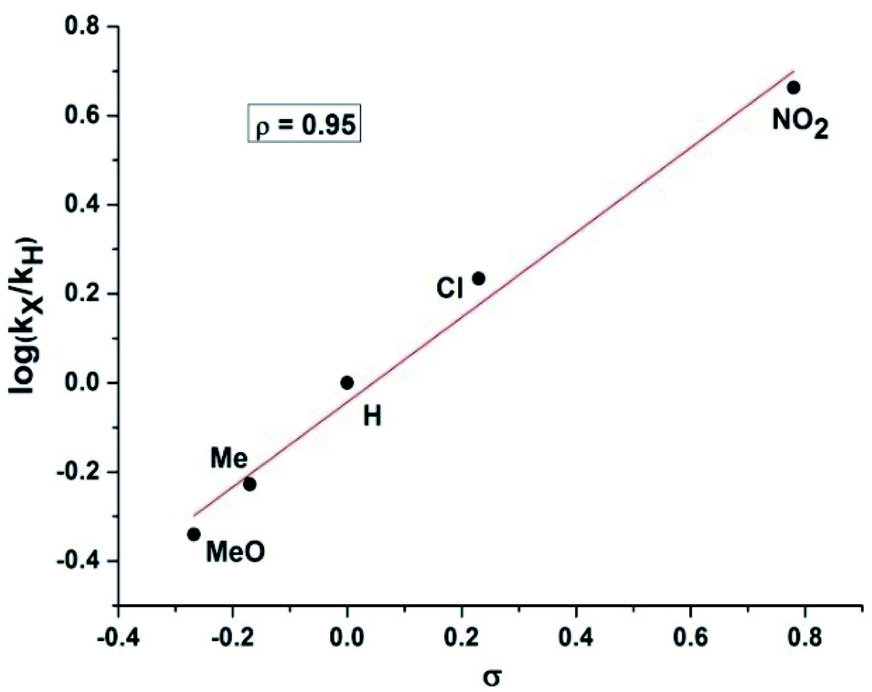 | ||
| Fig. 4 Hammett analysis of electronically disparate aldehydes for the direct synthesis of nitriles from aldehydes using standard reaction conditions. | ||
Based on the aforesaid investigations and literature precedence,10a,14a–c a plausible mechanistic pathway for this oxidative transformation is depicted in Scheme 3. At the outset, Cu(OAc)2 activates the carbonyl carbon to react with NH4OAc to form aldimine intermediate (A). Then the unstable aldimine intermediate (A) reacts with Cu(OAc)2 to form the iminylcuprate intermediate (B), which on subsequent oxidation forms the corresponding nitrile 2 with the liberation of CuOAc which further oxidized to Cu(OAc)2 in the presence of aerial oxygen. Here, Cu(OAc)2 serving as a Lewis acid and aerial oxygen acts as an eco-friendly oxidant towards this oxidative transformation.
 | ||
| Scheme 3 Plausible mechanism for the oxidative transformation of aldehydes to nitriles. | ||
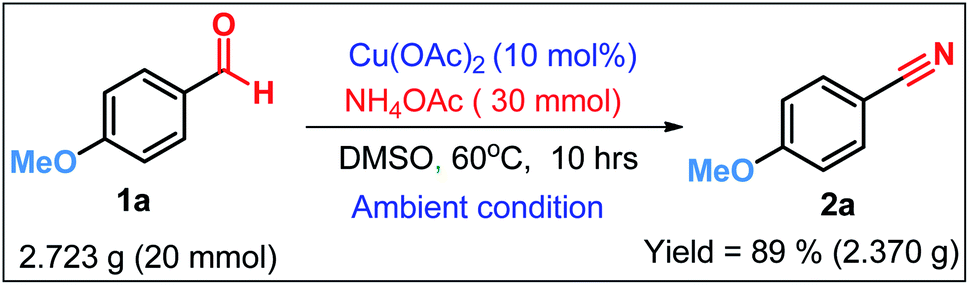
To ensure the synthetic scalability and practical applicability of our newly developed oxidative protocol, a gram scale reaction of 4-methoxybenzaldehyde 1a was performed (Scheme 4) the outcome of which was almost similar as that in the small scale reaction. The reaction mixture was extracted with EtOAc and the crude was further purified by column chromatography on a short column of silica gel using 1–5% ethyl acetate–hexane as eluent to obtain 2a. Therefore, Cu(OAc)2/NH4OAc catalyzed oxidative protocol can be readily scaled up to gram-scale, which bears a significant prospect for industrial application.
 | ||
| Scheme 4 Gram scale applicability of Cu(OAc)2/NH4OAc catalyzed oxidative protocol. | ||
Conclusions
We have developed a mild, operationally simple, cost-effective and eco-friendly protocol for the direct oxidative conversion of aldehydes to nitriles using commercially available relatively less toxic copper acetate as an inexpensive catalyst and ammonium acetate as the source of nitrogen in the presence of environmentally benign aerial oxygen as an eco-friendly oxidant under ligand-free and base-free condition. The kinetic experiments showed the first-order dependence of the substrate aldehyde towards the reaction rate. Moreover, Hammett analysis confirmed that the reaction was faster with the electron-deficient aldehydes. The synthetic utility and practical applicability of this newly developed protocol were demonstrated through a scale-up experiment. The salient features of the present protocol are procedural simplicity, ready accessibility and lower toxicity of the copper catalyst as well as the nitrogen source, sustainability in terms of using aerial oxygen as an eco-friendly oxidant, and tolerance of various sensitive moieties during the reaction.Experimental section
Materials and methods
All reactants were purchased from SRL, AVRA Chemicals, Alfa-aesar, Spectrochem, and Sigma Aldrich and used as received without further purification. 1H and 13C NMR spectra were obtained on a Bruker spectrometer (300 MHz and 400 MHz) and JEOL Spectrometer (500 MHz) in CDCl3 and DMSO-d6 solutions with TMS as an internal reference. Melting points were determined in open capillary on electrical bath which is uncorrected. Column chromatography was performed on silica gel (60–120 mesh) from SRL, India. Thin layer chromatographic separations were performed on pre-coated silica gel plates using silica gel G for TLC (E. Merck).General experimental procedure for the Cu(OAc)2 catalyzed oxidative transformation of aldehydes to nitriles
To a stirred suspension of aldehyde 1 (1.0 mmol) and ammonium acetate (1.5 mmol) in DMSO (3 mL), Cu(OAc)2 (10 mol%) was added. The reaction mixture was stirred for an appropriate time at 60 °C under ambient atmosphere. The progress of the reaction was monitored with TLC. Then the reaction mixture was cooled to room temperature, ethyl acetate (15 mL) was added to dissolve the product. The reaction mixture was repeatedly extracted with ethyl acetate (3 × 5 mL). The combined organic extracts were washed with water (3 × 5 mL) and dried over anhydrous Na2SO4. The crude product 2 was obtained by removal of the solvent under reduced pressure which was further purified by column chromatography on a short column of silica gel using 1–10% ethyl acetate–hexane as eluent.Procedure for the Cu(OAc)2 catalyzed gram-scale synthesis of nitrile (2a)
To a stirred suspension of 4-methoxybenzaldehyde 1a (20.0 mmol, 2.723 g) and ammonium acetate (30.0 mmol, 2.310 g) in DMSO (60 mL), Cu(OAc)2 (0.336 g, 10 mol%) was added and the reaction mixture was stirred for the appropriate time at 60 °C under ambient atmosphere. The progress of the reaction was monitored with TLC. Then the reaction mixture was cooled to room temperature, ethyl acetate (300 mL) was added to dissolve the product. The reaction mixture was repeatedly extracted with ethyl acetate (3 × 100 mL). The combined organic extracts were washed with water (3 × 100 mL). After drying with anhydrous sodium sulfate, the solvent was removed under reduced pressure to furnish the crude product 2a, which was further purified by column chromatography of silica gel using ethyl acetate–hexane as eluent. (Yield: 89%, 2.370 g).Conflicts of interest
The authors declare that there is no conflict of interest in this study.Acknowledgements
Financial assistance from RUSA-Programme and UGC-CAS-II programme in Chemistry at Jadavpur University are gratefully acknowledged. S. N. thanks DST, INSPIRE, New Delhi for senior research fellowship. Thanks to Mr M. Maji of IIT Kanpur for necessary assistance.Notes and references
- (a) C. Houben-Wiley Grundnann, in Methodender Organischen Chemie, E5, ed. J. Falbe, Georg Thieme Verlag, Stuttgart, 1985, pp.1313–1527 Search PubMed; (b) R. C. Larock, in Comprehensive Organic Transformations: a Guide to Functional Group Preparations, Wiley-VCH, Weinheim, Germany, 1989, pp. 819–995 Search PubMed; (c) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances: Syntheses, Patents, Applications, Georg Thieme, Stuttgart, 4th edn, 2001 Search PubMed.
- (a) M. N. Janakirman, K. D. Watenpaugh and K. T. Chong, Bioorg. Med. Chem. Lett., 1998, 8, 1237 CrossRef; (b) D. Dube, M. Blouin and C. Brideau, Bioorg. Med. Chem. Lett., 1998, 8, 1255 CrossRef CAS PubMed; (c) S. I. Murahashi, Sci. Synth., 2004, 19, 345–402 Search PubMed , Georg Thieme;; (d) S. J. Collier and P. Langer, Sci. Synth., 2004, 19, 403–425 Search PubMed , Georg Thieme;; (e) L. H. Jones, N. W. Summerhill, N. A. Swain and J. E. Mills, MedChemComm, 2010, 1, 309 RSC; (f) A. M. Sweeney, P. Grosche, D. Ellis, K. Combrink, P. Erbel, N. Hughes, F. Sirockin, S. Melkko, A. Bernardi, P. Ramage, N. Jarousse and E. Altmann, ACS Med. Chem. Lett., 2014, 5, 937–941 CrossRef PubMed.
- (a) T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 1633–1635 CrossRef; (b) T. Sandmeyer, Chem. Ber., 1884, 17, 2650–2653 CrossRef; (c) T. Sandmeyer, Chem. Ber., 1885, 18, 1492–1496 CrossRef; (d) T. Sandmeyer, Chem. Ber., 1885, 18, 1946–1948 Search PubMed; (e) K. W. Rosenmund and E. Struck, Chem. Ber., 1919, 2, 1749–1756 CrossRef.
- (a) L. Friedman and H. Shechter, J. Org. Chem., 1960, 25, 877–879 CrossRef CAS; (b) W. Wilting, M. Janssen, C. Muller, M. Lutz, A. L. Spek and D. Vogt, Adv. Synth. Catal., 2007, 349, 350–356 CrossRef; (c) G. Wang, X. Xie, W. Xu and Y. Liu, Org. Chem. Front., 2019, 6, 2037–2042 RSC.
- (a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779–794 CrossRef CAS; (b) D. W. Kim, C. E. Song and D. Y. Chi, J. Org. Chem., 2003, 68, 4281–4285 CrossRef CAS PubMed.
- (a) W. Zhou, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 7094–7097 CrossRef CAS PubMed; (b) D. Tsuchisya, Y. Kawagoe, K. Moriyama and H. Togo, Org. Lett., 2013, 15, 4194–4197 CrossRef PubMed; (c) Y. Kawagoe, K. Moriyama and H. Togo, Eur. J. Org. Chem., 2014, 4115–4122 CrossRef CAS; (d) S. Guo, G. Wan, S. Sun, Y. Jiang, J. T. Yu and J. Cheng, Chem. Commun., 2015, 51, 5085–5088 RSC.
- (a) C. Qin and N. Jiao, J. Am. Chem. Soc., 2010, 132, 15893–15895 CrossRef CAS PubMed; (b) W. Zhou, J. Xu, L. Zhang and N. Jiao, Org. Lett., 2010, 12, 2888–2891 CrossRef CAS PubMed; (c) Z. Guofu, Z. Guihua, L. Jie, L. Shasha, X. Shengjun, D. Chengrong and S. Shang, Chem. Res. Chin. Univ., 2016, 32, 586–593 CrossRef.
- (a) M. Shevlin, Tetrahedron Lett., 2010, 51, 4833–4836 CrossRef CAS; (b) P. Y. Yeung, C. M. So, C. P. Lau and F. Y. Kwong, Org. Lett., 2011, 13, 648–651 CrossRef CAS PubMed; (c) D. T. Cohen and S. L. Buchwald, Org. Lett., 2015, 17, 202–205 CrossRef CAS PubMed; (d) D. Ganapathy, S. S. Kotha and G. Sekar, Tetrahedron Lett., 2015, 56, 175–178 CrossRef CAS; (e) S. Xu, T. Cai and Z. Yun, Synlett, 2016, 221–224 CAS; (f) D. D. Beattie, T. Schareina and M. Beller, Org. Biomol. Chem., 2017, 15, 4291–4294 RSC.
- (a) H. Veisi, Synthesis, 2010, 15, 2631–2635 CrossRef; (b) Y. Z. Zhu and C. Cai, Monatsh. Chem., 2010, 141, 637–639 CrossRef CAS; (c) L. Wang, C. Shen, H. P. Wang, W. Y. Zhou, F. A. Sun, M. Y. He and Q. Chen, J. Chem. Res., 2012, 36, 460–462 CrossRef CAS; (d) Y. Z. Zhu, X. Q. Zhang, F. Liu, H. M. Gu and H. L. Zhu, Synth. Commun., 2013, 43, 2943–2948 CrossRef CAS; (e) C. B. Kelly, K. M. Lambert, M. A. Mercadante, J. M. Ovian, W. F. Bailey and N. E. Leadbeater, Angew. Chem., Int. Ed., 2015, 54, 4241–4245 CrossRef CAS PubMed; (f) C. Fang, M. Li, X. Hu, W. Mo, B. Hu, N. Sun, L. Jin and Z. Shen, Adv. Synth. Catal., 2016, 358, 1157–1163 CrossRef CAS; (g) Y. Zhao, G. Mei, H. Wang, G. Zhang and C. Ding, Synlett, 2019, 30, 1484–1488 CrossRef CAS.
- (a) J. Noh and J. Kim, J. Org. Chem., 2015, 80, 11624–11628 CrossRef CAS PubMed; (b) P. Nimnual, J. Tummatorn, C. Thongsornkle and S. Ruchirawat, J. Org. Chem., 2015, 80, 8657–8667 CrossRef CAS PubMed; (c) J. Nandi and N. E. Leadbeater, Org. Biomol. Chem., 2019, 17, 9182–9186 RSC; (d) F. Verma, P. Shukla, S. R. Bhardiya, M. Singh, A. Rai and V. K. Rai, Catal. Commun., 2019, 119, 76–81 CrossRef CAS; (e) H. Chen, S. Sun, H. Xi, K. Hu, N. Zhang, J. Qu and Y. Zhou, Tetrahedron Lett., 2019, 60, 1434–1436 CrossRef CAS; (f) L. M. Dornan, Q. Cao, J. C. A. Flanagan, J. J. Crawford, M. J. Cook and M. J. Muldoon, Chem. Commun., 2013, 49, 6030–6032 RSC.
- (a) S. Nandy, A. Ghatak, A. K. Das and S. Bhar, Synlett, 2018, 29, 2208–2212 CrossRef CAS; (b) M. Maji, K. Chakrabarti, D. Panja and S. Kundu, J. Catal., 2019, 373, 93–102 CrossRef CAS; (c) S. Nandy, A. K. Das and S. Bhar, Synth. Commun., 2020, 50, 3326–3336 CrossRef CAS.
- (a) P. Taboonpong and W. Chavasiri, Catal. Commun., 2018, 104, 9–12 CrossRef CAS; (b) J. Y. Zhang, W. H. Bao, F. H. Qin, K. W. Lei, Q. Li and W. T. Wei, Asian J. Org. Chem., 2019, 8, 2050–2053 CrossRef CAS; (c) J. Kim, S. Park, H. Kim and J. Kim, Tetrahedron Lett., 2020, 61, 152112 CrossRef CAS; (d) L. J. Zhou, K. Wang, H. R. Guan, A. Q. Zheng, H. T. Yang and C. B. Miao, J. Org. Chem., 2020, 85, 7925–7938 CrossRef CAS PubMed; (e) Q. Wang, F. Ye, J. Cao, Z. Xu, Z. J. Zheng and L. W. Xu, Catal. Commun., 2020, 138, 105950 CrossRef CAS; (f) E. M. Miller and M. A. Walczak, J. Org. Chem., 2020, 85, 8230 CrossRef CAS PubMed.
- (a) A. K. Das, N. Sepay, S. Nandy, A. Ghatak and S. Bhar, Tetrahedron Lett., 2020, 61, 152231 CrossRef CAS; (b) A. K. Das, S. Nandy and S. Bhar, Appl. Organomet. Chem., 2021, 35, e6282 CrossRef CAS.
- (a) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932 CrossRef CAS PubMed; (b) L. Zhang, G. Y. Ang and S. Chiba, Org. Lett., 2010, 12, 3682 CrossRef CAS PubMed; (c) B. B. Feng, J. Q. Liu and X. S. Wang, J. Org. Chem., 2017, 82, 1817 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07701e |
| This journal is © The Royal Society of Chemistry 2022 |