
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and spectroscopic studies of spontaneously formed crystalline Eu(III)–aliphatic dicarboxylates at room temperature†
Hee-Kyung Kim *,
Dong Woo Lee,
Sojeong Park,
Euo Chang Jung,
Sang Ho Lim,
Wansik Cha and
Hye-Ryun Cho
*,
Dong Woo Lee,
Sojeong Park,
Euo Chang Jung,
Sang Ho Lim,
Wansik Cha and
Hye-Ryun Cho
Nuclear Chemistry Research Team, Korea Atomic Energy Research Institute, Daejeon 34057, Republic of Korea. E-mail: hkim11@kaeri.re.kr
First published on 31st January 2022
Abstract
Complexation of actinides and lanthanides with carboxylic organic ligands is a critical issue affecting radionuclide migration from deep geological disposal systems of spent nuclear fuel. A series of Eu(III)–aliphatic dicarboxylate compounds, as chemical analogs of radioactive Am(III) species, Eu2(Ox)3(H2O)6, Eu2(Mal)3(H2O)6, and Eu2(Suc)3(H2O)2, were synthesized and characterized using X-ray crystallography and time-resolved laser fluorescence spectroscopy to examine the ligand-dependent binding modes and the corresponding changes in spectroscopic properties. Powder X-ray crystallography results confirmed that all of the compounds presented a crystalline polymer structure with a trigonal prism square-face tricapped polyhedron geometry centered on Eu(III) in a nine-coordinate environment involving nine oxygen atoms. This study captures the transition of the coordination modes of aliphatic dicarboxylate ligands from side-on to end-on binding as the carbon chain length increases. This transition is illustrated in malonate bindings involving a combination of side-on and end-on modes. Strongly enhanced luminescence, especially for the hypersensitive peak, indicates a low site symmetry in the formation of solid compounds. The number of remaining bound water molecules was estimated from the resultant increased luminescence lifetimes, which were in good agreement with crystal structures. The excitation–emission matrix spectra of these crystalline polymers suggest that Ox ligands promote the sensitized luminescence of Eu(III), especially in the UV region. In the case of Mal and Suc ligands, charge transfer occurs in the opposite direction from Eu(III) to the ligands under UV excitation, resulting in weaker luminescence.
Introduction
Safety assessment of deep geological disposal systems of spent nuclear fuel requires a thorough understanding of the chemical behavior of radionuclides in the geochemical environment.1 The migration of actinide (An) elements, a major group of long-term radiotoxic radionuclides, in groundwater is accelerated by complexation with natural organic matter (NOM).2,3 Cooperative interactions among polycarboxyl groups in NOM induce strong complexation with An.2,4 Trivalent An(III) and lanthanide (Ln(III)) complexes with a series of aliphatic dicarboxylates (−OOC–(CH2)n–COO−) have been studied as model systems to investigate their interactions with NOM.4–11The thermodynamic stabilities of these complexes are often explained in terms of the binding modes of the ligands. Fröehlich et al. proposed that oxalate (Ox) (n = 0), malonate (Mal) (n = 1), and succinate (Suc) (n = 2) prefer side-on bindings (Scheme 1(a)) in aqueous Cm(III) complexes based on serial studies using time-resolved laser fluorescence spectroscopy (TRLFS) and density functional theory.7–9 In contrast, our previous studies on aqueous Am(III) complexes anticipated that Ox and Suc bind to Am(III) in side-on (Scheme 1(a)) and end-on (Scheme 1(b)) modes, respectively, based on the UV-vis absorption and TRLFS results.11 However, the binding modes of these complexes have not been directly resolved.
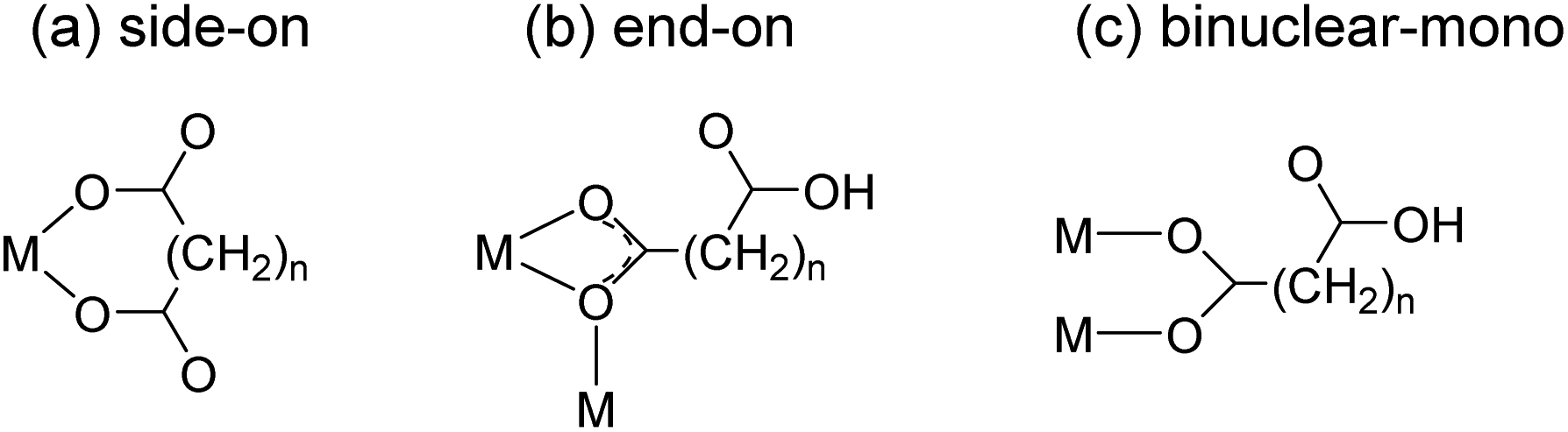 | ||
| Scheme 1 Schematic of representative coordination modes of dicarboxylate ligands, oxalate (Ox) (n = 0), malonate (Mal) (n = 1), and succinate (Suc) (n = 2). The binding modes of (a) side-on, (b) end-on and (c) binuclear-mono are named based on a single metal ion (M). | ||
Structural studies of solid compounds can provide insights into the coordination environments of aqueous complexes. There are numerous crystallographic studies on Ln(III)-aliphatic dicarboxylate frameworks owing to their potential applications in various areas, including the control of topological structures and optoelectronic devices.12–21 Among the many possible binding modes of dicarboxylates,15 rigid Ox ligands bind to Ln ions, producing well-defined structures wherein uniform side-on bidentate bonds (Scheme 1(a)) form stable 5-membered ring structures.13,14 The crystal structures of Pu(III)–Ox22 and Am(III)–Ox23 revealed that they are isomorphous with Ln(III) complexes. Meanwhile, Mal and Suc ligands are flexible linkers containing carbon chains between the two carboxyl groups and thus show diverse binding modes. In particular, Mal can adopt many different coordination modes, such as side-on binding of two carboxyl groups to form a six-membered ring (Scheme 1(a)) and end-on binding of one carboxyl group to form a four-membered ring (Scheme 1(b)).15–17,24 Consequently, diverse structures and temperature-dependent phase transitions have been reported for Ln(III)–Mal polymer frameworks.15,17,24 The binding modes of Suc are relatively less diverse. It frequently shows end-on (Scheme 1(b)) and mono-binding (Scheme 1(c)) modes.18–21,25
However, in the aforementioned structural studies, Ln(III)–aliphatic dicarboxylate compounds are frequently prepared under special conditions, such as hydrothermal processes involving high temperatures13,19,25 and with the use of template molecules16,19 and non-aqueous media.13,15,17,23,24 In addition, the majority of these studies investigated one type of ligand linker with a series of metal ions. To examine ligand-dependent structural and spectroscopic changes, a series of ligands should be systematically examined within the same metal ion under the same experimental conditions.
Here, we studied solid Eu–Ox, Eu–Mal, and Eu–Suc compounds in series to investigate the binding modes of aliphatic dicarboxylate ligands on Eu(III) ions under conditions similar to those used in studies on aqueous complexes, but with oversaturated conditions.10,11 In order to obtain spontaneously formed solid compounds, the samples were prepared in aqueous solutions at room temperature (23–27 °C) without additional template molecules. The molecular structures were characterized using powder X-ray diffraction (XRD) analysis. TRLFS was employed to examine the changes in the coordination environments and symmetries around the Eu(III) ion. Excitation and emission matrix (EEM) spectra were obtained to investigate the sensitized luminescence in relation to the binding mode and type of ligand.
Experimental
Sample preparation
Europium dioxide (Sigma-Aldrich, 99.9%) was dissolved in 1.1 M perchloric acid to make a Eu3+ solution, whose concentration was quantified to be 278 mM using ICP-AES (Ultima-2C, HORIBA Jobin Yvon). Sodium oxalate (Sigma-Aldrich, trace metals basis, ≥99.99%), sodium malonate dibasic monohydrate (Sigma-Aldrich, BioXtra), and sodium succinate dibasic hexahydrate (Sigma-Aldrich, ReagentPlus®, ≥ 99%) were dissolved in Millipore water to obtain concentrations of 125, 500, and 483 mM, respectively. All solutions were filtered through 0.45 μm membrane syringe filters (Millipore). Recrystallized NaClO4 (Sigma-Aldrich, 99%) was used to prepare a NaClO4 stock solution (4.99 M). NaOH (Sigma-Aldrich, semiconductor grade, 99.99%) was dissolved in slightly acidified (pH ∼5.3) Millipore water in an Ar-filled glove box to avoid CO2 dissolution.Eu–Ox, Eu–Mal, and Eu–Suc compounds were prepared by adding the Eu3+ solution to the organic ligand solutions in 0.1 M NaClO4 with stirring at room temperature. A range of stoichiometries and concentrations of Eu and ligands were tested (details in Table S1†) to obtain sufficient yields for XRD and spectroscopic measurements. To avoid precipitation of acidic forms of the organic ligands at low pH, a known amount of NaOH solution (1 M) was pre-mixed with the ligand solutions prior to addition of the Eu3+ solution so that pH values of the prepared solutions were in the range of 5–6. If necessary, the pH was further adjusted by adding dilute HClO4 (J. T. Baker, ULTREX II, Ultrapure reagent) or NaOH solutions (0.01–0.1 mM). Solutions were left at room temperature until solid powders were formed. Typically, solid Eu–Ox compounds formed instantly, while Eu–Mal and Eu–Suc compounds became visible after several hours to days. Eu–Ox and Eu–Suc solid compounds started precipitating out when millimolar concentrations were used, while the Eu–Mal solid compound was formed when tens of millimolar concentrations were used (Table S1†). The solid products were collected using 0.2 μm membrane filters, dried in air at room temperature, and examined without further treatment.
Powder X-ray diffraction
XRD data were obtained using a Bruker D8-Advance diffractometer with Cu Kα radiation at room temperature and 40 kV and 40 mA. Ground polycrystalline samples were mounted on sample holders and scanned in a 2θ range of 5–80° with a step size of 0.02° and a step time of 0.5 s. The obtained powder XRD patterns were compared to the simulated powder patterns from the single-crystal structures of Ln(III) compounds that were previously deposited at the Cambridge Crystallographic Database Centre (CCDC).Thermogravimetric analysis
A Sinco STA N-1500 thermogravimetric analyzer was used for thermogravimetric analysis (TGA). Polycrystalline samples of the Eu compounds were placed in alumina crucibles and heated at a rate of 10 °C min−1 from 30 to 1000 °C under an argon flow.TRLFS analysis
Time-resolved luminescence spectra were acquired using a nanosecond-pulsed Nd:YAG laser-pumped OPO (Vibrant B, Opotek Inc.) as an excitation source at 394 nm (0.4–1.0 mJ). Dried solid powders were placed in quartz cells (1 mm × 10 mm) and tilted by ∼30° to the incident excitation laser beam path. Emissions were collected using a fiber bundle connected to a spectrograph (SR-303i, Andor Technology). An intensified CCD (ICCD) (iStar, Andor Technology) was used as the detector and its insertion delay time was managed relative to the incident laser pulse.26 A long-path filter (500 nm cutoff) was placed in front of the detector to protect it from the scattered excitation light. The ICCD was set with a gate width of 5 ms and a gain of 1. Spectra were collected by accumulating luminescence signals from 100 excitation bursts. A grating of 600 lines per mm was mainly employed with a slit width of 10 μm.Luminescence lifetimes, the inverse of the decay rate (τ = 1/kobs), were measured using the kinetic mode of the ICCD with a series of delayed gate openings from the burst of the incident laser pulses. The inner-sphere hydration number (N(H2O)) of the Eu(III) ion was calculated based on its established relationship with the luminescence lifetime (τ), as expressed in eqn (1).27,28
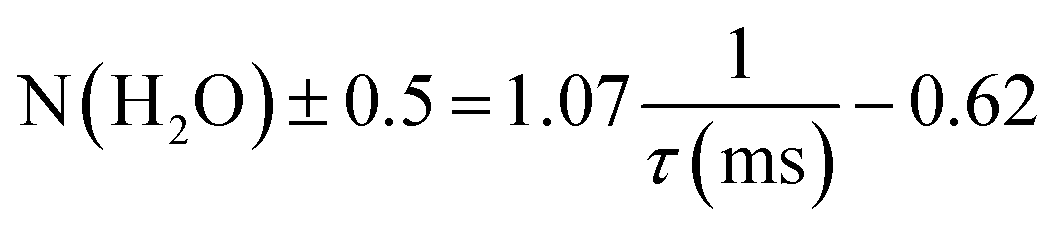 | (1) |
Excitation-emission matrix spectroscopy
EEM spectra were obtained using a spectrofluorometer equipped with a xenon arc lamp, an excitation monochromator, and a multichannel CCD detector (Aqualog®, Horiba sci.). The photoluminescence of the Eu(III) samples was collected in the range of 550–750 nm by scanning the excitation wavelength from 220 to 420 nm in 1 nm increments. A long-path cutoff filter at 420 nm was placed in front of the detector to block the excitation light from entering the detector. Excitation and emission spectra were extracted from the EEM dataset.Results and discussion
Crystal structure characterization
Powder XRD patterns were examined to confirm the phase purity and crystal structures of the synthesized Eu–Ox, Eu–Mal, and Eu–Suc compounds. Among the test samples with enough solid yield for XRD and spectroscopic measurements, solid compounds formed from lower bound concentrations of the reagents were subjected to powder XRD measurements. The tested samples for each ligand showed identical XRD patterns (Table S1†). Solid samples obtained from solutions of 2 mM Eu(III) + 3 mM Ox, 30 mM Eu(III) + 50 mM Mal, and 5 mM Eu(III) + 7.5 mM Suc were chosen for detailed examination. These samples were prepared with similar concentration ratios of organic ligand to Eu(III) (1.5–1.7) (see Table S1†) and provided good diffraction patterns.As shown in Fig. 1, the observed diffraction patterns matched well with the simulated patterns of the crystal structures that have been previously reported in the literature.13,16,19 In all three compounds, the unique Eu(III) cation is in a nine-coordinate environment with nine oxygen atoms from water molecules and ligands forming a trigonal prism square-face tricapped polyhedron. The ball-and-stick representations of the crystal structures are shown in Fig. S1,† and the corresponding two-dimensional structures are shown in Fig. 2 for clarity.
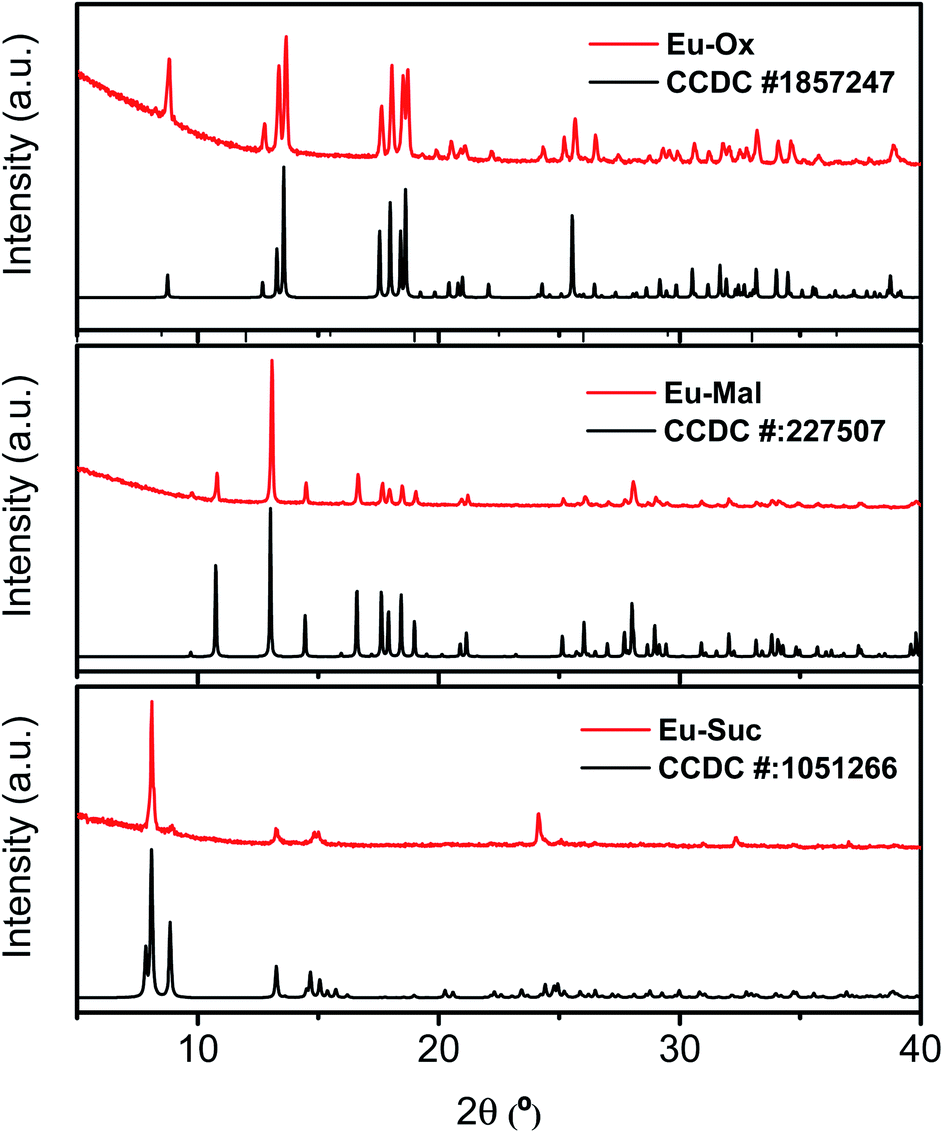 | ||
| Fig. 1 Powder XRD patterns of Eu–Ox, Eu–Mal, and Eu–Suc complexes acquired in this study (red) and the corresponding literature diffraction patterns (black).13,16,19 Bottom: the XRD pattern of Nd2(Suc)3(H2O)2 (CCDC # 1051266) is displayed for Eu–Suc, because there is no reported Eu–Suc crystal structure. | ||
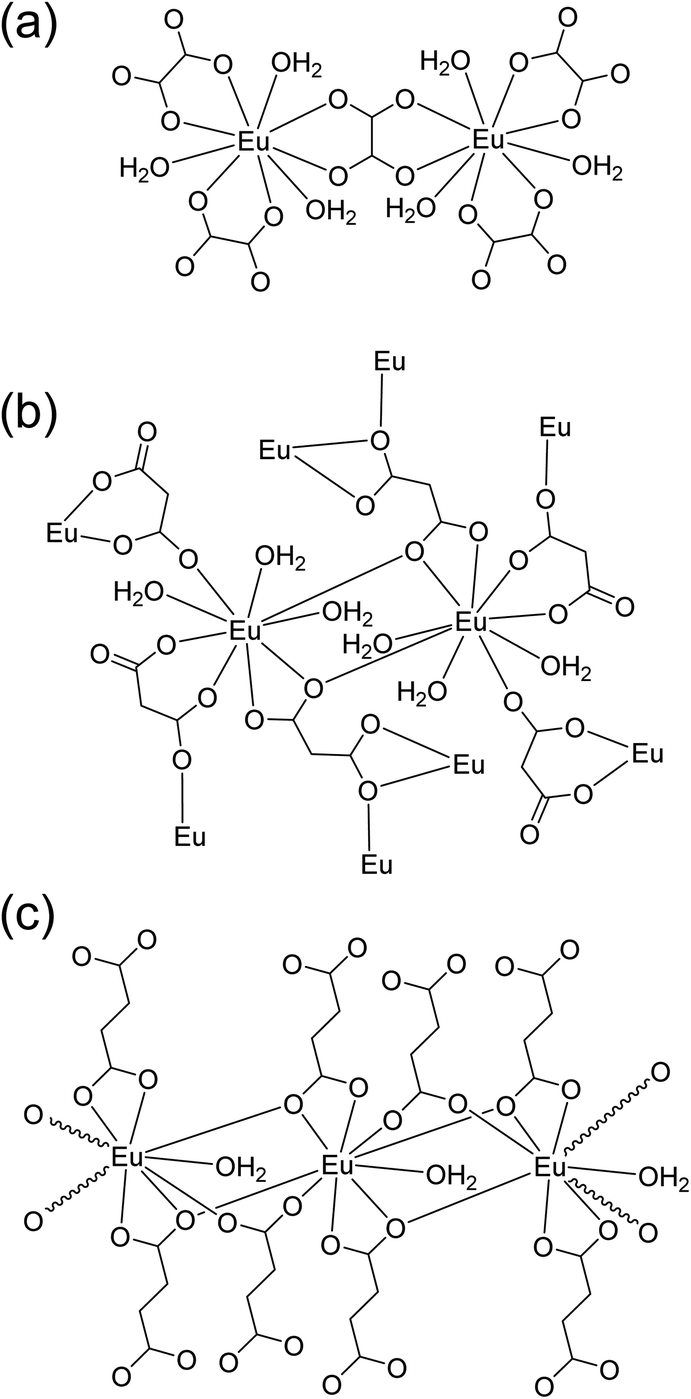 | ||
| Fig. 2 Schematic of two-dimensional coordination environments of the (a) Eu2(Ox)3(H2O)6, (b) Eu2(Mal)3(H2O)6, and (c) Eu2(Suc)3(H2O)2 frameworks according to the crystal structures shown in Fig. S1.†13,16,19 The structures are not drawn to scale to depict each bond clearly. | ||
Eu–Ox (Eu2(Ox)3(H2O)6) crystallizes in the monoclinic space group, P2/c.13 Three side-on coordinated Ox (Scheme 1(a)) and three water molecules are bonded to a Eu(III) cation through oxygen atoms (Fig. 2(a)). Eu–Mal (Eu2(Mal)3(H2O)6) crystallizes in the monoclinic space group, C2/c.16 Four coordinated Mal and three water molecules are connected to a Eu(III) cation through oxygen atoms (Fig. 2(b)). Among the four coordinated Mal, one is coordinated side-on (Scheme 1(a)), another is end-on (Scheme 1(b)), and the others are bonded in monodentate coordination modes, bridging the adjacent Eu(III) atoms.
Although numerous reports exist on the structures of other Ln(III) ions with the Suc ligand, a well-defined crystal structure of Eu–Suc has not been published. However, the observed powder XRD pattern of the Eu–Suc compound matches well with those of Ln(III)–Suc (Ln = La, Pr, Nd, Eu, Tb, and Gd) reported by D'Vries et al.19 They also reported that Ln2(Suc)3(H2O)2 (Ln = La, Pr, Nd, and Gd) compounds crystallize in the triclinic space group P-1. Based on those Ln(III) crystal structures, we infer that six coordinated Suc and one water molecule are connected to a Eu(III) cation through oxygen atoms (see Fig. 2(c)). Among the six coordinated Suc, two Suc are coordinated end-on (Scheme 1(b)), and the oxygen atoms of the carboxylate groups in the other two Suc ligands are shared in a binuclear monodentate coordination mode with adjacent Eu(III) cations, as shown in Scheme 1(c). The remaining two Suc ligands are end-on, bridging the adjacent Eu(III) cations.
Overall, these results confirm that the coordination modes of aliphatic dicarboxylate ligands transit from side-on to end-on binding as the carbon chain length increases. Malonate binding illustrates this transition involving a combination of side-on and end-on modes. These results are consistent with those of our previous studies, where results from various spectroscopic studies indicated a close correlation between the thermodynamic stability and electronic absorption strength of the Am(III) complexes.10,11 Ox forms stable complexes with Am(III) with increased molar absorption coefficients, while Suc complexes are less stable with decreased molar absorption coefficients. Mal, with a carbon chain length between that of Ox and Suc, resulted in a binding stability and optical properties that were intermediate with respect to those of the two ligands.
The bond lengths are shown in Fig. S2,† and the average bond lengths in each coordination mode are summarized in Table S2.† The bond lengths of the side-on bound Eu–O are shorter than those of the end-on bound Eu–O. The same trends were observed for aqueous complexes of U(VI)O2 (ref. 29 and 30) and Am(III) systems.11 The FTIR and Raman spectra of these compounds are shown in Fig. S3 and S4.† The observed split peak patterns in the FTIR and Raman spectra of the Eu–Mal and Eu–Suc compounds are explained by the presence of the inequivalent binding modes of the ligands.
TGA analysis
The thermal behavior of the synthesized compounds was analyzed by TGA. The TGA plots for all three materials are shown in Fig. S5.† Eu2(Ox)3(H2O)6 was stable up to 100 °C; above 100 °C, loss of water was observed corresponding to a total weight loss of 17.31% (calcd 15.94% for the six water molecules). Decomposition commenced above 150 °C due to the decomposition of Ox. Eu2(Mal)3(H2O)6 exhibited a weight loss of 13.32% between room temperature and 150 °C, which was attributed to the loss of occluded water molecules (calcd 15.01% for the six water molecules). This was followed by complete decomposition due to the loss of Mal. Eu2(Suc)3(H2O)2 shows an initial weight loss of 7.35% between room temperature and 200 °C, arising from the loss of two occluded water molecules (calcd 5.32%). The dehydrated material was thermally stable at temperatures up to 300 °C. Above 300 °C, decomposition resulting from the elimination of Suc occurred. The TGA results were in good agreement with the expected water content inferred from the crystal structures.TRLFS analysis
The luminescence spectra of these compounds have been reported previously,13,14,17,19,21,24 but detailed features were not described. Time-resolved luminescence properties can provide information on the inner-sphere coordination environments of Eu(III), but have never been investigated. Thus, we examined luminescence properties of these compounds in detail by using TRLFS.The Eu–Ox, Eu–Mal, and Eu–Suc compounds are strongly luminescent in the visible region, as shown in their luminescence spectra in Fig. 3. The observed luminescence results from the electronic transitions of 5D0 → 7FJ (J = 0–4). The 5D0 → 7F1 and 5D0 → 7F2 transitions are referred to as the magnetic dipole transition (MDT) and hypersensitive electric dipole transition (EDT), respectively.31 These transitions are sensitive to site symmetry and particularly the center of inversion. A higher asymmetric ratio, determined from the EDT to MDT intensity ratio, indicates a higher degree of deviation from the center of inversion symmetry.31
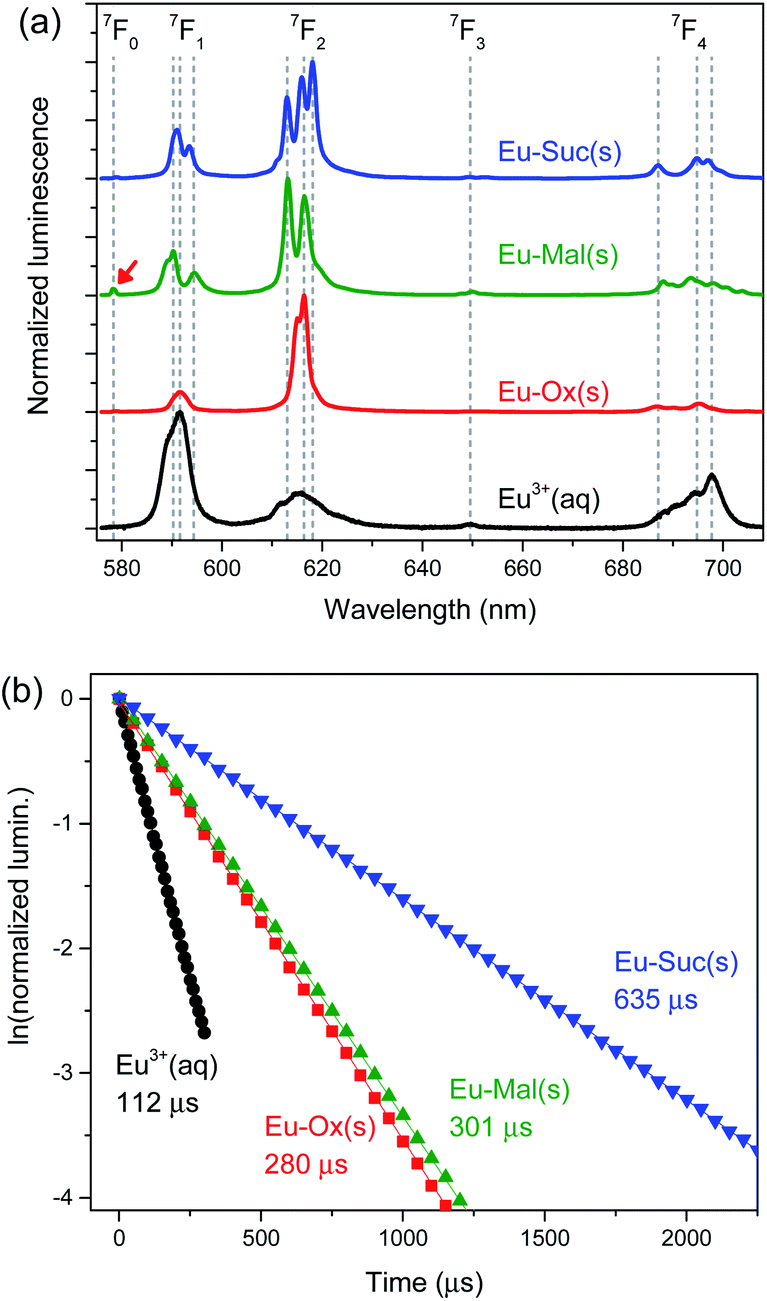 | ||
| Fig. 3 Luminescence from 5D0 → 7FJ (J = 0–4) transitions of solid Eu–Ox, Eu–Mal, and Eu–Suc compounds: (a) luminescence spectra and (b) luminescence decay plots. The samples were the same as those used for the powder XRD measurements. The red arrow indicates 5D0 → 7F0 transition peak in the Eu–Mal luminescence. | ||
The luminescence properties of aqueous Eu3+ are well characterized with an asymmetric ratio of ∼0.3, as shown in Fig. 3(a) (black). The luminescence lifetime of Eu3+ was determined to be 112 ± 1 μs, and the corresponding coordination number was estimated to be 8.9 ± 0.5, according to eqn (1), which is in good agreement with the reported value of 9.0 ± 0.5.27,32 In comparison to the luminescence spectrum of aqueous Eu3+, solid Eu–Ox, Eu–Mal, and Eu–Suc compounds show substantially enhanced hypersensitive peak (J = 2) intensity and narrower spectral widths (Fig. 3(a)). Such increased asymmetric ratios indicate a loss of inversion symmetry in the formation of solid Eu–Ox, Eu–Mal, and Eu–Suc compounds. Another feature in particular in the luminescence of Eu–Mal(s) is the apparent appearance of the 5D0 → 7F0 transition peak at approximately 575 nm as indicated with a red arrow. It is indicative of higher degree of losing site symmetry around the Eu3+ ion in the Eu–Mal compound.
The luminescence spectral shapes of these compounds remained the same throughout the detection time window of 1 μs to 2.5 ms after the incident laser pulse. In addition, the luminescence decay plots were well fitted to single exponential decay curves, as shown in Fig. 3(b). These observations indicate that each sample is likely composed of an unique Eu species located in an identical coordination environment. Thus, the observed peak splittings in Fig. 3(a) are ascribed to the crystal field effects.31 The splitting patterns in the luminescence spectra of the Eu(III) compounds differed depending on the ligand (Fig. 3(a)), which supports the distinct geometries of each compound, as revealed by the crystal structures in Fig. 2.
The luminescence lifetimes of Eu–Ox, Eu–Mal, and Eu–Suc were 280 ± 7, 301 ± 2, and 635 ± 9 ns, respectively (Fig. 3(b) and Table 1). According to eqn (1), the corresponding numbers of water molecules bound in the inner sphere of the Eu(III) centers of Eu–Ox, Eu–Mal, and Eu–Suc were estimated to be 3.1 ± 0.5, 2.9 ± 0.5, and 1.0 ± 0.5, respectively (Table 1). These N(H2O) values are in good agreement with the powder XRD results described above: there are three H2O molecules per Eu(III) in Eu2(Ox)3(H2O)6 and Eu2(Mal)3(H2O)6, and one in Eu2(Suc)3(H2O)2. In contrast to the proposed reduction of the Eu(III) coordination number to eight upon binding of two or more Ox ligands,33 the current results show that the Eu(III) coordination number remains nine in solid compounds as well as in aqueous complexes.10,11
| Sample | t (μs) | N(H2O) |
|---|---|---|
| Eu3+ (aq) | 112 ± 1 | 8.9 ± 0.5 |
| Eu–Ox(cr) | 280 ± 7 | 3.1 ± 0.5 |
| Eu–Mal(cr) | 301 ± 2 | 2.9 ± 0.5 |
| Eu–Suc(cr) | 635 ± 9 | 1.0 ± 0.5 |
EEM
The excitation wavelength from 220 to 420 nm was scanned while collecting the luminescence from 550 to 750 nm to acquire the EEM spectra of the Eu compounds, as shown in Fig. S6.† For all three compounds, the normalized emission spectral shapes remained the same regardless of the excitation wavelength (Fig. S7†). Excitation spectra in Fig. 4(a) were extracted from the EEM measurements by collecting 5D0 → 7FJ (J = 0–4) emission signals in the region of 550–750 nm. Regarding the peak positions, the overall patterns of the excitation spectra of the Eu(III) compounds closely followed the absorption pattern of the aqueous Eu3+ ion (Fig. 4(b)). However, the relative luminescence intensities were enhanced at excitation wavelengths above 310 nm. These observations indicate changes in the relaxation pathways of the excited Eu(III) compounds compared to those of the Eu3+ ion. | ||
| Fig. 4 (a) Excitation spectra of Eu–Ox, Eu–Mal, Eu–Suc solid compounds obtained by collecting luminescence signals between 550 and 750 nm via EEM measurements. The pink box highlights probable charge transfer between metal and ligand. (b) An absorption spectrum of aqueous Eu3+ (41 mM in 0.16 M HClO4) acquired using a spectrophotometer is displayed for comparison. | ||
Interestingly, a broad band is observed in the excitation region of 220–270 nm only for the Eu–Ox compound, as indicated by the pink box in Fig. 4. This broad band is attributed to the charge transfer (CT) transition, which involves chiefly the electron transfer from O → Eu.34 The strong absorption band of the Ox ligand in this region (Fig. S8†) is ascribed to ligand-to-metal CT, resulting in sensitization.35 In contrast, the Mal and Suc complexes do not exhibit absorption in this region, and their luminescence is comparatively quenched at excitation wavelengths below 290 nm. This is likely because a metal-to-ligand CT is dominant in these two complexes.34
Conclusions
Eu–Ox, Eu–Mal, and Eu–Suc solid complexes were spontaneously formed under oversaturated conditions and were structurally and spectroscopically characterized. Powder XRD results confirmed the formation of crystalline binuclear polymers, Eu2(Ox)3(H2O)6, Eu2(Mal)3(H2O)6, and Eu2(Suc)3(H2O)2. The time-resolved luminescence spectra illustrate the lower symmetries of the Mal compound, compared to that of the Ox and Suc compound. The number of bound water molecules calculated using the measured luminescence lifetimes was 3.1, 2.9, and 1.1 for Ox, Mal, and Suc derivatives, respectively, which is in good agreement with the water content indicated in the crystal structures. Our results, together with our previous findings on analogous aqueous complexes, show that the coordination number around Eu(III) remains constant for both solid and aqueous complexes. Ox shows a well-defined side-on coordination mode, and Suc prefers end-on binding and binuclear monodentate bridges. In contrast, the Mal ligands show a mixture of side-on and end-on binding of the dicarboxylic groups. These results explain the intermediate-like binding stability and spectroscopic properties of Am(III)–Mal complexes with respect to those of Ox and Suc complexes in our previous works.10,11 The EEM results suggest that, in the UV excitation region, Ox ligands provide sensitized luminescence via charge transfer from the ligand to the metal ion, while Mal and Suc ligands induce charge transfer in the opposite direction from the metal to the ligand, resulting in decreased luminescence.Author contributions
The manuscript was written through the contributions of all the authors. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Nuclear Research and Development Program of the National Research Foundation of Korea (NRF) (No. 2017M2A8A-5014719) and by the Institute for Korea Spent Nuclear Fuel and NRF of Korea (No. 2021M2E1A1085202).References
- J. I. Kim, Nucl. Eng. Technol., 2006, 38, 459–482 CAS.
- S. A. Wood, Eng. Geol., 1993, 34, 229–259 CrossRef.
- J. F. McCarthy, W. E. Sanford and P. L. Stafford, Environ. Sci. Technol., 1998, 32, 3901–3906 CrossRef CAS.
- P. Thakur, P. N. Pathak, T. Gedris and G. R. Choppin, J. Solution Chem., 2009, 38, 265 CrossRef CAS.
- Z.-M. Wang, L. J. van de Burgt and G. R. Choppin, Inorg. Chim. Acta, 2000, 310, 248–256 CrossRef CAS.
- T. Sasaki, S. Kubo, T. Kobayashi, I. Takagi and H. Moriyama, J. Nucl. Sci. Technol., 2005, 42, 724–731 CrossRef CAS.
- A. Skerencak-Frech, M. Maiwald, M. Trumm, D. R. Fröehlich and P. J. Panak, Inorg. Chem., 2015, 54, 1860–1868 CrossRef CAS PubMed.
- D. R. Fröehlich, M. Trumm, A. Skerencak-Frech and P. J. Panak, Inorg. Chem., 2016, 55, 4504–4511 CrossRef PubMed.
- A. Skerencak-Frech, M. Trumm, D. R. Fröhlich and P. J. Panak, Inorg. Chem., 2017, 56, 10172–10180 CrossRef CAS PubMed.
- H. K. Kim, K. Jeong, H. R. Cho, E. C. Jung, K. Kwak and W. Cha, Dalton Trans., 2019, 48, 10023–10032 RSC.
- H.-K. Kim, K. Jeong, H.-R. Cho, K. Kwak, E. C. Jung and W. Cha, Inorg. Chem., 2020, 59, 13912–13922 CrossRef CAS PubMed.
- R. Janicki, A. Mondry and P. Starynowicz, Coord. Chem. Rev., 2017, 340, 98–133 CrossRef CAS.
- D. Alexander, K. Thomas, M. Joy, P. R. Biju, N. V. Unnikrishnan and C. Joseph, Acta Crystallogr., Sect. C: Struct. Chem., 2019, 75, 589–597 CrossRef CAS PubMed.
- D. Alexander, K. Thomas, S. Sisira, L. A. Jacob, S. Gopi, A. Kumar S, P. R. Biju, N. V. Unnikrishnan and C. Joseph, Opt. Mater., 2018, 86, 366–375 CrossRef CAS.
- F. S. Delgado, P. Lorenzo-Luís, J. Pasán, L. Cañadillas-Delgado, O. Fabelo, M. Hernández-Molina, A. D. Lozano-Gorrín, F. Lloret, M. Julve and C. Ruiz-Pérez, CrystEngComm, 2016, 18, 7831–7842 RSC.
- C.-Z. Zhang, H.-Y. Mao, Y.-L. Wang, H.-Y. Zhang and J.-C. Tao, J. Phys. Chem. Solids, 2007, 68, 236–242 CrossRef CAS.
- S. Hussain, X. Chen, W. T. A. Harrison, S. Ahmad, M. R. J. Elsegood, I. U. Khan and S. Muhammad, Front. Chem., 2019, 7, 728 CrossRef CAS PubMed.
- C.-G. Wang, Y.-H. Xing, Z.-P. Li, J. Li, X.-Q. Zeng, M.-F. Ge and S.-Y. Niu, J. Mol. Struct., 2009, 921, 126–131 CrossRef CAS.
- R. F. D'Vries, I. Camps and J. Ellena, Cryst. Growth Des., 2015, 15, 3015–3023 CrossRef.
- N. Rahahlia, B. Benmerad, A. Guehria-Laïdoudi, S. Dahaoui and C. Lecomte, J. Mol. Struct., 2007, 833, 42–48 CrossRef CAS.
- G.-H. Cui, J.-R. Li, R.-H. Zhang and X.-H. Bu, J. Mol. Struct., 2005, 740, 187–191 CrossRef CAS.
- W. Runde, L. F. Brodnax, G. Goff, A. C. Bean and B. L. Scott, Inorg. Chem., 2009, 48, 5967–5972 CrossRef CAS PubMed.
- C. Tamain, B. Arab-Chapelet, M. Rivenet, X. F. Legoff, G. Loubert, S. Grandjean and F. Abraham, Inorg. Chem., 2016, 55, 51–61 CrossRef CAS PubMed.
- M. Hernández-Molina, P. Lorenzo-Luis, C. Ruiz-Pérez, T. López, I. R. Martín, K. M. Anderson, A. G. Orpen, E. H. Bocanegra, F. Lloret and M. Julve, J. Chem. Soc., Dalton Trans., 2002, 3462–3470, 10.1039/B202649J.
- S. C. Manna, E. Zangrando, A. Bencini, C. Benelli and N. R. Chaudhuri, Inorg. Chem., 2006, 45, 9114–9122 CrossRef CAS PubMed.
- E. C. Jung, H. R. Cho, M. H. Baik, H. Kim and W. Cha, Dalton Trans., 2015, 44, 18831–18838 RSC.
- T. Kimura and G. R. Choppin, J. Alloys Compd., 1994, 213, 313–317 CrossRef.
- W. D. Horrocks and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef CAS.
- V. Vallet, H. Moll, U. Wahlgren, Z. Szabó and I. Grenthe, Inorg. Chem., 2003, 42, 1982–1993 CrossRef CAS PubMed.
- L. Rao, J. Jiang, P. Zanonato, P. Di Bernardo, A. Bismondo and Y. Garnov Alexander, Radiochimica Acta, 2002, 90, 581 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- H.-K. Kim, S. Choi, E. C. Jung, H.-R. Cho, J.-I. Yun and W. Cha, J. Lumin., 2018, 202, 469–474 CrossRef CAS.
- D. Xian, W. Zhou, J. Wang, D. Pan, X. Li, Y. Li, Y. Shi, W. Wu, Z. Tan and C. Liu, Dalton Trans., 2021, 50, 9388–9398 RSC.
- S. K. Gupta, C. Reghukumar and R. M. Kadam, RSC Adv., 2016, 6, 53614–53624 RSC.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07565a |
| This journal is © The Royal Society of Chemistry 2022 |