
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMitoxantrone dihydrochloride, an FDA approved drug, binds with SARS-CoV-2 NSP1 C-terminal†
Prateek Kumar‡
,
Taniya Bhardwaj‡ and
Rajanish Giri *
*
Indian Institute of Technology Mandi, School of Basic Sciences, VPO Kamand, Himachal Pradesh 175005, India. E-mail: rajanishgiri@iitmandi.ac.in; Fax: +91-01905-267138; Tel: +91-01905-267134
First published on 16th February 2022
Abstract
One of the major virulence factors of SARS-CoV-2, NSP1, is a vital drug target due to its role in host immune evasion through multiple pathways. NSP1 protein is associated with inhibiting host mRNA translation by binding to the small subunit of ribosome through its C-terminal region. Previously, we have shown the structural dynamics of the NSP1 C-terminal region (NSP1-CTR) in different physiological environments. So, it would be very interesting to investigate the druggable compounds that could bind with NSP1-CTR. Here, in this article, we have performed different spectroscopic technique-based binding assays of an anticancer drug mitoxantrone dihydrochloride (MTX) against the NSP1-CTR. We have also performed molecular dynamics simulations of the docked complex with two different force fields up to one microsecond. Overall, our results have suggested good binding between NSP1-CTR and MTX and may have implications in developing therapeutic strategies targeting the NSP1 protein of SARS-CoV-2.
Introduction
Non-structural proteins (NSPs) of coronaviruses exclusively carry out replication and translation of the viral genome, hence they are considered to be one of the prime targets for drug discovery.1 SARS-CoV-2 NSP1 is the first protein to be translated as a part of polyprotein 1a and 1ab, and is considered important as it works to suppress the host immune system.2 It restricts the host mRNA binding to ribosome by associating with the 40S subunit of the host cell using its C-terminal region (CTR).3,4 Upon interaction with the ribosome, the NSP1-CTR partially gains α-helical structure propensity.3,4 In our previous study, we observed that NSP1-CTR remains in disordered conformation in isolation and is found to adopt a secondary structure in different environments.5 It also interacts with mRNA nuclear exporter heterodimer NXF1–NXT1 and hampers the association of mRNA with NXF1, resulting in the abolishment of NXF1 docking on nuclear pore complex.6 Recently, a study exposed the detailed insights of host translation inhibition where the eukaryotic translation initiation factors (eIF3 and eIF4F) allosterically control the NSP1 association with ribosome in the early stages of translation in absence of mRNA.7 Moreover, mutagenesis-based studies have revealed several key residues in its CTR that are responsible for its interaction with ribosome and abrogation of the translational activity.4,6 All these facts make NSP1-CTR an essential drug target that can lead to ultimate blockage of NSP1 activity.Many studies for drug discovery have been performed in the last few months of pandemic, including the drug repurposing as a time and cost-effective approach.8,9 Drug repurposing is the most effective approach to identifying potential drugs against this deadly pandemic-causing disease. The scientific community has tried several existing FDA-approved drugs including different antivirals, antibiotics, steroids, to name a few, against the SARS-CoV-2 infection.9 Drugs like remdesivir, tocilizumab, and favipiravir are being given to the infected patients.8,10–12 In addition, various druggable compounds, including natural compounds, have been reported to be effective against the virus.13,14
One such drug mitoxantrone dihydrochloride (MTX), has been repurposed and is demonstrated to inhibit the SARS-CoV and SARS-CoV-2 virus entry into cells.15 Previously, the antiviral activity of MTX has been established against many other viruses such as Herpes Simplex Virus 1 (HSV-1), and Vaccinia virus.16,17 Additionally, the anticancer activity of this drug against lymphoma, breast cancer, and prostate cancer is very well-known.18–21 Overall, these properties make it a potential candidate to be tested for its specific binding target with SARS-CoV-2 proteins.
In this report, we have demonstrated the binding of anticancer compound MTX with the C-terminal region of NSP1 (NSP1-CTR) using spectroscopy-based techniques and extensively long computational simulations. According to the finding, MTX binds with NSP1-CTR in its disordered conformation and can be developed into a drug against the virus. Furthermore, this study may provide a useful insight on interacting residues of the C-terminal region for developing antiviral against SARS-CoV-2 NSP1 protein which could prevent its activity inside the host.
Results
Given the functional importance of NSP1 protein and its disordered C-terminal region, the druggability of NSP1 must be explored to identify novel inhibitors or repurposed drugs against the SARS-CoV-2. Furthermore, the blocking of NSP1-CTR may possibly restrict the binding of NSP1 with ribosomes inside host cells. Following this, we have investigated the binding of an antineoplastic drug MTX with the C-terminal tail of NSP1. Herein, we have performed binding kinetics of MTX with NSP1-CTR using fluorescence and CD spectroscopic techniques. Further, the fluorescence lifetime and anisotropy decay have been measured in the presence and absence of the drug. We have also confirmed the binding through computational modelling and docking studies.Fluorescence intensity analysis
The binding of MTX with NSP1-CTR is studied experimentally by monitoring the changes in intrinsic fluorescence of NSP1 tryptophan (Trp161) residue. As shown in Fig. 1A, the fluorescence intensity of tryptophan residue of the peptide is observed to be decreasing gradually on increasing the concentration of MTX in the solution. The addition of MTX has substantially quenched the tryptophan fluorescence intensity which indicates the possible binding of MTX with NSP1-CTR. As discussed in previous report on structural dynamics of NSP1-CTR, the λmax of the tryptophan residue occurs at 346 nm. Therefore, the dissociation constant (Kd) is obtained by considering the maximum fluorescence intensity at 346 nm using the non-linear fitting of one site-specific binding. The Kd for MTX–NSP1-CTR interaction is calculated to be 31.83 μM (R2 = 0.9) (Fig. 1B). | ||
| Fig. 1 Tryptophan (Trp161) fluorescence emission analysis of NSP1-CTR peptide with MTX. (A) The decreasing intrinsic fluorescence intensity of NSP1-CTR upon addition of MTX in increasing concentrations (0 to 300 μM). (B) The fluorescence intensity at 346 nm is plotted against the concentration of MTX. Inset: shows the non-linear fitting of one-site specific binding with the calculated dissociation constant. | ||
Fluorescence lifetime measurements
With conformational changes in proteins due to variation in temperature, viscosity, and surrounding solvent, the fluorescence lifetime changes.22 Therefore, the binding and conformational changes in the NSP1-CTR–MTX complex are observed through fluorescence lifetime measurement, where the decay in fluorescence of tryptophan fluorophore was recorded. As observed in graphs depicted in Fig. 2A and B, the average fluorescence lifetime of Trp161 in NSP1-CTR shows a decreasing trend compared to the fluorescence lifetime in unbound conformation (see Table 1). The average lifetime of unbound protein is measured to be 1.98 ns with a chi-square (χ2) value of 1.11, whereas, in presence of 200 μM MTX, the average lifetime decreased to 1.66 ns (χ2 = 1.08). The changes in components of Trp161 lifetime are given in detail in ESI Table 1.† The declining fluorescence lifetime is evident of binding of MTX with NSP1-CTR peptide. | ||
| Fig. 2 Assessment of Trp161 by using fluorescence lifetime measurement. (A) Fluorescence lifetime decay curve of NSP1-CTR peptide in absence and presence of drug MTX at four increasing concentrations. (B) Illustration of decrease in average fluorescence lifetime of Trp161 with increasing concentration of MTX. | ||
| Mitoxantrone concentration (μM) | τ (average lifetime, ns) | χ2-Value |
|---|---|---|
| 0 (only NSP1-CTR; 7.5 μM) | 1.98 | 1.11 |
| 25 | 1.81 | 1.06 |
| 50 | 1.77 | 1.13 |
| 100 | 1.76 | 1.24 |
| 200 | 1.66 | 1.08 |
Time-resolved anisotropy decay analysis
As aforementioned, tryptophan fluorescence is susceptible to changes in its local environment. Its global and local rotations are affected by the changes in the surroundings. Therefore, to further observe the binding of MTX with NSP1-CTR, we have performed an anisotropy decay analysis of Trp161 residue. Here, the fluorescence decays of tryptophan residues in perpendicular and parallel emission polarization of light are recorded. The decay curve is fitted by using a bi-exponential decay function which defines the fast (θ1) and slow (θ2) rotational correlational time where θ1 is the local rotation of Trp161 while the θ2 is the global rotation of the peptide.23 In unbound state, NSP1-CTR (30 μM) has an average rotational correlation time (τr) equals to 4.46 ns (χ2 = 1.19) (Fig. 3A and B). Interestingly, after binding with MTX (25 μM), its average rotational correlation time decreases to 962.1 ns (χ2 = 1.14) (Fig. 3C and D). θ1 and θ2 for NSP1-CTR have been calculated to be 1.13 ns and 15.6 ns respectively, while in presence of MTX, the θ1 and θ2 are observed to be 1.13 ns and 1070.1 ns respectively. These results correspond to the interaction of the drug with the peptide as binding of MTX has slowed the global rotation of the peptide. | ||
| Fig. 3 Fluorescence anisotropy decay measurements of NSP1-CTR. (A) The conformational dynamics in unbound state of NSP1-CTR (30 μM), and (C) in the bound state with compound MTX (25 μM). (B) and (D) depict the residual of the fitting curve corresponding to unbound and bound state of NSP1-CTR, respectively. | ||
Circular dichroism-based conformational studies
The far-UV CD spectra of NSP1-CTR were recorded in the presence and absence of MTX. As reported before, NSP1-CTR remains in a disordered state in isolation.5 Therefore, in the unbound state, the negative ellipticity at ∼200 nm wavelength represents a typical spectrum corresponding to the disordered state of NSP1-CTR. However, the presence of MTX at a concentration of 25 μM, has caused minimal fluctuations in the negative ellipticity at ∼200 nm (Fig. 4). Overall, these CD results demonstrate that the drug MTX does not cause any noticeable structural changes in the secondary structure of the peptide, but MTX may bind with NSP1-CTR in the latter's disordered form.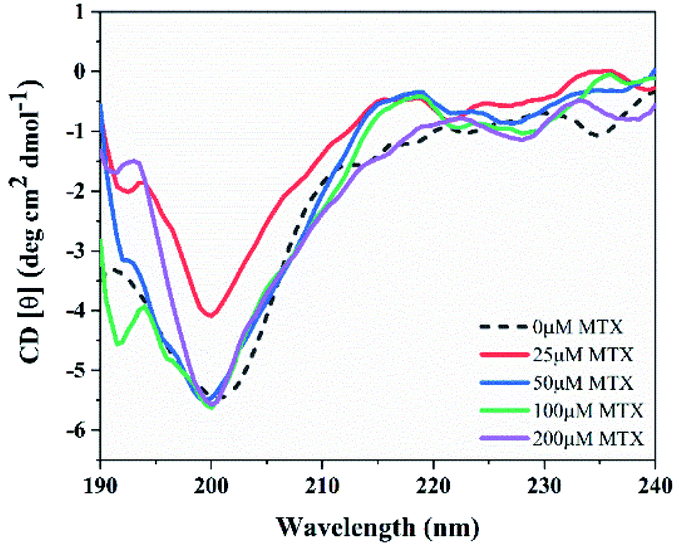 | ||
| Fig. 4 Circular dichroism spectra of NSP1 C-terminal upon interaction with compound MTX on varying concentrations from 0 to 200 μM in comparison with the unbound state of NSP1-CTR at 25 μM. | ||
Molecular docking analysis
As previously reported by our group, the NSP1-CTR is mainly in disordered conformation and contains helical structure forming tendencies in different environmental conditions.5 This depicts that it gains alpha helix upon interacting with its binding partner i.e., 40S ribosomes (PDB ID: 6ZOJ). MD simulations of its modelled structure have also revealed the α-helical structure, therefore, we have selected the structure at last frame (at 500 ns; described as NSP1–F1 in our previous study) of NSP1-CTR from the simulation trajectory in aqueous conditions.5The MTX drug is docked against NSP1-CTR by enclosing the whole structure as a receptor grid, as the structure constitutes no proper binding pocket. Glide scoring function of the Schrodinger suite is employed for docking and Prime module for MM/GBSA calculation. The MTX has scored a docking score of −5.335 kcal mol−1 and binding energy of −47.49 kcal mol−1. Besides, the strong binding with six hydrogen bonds and two salt bridge interactions has been observed between NSP1-CTR and MTX (Fig. 5). According to binding pose and ligand interaction diagram, the residues involved are Ser142, Asp144, Glu155, and Arg171 (Fig. 5).
 | ||
| Fig. 5 In silico interaction analysis through molecular docking: representation of binding poses and interactions of NSP1-CTR and ligand complexes calculated by enclosing whole structure in the grid. (A) The surface representation of NSP1-CTR with docked drug MTX, (B) 2D interaction showing interacting residues of NSP1-CTR with MTX, (C and D) surface and cartoon views of superimposed binding poses of mitoxantrone (blue) and top ranked similar compound (PubChem ID: 88654295; in orange) and conformer (PubChem ID: 9543511; in light green), respectively. | ||
Besides, we have also performed docking calculations on 665 compounds from the PubChem database, similar to mitoxantrone or having anthracene as their parent molecule (referred as ‘SC’, top selected compounds shown in ESI Fig. 1A and B†). As per our observations, a total of 32 compounds have been identified with docking scores more than mitoxantrone (−5.3 kcal mol−1). The SC1 compound with PubChem ID: 88654295 ((2R)-2-amino-N-[2-[[4-[2-[[(2R)-2-amino-3-sulfanylpropanoyl]amino]ethylamino]-5,8-dihydroxy-9,10-dioxoanthracen-1-yl]amino]ethyl]-3-sulfanylpropanamide), has highest docking score of −6.9 kcal mol−1 amongst all other compounds and have multiple interactions with Ser142, Asp144, Asp147, Glu148, Glu155, Gln158, Thr170, and Arg171. The 3D binding pose depicting multiple interactions is shown in the Fig. 5C, superimposed with binding pose of MTX. The other resultant compounds with docking scores and binding energies are shown in ESI Table 2.† Majority of the compounds have shown interactions with the aforementioned residues which are similar to the interacting residues of MTX. Out of 32 selected compounds, top 5 best scoring compounds (SC1–SC5) are depicted in ESI Fig. 3A and B† with their 3D binding poses and 2D interaction diagrams. Among these binding poses, it has been observed that the residues like Ser142, Asp144, Glu155, Thr170 and Arg171 are commonly interacting with the compounds.
Furthermore, we have also checked the interaction of around 160 similar conformers (referred as ‘SF’, top selected compounds are shown in ESI Fig. 2†) of mitoxantrone from the PubChem database. All these conformers are anthracene molecules with the same backbone and different functional groups. In molecular docking of these compounds, most have shown similar interactions and docking score of maximum −6.1 kcal mol−1. As observed earlier in the docking study with MTX and its similar compounds, the SF molecules also have interactions with residues like Ser142, Asp144, Asp147, Glu148, Glu155, Gln158, Thr170, and Arg171. The binding poses and interacting residues of top 5 best scoring compounds (SF1–SF5) are shown in ESI Fig. 4A and B.† In ESI Table 3,† we have listed the 15 identified compounds with docking score more than −5.3 kcal mol−1 along with their binding energies.
MD simulations analysis of NSP1-CTR and MTX complex
 | ||
| Fig. 6 Molecular dynamics simulation analysis of MTX bound NSP1-CTR using OPLS 2005 forcefield. (A) Histogram plot for representation of the fraction of interaction with residues throughout the simulations where hydrogen bonds (green), water bridges (blue), hydrophobic (purple), and ionic (red) are shown, and one bar with multiple colors shows multiple interactions with that specific residue and (B) timeline representation of interacting residues where dark color depicts greater number of interactions. | ||
Discussion
Initially, MTX was developed as an analog of doxorubicin, an anthracycline, and is currently being used as an anticancer drug since the 1980s due to its better anti-tumor activity and less cardiotoxic properties.19 The antiviral activity of this drug has been also established for several viruses. Recently, Zhang et al. has observed the role of heparan sulfate, which is present on the cell surface, as a binding mediator of ACE-2 assisted attachment of SARS-CoV and SARS-CoV-2 in host cells. The authors have abolished this interaction using the drug MTX which targets the heparan sulfate on host cells ultimately preventing the viral entry.15 In Vaccinia virus, it prevents viral maturation and assembly by blocking the processing of mature structural proteins.17 Another study on Herpes Simplex Virus 1 (HSV-1) reported the binding of MTX has suppressed the viral gene expression.16 Moreover, a report based on computational docking has shown that MTX has a higher docking score against SARS-CoV-2 main protease among various other drugs.24In this manuscript, we have performed experimentations on NSP1-CTR with literature identified anticancer drug, mitoxantrone. The synthetic peptide of NSP1-CTR has a single Trp residue at 161st position, monitored for fluorescence quenching and lifetime measurements in presence of the chosen compound MTX. Previously, it has been reported that the MTX drug form dimer and aggregates at higher concentrations more than 100 μM. According to absorbance spectra of MTX at varying concentrations, it has been shown that the maxima peaks around 660 nm, 610 nm, and 560 nm signifies the monomer, dimer, and higher aggregates, respectively.25 Therefore, we have also checked the absorbance of MTX at two different concentrations including its identified Kd concentration against NSP1-CTR and higher concentration used in this study (ESI Fig. 10†). As per our absorbance spectra, we have observed two significant peaks at 660 nm and 610 nm which points towards the monomeric and dimeric forms of MTX. There is no peak (around 560 nm) observed which depicts the formation of higher aggregates. Based on these observations, it can be inferred that the drug MTX may have formed dimer during interaction with NSP1-CTR.
Further, the compound MTX has exerted a docking score of more than −5.0 kcal mol−1 and significantly good binding energy (MM-GBSA score). Its interaction with key residues like Ser142, Asp144, Glu155, Arg171 is also observed. The docked complex has been investigated with extensive MD simulations up to microsecond time where some interactions with residues Asp144 and Tyr154 are in contact for long simulation time. Due to structural flexibility of NSP1-CTR, some of the interactions are not retained in any of the simulation. However, the simulation system has been stabilized by some other contacts formed by residues with MTX. Taking these results together, it is evident that MTX binds NSP1-CTR with excellent efficiency. To further check the structural changes in NSP1-CTR upon binding with MTX, we performed CD spectroscopy at different concentrations of MTX. The observation suggests that MTX does not change any conformation of NSP1-CTR even at higher concentrations up to 200 mM. Based on simulations with two different forcefields, it is also clearly evident that the NSP1-CTR interacts with compound MTX in its disordered state as no structure gain property is observed in virtual simulations similar to experimental observation through CD spectroscopy. In fact, the loss of secondary structure element is observed in the simulations. Previously, we have shown the similar case of disorder-based protein–ligand interaction of a cancer regulatory transcription factor c-Myc upon binding with a natural molecule salvianolic acid b where no structure gain is observed in peptide upon interaction with the compound at varying concentrations.26 However, due to the unavailability of full-length NSP1 or the N-terminal region, the binding experiments couldn't be performed, which may or may not have any implications based on the current study.
To get an insightful detail about parent backbone and conformation of MTX, we have also checked the interactions of more than 800 similar compounds or conformers (obtained from PubChem database) to MTX. The docking calculations are performed with similar compounds and conformations to MTX against NSP1-CTR which has anthracene as backbone. With similar backbone and different functional groups or side chains, these compounds have shown similar interactions like MTX with NSP1-CTR. Earlier, one of the reports on NSP1 has shown the functional importance in binding with ribosomal subunit and host cellular translation suppression by few amino acids at its CTR by performing mutagenesis studies.4 As shown by Schubert et al., residues 154–157, 164–165, and 171–175 of NSP1-CTR are functionally relevant to NSP1 which showed loss of function. Interestingly, the interactions of MTX and other similar compounds with some of these key residues like Tyr154, Glu155, and Arg171 of NSP1-CTR also confer that it may inhibit the functions of NSP1 and restrict its association with the ribosomal subunit.
Conclusion
A lot of efforts are being made by researchers worldwide to curb the spread and develop some therapeutic strategy against SARS-CoV-2 infection. In this regard, we have also performed a drug repurposing-based study to get insight on druggability of a known anticancer drug mitoxantrone against a druggable target protein of SARS-CoV-2, NSP1. Based on the functional importance of NSP1 and specifically its C-terminal region, we have experimented on its binding with the drug MTX. Previously, we have found its structural dynamicity using various spectroscopic techniques in presence of different solvents. By continuing our investigation on this functionally relevant region in this manuscript, we have found the binding of known anticancer drug MTX with NSP1-CTR. Also, we have shown the interaction of other compounds with similar parental backbone (i.e., anthracene) and conformation with key residues of NSP1-CTR. Therefore, in the future, based on this study, it is highly possible to target NSP1 and, specifically, its CTR for developing therapeutic strategies, and further experimentation can be done using MTX as a lead or parent molecule. Based on above observations, it is also highly plausible that the compounds like MTX and others with anthracene moiety could be explored further for experimental investigation against different targets of SARS-CoV-2.Material and methods
Materials
NSP1 peptide: the sequence of NSP1-CTR (residues 131–180) “NH2-AGGHSYGADLKSFDLGDELGTDPYEDFQENWNTKHSSGVTRELMRELNGG-COOH” was retrieved from the UniProt (ID: P0DTC1.1). The peptide of purity >81% was ordered from GenScript USA.Compound: mitoxantrone (1,4-dihydroxy-5,8-bis[2-(2-hydroxyethylamino)ethylamino]anthracene-9,10-dione; dihydrochloride) was purchased from Sigma-Aldrich, USA.
Fluorescence measurement
Intrinsic tryptophan fluorescence of NSP1-CTR (Trp161) was monitored using a spectrofluorometer (Horiba Scientific Fluorolog, model no. 1073) with an excitation wavelength of 284 nm from 300 nm to 450 nm wavelength range at 25 °C. The slits bandwidths for both excitation and emission were kept at 4 nm. Binding of MTX with NSP1-CTR was performed by titration experiment where protein concentration was kept constant at 8 μM in presence of 50 mM sodium phosphate (pH 7.4) buffer. The inner filter effect in fluorescence data was corrected using the following equation:22| Fcor = Fobs × antilog[(Aex + Aem)/2] |
Fluorescence lifetime measurement
The fluorescence lifetime of tryptophan residue (Trp161) in NSP1-CTR in absence and presence of MTX was measured using the Horiba Scientific DeltaFlex TCSPC system at 25 °C. The samples were excited at 284 nm using the nanoLED as a light source. The wavelength of the emission monochromator was set at 346 nm. The measurement range was adjusted to 400 ns with bandpass of 32 nm and peak preset of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts. Ludox was used to correct the instrument response factor (IRF) at 284 nm. The concentration of NSP1-CTR was kept constant at 8 μM and in presence of 50 mM sodium phosphate (pH 7.4) buffer. Finally, the three exponential decay function fitting was used to obtain the tryptophan lifetimes.
000 counts. Ludox was used to correct the instrument response factor (IRF) at 284 nm. The concentration of NSP1-CTR was kept constant at 8 μM and in presence of 50 mM sodium phosphate (pH 7.4) buffer. Finally, the three exponential decay function fitting was used to obtain the tryptophan lifetimes.
Circular dichroism (CD) measurement
Far-UV CD spectra of NSP1-CTR in 50 mM sodium phosphate (pH 7.4) buffer were recorded using Jasco-1500 spectrophotometer from 190–240 nm, using 1 mm quartz cuvette (Hellma, Plainview, NY, USA) at 25 °C. The bandwidth was kept at 4 nm for all measurements. For CD spectra of NSP1 C-terminal peptide, a control spectrum consisting of only buffer was subtracted from the peptide spectrum. To analyze the CD spectra of peptide in presence of compound, CD spectra of compound were subtracted from the peptide spectrum. For all measurements, the protein concentration was kept at 25 μM. All the measurements were smoothened by 5 points through the adjacent-averaging method in Origin software.Time-resolved anisotropy decay measurement
To confirm the binding of NSP1-CTR and MTX, time-resolved anisotropy decay was measured using DeltaFlex TCSPC system (Horiba Scientific) at 25 °C. A 1 ml pathlength quartz cuvette was used to record the measurements where the measurement range was set up to 200 ns. The bandpass of 16 nm, peak preset of 1000 counts, and repetition rate of 1 MHz were used during experimentation. The protein concentration was kept constant at 30 μM. At 284 nm wavelength, Ludox was used for adjusting the instrument response factor (IRF). Using the data analysis software DAS6 (Horiba Scientific), the resultant anisotropy decay curves were fitted into a bi-exponential decay function:22where, anisotropy at time t is denoted by r(t) and initial anisotropy is denoted by r0, and A1 and A2 defines the rotational correlation time θ1 and θ2, respectively.

Molecular docking
The structure of MTX (PubChem ID: 4212) and its similar compounds and conformers were retrieved from PubChem in sdf file format. These similar conformers (referred as ‘SF’) and compounds (referred as ‘SC’) either have similar or exact parent backbone with changes in their functional groups. The docking of all compounds against the simulated structure of NSP1-CTR was performed and the molecular interactions between the complexes were analyzed using the Glide XP (Extra Precision) program in the Schrodinger suite.27 The parameters for protein structure and ligand preparation are kept as previously reported.28 Briefly, Epik program of Schrodinger suite's Ligprep module was used for generating 3D conformations with proper ionization and tautomeric states at pH 7. For all ligands, a total number of possible stereoisomers was set to 32. Further, these ligands were docked against NSP1-CTR with the defined grid coordinates as 28.0, 24.0, and 33.18 Å for X, Y, and Z, respectively. For proper and detailed interaction analysis of each ligand, Glide XP was set to generate possible 10 binding poses. The binding energy of these complexes was calculated using Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) approach embedded in Prime module of Schrodinger.29 It utilizes VSGB solvation model and OPLS 2005 forcefield to calculate energy parameters.Molecular dynamics simulations
The MD simulations were performed to observe the binding of MTX with NSP1-CTR using two different forcefields viz. OPLS 2005 forcefield in Desmond simulation package (v2018.4)30 and GROMOS54A7 forcefield in Gromacs v5.1.2,31 utilizing the in-house cluster facility.We have used our previously reported protocol for simulations of protein–ligand complex using Desmond.26 Briefly, we have utilized TIP3P water model along with counterions present in the system for charge neutralization in an orthorhombic box with an edge distance of 10 Å. For energy minimization, a total 5000 iterations of 10 steepest-descent steps each was run for the simulation setup. Nose–Hoover and Martina–Thomas–Klein methods were used for temperature and pressure controlling in equilibration (of 100 ps) and final production runs. In last, a total of one microsecond (μs) simulation was performed.
For MD simulations in Gromacs, the SPC water model was added and charge neutralization was done by adding counterions in the simulation box of volume 186.95 nm3. The energy minimization was performed by implementing the 50000 steps of steepest-descent algorithm with Verlet cut-off scheme to calculate the neighboring interactions. Further, equilibration process was carried out under NPT and NVT conditions for 1 ns. Parrinello–Rahman and V-rescale methods were utilized for pressure and temperature coupling, respectively. The Gromacs topology of the compound was generated using PRODRG server.32 LINCS algorithm was used for calculating bond parameters.33 Final production run was performed for 500 ns by generating frames at every 2 fs. Furthermore, the simulation analysis parameters are analyzed using gmx toolbox, including rms, rmsf, gyrate, hbond, covar, and anaeig commands. For visualization, UCSF Chimera34 and Schrodinger Maestro were used.
Author contributions
RG: conception, design, and review of the manuscript. PK and TB: acquisition and interpretation of data, writing of the manuscript. PK and TB have contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All the authors would like to thank IIT Mandi and HPC utility for providing facilities. RG would like to acknowledge the Department of Biotechnology, Govt. of India (BT/11/IYBA/2018/06) and SERB-India (CRG/2019/005603). TB is grateful to the Department of Science and Technology, Govt. of India for her INSPIRE fellowship for Funding.Notes and references
- E. J. Snijder, Y. van der Meer, J. Zevenhoven-Dobbe, J. J. M. Onderwater, J. van der Meulen, H. K. Koerten and A. M. Mommaas, J. Virol., 2006, 80, 5927–5940 CrossRef CAS PubMed.
- Y. Q. Min, Q. Mo, J. Wang, F. Deng, H. Wang and Y. J. Ning, Front. Microbiol., 2020, 11, 1–12 CrossRef PubMed.
- M. Thoms, R. Buschauer, M. Ameismeier, L. Koepke, T. Denk, M. Hirschenberger, H. Kratzat, M. Hayn, T. Mackens-Kiani, J. Cheng, C. M. Stürzel, T. Fröhlich, O. Berninghausen, T. Becker, F. Kirchhoff, K. M. J. Sparrer and R. Beckmann, Science, 2020, 369, 1249–1255 CrossRef CAS PubMed.
- K. Schubert, E. D. Karousis, A. Jomaa, A. Scaiola, B. Echeverria, L. A. Gurzeler, M. Leibundgut, V. Thiel, O. Mühlemann and N. Ban, Nat. Struct. Mol. Biol., 2020, 27, 959–966 CrossRef CAS PubMed.
- A. Kumar, A. Kumar, P. Kumar, N. Garg and R. Giri, Curr. Res. Virol. Sci., 2021, 2, 100007 CrossRef PubMed.
- K. Zhang, L. Miorin, T. Makio, I. Dehghan, S. Gao, Y. Xie, H. Zhong, M. Esparza, T. Kehrer, A. Kumar, T. C. Hobman, C. Ptak, B. Gao, J. D. Minna, Z. Chen, A. García-Sastre, Y. Ren, R. W. Wozniak and B. M. A. Fontoura, Sci. Adv., 2021, 7, eabe7386 CrossRef CAS PubMed.
- C. P. Lapointe, R. Grosely, A. G. Johnson, J. Wang, I. S. Fernández and J. D. Puglisi, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(6) DOI:10.1073/pnas.2017715118.
- X. Wang and Y. Guan, Med. Res. Rev., 2021, 41, 5–28 CrossRef CAS PubMed.
- W. D. Jang, S. Jeon, S. Kim and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(30) DOI:10.1073/pnas.2024302118.
- P. Luo, Y. Liu, L. Qiu, X. Liu, D. Liu and J. Li, J. Med. Virol., 2020, 92, 814–818 CrossRef CAS PubMed.
- Q. Cai, M. Yang, D. Liu, J. Chen, D. Shu, J. Xia, X. Liao, Y. Gu, Q. Cai, Y. Yang, C. Shen, X. Li, L. Peng, D. Huang, J. Zhang, S. Zhang, F. Wang, J. Liu, L. Chen, S. Chen, Z. Wang, Z. Zhang, R. Cao, W. Zhong, Y. Liu and L. Liu, Engineering, 2020, 6, 1192–1198 CrossRef CAS PubMed.
- J. Grein, N. Ohmagari, D. Shin, G. Diaz, E. Asperges, A. Castagna, T. Feldt, G. Green, M. L. Green, F.-X. Lescure, E. Nicastri, R. Oda, K. Yo, E. Quiros-Roldan, A. Studemeister, J. Redinski, S. Ahmed, J. Bernett, D. Chelliah, D. Chen, S. Chihara, S. H. Cohen, J. Cunningham, A. D'Arminio Monforte, S. Ismail, H. Kato, G. Lapadula, E. L'Her, T. Maeno, S. Majumder, M. Massari, M. Mora-Rillo, Y. Mutoh, D. Nguyen, E. Verweij, A. Zoufaly, A. O. Osinusi, A. DeZure, Y. Zhao, L. Zhong, A. Chokkalingam, E. Elboudwarej, L. Telep, L. Timbs, I. Henne, S. Sellers, H. Cao, S. K. Tan, L. Winterbourne, P. Desai, R. Mera, A. Gaggar, R. P. Myers, D. M. Brainard, R. Childs and T. Flanigan, N. Engl. J. Med., 2020, 382, 2327–2336 CrossRef CAS PubMed.
- S. A. Ayatollahi, J. Sharifi-Rad, P. V. Tsouh Fokou, G. B. Mahady, H. Ansar Rasul Suleria, S. Krishna Kapuganti, K. Gadhave, R. Giri, N. Garg, R. Sharma, D. Ribeiro, C. F. Rodrigues, Ž. Reiner, Y. Taheri and N. Cruz-Martins, Front. Pharmacol., 2021, 12, 575877 CrossRef CAS PubMed.
- S. Verma, D. Twilley, T. Esmear, C. B. Oosthuizen, A. M. Reid, M. Nel and N. Lall, Front. Pharmacol., 2020, 11, 1514 Search PubMed.
- Q. Zhang, C. Z. Chen, M. Swaroop, M. Xu, L. Wang, J. Lee, A. Q. Wang, M. Pradhan, N. Hagen, L. Chen, M. Shen, Z. Luo, X. Xu, Y. Xu, W. Huang, W. Zheng and Y. Ye, Cell Discovery, 2020, 6, 1–14 Search PubMed.
- Q. Huang, J. Hou, P. Yang, J. Yan, X. Yu, Y. Zhuo, S. He and F. Xu, BMC Microbiol., 2019, 19, 1–9 CrossRef PubMed.
- L. Deng, P. Dai, A. Ciro, D. F. Smee, H. Djaballah and S. Shuman, J. Virol., 2007, 81, 13392–13402 CrossRef CAS PubMed.
- D. H. Bell, Biochim. Biophys. Acta, Gene Struct. Expression, 1988, 949, 132–137 CrossRef CAS.
- E. J. Fox, Neurology, 2004, 63, S15–S18 CrossRef CAS PubMed.
- L. J. Scott and D. P. Figgitt, CNS Drugs, 2004, 18, 379–396 CrossRef CAS PubMed.
- M. Enache, A. M. Toader and M. I. Enache, Molecules, 2016, 21, 1–17 CrossRef PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, Spain, 2006 Search PubMed.
- C. A. Royer and S. F. Scarlata, Fluorescence Approaches to Quantifying Biomolecular Interactions, Elsevier Inc., 1st edn, 2008, vol. 450 Search PubMed.
- K. B. Lokhande, S. Doiphode, R. Vyas and K. V. Swamy, J. Biomol. Struct. Dyn., 2020, 1–12 Search PubMed.
- M. Enache and E. Volanschi, Rev. Roum. Chim., 2010, 55, 255–262 CAS.
- A. Singh, A. Kumar, P. Kumar, T. Bhardwaj, R. Giri and N. Garg, Future Med. Chem., 2021, 13, 1341–1352 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- P. Kumar, T. Bhardwaj, A. Kumar, B. R. Gehi, S. K. Kapuganti, N. Garg, G. Nath and R. Giri, J. Biomol. Struct. Dyn., 2020, 1–15 Search PubMed.
- M. P. Jacobson, D. L. Pincus, C. S. Rapp, T. J. F. Day, B. Honig, D. E. Shaw and R. A. Friesner, Proteins: Struct., Funct., Bioinf., 2004, 55, 351–367 CrossRef CAS PubMed.
- K. J. Bowers, K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, ACM/IEEE SC 2006 Conf., 2006, p. 43 Search PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- D. M. F. Van Aalten, J. Comput.-Aided Mol. Des., 1996, 10, 255–262 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07434b |
| ‡ Authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |