
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multi pathway coupled domino strategy: I2/TBHP-promoted synthesis of imidazopyridines and thiazoles via sp3, sp2 and sp C–H functionalization†
Yishou Wang,
Shichen Li,
Xinfeng Wang,
Yiming Yao,
Lei Feng* and
Chen Ma *
*
School of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, P. R. China. E-mail: chenma@sdu.edu.cn
First published on 17th February 2022
Abstract
I2/TBHP-promoted, one-pot, multi pathway synthesis of imidazopyridines and thiazoles has been achieved through readily available ethylarenes, ethylenearenes and ethynearenes. I2/TBHP as an initiator and oxidant is used to realize the C–H functionalization of this domino reaction. Simple and available starting materials, wide range of functional group tolerance, high potential for drug activity of the products and application in production are the advantageous features of this method.
Introduction
Nitrogen- and sulfur-containing heterocyclic scaffolds generally exhibit biological activities such as anti-inflammatory,1a anticancer,1b antibacterial,1c antiviral1d or signal conditioning activity.1e Such scaffolds can be found in many drug skeletons and natural products.2 For example, zolimidine, zolpidem, olprinone and saripidem are commercialized drugs, and YM-11124 and amiphenazole play an important signal regulation role in life activities.3 Therefore, exploring and developing new methods of synthesizing these compounds has very important commercial and application value (Scheme 1).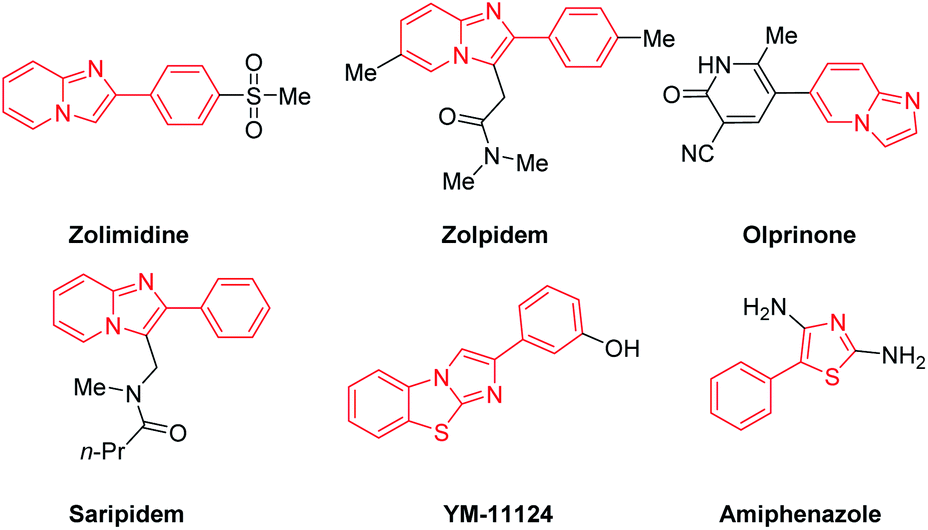 | ||
| Scheme 1 The drugs and biologically active compounds containing imidazopyridines or thiazoles units. | ||
To date, there are many ways to synthesize imidazole or thiazole rings. In general, the usual synthetic strategy is the direct condensation and cyclization of α-halo acetophenone and 2-aminopyridine (scheme 2a).4 Alternatively, acetophenone as a raw material, first completes the ortho-halogenation of the carbonyl group, and then condenses and cyclizes with 2-aminopyridine (Scheme 2b).5 In recent years, some new synthetic methods have been developed. For instance, when ethylbenzene6 and styrene7 act as starting materials, α-halo acetophenone is synthesized by adding an oxidant and a halogenating reagent (Scheme 2c and d). In addition, transition metal-catalyzed C–H bond activation methods for phenylacetylene8 have been reported (Scheme 2e).
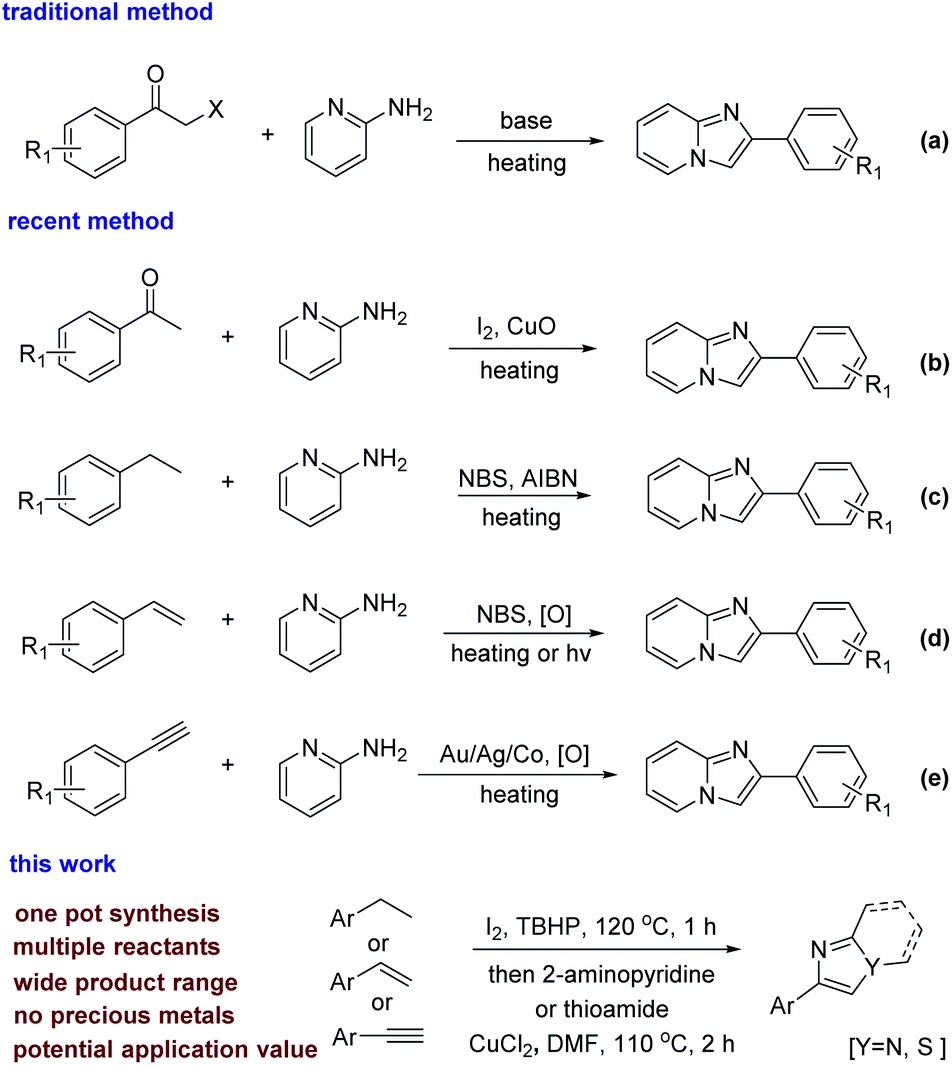 | ||
| Scheme 2 Approach for the synthesis of imidazo[1,2-a]pyridine and thiazole. | ||
In the literature reported in the past, α-bromo acetophenone has been used more often, and the reaction of α-bromo acetophenone is more mature.9 But α-bromo acetophenone, as a chemical weapon used for tearing, can cause great harm to our scientific researchers and the environment.10 Iodine compounds have better reactivity due to the easy removal of iodine, the reaction process is milder and greener, and the post-reaction treatment is also simple and convenient. Meanwhile, it is rare that ethylbenzene is directly functionalized as a raw material. Styrene mainly reacts by the addition of unsaturated bonds. Phenylacetylene yields the corresponding products through the coordinated activation of acetylene bonds by transition metals. While the reaction systems were not been reported which were simultaneously suitable for ethylbenzene, styrene and phenylacetylene. This has application value in the petrochemical industry, because the boiling points of ethylbenzene, styrene, and phenylacetylene are similar (136–145 °C). Their mixtures are widely present in industrial production,11 for example, the process of cracking of petroleum and the process of dehydrogenating ethylbenzene to synthesize styrene.12 The fact means that the system suitable for ethylbenzene, styrene and phenylacetylene has very cheap and readily available raw materials in practical applications. In this context, we developed a strategy that is applicable to them. This strategy can be used to synthesize imidazopyridines and thiazoles in one pot, and the source of raw materials is wide. α-Iodo acetophenone is an intermediate in this system, which means it is more environmentally friendly. We also explored the mixed-amplified experiment of the system, which proves it has potential industrial application value.
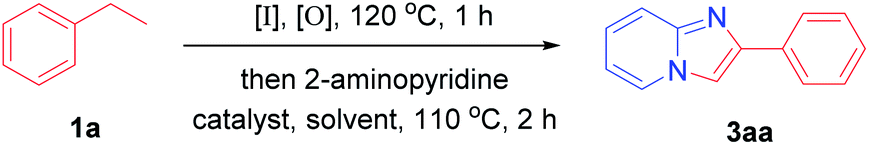
We chose ethylbenzene and 2-aminopyridine as standard substrates to screen the most suitable reaction conditions (Table 1). The product 3aa was obtained with a yield of 36% when CuI was used as a catalyst (entry 1). First, we studied the influence of different iodine sources on the reaction yield. When no iodine source, the target product can not be obtained, which proved iodine source necessary in the reaction process (entry 2). When N-iodosuccinimide (NIS), tetrabutylammonium iodide (TBAI) and KI were involved in the system, only trace products were formed (entry 3, 4 and 5). I2 was used as the iodine source, the product was obtained with a considerable yield of 76% (entry 6). The oxidant was also crucial for this reaction. When Na2S2O8, dibenzoyl peroxide (BPO) and O2 were used as oxidant in the reaction system, the yield of the product was 57%, 65% and a trace (entry 7, 8 and 9). Experiments showed that tert-butyl hydroperoxide (TBHP) is a better oxidant in the reaction system (entry 6). Then, the catalyst was optimized and screened. When no catalyst, the target product can be obtained with a yield of 30% (entry 10). When NH4Cl and FeCl3 was added, the yield of the product was 27% and 34% (entry 11 and 12). The product can not be obtained when AlCl3 and K2CO3 were used as catalysts (entry 13 and 14). Other transition metal salts also were involved as catalysts, such as Cu(OAc)2 and AgNO3, and the yield of 3aa was not as good as when CuCl2 was used (entry 15 and 16). Then, we tried different solvents and found DMF was best (entry 17, 18 and 19). Finally, we determined that in the presence of I2 (0.4 eq.), TBHP (3.0 eq.) and CuCl2 (0.5 eq.), DMF as solvent, the product was obtained with a yield of 82%.
|
|||||
|---|---|---|---|---|---|
| Entry | [I] | [O] | Catalyst | Solvent | Yieldb, % |
| a Reaction conditions: 1a (1 mmol), [I] (0.4 mmol), [O] (3 mmol), 120 °C, 1 h then 2-aminopyridine (2.0 mmol), CuCl2 (0.5 mmol), solvent (3 mL), 110 °C, 2 h.b Isolated yield. | |||||
| 1 | CuI | TBHP | — | DMSO | 36 |
| 2 | — | TBHP | CuCl2 | DMSO | None |
| 3 | NIS | TBHP | CuCl2 | DMSO | Trace |
| 4 | TBAI | TBHP | CuCl2 | DMSO | Trace |
| 5 | KI | TBHP | CuCl2 | DMSO | Trace |
| 6 | I2 | TBHP | CuCl2 | DMSO | 76 |
| 7 | I2 | Na2S2O8 | CuCl2 | DMSO | 57 |
| 8 | I2 | BPO | CuCl2 | DMSO | 65 |
| 9 | I2 | O2 | CuCl2 | DMSO | Trace |
| 10 | I2 | TBHP | — | DMSO | 30 |
| 11 | I2 | TBHP | NH4Cl | DMSO | 27 |
| 12 | I2 | TBHP | FeCl3 | DMSO | 34 |
| 13 | I2 | TBHP | AlCl3 | DMSO | None |
| 14 | I2 | TBHP | K2CO3 | DMSO | None |
| 15 | I2 | TBHP | Cu(OAc)2 | DMSO | 52 |
| 16 | I2 | TBHP | AgNO3 | DMSO | 54 |
| 17 | I2 | TBHP | CuCl2 | DCE | 65 |
| 18 | I2 | TBHP | CuCl2 | DMF | 82 |
| 19 | I2 | TBHP | CuCl2 | MeCN | 78 |
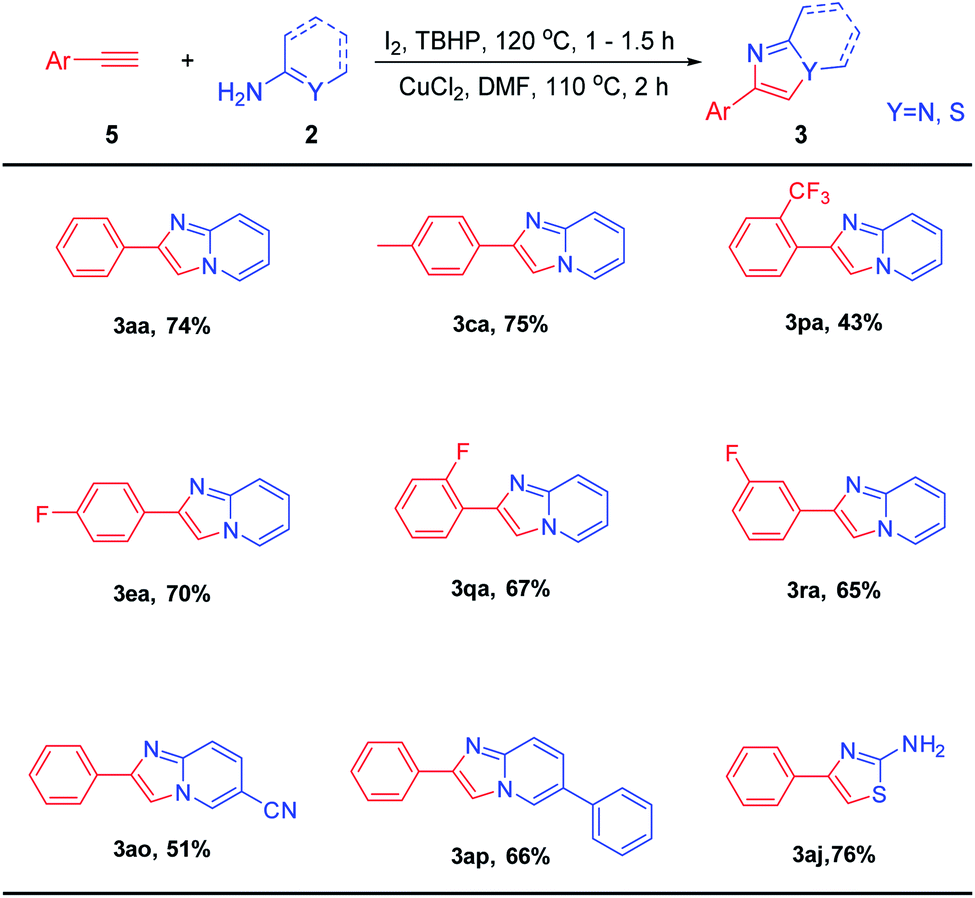
After determining the optimal reaction conditions, we explored the scope and limitations of this transformation by using substituted ethylarenes (Table 2). We found both the electron-deficient and electron-rich ethylarenes were suitable substrates to provide the desired products in moderate to excellent yields (3aa–3ea, 3ia–3ka). This proved that the reaction has a wide range of application to ethylarenes connected with various groups. The reaction had a higher yield when the ethylarenes had a larger conjugated structure (3ca, 3fa, 3ia). Meanwhile, substrates with large steric hindrance like 2-methyl ethylbenzene and 1,2-diphenylethane were also tolerant to this reaction to afford the desired products in moderate yields (3ba, 3ga). It was worth mentioning that 3ha zolimidine can also be synthesized by this reaction with a relatively good yield. Zolimidine is a common gastroprotective drug which was used to treat stomach ulcers and gastroesopha geal reflux disease. Excitingly, heteroarylethanes such as 2-ethylfuran and 2-ethylthiophene were also suitable for this reaction, and the products were obtained with relatively good yields (3la, 3ma). The scope of 2-aminopyridines was also studied. To our satisfaction, when electron-donating and electron-withdrawing groups substituted 2-aminopyridines were subjected to this reaction, the corresponding products (3ab–3ai) were isolated in 57–75% yields. To our delight, structures with carbon–sulfur unsaturated double bonds like thiourea and benzothioamide, thiazole hybrid structures like α-aminothiazole were also suitable for this reaction system, and the target products (3aj–3al) were furnished with good yields.
| a Reaction conditions: 1a (1 mmol), I2 (0.4 mmol), TBHP (3 mmol), 120 °C, 1 h then 2-aminopyridine (2.0 mmol), CuCl2 (0.5 mmol), solvent (3 mL), 110 °C, 2 h. |
|---|
 |
Having obtained encouraging results with ethylarenes, we turned our attention to C–H functionalization of ethylenearenes followed C–N bond construction coupling with 2-aminopyridines (Table 3). Excitingly, under optimized conditions, the corresponding product (3aa) can be obtained with a yield of 79%. Furthermore, whether styrenes were attached with electron-rich group such as –Me, or attached with electron-withdrawing groups such as –NO2 and –Cl at the meta or para positions, the corresponding products can be obtained in good yields (3ca–3na). The heterocycles, such as 2-bromo thiophene, did not affect the overall efficiency, and the corresponding products 3oa were furnished in moderate yields. 2-Aminopyridines with halogen (5-Br, 4-Br), electron-deficient (5-COOMe), and electron-rich (4-OMe) groups reacted smoothly to afford the corresponding products (3ag–3an) in moderate to good yields. Finally, as expected, both benzothioamide and 2-aminothiazole were tolerant to this system, and the corresponding products (3ak, 3al) can be obtained with good yields.
| a Reaction conditions: 1a (1 mmol), I2 (0.4 mmol), TBHP (3 mmol), 120 °C, 1–1.5 h then 2-aminopyridine (2.0 mmol), CuCl2 (0.5 mmol), solvent (3 mL), 110 °C, 2 h. |
|---|
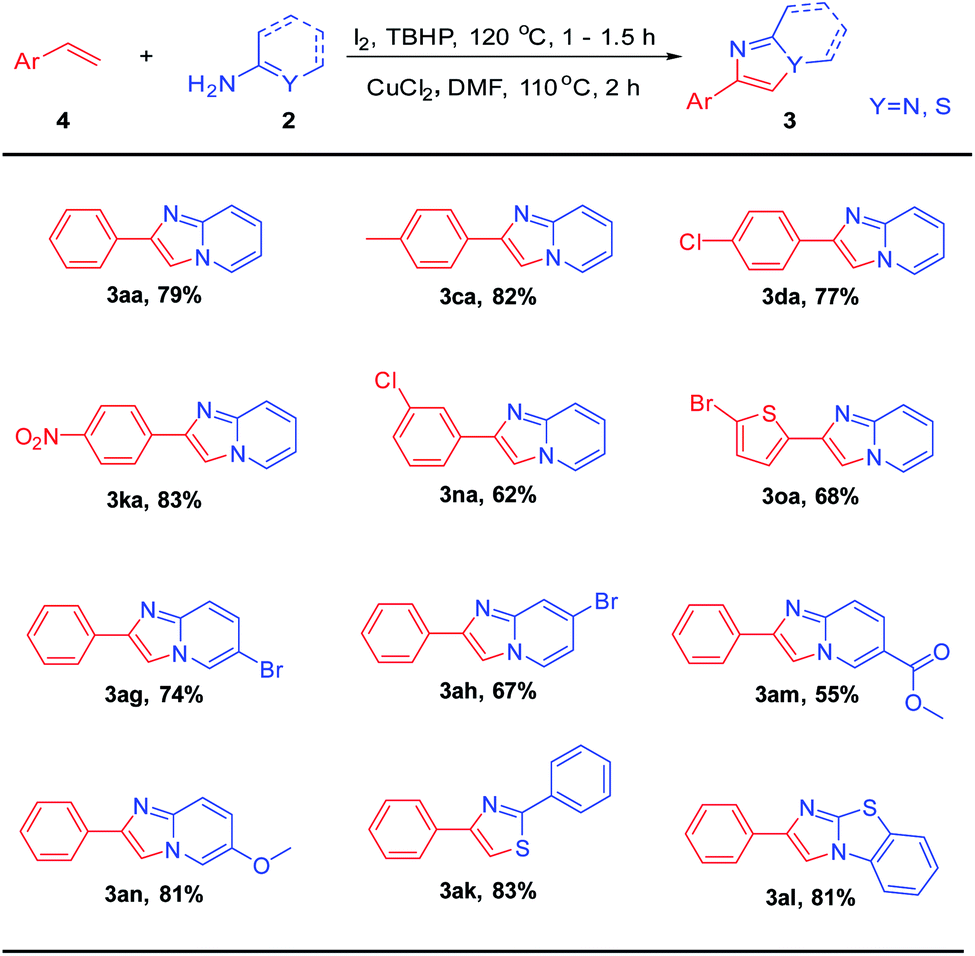 |
With results of ethylarenes and ethylenearenes in hand, we tried the C–H functionalization of phenylacetylene and then reacted with 2-aminopyridines under the optimal reaction conditions (Table 4). Excitingly, 3aa was provided with a yield of 74%. The substituted phenylacetylene (4-Me) afforded the corresponding imdazo[1,2-a]pyridine in good yield. Meanwhile, substrates with large steric hindrance like 2-trifluoromethyl phenylacetylene was also tolerant to this reaction to afford 3pa in moderate yields. The fluorine groups on different substitution positions (2-F, 3-F, 4-F) reacted smoothly to afford the corresponding products (3ea, 3qa, 3ra) in moderate to good yields. Finally, when nitrile or phenyl substituted 2-aminopyrides were subjected to this reaction, the corresponding products (3ao, 3ap) were isolated in good yields. Thiourea also can react to get 3aj in 76% yield.
| a Reaction conditions: 1a (1 mmol), I2 (0.4 mmol), TBHP (3 mmol), 120 °C, 1–1.5 h then 2-aminopyridine (2.0 mmol), CuCl2 (0.5 mmol), solvent (3 mL), 110 °C, 2 h. |
|---|
 |
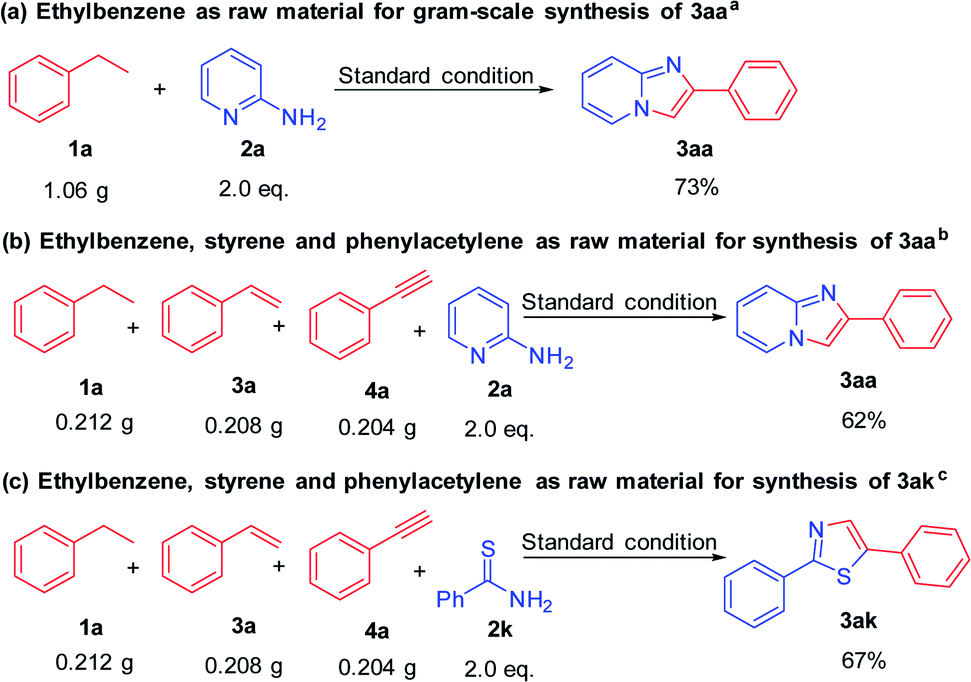 | ||
| Scheme 3 Gram-scale synthesis of 3aa and 3ak. aReaction conditions: 1a (10 mmol), I2 (4 mmol), TBHP (30 mmol), 120 °C, 1 h 10 min then 2-aminopyridine (20 mmol), CuCl2 (5 mmol), solvent (20 mL), 110 °C, 4 h. bReaction conditions: 1a (2 mmol), 3a (2 mmol), 4a (2 mmol), I2 (2.4 mmol), TBHP (18 mmol), 120 °C, 1 h 20 min then 2-aminopyridine (12 mmol), CuCl2 (3 mmol), solvent (10 mL), 110 °C, 2.5 h. cReaction conditions: 1a (2 mmol), 3a (2 mmol), 4a (2 mmol), I2 (2.4 mmol), TBHP (18 mmol), 120 °C, 1 h 20 min then 2k (12 mmol), CuCl2 (3 mmol), solvent (10 mL), 110 °C, 2 h. | ||
In order to evaluate the practical application value and potential of this reaction system, we carried out a gram-scale amplification experiment under standard reaction conditions (Scheme 3). When the scale of ethylbenzene reached 10 mmol (1.06 g), this transformation proceeded smoothly and imidazo[1,2-a]pyridine 3aa was obtained in 73% yield. Next, we made a bolder attempt, ethylbenzene (2 mmol), styrene (2 mmol) and phenylacetylene (2 mmol) were mixed together to examine this system, and the target product 3aa can also be obtained with good yield. 3ak can also be obtained from this mixed-amplified experiment in 67% yield. This proved it has great value in actual production applications.
In order to carefully explore the process of the mechanism in the reaction, some control experiments were performed (Scheme 4). It was found that ethylbenzene 1a can be converted to acetophenone 1aa under oxidant-promoted conditions.13 1aa can be confirmed by HRMS and GC-MS. Acetophenone 1aa can further react to obtain α-iodo ketone 1ab under the conditions of I2/TBHP catalysis. 1ab reacted with 2-aminopyridine smoothly to afford product 3aa in 90% yield in CuCl2/DMF conditions (Scheme 4a). α-Iodo ketone 1ab was an important intermediate in the reaction process, which was also obtained from compound 2a and 3a (Scheme 4b). α-Iodo ketone 1ab was easily converted to phenylglyoxal 1ac in DMSO,14 and further condensed with 2-aminopyridine to form 1ad, which also explained why the yield of 3aa in DMSO was not as good as in DMF during the screening of reaction conditions (Scheme 4c).
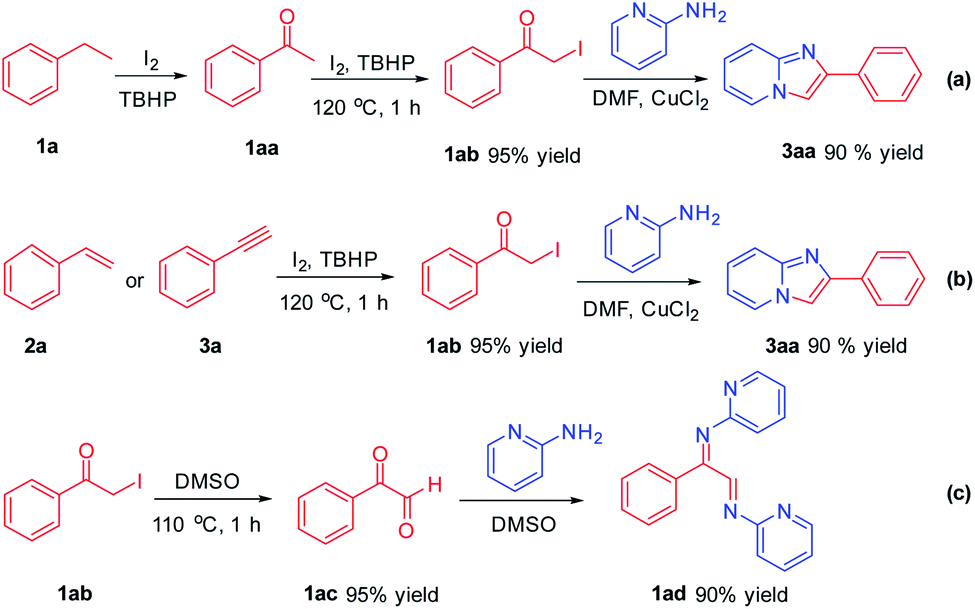 | ||
| Scheme 4 The control experiments. | ||
On the basis of the control experiments described above and reported literature,15 a possible mechanism for multiple C–H activation followed by circularization cross-coupling is outlined in Scheme 5.
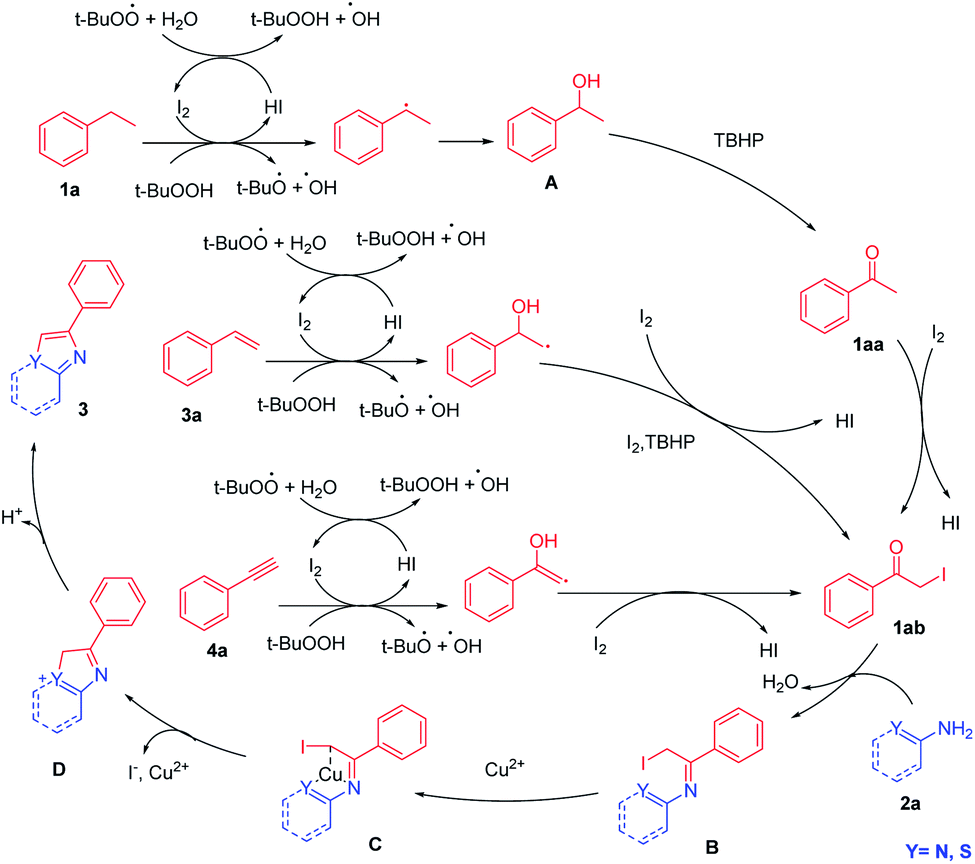 | ||
| Scheme 5 The proposed reaction mechanism. | ||
Initially, ethylbenzene was initiated by I2/TBHP and underwent benzylic radical of ethylbenzene process to obtain 1-phenyl ethanol (A). Then, 1-phenyl ethanol (A) was converted to acetophenone (1aa) via TBHP oxidation, and 1aa can be confirmed by TLC, HRMS and GC-MS. Acetophenone (1aa) on iodination offered α-iodo acetophenone (1ab). Styrene (3a) and phenylacetylene (4a) can also be converted into this key intermediate (1ab) through the addition of hydroxyl radicals to unsaturated bonds and the catalysis of I2/TBHP. 1ab can be confirmed by TLC and HRMS. α-Iodo acetophenone (1ab) and 2a condensed to obtain intermediate (B). Cu2+ coordinated with the two atoms in the C to activate, and the pair of electrons on the Y atom (Y = N, S) carried out a nucleophilic attack on the C–I bond. I− left and C–Y was built to get intermediate D. The deprotonation of D provided the desired product 3aa.
Conclusions
In conclusion, we have developed a multi pathway, environmentally friendly domino synthesis strategy promoted by I2/TBHP. This strategy is applicable to ethylarenes, ethylenearenes and ethynearenes that can react with 2-aminopyridine, 2-aminothiazole and benzothioamide to obtain the corresponding imidazole or thiazole structure. Its resistance to various substituent groups is also excellent. Its mixed-amplified experiment indicates the potential application in industrial production. Owing to the generality of the reaction, this protocol should be of great utility in organic methodology and industrial application.Experimental
General information
The Reagents and solvents are commercially purchased and used directly unless otherwise mentioned. All reactions were carried out in a sealed Schlenk tubes and monitored by TLC. Flash column chromatography was performed with silica gel (300–400 mesh). 1H and 13C NMR spectra were recorded on a Bruker Avance 400 instrument (400 MHz for 1H and 101 MHz for 13C NMR spectroscopy). Data were reported as chemical shifts in ppm relative to TMS (0.00 ppm) for 1H and CDCl3 (77.16 ppm) or DMSO-d6 (39.52 ppm) for 13C. HRMS spectra were measured on a Q-TOF instrument in positive-ion mode with an ESI ion source.General procedure for substrates
Ethyl arenes or ethylene arenes or ethyne arenes (1.0 mmol), I2 (0.40 mmol), tert-butyl hydroperoxide (TBHP, 3.5 mmol, 70%, aq. solution) were placed in a sealed tube equipped with a magnetic stirring bar. These mixtures were stirred and reacted at 120 °C for 1–1.5 h. After disappearance of the reactant monitored by TLC, 2-aminopyridine (2.0 mmol) and CuCl2 (0.5 mmol) in DMF (3 mL) was added to the sealed tube at 110 °C and heated for 2 h. After the reaction, 30 mL of saturated Na2S2O3 solution was added to the reaction solution to quench the remaining iodine. Then the solution was extracted with ethyl acetate (3 × 30 mL), dried over anhydrous Na2SO4. The solid was removed by filtration, and the solution was concentrated under vacuum. The residue was purified through silica gel eluting with ethyl acetate/hexanes to give the product.Characterization data for the products
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Science Foundation of China (no. 21572117) and the Shandong Key Research Program (no. 2019JZZY021015 and 2019GHY112053) for their financial support. We are also grateful to the Analytical Center for Structural Constituent and Physical Property of Core Facilities Sharing Platform, Shandong University for their technology and service support.Notes and references
- (a) I. B. Sayeed, V. Lakshma Nayak, M. A. Shareef, N. K. Chouhan and A. Kamal, MedChemComm, 2017, 8, 1000–1006 RSC; (b) R. Lefin, M. M. van der Walt, P. J. Milne and G. Terre'Blanche, Bioorg. Med. Chem. Lett., 2017, 27, 3963–3967 CrossRef CAS PubMed; (c) B. S. Santaniello, M. J. Price and J. K. Murray Jr, J. Chem. Educ., 2017, 94, 388–391 CrossRef CAS; (d) A. R. Katritzky, Y. J. Xu and H. J. Tu, Org. Chem., 2003, 68, 4935–4937 CrossRef CAS PubMed; (e) R. Martin, M. Rodriguez Rivero and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 7079–7082 CrossRef CAS PubMed.
- A. J. Stasyuk, M. Banasiewicz, M. K. Cyrań ski and D. T. Gryko, J. Org. Chem., 2012, 77, 5552–5558 CrossRef CAS PubMed.
- (a) S. Z. Langer, S. Arbilla, J. Benavides and B. Scatton, Adv. Biochem. Psychopharmacol., 1990, 46, 61 CAS; (b) K. Mizushige, T. Ueda, K. Yukiiri and H. Suzuki, Cardiovasc. Drug Rev., 2002, 20, 163 CrossRef CAS PubMed; (c) L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305 CrossRef CAS PubMed; (d) R. J. Boerner and H. J. Moller, Psychopharmakother., 1997, 4, 145 Search PubMed; (e) N. Gunaganti, A. Kharbanda, N. R. Lakkaniga, L. Zhang, R. Cooper, H. Y. Li and B. Frett, Chem. Commun., 2018, 54, 12954–12957 RSC; (f) T. Guo, J. J. Liang, S. Yang, H. Chen, Y. N. Fu, S. L. Han and Y. H. Zhao, Org. Biomol. Chem., 2018, 16, 6039–6046 RSC.
- (a) S. Ulloora, A. V. Adhikari and R. Shabaraya, Chin. Chem. Lett., 2013, 24, 853–856 CrossRef CAS; (b) A. Herath, R. Dahl and N. D. P. Cosford, Org. Lett., 2010, 12, 412–415 CrossRef CAS PubMed.
- (a) M.-L. Feng, S.-Q. Li, S.-Y. Chen and X.-Q. Yu, Green Chem., 2019, 21, 1619–1624 RSC; (b) D. C. Mohan, S. N. Rao, C. Ravi and S. Adimurthy, Asian J. Org. Chem., 2014, 3, 609–613 CrossRef CAS; (c) W. Ge, X. Zhu and Y. Wei, Eur. J. Org. Chem., 2013, 27, 6015–6020 CrossRef.
- L. Chen, H. Zhu, J. Wang and H. Liu, Molecules, 2019, 24(5), 893/1–893/13 CAS.
- (a) H. Shinde Mahesh and A. Kshirsagar Umesh, Green Chem., 2016, 18(6), 1455–1458 RSC; (b) S. Samanta, S. Jana, S. Mondal, K. Monir, S. K. Chandraa and A. Hajraa, Org. Biomol. Chem., 2016, 14(22), 5073–5078 RSC; (c) A. Das and K. R. Justin Thomas, Asian J. Org. Chem., 2020, 9(11), 1820–1825 CrossRef CAS.
- (a) S. k. Rasheed, D. N. Rao and P. Das, Asian J. Org. Chem., 2016, 5(10), 1213–1218 CrossRef; (b) S. K. Samantaa and M. K. Bera, Org. Biomol. Chem., 2019, 17(26), 6441–6449 RSC; (c) E. P. A. Talbot, M. Richardson, J. M. McKenna and F. D. Toste, Adv. Synth. Catal., 2014, 356(4), 687–691 CrossRef CAS PubMed; (d) C. He, J. Hao, H. Xu, Y. Mo, H. Liu, J. Han and A. Lei, Chem. Commun., 2012, 48(90), 11073–11075 RSC; (e) P. Starkov, F. Rota, J. M. D'Oyley and T. D. Sheppard, Adv. Synth. Catal., 2012, 354(17), 3217–3224 CrossRef CAS; (f) J. Zeng, Y. J. Tan and X.-W. Liu, Org. Lett., 2012, 14, 4386–4389 CrossRef CAS PubMed; (g) Y. Gao, M. Yin and H. Jiang, Adv. Synth. Catal., 2013, 355, 2263–2273 CrossRef CAS.
- (a) B. Hoi and P. H. Ng, J. Org. Chem., 1954, 19, 1370–1375 CrossRef; (b) E. S. Hand and W. W. Paudler, J. Org. Chem., 1978, 43(4), 658–663 CrossRef CAS.
- (a) C. S. Kerem, G. Sertac and E. Erhan, J. Forensic Sci., 2016, 61, 551–554 CrossRef PubMed; (b) M. M. Roberto, Med. Sci. Law, 2020, 39, 79–84 Search PubMed; (c) L. Annie, S. Andrew and S. Hoffman Robert, J. Emerg. Med., 2007, 32, 309–310 Search PubMed.
- (a) T.-K. Ariana, H. Jurn and K. Rajamani, Langmuir, 2015, 12, 3771–3778 Search PubMed; (b) B. Cao, H. Hinode and T. Kajiuchi, J. Membr. Sci., 1999, 156, 43–47 CrossRef CAS; (c) M. Michael, V. Frederik and A. Luc, J. Am. Chem. Soc., 2010, 132(43), 15277–15285 CrossRef PubMed.
- (a) C. Yu and H. Xu, Sep. Purif. Technol., 2011, 78, 249–252 CAS; (b) V. Zhyznevskiy, R. Tsybukh and V. Gumenetskiy, React. Kinet. Catal. L, 2000, 71, 209–215 CrossRef CAS; (c) R. Watanabe, S. Yasushi and M. Masahiko, Catal. Lett., 2009, 131, 54–58 CrossRef CAS.
- (a) K.-P. Shing, B. Cao and C.-M. Che, J. Am. Chem. Soc., 2018, 140, 7032–7042 CrossRef CAS PubMed; (b) G. Urgoitia, R. SanMartin and E. Domínguez, Chem. Commun., 2015, 51, 4799–4802 RSC; (c) M. Munkir Hossain and S.-G. Shyu, Tetrahedron, 2016, 72(29), 4252–4257 CrossRef.
- (a) K. K. Durga Rao Viswanadham, M. Prathap Reddy, P. Sathyanarayana, O. Ravi, R. Kant and S. Reddy Bathula, Chem. Commun., 2014, 50(88), 13517–13520 RSC; (b) S. Guha, K. Imran, P. Mukherjeea and G. Sekar, Chem. Commun., 2017, 53(79), 10942–10945 RSC; (c) Y.-p. Zhu, F.-c. Jia, M.-c. Liu and A.-x. Wu, Org. Lett., 2012, 14(17), 4414–4417 CrossRef CAS PubMed.
- (a) Q. Cai, M.-C. Liu, B.-M. Mao, X. Xie, F.-C. Jia, Y.-P. Zhu and A.-X. Wu, Chin. Chem. Lett., 2015, 26, 881–884 CrossRef CAS; (b) K. S. Vadagaonkar, H. P. Kalmode, K. Murugan and A. C. Chaskar, RSC Adv., 2015, 5, 5580–5590 RSC; (c) H. P. Kalmode, K. S. Vadagaonkar and A. C. Chaskar, RSC Adv., 2014, 4, 60316–60326 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data and copies of NMR spectra. See DOI: 10.1039/d1ra07438e |
| This journal is © The Royal Society of Chemistry 2022 |