
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging the 1,3-azadiene-anhydride reaction for the synthesis of functionalized piperidines bearing up to five contiguous stereocenters†
Jorge Garcia,
Jane Eichwald,
Jayme Zesiger and
Timothy K. Beng *
*
Department of Chemistry, Central Washington University, Ellensburg, WA 98926, USA. E-mail: Timothy.beng@cwu.edu
First published on 20th December 2021
Abstract
A modular and scalable strategy, which remodels 3-methylglutaric anhydride to 2-oxopiperidines bearing at least three contiguous stereocenters is described. The approach relies on the chemoselective and stereocontrolled annulation of 1,3-azadienes with the anhydride component. The resulting acid-tethered allylic 2-oxopiperidines are then engaged in several selective fragment growth processes, including catalytic denitrative alkenylation, halolactonization, and Vilsmeier–Haack functionalization.
Introduction
An overarching and intrinsic goal when designing synthetic methodology is to identify approaches whereby simple commercially available precursors or easily accessible synthetic intermediates are converted into a broad array of products, which efficiently tap into uncharted chemical space. Moreover, there are high incentives for the construction of architecturally complex sp3-enriched azaheterocyclic scaffolds,1 especially the piperidine motif, which is arguably the most prevalent.2 As such, synthetic methods for its construction and diversification are highly coveted. In particular, polysubstituted piperidines and 2-oxopiperidines are increasingly attractive fragments for potential drug discovery since the strategic placement of substituents about this three-dimensional scaffold is ideally suited for structure–activity relationship studies.3 Although the synthesis of functionalized piperidines remains daunting,4 recent advances5 are starting to bring these once elusive building blocks into the mainstream. However, most of these advances are predicated on taking an existing piperidine scaffold and installing substituents by functionalization (e.g., through cross-coupling).6 There is also a relative paucity in stereo- and regioselective approaches to alkenylated and methylated piperidines, despite their prevalence in natural products (e.g., GB 17, arboflorine, spirafine D, caulophyllumine B, dienomycin C, and pinidine, Fig. 1) and their frequent use as versatile building blocks for the preparation of medicinal agents.7 For instance, the introduction of methyl groups has the potential to drastically improve the biological activities of a drug candidate by altering its binding affinity, solubility, and metabolism.8 Methyl branches are also favorable structural units in tailor-made fuel components with advanced combustion properties.9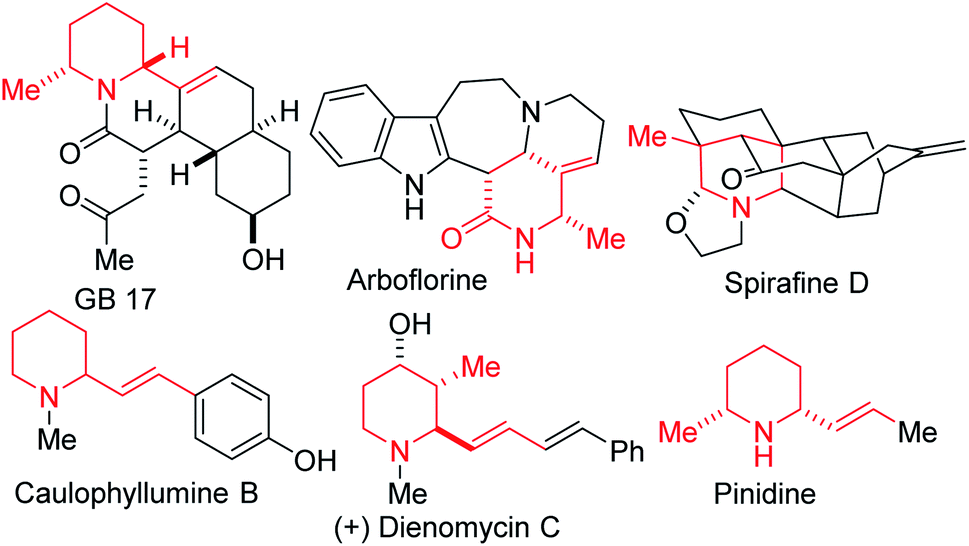 | ||
| Fig. 1 Examples of biologically active methylated and/or alkenylated piperidines. | ||
A complementary strategy to these versatile piperidine scaffolds is to directly forge the azaheterocycle by an annulation protocol. We were therefore motivated by current methodological limitations as well as the relevance of methylated and alkenylated piperidines to develop a step-economical, diastereoselective, and scalable method for the preparation of highly decorated sp3-rich piperidines bearing at least three contiguous stereocenters. Previously, we disclosed that glutaric anhydride (1a) is a more competent substrate than 3,3-dimethylglutaric anhydride (1b) in the 1,3-azadiene-anhydride reaction (Fig. 2).10 Indeed, sterically challenged 1b failed to react even at elevated temperatures (up to 150 °C) even though constitutionally isomeric 2,2-dimethylglutaric anhydride reacted efficiently with 1,3-azadienes of type 2 at 110 °C.10 In the current study, we seek to evaluate the performance (with respect to reactivity, diastereoselectivity, and chemoselectivity) of 3-methylglutaric anhydride (1c) toward 1,3-azadiene partners, and elaborate the product (i.e., 3c) to several synthetically and pharmaceutically attractive topologies (see 4–7). Since many existing cyclization strategies for piperidine synthesis tend to exhibit poor generality, suffer from low regio- and stereochemical control, require protecting group manipulations, or depend on a specific directing group,11 we anticipate that the current approach, which side-steps these aforementioned limitations, would greatly expand the structural space for the discovery of new piperidine pharmacophores. Indeed, this anhydride-imine reaction is known to be a powerful tool for the synthesis of combinatorial and targeted compound libraries as well as for producing building blocks in multigram (up to 100 g) scale.12
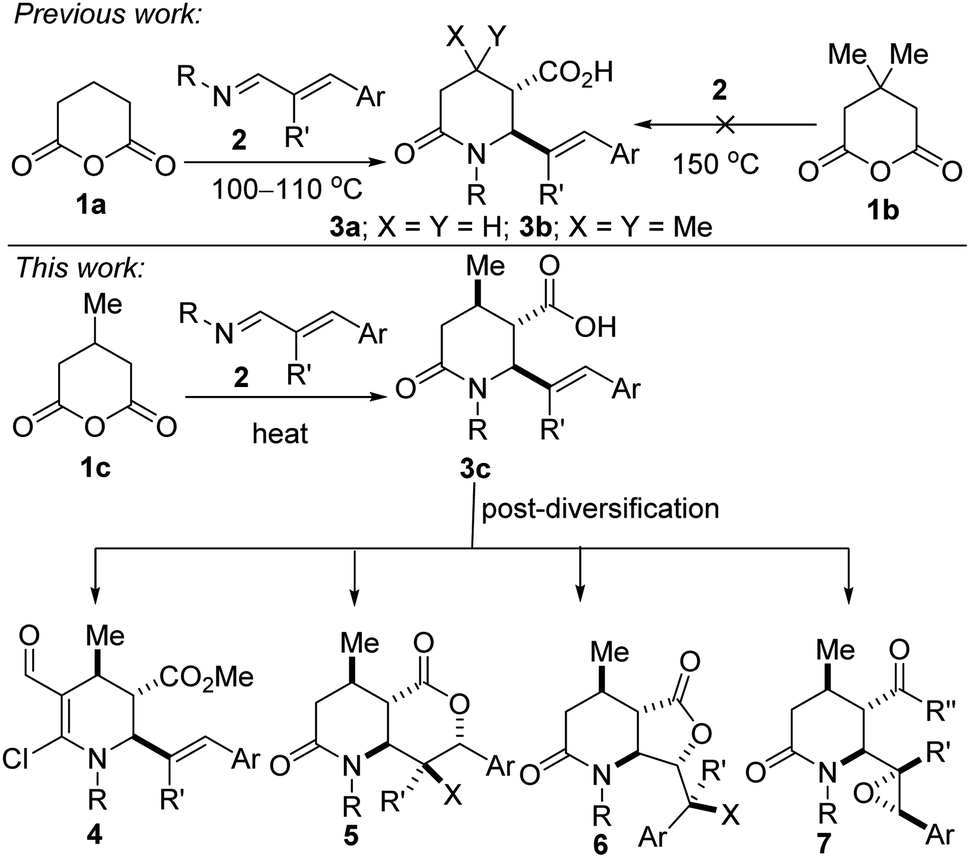 | ||
| Fig. 2 Proposed plan for the synthesis of methylated allylic piperidines and potential synthetic applications. | ||
Results and discussion
We initiated studies toward the construction of methylated and alkenylated piperidine derivatives and subsequent fragment growth by attempting to find suitable directing group-free conditions for the annulation reaction between 1c and model azadiene 2a (Table 1). Although we were cognizant of the promiscuous reactivity of 2 with enolizable cyclic anhydrides (which leads to a plethora of products, including Tamura-type, anhydride-Mannich-type, and Perkin-type products),13 we surmised that just like 1a, the mitigated reactivity of 1c would provide an ideal balance of selectivity and reactivity. Reaction optimization was eventually carried out and it was established that 2-methyltetrahydrofuran (2-MeTHF) out-performs other reaction media (e.g., benzene, o-xylene, chlorobenzene, toluene, diphenyl ether, and 1,4-dioxane). The chemoselective formation of 2-oxopiperidine 3c1 indicates a scenario whereby the anhydride-imine pathway (i.e., Castagnoli–Cushman selectivity) out-competes the anhydride-alkene pathway (i.e., Tamura selectivity). Strikingly, the reaction proceeds with impeccable stereochemical control at the C4 stereocenter, leading to the formation of predominantly one stereoisomer (as judged by GC-MS and 1H NMR analyses of the crude mixture). The atom efficiency of this transformation is commendable given that with the exception of the water formed during the formation of the 1,3-azadiene (i.e., 2), all atoms originating from the enal, primary amine, and anhydride 1c are incorporated into the product structure.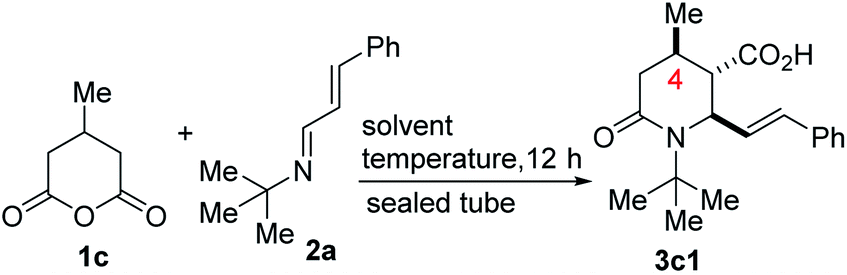
The scope of the reaction with respect to the alkenyl moiety as well as the N-substituent has been explored. For purposes of easier isolation and purification, the initially formed carboxylic acid has been converted to the corresponding methyl ester. These studies have revealed that 1,3-azadienes harboring both electron-neutral, electron-rich, and electron-deficient styryl groups react competently. However, there is a slight preference for electron-rich substituents (3c1 vs. 3c2 vs. 3c3), which is not surprising. Indeed, we are not aware of any reports on the use of aldimines derived from strongly deactivating p-nitrobenzaldehyde in the anhydride-Mannich reaction.14 It is therefore a testament to the enhanced reactivity of 2 that it undergoes satisfactory annulation with 1c. Understandably, ortho-substituted styrenes react with a slight decrease in efficiency (3c2 vs. 3c4 or 3c3 vs. 3c5). N-Aryl piperidines and 2-oxopiperidines are embedded in several pharmacologically pertinent targets, including Factor Xa inhibitors and phosphodiesterase-10 inhibitors.15 N-aryl imines are typically reluctant substrates for the CCR,16 presumably due to the hydrolytic instability (and subsequent proneness of the resulting amine to undergo acylation with the anhydride) as well as the relatively reduced nucleophilicity of the nitrogen atom. The reluctance is further exacerbated when electron-withdrawing anilines are employed. These challenges notwithstanding, we have demonstrated that N-aryl-1,3-azadienes react with varying degrees of success (3c7–3c16). It is once again a testament to the enhanced reactivity of N-aryl-α,β-unsaturated imines of type 2 (compared to the N-aryl aldimine congeners) that it undergoes satisfactory annulation with 1c. In these cases, it was even more critical to maintain an anhydrous medium. The use of molecular sieves was advantageous.
There is no compromise in the E/Z stereoselectivity of the alkene even when trisubstituted alkenes are employed (3c17–3c22). The synthesis of metathesis-suitable bis-allylic 2-oxopiperidine 3c17 sets the stage for the construction of N-fused bicyclic amines such as indolizidines. Halogenated styrenes are well tolerated (see 3c10, 3c13, and 3c14), which bodes well for late-stage diversification as the halogen group may be utilized as a functional handle for cross-coupling purposes. The incorporation of a fluorinated moiety into organic molecules generally increases the solubility, lipophilicity and metabolic stability of the parent molecules, thus, explaining why ∼25% of existing preclinical drugs and 40% of agrochemicals contain at least one fluorine atom.17 Specifically, the CF3 group enjoys a privileged role because its incorporation often enhances efficacy by promoting electrostatic interactions with targets, improving cellular membrane permeability, and increasing robustness toward oxidative metabolism of the drug.18 It is therefore noteworthy that fluorinated products 3c9, 3c10, and 3c16 are obtainable in satisfactory yields.
In the synthesis of potential drug candidates, scalability is often a significant factor as it serves to provide sufficient amounts for clinical tests. A potentially beneficial aspect of this methodology is therefore the scalable nature of the reactions given that products such as 3c13 have been prepared in gram scale, with little to no compromise in efficiency and diastereoselectivity. Of note, anhydride 1b still does not react with 2 under these identified reaction conditions, presumably due to steric congestion.
This 1,3-azadiene-anhydride reaction is thought to proceed via either of two possible paths: (a) intermolecular imine acylation followed by intramolecular Mannich reaction and/or (b) intermolecular Mannich reaction followed by intramolecular acylation or anhydride aminolysis. In the current scenario, we propose that thermally-assisted tautomerization of anhydride 1c affords enol 1c′ (Fig. 3), which undergoes Mannich-type addition with 1,3-azadiene 2a to afford formal hydroalkylation product 8. Subsequent intramolecular aminolysis of the anhydride by the pendant secondary amine gives rise to lactam 3c1. The iminolysis pathway (see 9 and 10) has not been completely ruled out at this point.
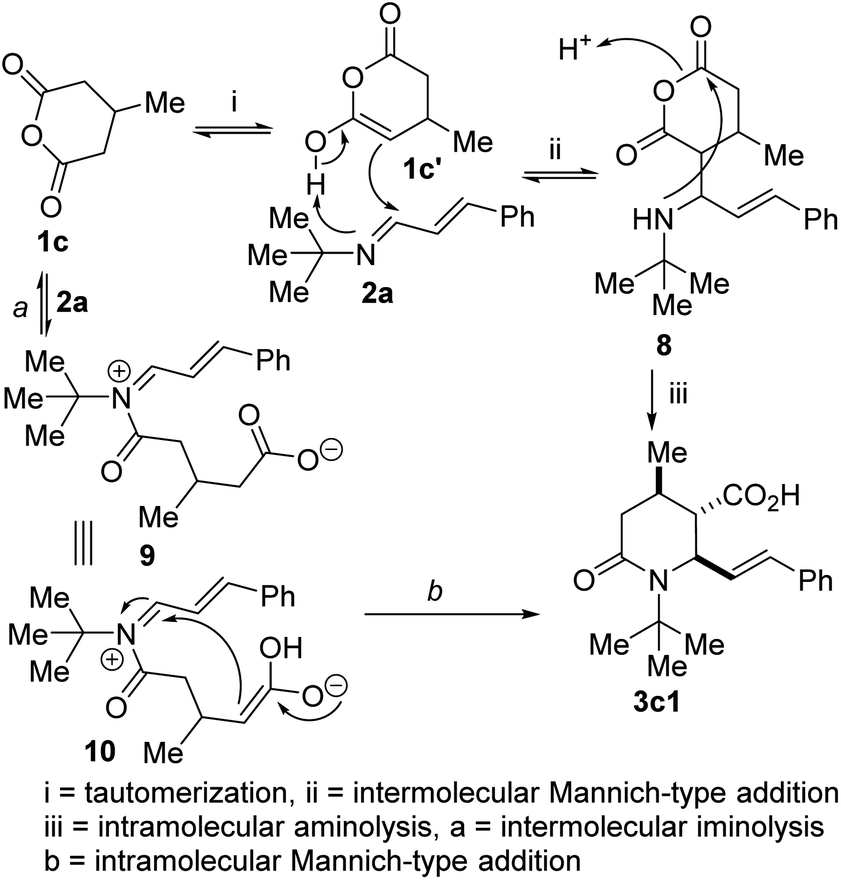 | ||
| Fig. 3 Proposed mechanism for the formation of 3c1. | ||
One of the key steps in drug discovery is to ‘grow’ fragment hits from potentially any position. Accordingly, we have explored the amenability of 3c to fragment growth. Enamines represent a fundamental cornerstone of the organic synthetic toolbox, owing to their exceptional nucleophilicity. However, the unique reactivity of enamines is often accompanied by a high propensity to undergo hydrolysis, leading to considerable difficulties in the handling of these versatile synthons. These challenges notwithstanding, our studies have revealed that stock precursor 3c undergoes productive lactam-selective Vilsmeier–Haack functionalization to furnish the activated enamines depicted in Scheme 2. The dehydropiperidines were obtained in satisfactory yields (see 4a-p).
 | ||
| Scheme 1 Chemoselective and diastereoselective hexannulation of 1,3-azadienes with anhydride 1c. | ||
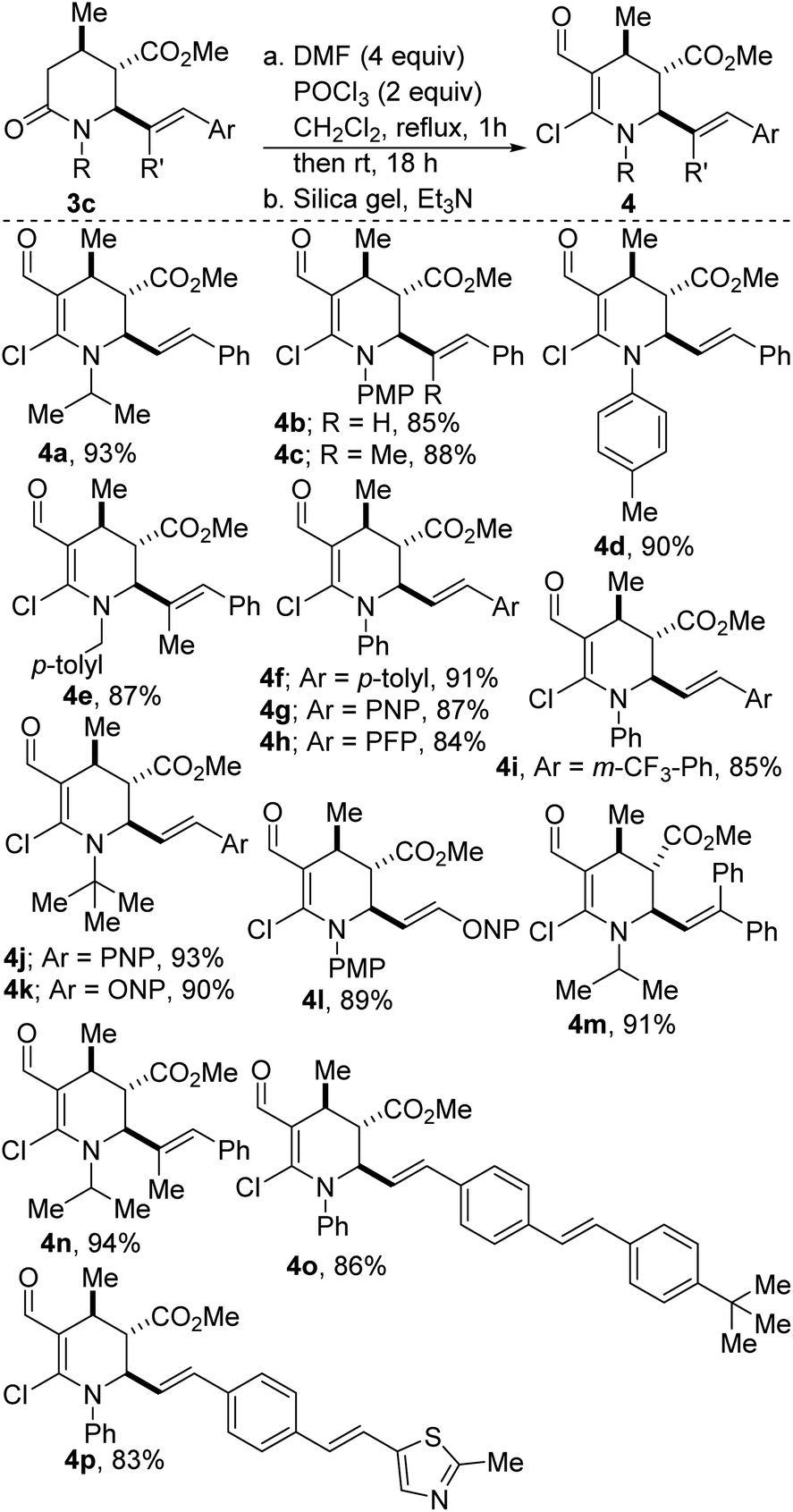 | ||
| Scheme 2 Construction of fully substituted dehydropiperidines. | ||
These studies have revealed that solvent-controlled halolactonization of 3c furnishes lactam–lactones such as 5/6 (Scheme 3). When dichloromethane is employed as the reaction medium, 6-endo cyclization proceeds cleanly to furnish [6-6]-bicycles of type 5, which harbor five contiguous stereocenters. Conversely, the use of N,N-dimethylformamide (DMF) leads to predominant 5-exo cyclization and furnishes [6-5]-bicycles of type 6. The construction of these sp3-rich bicyclic lactam–lactones is noteworthy since medicinal chemists are becoming increasingly keen on escaping flatland in view of exploring 3D-structural space.
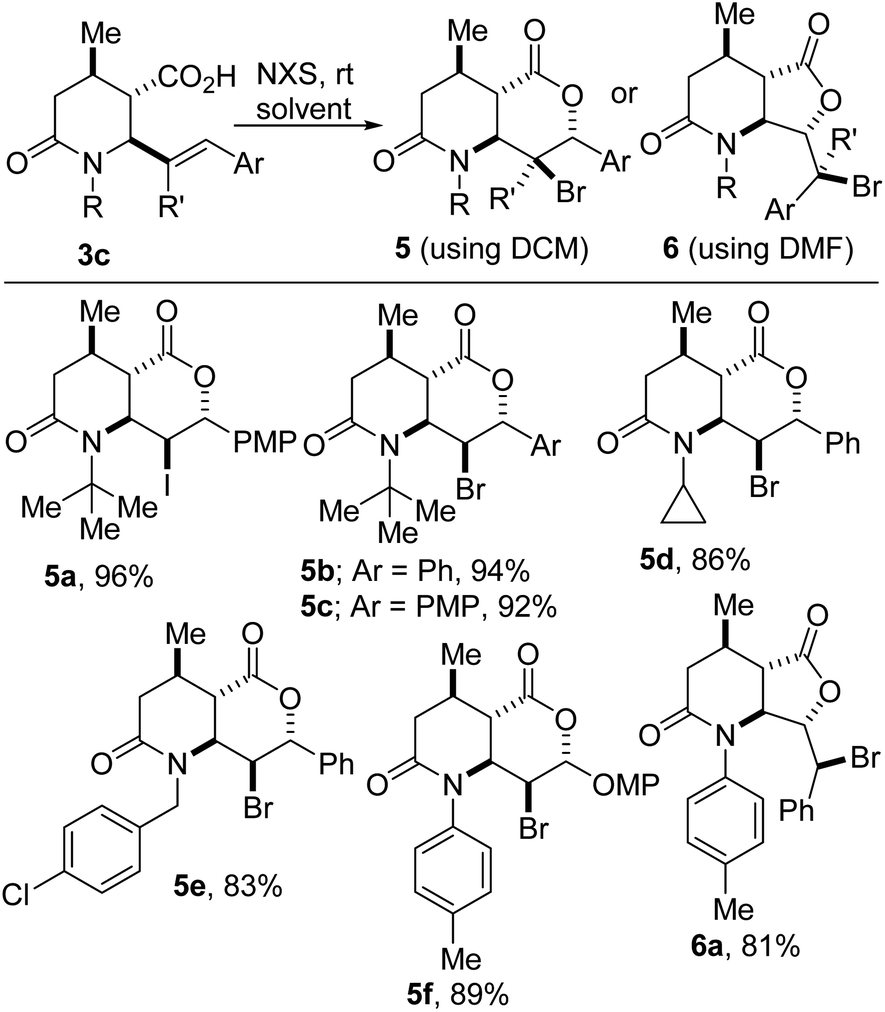 | ||
| Scheme 3 Halolactonization of lactam-tethered γ-alkenoic acids. | ||
We have found that fused bicyclic lactam–halolactones of type 5 undergo cascade deconstructive epoxy-amidation and epoxy-esterification to afford epoxylactam carboxamides (Scheme 4, see 7a–c) or epoxylactam esters such as 7d–f. This modular three-step sequence (starting from 1c and 2) proceeds with the installation of five contiguous stereocenters.
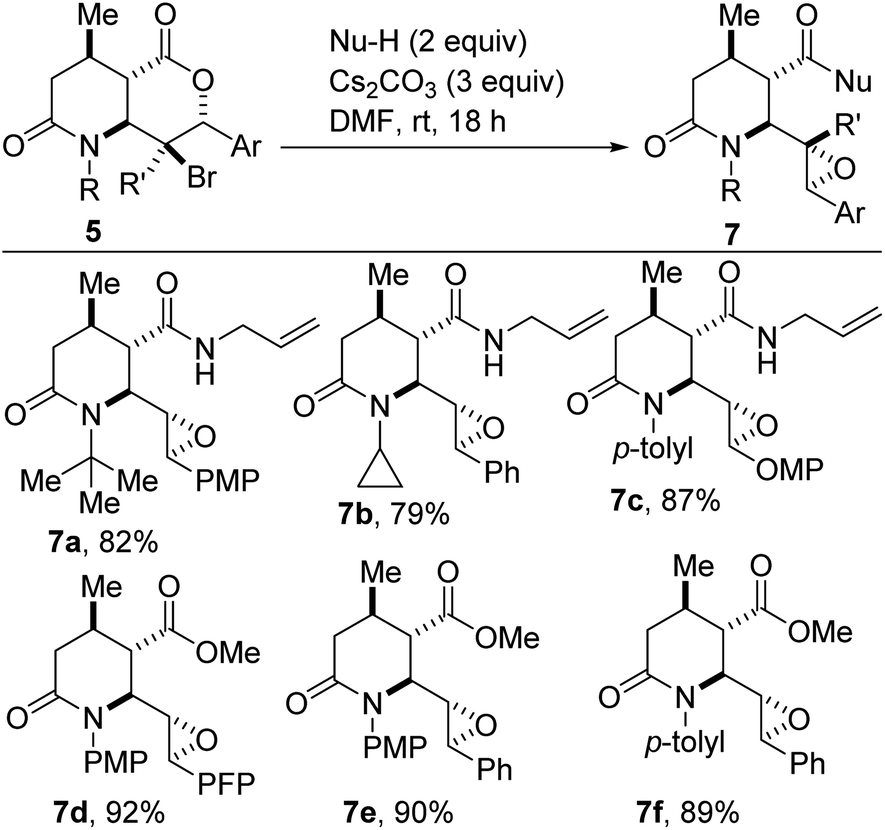 | ||
| Scheme 4 Deconstructive epoxy-amidation and epoxy-esterification of 5. | ||
One of the limitations of the anhydride-imine reaction described herein is the futility of imines derived from enolizable aldehydes such as p-nitrohydrocinnamaldehyde given that their enamine tautomers readily undergo acylation reactions with the anhydride component. For similar reasons, the azadiene component cannot possess any unprotected primary amine functionality. Using the current approach, both limitations can be side-stepped through catalytic hydrogenation of both the alkenyl and nitro motifs resident in lactam esters such as 3c3 and 3c11 (Scheme 5, see 11a,b). The unveiling of the aniline functionality allows for the late-stage synthesis of versatile 1,3-azadienes, which undergo another anhydride-imine reaction with phenylsuccinic anhydride (12) followed by bromolactonization to arrive at sp3-rich architectures such as 13.
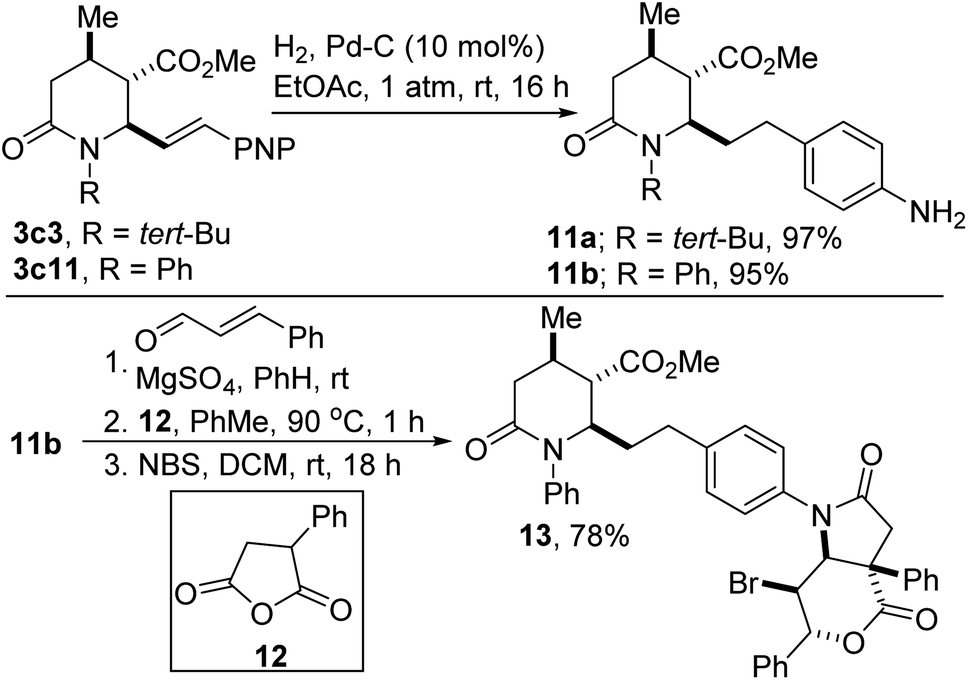 | ||
| Scheme 5 Post-modification of 4-nitrostyrenyl lactams. | ||
Denitrative couplings have recently emerged for the selective formation of C–C and C–X bonds.19 This blossoming strategy of employing nitroaromatics as aryl precursors in cross-couplings is quite attractive. The transformation is however plagued by several challenges, notably the difficulty of oxidative addition of Ar–NO2 to a metal center. If the scope of this reaction is expanded to include substrates with epimerizable stereocenters, remote basic amines, heteroaromatics, and halogenated aromatics, this would undeniably popularize its use in late-stage functionalization. Within this context, we have found that 2-oxopiperidine 3c11 undergoes productive catalytic denitrative alkenylation with styrenes and acrylamides to afford the highly conjugated polyenes depicted in Scheme 6. An electron-deficient vinyl thiazole derivative also couples satisfactorily (see 14d).
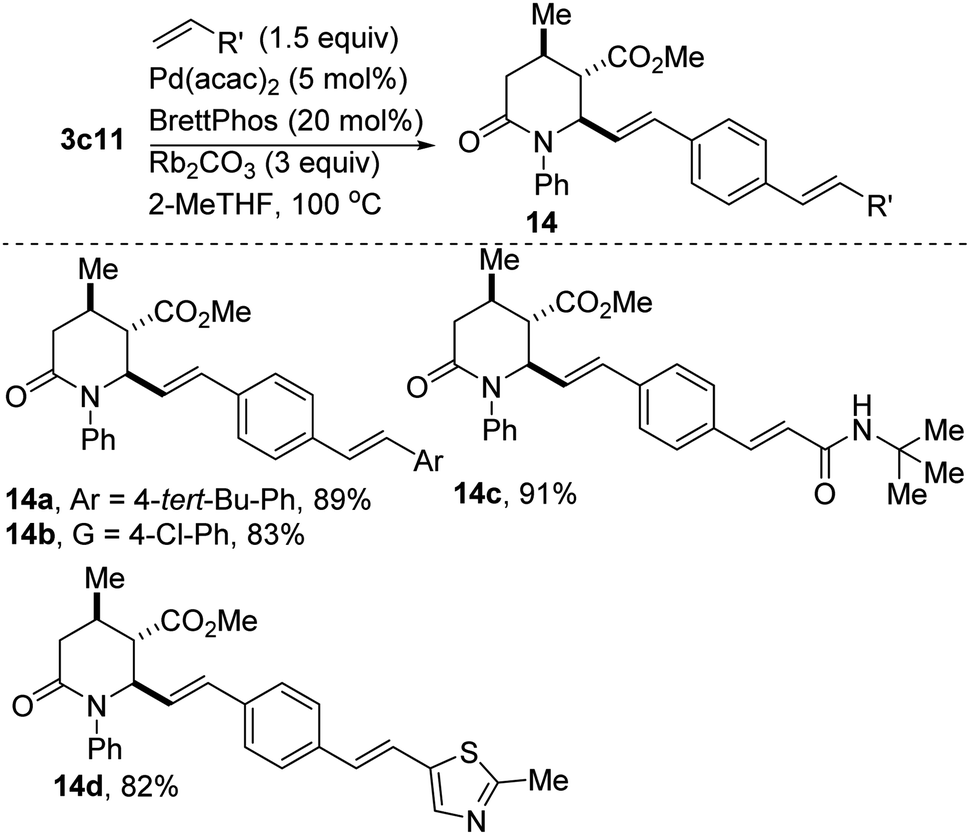 | ||
| Scheme 6 Pd-catalyzed alkenylation of lactam-tethered nitrostyrenes with electronically diverse alkenes. | ||
Transition metal-catalyzed cross-coupling reactions, especially those employing traceless activating groups have revolutionized almost all areas of chemical synthesis.20 However, efforts toward improving cost, versatility, and operational simplicity of these methods continually necessitate the development of simple protocols that rely on feedstock organic, native functionality such as vinyl chlorides as requisite handles for uniting complex fragments.21 Alkynes are frequently used as building blocks in organic synthesis and as catalysts in asymmetric catalysis owing to their rich reactivity profiles.22 Since the seminal work of Sonogashira and co-workers,23 Sonogashira-type cross-couplings have become one of the most reliable methods for the construction of internal alkynes with differential substitution.24 We were therefore pleased to find that vinyl chlorides of type 4 undergo efficient alkynylation with triisopropylsilylacetylene, in the presence of catalytic amounts of PdCl2(PhCN)2 and CuI (Scheme 7). In this protocol, 2,6-lupetidine serves as the base and 1,4-dioxane serves as the solvent. The ability to install a triisopropylsilyl motif bodes well for further diversification since its robust yet removable nature25 allows it to be employed as a terminal acetylene surrogate. Efforts to extend this alkynylation protocol to other alkyne coupling partners are underway.
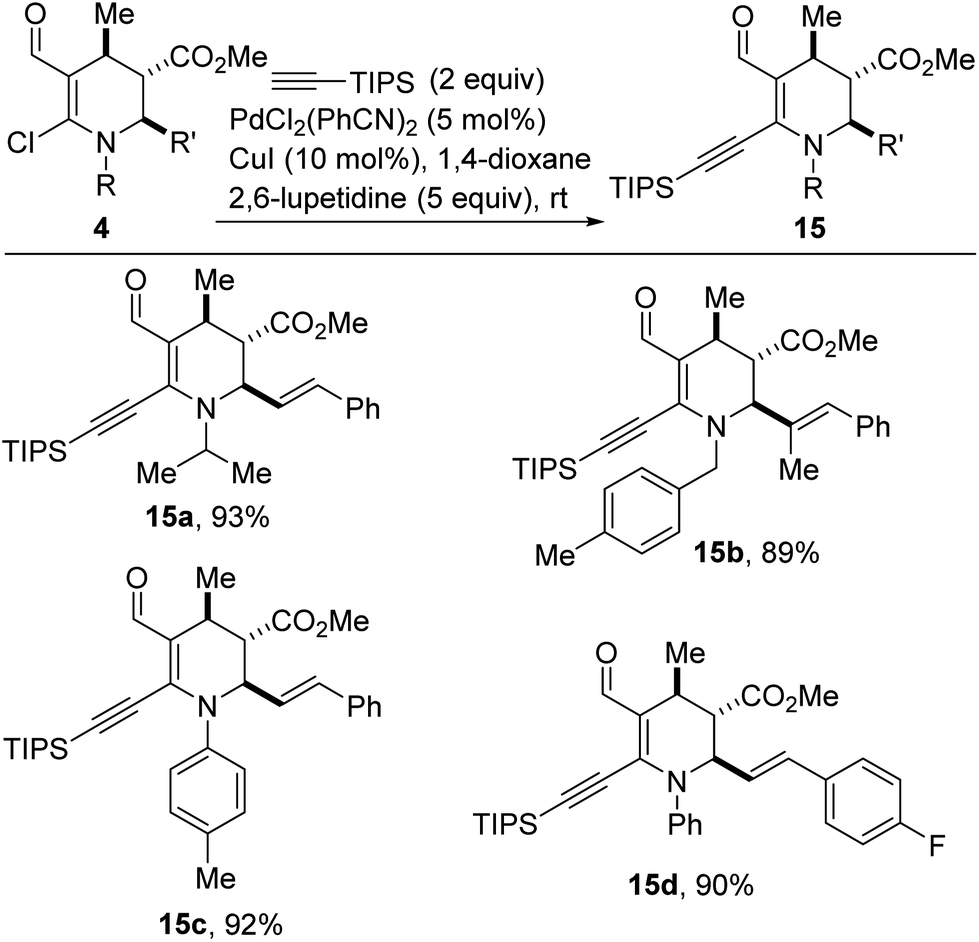 | ||
| Scheme 7 Catalytic alkynylation of α-chloroenamines. | ||
Conclusions
In summary, the modular construction of functionalized 2-oxopiperidines bearing at least three contiguous stereocenters has been accomplished. The approach hinges on the chemoselective and stereocontrolled annulation of 3-methylglutaric anhydride with 1,3-azadienes. The scalable nature of the reactions offers the opportunity for post-modification by incorporation of motifs with either known pharmaceutical value or that permit subsequent conversion (halogens, carboxylic acids, esters, alkenes, nitroarenes, aldehydes, and alkynes) to medicinally relevant entities. The criteria of efficiency, versatility, and pot-atom-step economy are of paramount importance in modern day synthetic chemistry and these studies have met these benchmarks. The amenability of these functionalized 2-oxopiperidines to C–C bond forming transformations such as denitrative and Sonagashira cross-couplings bodes well for future late-stage assembly of complex bioactive piperidines. We anticipate that the aforementioned merits will endear this methodology to both the synthesis and medicinal chemistry communities.Experimental
All experiments involving air and moisture sensitive reagents were carried out under an inert atmosphere of nitrogen and using freshly distilled solvents. Column chromatography was performed on silica gel (230–400 mesh). Thin-layer chromatography (TLC) was performed using Silicycle SiliaplateTM glass backed plates (250 μm thickness, 60 Å porosity, F-254 indicator) and visualized using UV (254 nm) or KMnO4 stain. Unless otherwise indicated, 1H, 13C, and DEPT-135 NMR, and NOESY spectra were acquired using CDCl3 solvent at room temperature. Chemical shifts are quoted in parts per million (ppm). HRMS-EI+ data were obtained using either electronspray ionization (ESI) or electron impact (EI) techniques. High-resolution ESI was obtained on an LTQ-FT (ion trap; analyzed using Excalibur). High resolution EI was obtained on an Autospec (magnetic sector; analyzed using MassLynx). Brine solutions are saturated solutions of aqueous sodium chloride. Representative GC-MS traces are provided.General procedure A
General procedure B
General procedure C
General procedure D
General procedure E
General procedure F
General procedure G
General procedure H
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30). Oily substance. Yield = 263.6 mg, 80%, 95:5 dr. 1H NMR (400 MHz, CDCl3) δ 7.37–7.23 (m, 5H), 6.52 (d, J = 15.9 Hz, 1H), 6.17 (dd, J = 16.0, 5.6 Hz, 1H), 4.85 (dd, J = 5.6, 2.7 Hz, 1H), 3.74 (s, 3H), 2.74 (dd, J = 4.3, 2.7 Hz, 1H), 2.55–2.31 (m, 3H), 1.47 (s, 9H), 1.07 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.0, 170.7, 135.9, 131.8, 131.0, 128.8, 128.1, 126.4, 58.1, 57.6, 51.7, 50.0, 39.2, 28.5, 25.1, 18.3. HRMS-EI+ (m/z): calc. for C20H27NO3, 329.1991, found 329.1996. FTIR (KBr): 2976.0, 2927.2, 1721.7, 1650.1, 1492.0, 1438.4, 1362.2, 1320.5, 1290.1, 1206.3, 1180.3, 1146.7, 1132.3, 995.8, 918.8, 700.1.
:30). Oily substance. Yield = 263.6 mg, 80%, 95:5 dr. 1H NMR (400 MHz, CDCl3) δ 7.37–7.23 (m, 5H), 6.52 (d, J = 15.9 Hz, 1H), 6.17 (dd, J = 16.0, 5.6 Hz, 1H), 4.85 (dd, J = 5.6, 2.7 Hz, 1H), 3.74 (s, 3H), 2.74 (dd, J = 4.3, 2.7 Hz, 1H), 2.55–2.31 (m, 3H), 1.47 (s, 9H), 1.07 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.0, 170.7, 135.9, 131.8, 131.0, 128.8, 128.1, 126.4, 58.1, 57.6, 51.7, 50.0, 39.2, 28.5, 25.1, 18.3. HRMS-EI+ (m/z): calc. for C20H27NO3, 329.1991, found 329.1996. FTIR (KBr): 2976.0, 2927.2, 1721.7, 1650.1, 1492.0, 1438.4, 1362.2, 1320.5, 1290.1, 1206.3, 1180.3, 1146.7, 1132.3, 995.8, 918.8, 700.1.Note: All other allylic lactams depicted in Scheme 1 were prepared as described above. Spectroscopic data can be found in the ESI.†
:15). Oily substance. Yield = 168.3 mg, 93%. 1H NMR (400 MHz, CDCl3) δ 9.74 (s, 1H), 7.39–7.20 (m, 5H), 6.62 (d, J = 15.9 Hz, 1H), 5.92 (dd, J = 15.9, 8.5 Hz, 1H), 4.54 (t, J = 9.0 Hz, 1H), 4.35 (hept, J = 6.9 Hz, 1H), 3.65 (s, 3H), 3.35 (qd, J = 6.9, 4.6 Hz, 1H), 2.74 (dd, J = 9.6, 4.7 Hz, 1H), 1.39 (dd, J = 16.2, 6.9 Hz, 6H), 1.00 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 186.4, 171.3, 151.1, 135.9, 133.5, 128.7, 128.3, 128.1, 126.6, 114.6, 59.1, 53.8, 51.9, 50.3, 27.8, 22.2, 21.3, 14.8. HRMS-EI+ (m/z): calc. for C20H24ClNO3, 361.1445, found 361.1449. FTIR (KBr): 2924.8, 1642.2, 1494.9, 1448.8, 1427.0, 1393.4, 1361.6, 1328.7, 1289.7, 1223.6, 1198.9, 1130.0, 1074.1, 1030.4, 988.5, 966.1, 925.5, 741.6, 693.4.Note: All other enamines depicted in Scheme 2 were prepared as described above. Spectroscopic data can be found in the ESI.†
:40). Oily substance. Yield = 185.3 mg, 94%. 1H NMR (400 MHz, CDCl3) δ 7.48–7.38 (m, 5H), 5.75 (d, J = 3.7 Hz, 1H), 4.86 (t, J = 3.5 Hz, 1H), 3.96 (dd, J = 12.1, 3.4 Hz, 1H), 3.21 (ddd, J = 12.1, 2.9, 1.0 Hz, 1H), 2.67–2.54 (m, 2H), 2.42–2.32 (m, 1H), 1.34 (s, 9H), 1.13 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.2, 170.0, 137.8, 129.3, 129.2, 125.5, 84.9, 60.8, 58.6, 50.5, 43.8, 42.0, 29.2, 23.6, 13.4. HRMS-EI+ (m/z): calc. for C19H24BrNO3, 393.0940, found 393.0944.Note: All other lactam–lactones depicted in Scheme 3 were prepared as described above. Spectroscopic data can be found in the ESI.†
:75). Oily substance. Yield = 164 mg, 82%. 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 7.7 Hz, 2H), 6.87 (d, J = 7.7 Hz, 2H), 6.00 (dd, J = 5.7, 3.0 Hz, 1H), 5.90–5.80 (m, 1H), 5.26–5.12 (m, 2H), 4.22 (dd, J = 7.0, 3.3 Hz, 1H), 4.02–3.84 (m, 2H), 3.80 (s, 3H), 3.72 (d, J = 2.1 Hz, 1H), 3.00 (dd, J = 7.0, 2.1 Hz, 1H), 2.75 (dd, J = 5.9, 3.3 Hz, 1H), 2.56–2.44 (m, 1H), 2.44–2.28 (m, 2H), 1.55 (s, 9H), 0.97 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.7, 160.0, 133.9, 127.9, 126.9, 117.2, 114.2, 66.5, 58.3, 56.9, 55.4, 55.3, 48.8, 42.1, 40.7, 28.7, 27.9, 16.7. HRMS-EI+ (m/z): calc. for C23H32N2O4, 400.2362, found 400.2369. FTIR (KBr): 3384.3, 3009.7, 2933.6, 1647.7, 1607.2, 1577.1, 1512.0, 1454.2, 1427.6, 1359.8, 1299.2, 1250.9, 1176.0, 1151.5, 1119.6, 1031.3, 990.3, 927.8, 825.4, 765.0, 749.7.Note: All other epoxide-tethered lactam carboxamides and lactam-ester depicted in Scheme 4 were prepared as described above. Spectroscopic data can be found in the ESI.†
:1). Oily substance. Yield = 286.9 mg, 78%. 1H NMR (400 MHz, CDCl3) δ 7.62–7.21 (m, 16H), 7.11–7.07 (m, 3H), 5.54 (s, 1H), 4.80 (dd, J = 10.9, 2.8 Hz, 1H), 4.36 (d, J = 10.8 Hz, 1H), 4.28–4.17 (m, 1H), 3.85–3.73 (m, 4H), 3.49–3.31 (m, 1H), 3.07–2.97 (m, 1H), 2.80–2.48 (m, 5H), 2.08–1.98 (m, 1H), 1.96–1.82 (m, 1H), 1.20 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 176.1, 172.4, 169.9, 169.8, 169.3, 141.2, 139.7, 136.1, 133.1, 129.9, 129.4, 129.0, 128.9, 128.0, 127.9, 126.2, 125.0, 124.9, 81.7, 68.6, 60.1, 53.0, 52.1, 50.7, 46.5, 44.5, 38.0, 34.3, 34.2, 31.0, 29.3, 26.7, 17.4, 17.4. HRMS-EI+ (m/z): calc. for C41H39BrN2O6, 734.1991, found 734.1998.:25). Oily substance. Yield = 225.9 mg, 89%. 1H NMR (400 MHz, CDCl3) δ 7.47–7.20 (m, 13H), 7.01 (s, 2H), 6.43 (dd, J = 15.8, 1.1 Hz, 1H), 6.11 (dd, J = 15.8, 7.2 Hz, 1H), 4.78–4.71 (m, 1H), 3.75 (s, 3H), 2.90 (t, J = 4.1 Hz, 1H), 2.72 (dd, J = 16.9, 5.4 Hz, 1H), 2.66–2.56 (m, 1H), 2.59–2.46 (m, 1H), 1.31 (s, 9H), 1.18 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.9, 169.4, 150.9, 141.1, 136.5, 135.9, 134.5, 133.5, 129.0, 128.7, 128.2, 128.0, 127.9, 127.2, 127.1, 126.6, 126.3, 125.6, 63.3, 52.0, 49.4, 38.1, 34.7, 31.3, 26.8, 17.8. HRMS-EI+ (m/z): calc. for C34H37NO3, 507.2773, found 507.2779.Note: All other denitrative coupling products depicted in Scheme 6 were prepared as described above. Spectroscopic data can be found in the ESI.†
:10). Oily substance. Yield = 118.0 mg, 93%. 1H NMR (400 MHz, CDCl3) δ 9.84 (s, 1H), 7.36–7.24 (m, 5H), 6.59 (d, J = 15.9 Hz, 1H), 5.93 (dd, J = 15.9, 8.2 Hz, 1H), 4.61 (hept, J = 7.0 Hz, 1H), 4.42 (t, J = 8.4 Hz, 1H), 3.64 (s, 3H), 3.27–3.17 (m, 1H), 2.72 (dd, J = 8.7, 5.0 Hz, 1H), 1.36 (dd, J = 9.2, 6.9 Hz, 6H), 1.15–1.05 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 187.8, 171.6, 143.5, 136.1, 133.0, 128.8, 128.7, 128.1, 126.5, 121.3, 104.2, 97.0, 55.8, 53.3, 51.6, 49.3, 26.0, 22.7, 20.6, 18.6, 15.3, 11.3. HRMS-EI+ (m/z): calc. for C31H45NO3Si, 507.3169, found 507.3175.Note: All other alkynyl enamines depicted in Scheme 7 were prepared as described above. Spectroscopic data can be found in the ESI.†
Author contributions
J. G. – investigation, data curation, validation; J. E. – investigation, methodology; J. Z. – investigation, methodology; T. K. B. – conceptualization, project administration, supervision, writing – original draft, internal funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Central Washington University (CWU) for continued, generous support of our research endeavors. The School of Graduate Studies and Research is thanked for partial support of this work through a Faculty Research Award to T. K. B. J. G. thanks Provost Denbeste for a research fellowship. The Office of Undergraduate Research (OUR) is thanked for support provided to J. G., J. E., and J. Z.Notes and references
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- For reviews on the alpha functionalization of unsubstituted heterocycles see: (a) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoal and B. U. W. Maes, Chem.–Eur. J., 2012, 18, 10092–10142 CrossRef CAS PubMed; (b) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC; (c) C. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS PubMed.
- (a) Z. Amara, J. Caron and D. Joseph, Nat. Prod. Rep., 2013, 30, 1211–1225 RSC; (b) T. K. Beng, H. Takeuchi, M. Weber and R. Sarpong, Chem. Commun., 2015, 51, 7653–7656 RSC; (c) K. Kubota, Y. Watanabe, K. Hayama and H. Ito, J. Am. Chem. Soc., 2016, 138, 4338–4341 CrossRef CAS PubMed; (d) B. Qu, H. P. R. Mangunuru, X. Wei, K. R. Fandrick, J.-N. Desrosiers, J. D. Sieber, D. Kurouski, N. Haddad, L. P. Samankumara, H. Lee, J. Savoie, S. Ma, N. Grinberg, M. Sarvestani, N. K. Yee, J. J. Song and C. H. Senanayake, Org. Lett., 2016, 18, 4920–4923 CrossRef CAS PubMed; (e) J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220–224 CrossRef CAS PubMed; (f) T. Sandmeier, S. Krautwald and E. M. Carreira, Angew. Chem., 2017, 129, 11673–11677 CrossRef; (g) W. Chen, L. Ma, A. Paul and D. Seidel, Nat. Chem., 2018, 10, 165–169 CrossRef CAS PubMed; (h) J. B. Roque, Y. Kuroda, L. T. Göttemann and R. Sarpong, Nature, 2018, 564, 244–248 CrossRef CAS PubMed; (i) A. Paul and D. Seidel, J. Am. Chem. Soc., 2019, 141, 8778–8782 CrossRef CAS PubMed; (j) I. S. Hassan, A. N. Ta, M. W. Danneman, N. Semakul, M. Burns, C. H. Basch, V. N. Dippon, B. R. McNaughton and T. Rovis, J. Am. Chem. Soc., 2019, 141, 4815–4819 CrossRef CAS PubMed; (k) A. Millet, P. Larini, E. Clot and O. Baudoin, Chem. Sci., 2013, 4, 2241–2247 RSC; (l) C. J. Cordier, R. J. Lundgren and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 10946–10949 CrossRef CAS PubMed; (m) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed; (n) T. K. Beng and D. P. Bassler, Tetrahedron Lett., 2014, 55, 6662–6664 CrossRef CAS; (o) D. P. Bassler, A. Alwali, L. Spence, O. Beale and T. K. Beng, J. Organomet. Chem., 2015, 780, 6–12 CrossRef CAS; (p) T. K. Beng, S. Langevin, H. Braunstein and M. Khim, Org. Biomol. Chem., 2016, 14, 830–834 RSC; (q) T. K. Beng, A. W. V. Silaire, A. Alwali and D. P. Bassler, Org. Biomol. Chem., 2015, 13, 7915–7919 RSC; (r) T. K. Beng, N. Fox, D. P. Bassler, A. Alwali, K. Sincavage and A. W. V. Silaire, Org. Biomol. Chem., 2015, 13, 8647–8651 RSC.
- For selected examples, see: (a) J. Ding, T. Rybak and D. G. Hall, Nat. Commun., 2014, 5, 5474 CrossRef PubMed; (b) M. J. Dearden, C. R. Firkin, J.-P. R. Hermet and P. O'Brien, J. Am. Chem. Soc., 2002, 124, 11870–11871 CrossRef CAS PubMed; (c) C. J. Cordier, R. J. Lundgren and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 10946–10949 CrossRef CAS PubMed.
- (a) P. R. Krishna and B. K. Reddy, Tetrahedron Lett., 2010, 51, 6262–6264 CrossRef; (b) D. L. Comins and G. M. Green, Tetrahedron Lett., 1999, 40, 217–218 CrossRef CAS; (c) M. A. Arai, N. Ishikawa, M. Tanaka, K. Uemura, N. Sugimitsu, A. Suganami, Y. Tamura, T. Koyano, T. Kowithayakorn and M. Ishibashi, Chem. Sci., 2016, 7, 1514–1520 RSC; (d) S. Hong, A. M. Kawaoka and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 15878–15892 CrossRef CAS PubMed; (e) C. Welter, A. Dahnz, B. Brunner, S. Streiff, P. Dübon and G. Helmchen, Org. Lett., 2005, 7, 1239–1242 CrossRef CAS PubMed; (f) D. Berthold, A. G. A. Geissler, S. Giofré and B. Breit, Angew. Chem., Int. Ed., 2019, 58, 9994–9997 CrossRef CAS PubMed; (g) X. Ma, I. R. Hazelden, T. Langer, R. H. Munday and J. F. Bower, J. Am. Chem. Soc., 2019, 141, 3356–3360 CrossRef CAS PubMed; (h) S. H. Lim, S. Ma and P. Beak, J. Org. Chem., 2001, 66, 9056–9062 CrossRef CAS PubMed; (i) T. K. Beng and R. E. Gawley, J. Am. Chem. Soc., 2010, 132, 12216–12217 CrossRef CAS PubMed; (j) T. K. Beng and R. E. Gawley, Org. Lett., 2011, 13, 394–397 CrossRef CAS PubMed; (k) T. K. Beng, S. M. Wilkerson-Hill and R. Sarpong, Org. Lett., 2014, 16, 916–919 CrossRef CAS PubMed; (l) D. P. Bassler, A. Alwali, L. Spence, O. Beale and T. K. Beng, J. Organomet. Chem., 2015, 780, 6–12 CrossRef CAS; (m) D. P. Bassler, L. Spence, A. Alwali, O. Beale and T. K. Beng, Org. Biomol. Chem., 2015, 13, 2285–2292 RSC.
- (a) E. J. Barreiro, A. E. Kummerle and C. A. Fraga, Chem. Rev., 2011, 111, 5215 CrossRef CAS PubMed; (b) H. Schçnherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed; (c) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- W. Leitner, J. Klankermayer, S. Pischinger, H. Pitsch and K. Kohse, Angew. Chem., Int. Ed., 2017, 56, 5412–5452 CrossRef CAS PubMed.
- H. Braunstein, S. Langevin, M. Khim, J. Adamson, K. Hovenkotter, L. Kotlarz, B. Mansker and T. K. Beng, Org. Biomol. Chem., 2016, 14, 8864–8872 RSC.
- M. M. Nebe and T. Opatz, Synthesis of Piperidines and Dehydropiperidines: Construction of the Six-Membered Ring, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2017, vol. 122, ch. 5, pp. 191–244 Search PubMed.
- (a) M. Krasavin and D. Dar'in, Tetrahedron Lett., 2016, 57, 1635–1640 CrossRef CAS; (b) M. Gonzalez-Lopez and J. T. Shaw, Chem. Rev., 2009, 109, 164–189 CrossRef CAS PubMed.
- (a) M. Krasavin and D. Dar'in, Tetrahedron Lett., 2016, 57, 1635–1640 CrossRef CAS; (b) M. Gonzalez-Lopez and J. T. Shaw, Chem. Rev., 2009, 109, 164–189 CrossRef CAS PubMed.
- N. A. Sorto, M. J. Di Maso, M. A. Munoz, R. A. Dougherty, J. C. Fettinger and J. T. Shaw, J. Org. Chem., 2014, 79, 2601–2610 CrossRef CAS PubMed.
- (a) X. Qiao, D. L. Cheney, R. S. Alexander, A. M. Smallwood, S. R. King, K. He, A. R. Rendina, J. M. Luettgen, R. M. Knabb, R. R. Wexler and P. Y. S. Lam, Bioorg. Med. Chem. Lett., 2008, 18, 4118–4123 CrossRef PubMed; (b) M. A. Larsen, E. T. Hennessy, M. C. Deem, Y. Lam, J. Sauri and A. C. Sather, J. Am. Chem. Soc., 2020, 142(2), 726–732 CrossRef CAS PubMed.
- D. Dar’in, O. Bakulina, S. Nikolskaya, I. Gluzdikov and M. Krasavin, RSC Adv., 2016, 6, 49411–49415 RSC.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- (a) B. Feng, Y. Yang and J. You, Chem. Commun., 2020, 56, 790 RSC; (b) M. R. Yadav, M. Nagaoka, M. Kashihara, R.-L. Zhong, T. Miyazaki, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2017, 139, 9423 CrossRef CAS PubMed; (c) T. Okita, K. K. Asahara, K. Muto and J. Yamaguchi, Org. Lett., 2020, 22, 3205 CrossRef CAS PubMed; (d) R.-L. Zhong, M. Nagaoka, Y. Nakao and S. Sakaki, Organometallics, 2018, 37, 3480 CrossRef CAS; (e) F. Inoue, M. Kashihara, M. R. Yadav and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 13307 CrossRef CAS PubMed; (f) M. Kashihara, M. R. Yadav and Y. Nakao, Org. Lett., 2018, 20, 1655 CrossRef CAS PubMed; (g) W. Chen, K. Chen, W. Chen, M. Liu and H. Wu, ACS Catal., 2019, 9, 8110 CrossRef CAS; (h) K. Chen, W. Chen, X. Yi, W. Chen, M. Liu and H. Wu, Chem. Commun., 2019, 55, 9287 RSC; (i) M. Kashihara, R.-L. Zhong, K. Semba, S. Sakaki and Y. Nakao, Chem. Commun., 2019, 55, 9291 RSC; (j) K. K. Asahara, T. Okita, A. N. Saito, K. Muto, Y. Nakao and J. Yamaguchi, Org. Lett., 2019, 21, 4721 CrossRef CAS PubMed; (k) B. Feng, Y. Yang and J. You, Chem. Sci., 2020, 11, 6031 RSC.
- (a) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (b) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed.
- (a) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC; (b) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654–2672 CrossRef CAS PubMed; (c) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed; (d) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed; (e) D. A. Everson, J. A. Buonomo and D. J. Weix, Synlett, 2014, 25, 233–238 CAS; (f) C. E. I. Knappke, S. Grupe, D. Gaertner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed.
- (a) H. Ehrhorn and M. Tamm, Chem.–Eur. J., 2019, 25, 3190 CAS; (b) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS PubMed; (c) U. H. F. Bunz, K. Seehafer, M. Bender and M. Porz, Chem. Soc. Rev., 2015, 44, 4322 RSC; (d) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783 CrossRef CAS PubMed; (e) X. Ren, G. Li, S. Wei and H. Du, Org. Lett., 2015, 17, 990 CrossRef CAS PubMed; (f) A. Ahlers, T. de Haro, B. Gabor and A. Furstner, Angew. Chem., Int. Ed., 2016, 55, 1406 CrossRef CAS PubMed; (g) P. M. Cromm, S. Schaubach, J. Spiegel, A. Furstner, T. N. Grossmann and H. Waldmann, Nat. Commun., 2016, 7, 11300 CrossRef CAS PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- For recent and representative examples, see: (a) E. Fernandez, M. Rivero Crespo, A. I. Dominguez, P. Rubio-Marques, J. Oliver-Meseguer, L. Liu, M. Cabrero-Antonino, R. Gavara, J. C. Hernandez-Garrido, M. Boronat, A. Leyva-Perez and A. Corma, J. Am. Chem. Soc., 2019, 141, 1928 CrossRef CAS PubMed; (b) T. Wang, J. Huang, H. Lv, Q. Fan, L. Feng, Z. Tao, H. Ju, X. Wu, S. L. Tait and J. Zhu, J. Am. Chem. Soc., 2018, 140, 13421 CrossRef CAS PubMed; (c) W. Liu, L. Li and C.-J. Li, Nat. Commun., 2015, 6, 6526 CrossRef CAS PubMed.
- T. W. Greene, Protective Groups in Organic Synthesis, Wiley Interscience: New York, 1981 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/d1ra07390g |
| This journal is © The Royal Society of Chemistry 2022 |