
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConjugation with gold nanoparticles improves the stability of the KT2 peptide and maintains its anticancer properties†
Pornsuda Maraminga,
Jureerut Daduang *a and
James Chen Yong Kah*b
*a and
James Chen Yong Kah*b
aCentre for Research and Development of Medical Diagnostic Laboratories, Faculty of Associated Medical Sciences, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: jurpoo@kku.ac.th
bDepartment of Biomedical Engineering, National University of Singapore, 4 Engineering Drive 3, Blk E4, #04-08, Singapore 117583. E-mail: biekahj@nus.edu.sg
First published on 20th December 2021
Abstract
One of the major weaknesses of therapeutic peptides is their sensitivity to degradation by proteolytic enzymes in vivo. Gold nanoparticles (GNPs) are a good carrier for therapeutic peptides to improve their stability and cellular uptake in vitro and in vivo. We conjugated the anticancer KT2 peptide as an anticancer peptide model to PEGylated GNPs (GNPs-PEG) and investigated the peptide stability, cellular uptake and ability of the GNPs-KT2-PEG conjugates to induce MDA-MB-231 human breast cancer cell death. We found that 11 nm GNPs protected the conjugated KT2 peptide from trypsin proteolysis, keeping it stable up to 0.128% trypsin, which is higher than the serum trypsin concentration (range 0.0000285 ± 0.0000125%) reported by Lake-Bakaar, G. et al., 1979. GNPs significantly enhanced the cellular uptake of KT2 peptides after conjugation. Free KT2 peptides pretreated with trypsin were not able to kill MDA-MB-231 cells due to proteolysis, while GNPs-KT2-PEG was still able to exert effective cancer cell killing after trypsin treatment at levels comparable to GNPs-KT2-PEG without enzyme pretreatment. The outcome of this study highlights the utility of conjugated anticancer peptides on nanoparticles to improve peptide stability and retain anticancer ability.
1. Introduction
An increasing number of studies have shown the potential of peptides as new candidates for drug discovery and development to combat cancer.1–3 Peptides have low immunogenicity, excellent cell and tissue penetration, and low production cost and can be easily modified to improve their functionality and stability in vivo, which make them ideal for cancer treatment.4 Therapeutic peptides for cancer treatment have been classified into three main groups: antimicrobial/pore-forming peptides, cell-penetrating peptides, and tumor-targeting peptides.4Anticancer peptides often function as molecular targeting peptides, especially those with α-helical structures that are able to penetrate the plasma membrane, nuclear membrane and/or mitochondrial membrane of cancer cells to induce membrane disruption, apoptosis, autophagy, necrosis, and alteration of gene expression involving DNA synthesis or cell division.3,5–7 Such selectivity for cancer cells is possible due to the net negative charge on the outer membrane leaflet of the cancer cells interacting with the cationic anticancer peptides leading to cell death; in contrast, the outer membrane leaflet of healthy cells with neutral net charge interacts minimally with these anticancer peptides.3
The cell-penetrating KT2 peptide NGVQPKYKWWKWWKKWW-NH2 is a cationic amphipathic peptide (17 amino acids in length, 53% hydrophobicity, and seven positive charges), with the first 7 amino acids derived from identified peptides in the white blood cell extract of Crocodylus siamensis. Tryptophan (W) was included to increase the hydrophobicity, and lysine (K) increased the hydrophilicity and peptide charges to improve peptide stability and enhance bacterial killing.8 KT2 peptides exhibited antibacterial activities against both Gram-positive and Gram-negative bacteria but were not toxic to normal cells.8 We found that KT2 was toxic against cervical cancer HeLa cells and colon cancer HCT 116 cells, as it induced apoptotic cell death in these cells both in vitro and in vivo.9–12 Moreover, KT2 suppressed the migration of HCT 116 cells.10
Despite their efficacy, therapeutic peptides are often prone to aggregation, susceptibility to proteolytic degradation in biological environments, and short circulating half-lives resulting in the requirement for frequent administrations to maintain their efficacy in vivo.13 Apart from peptide modifications to improve their delivery, their surface conjugation to nanoparticle carriers serves as another promising strategy that also offers selective cancer targeting through the enhanced permeability and retention (EPR) effect that nanoparticulate systems could afford in vivo as a therapeutic carrier.14–16 However, the ability of nanoparticles to improve the stability of conjugated peptides has not been systematically demonstrated to date. In the last few decades, gold nanoparticles (GNPs) have emerged as one of the most active field for researchers. The unique properties and various surface functionalities of GNPs have provided them potential applications in bionanotechnology including drug delivery, sensing, and imaging.17–20 Here, we utilized GNPs as a peptide carrier due to their facile synthesis over a broad size range, good biocompatibility, and ease of surface functionalization, such as PEGylation.21 We conjugated KT2 peptides on PEGylated GNPs (GNPs-PEG) and examined the improved peptide stability, cellular uptake, and cell cytotoxicity of GNPs-PEG-KT2 conjugates against MDA-MB-231 human breast cancer cells (Fig. 1).
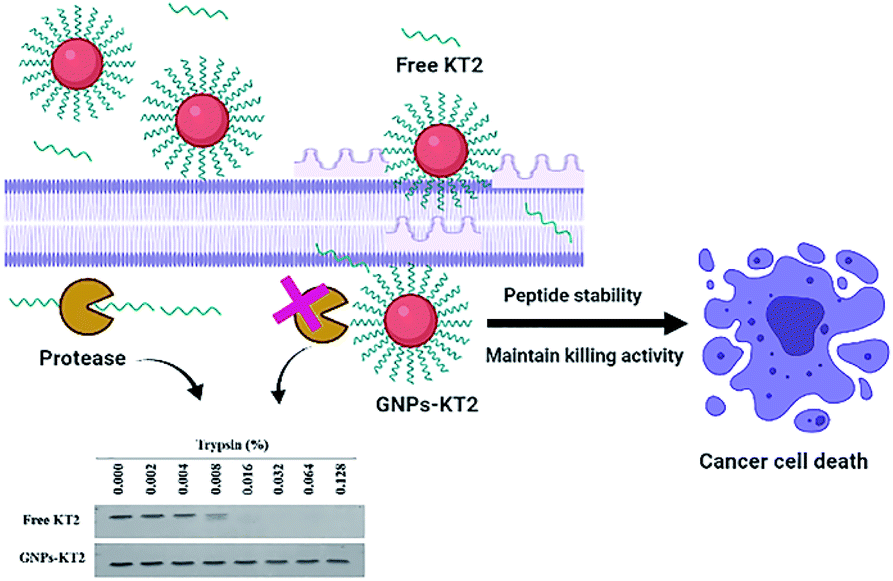 | ||
| Fig. 1 Schematic showing the use of GNPs to increase the stability of the KT2 peptide while maintaining the anticancer effect of KT2. | ||
2. Materials and method
2.1 Synthesis of gold nanoparticles
GNPs were synthesized following the citrate reduction method of Turkevich.22 Briefly, 100 mL of 1 mM HAuCl4 was added to an Erlenmeyer flask and boiled on a hotplate. While boiling, 15 mL of a 1% solution of trisodium citrate dihydrate was quickly added with stirring and allowed to turn deep red for 15 min. The solution was allowed to reach room temperature before centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min to remove excess citrate. The GNPs pellet was resuspended in ultrapure water (18.2 MΩ cm) to a concentration of 10 nM based on the absorbance measurement and was stored at 4 °C until further use.
000 rpm for 20 min to remove excess citrate. The GNPs pellet was resuspended in ultrapure water (18.2 MΩ cm) to a concentration of 10 nM based on the absorbance measurement and was stored at 4 °C until further use.
2.2 GNPs-KT2 conjugation
KT2 peptide (GL Biochem Ltd., Shanghai, China) with an additional cysteine residue at the N-terminus was used for conjugation on the GNPs. The cysteine contained a thiol group that could form the gold–sulfur (Au–S) coordinate bond on the surface of GNPs. KT2 was conjugated on GNPs at a low pH and a high peptide-to-GNPs ratio following established protocols.23 In one reaction, 440 μL of ultrapure water, 30 μL of 1 M HCl, and 30 μL of 1 mM KT2 were mixed together in a microcentrifuge tube followed by hydrolysis on a platform shaker for 10 min. Then, 500 μL of 10 nM GNPs was added to the tube while vortexing. The mixture was mixed continuously using a platform shaker for 3 h at room temperature. The resulting GNPs-KT2 was washed three times with ultrapure water and centrifuged at 10000 rpm for 20 min to remove unbound KT2. The pellet was resuspended in ultrapure water. GNPs-KT2-PEG was also prepared by adding the same protocol as GNPs-KT2, KT2 peptides were added to first form GNPs-KT2, followed by the further addition of 20 μL of 2 mM mPEG-SH (MW = 1 K) to form GNPs-KT2-PEG.
The absorbance spectra of GNPs, GNPs-PEG, GNPs-KT2, and GNPs-KT2-PEG were measured using UV-vis spectroscopy (UV-2450, Shimadzu, Japan) over wavelengths of 400 to 900 nm. The hydrodynamic diameter and zeta potential of all nanoparticles were also measured using a Zetasizer (Nano ZS, Malvern, UK). The core size of GNPs was visualized under a transmission electron microscope (TEM) (TecnaiG2 20 FEI, USA).
2.3 Semiquantitative measurement of the amount of KT2 conjugated on gold nanoparticles
A mixture consisting of 20 μL of 16 nM GNPs-KT2, 8 μL of 5× loading buffer, 10 μL of 1% SDS, and 2 μL of 0.5 M DTT was prepared in triplicate and heated at 100 °C for 10 min to release the conjugated KT2 from GNPs. Fifteen microliters of heated samples were loaded for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The gel was run at a voltage of 85 V for 10 min and at 120 V for 45 min, rinsed with ultrapure water 3 times, and fixed with 10% acetic acid plus 30% methanol in ultrapure water for 30 min. Peptide bands were stained with Bio-Safe Coomassie stain (Bio-Rad, USA) and analyzed against a standard curve of known KT2 concentrations using ImageJ software to determine the number of conjugated KT2 per GNP.2.4 Stability of conjugated KT2 peptide on gold nanoparticles
The proteolytic degradation of free KT2 peptide and conjugated KT2 on GNPs was compared using trypsin as a model protease. Ten microliters of 25 nM GNPs-KT2 or 37.5 μM free KT2 peptide at equivalent amounts as determined previously was added to 10 μL of different percentages of trypsin (0.000, 0.002, 0.004, 0.008, 0.016, 0.032, 0.064, and 0.128% W/V) in a microcentrifuge tube to enable proteolytic degradation by trypsin. The mixtures were incubated at 37 °C for 5 min and kept on ice before 5 μL of 5× loading buffer was added. The mixture of trypsin and GNPs-KT2 or free KT2 was then heated at 100 °C for 5 min to heat-inactivate the trypsin and denature the peptide samples before running them on 12% SDS-PAGE and staining the peptide fragments with Coomassie blue. The peptide bands were analyzed using ImageJ software and compared against the protein control bands.2.5 Cell culture
MDA-MB-231 human breast cancer cells were kindly gifted by Dr James Chen Yong Kah from National University of Singapore. The cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin and 100 mg mL−1 streptomycin at 37 °C in a 5% CO2 humidified atmosphere. MDA-MB-231 cells were subcultured when 80–100% confluent to carry out passage of the cells.2.6 Cellular uptake study
The qualitative uptake of GNPs-KT2 was visualized using dark-field imaging, where the GNPs exhibited a strong optical scattering signal against a dark background. MDA-MB-231 cells (1.0 × 105 cells per well) were seeded onto cover slips in a 24-well plate for 24 h before being treated with 33.33 nM GNPs-KT2-PEG for 6 h at 37 °C, with 33.33 nM GNPs-PEG and 500 μL of 50 μM KT2 as controls. After washing thrice with 1× PBS, the cells were fixed with 3.7% formalin solution for 10 min, and the nuclei were stained with 300 nM DAPI solution for 10 min at room temperature. Dark-field and fluorescence signals were imaged using a Nikon Ci-L Fluorescence Upright Microscope (Nikon Instruments, Japan) with an oil immersion 60× objective.2.7 Cell cytotoxicity test
PrestoBlue reagent was used to evaluate the viability of MDA-MB-231 cells after treatment with KT2 or GNPs-KT2. The cells were seeded at 1 × 104 cells per well into 96-well plates and incubated for 24 h before being treated with 6.67, 13.33, 20.00, 26.67, and 33.33 nM GNPs-KT2 or 10, 20, 30, 40, and 50 μM free KT2 (at an equivalent concentration as that conjugated on GNPs) for 24 h. The excess GNPs-KT2 and free KT2 were removed, and the cells were rinsed three times with 1× PBS (pH 7.4) after treatment. Then, 100 μL of 1× PrestoBlue solution was added to each well and incubated for 1 h at 37 °C. The PrestoBlue reagent was quickly reduced by metabolically active cells, which turned red and became highly fluorescent (λEx/λEm = 560/590 nm) to enable a quantitative measure of viability using a microplate reader (Tecan, Switzerland).Cell viability was also assessed after MDA-MB-231 cells were treated with a mixture of inactivated trypsin with GNPs-conjugated peptides or free peptides to examine for any reduced treatment efficacy by KT2 peptide postproteolytic degradation. Forty microliters of 333.3 nM GNPs-PEG, GNPs-KT2 and GNPs-KT2-PEG or 500 μM KT2 were mixed with 40 μL of 0.032% trypsin or ultrapure water in a microcentrifuge tube. The mixtures were incubated at 37 °C for 5 min before 320 μL of FBS-containing medium was added to each tube, and the resulting mixture was added to previously seeded cells (1 × 104 cells per well) for 24 h.
2.8 Statistical analysis
The data are expressed as the mean ± SD. All data were analyzed using SPSS® software version 19.0, and the differences between all groups were established by one-way ANOVA. Intergroup comparisons were performed using the unpaired t test, and p values of <0.05 were considered statistically significant.3. Results and discussion
3.1 Characterization of KT2-conjugated GNPs
The synthesized GNPs were generally spherical and monodispersed, with an average diameter of 11.39 ± 1.71 nm, as observed under TEM (Fig. 2a and b). The acidic conditions during peptide conjugation and a high peptide-to-GNPs ratio (6000:1) allowed GNPs-KT2 and GNPs-KT2-PEG to maintain colloidal stability after peptide conjugation since the absence of such acid conditions caused GNPs to aggregate when KT2 peptides were added (Fig. 3). Despite a KT2/GNP ratio of 6000, the peptide conjugation ratio was unlikely as a previous study has shown that a much higher peptide/GNP incubation ratio of 20000,23 was sufficient to maintain the colloidal stability of the GNPs.
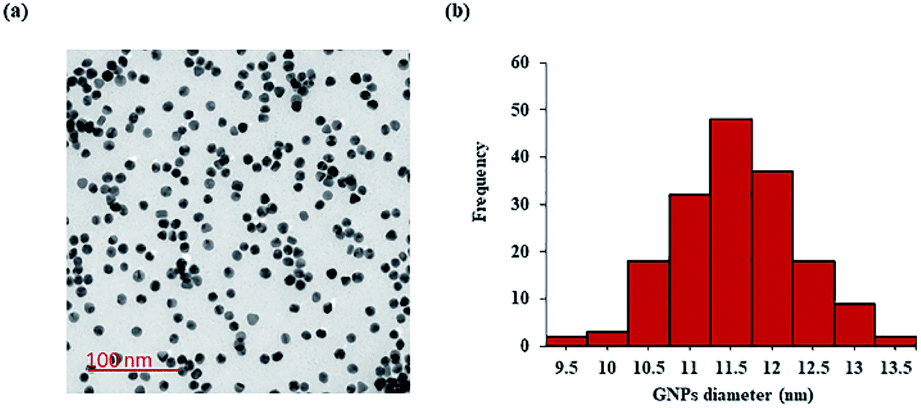 | ||
| Fig. 2 Characterization of synthesized citrate-capped GNPs. (a) Citrate-capped GNPs were monodispersed, as imaged under TEM, and (b) had an average diameter of 11.39 ± 1.71 nm, as determined by ImageJ analysis. | ||
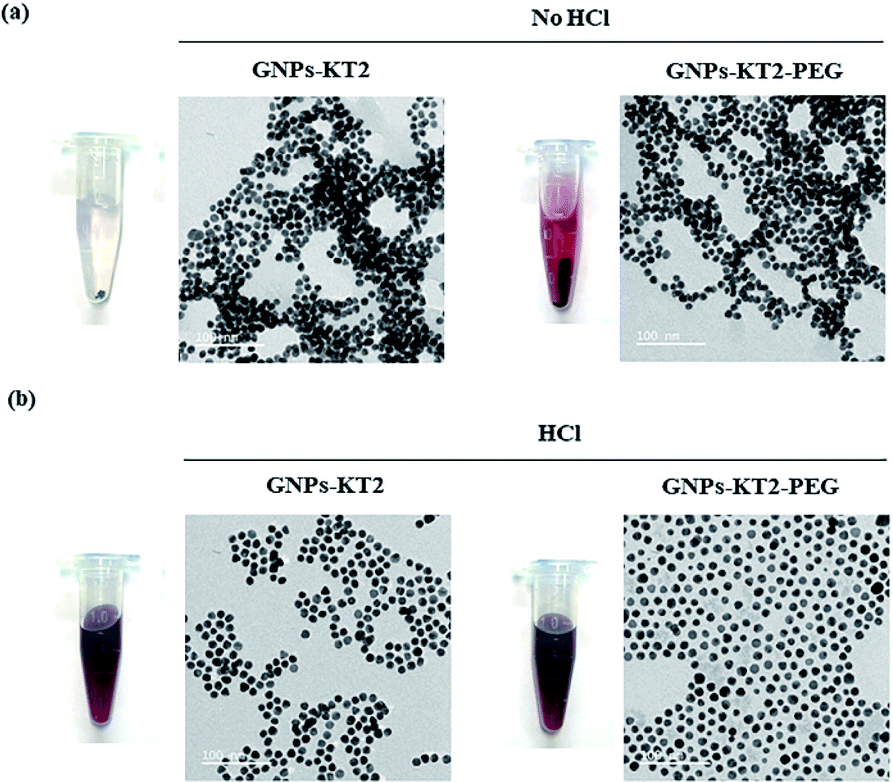 | ||
| Fig. 3 The results of KT2 peptides conjugated on GNPs under with or without acid conditions. (a) GNPs-KT2 and GNPs-KT2-PEG in the absence of acid condition showed aggregations. (b) GNPs-KT2 and GNPs-KT2-PEG in acid condition were monodispersed, as imaged under TEM. | ||
GNPs-PEG had a surface plasmon resonance (SPR) peak at 523 nm, while GNPs-KT2 and GNPs-KT2-PEG had SPR peaks at 530 and 525 nm, respectively. The slight redshift in peak absorbance over citrate-capped GNPs (520 nm) was due to the presence of PEG and KT2 on the surface of GNPs, which changed the local refractive index and was indicative of successful conjugation (Fig. 4a). The presence of peptide molecules capped on the GNP surface also caused the hydrodynamic diameter (DH) to increase from 15.62 ± 0.90 nm (citrate-capped GNPs) to 20.65 ± 0.03 (GNPs-PEG), 25.72 ± 2.22 (GNPs-KT2) and 22.63 ± 0.09 nm (GNPs-KT2-PEG) after peptide conjugation (Fig. 4b). The particle size distribution histogram (Fig. 4b) also confirmed the absence of large aggregations in GNPs-KT2 and GNPs-KT2-PEG after peptide conjugation.
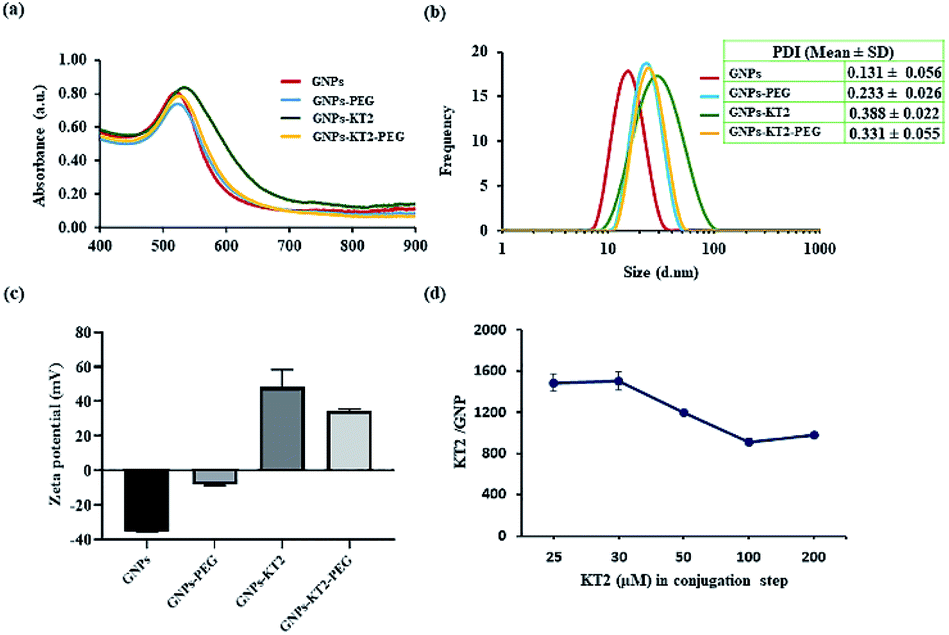 | ||
| Fig. 4 (a) Absorbance spectra of GNPs measured using UV-vis spectroscopy. (b) Hydrodynamic diameter (DH) of GNPs as determined by dynamic light scattering (DLS). (c) Zeta potential of GNPs and (d) number of peptides conjugated to each GNP in GNPs-KT2. The polydispersity index (PDI) represents the size dispersity of the nanoparticle population. Error bars are expressed as the mean ± SD. | ||
Successful peptide conjugation was also confirmed from the flip in zeta potential of citrate-capped GNPs from −35.13 ± 0.32 mV due to negatively charged citrate to +34.56 ± 1.15 mV (GNPs-KT2-PEG) and +48.32 ± 1.15 mV (GNPs-KT2) due to the positive charge of KT2 (Fig. 4c). By disrupting the Au–S bond and performing SDS-PAGE to quantify the released peptides that migrated through the separating gel and were stained by Coomassie blue, we found ∼1500 ± 125 KT2 peptides conjugated per GNP (Fig. 4d) when 30 μM of KT2 was used in conjugation. Chan et al. reported 540 ± 26 molecules of p53 peptides conjugated on each GNP when used a higher peptide/GNP incubation ratio (20000:1) with similar particle size and peptide length compared to our study.23 The number of peptides per GNP was reduced when the KT2 concentration was increased between 50–200 μM, as the high peptide concentration could cause possible steric hindrance during peptide attachment.24
3.2 Improved stability of the KT2 peptide with conjugation
Free peptides are highly susceptible to proteolytic degradation, which consequently limits their therapeutic efficacy at the target site. In this study, we compared the stability of conjugated KT2 peptides against free KT2 peptides under proteolysis with trypsin as our model protease. Trypsin is a serine protease produced in the pancreas of many vertebrates, with its cleavage site on the C-terminal ends of lysine and arginine amino acid residues. It is capable of cleaving the five lysine amino acids found in the KT2 peptide.25,26Free KT2 peptide at the same concentration as that conjugated on GNPs was stable at up to 0.004% trypsin before degradation became observable from 0.008% trypsin onwards, as evidenced by the decreasing band intensity. The free peptides were completely degraded by 0.032% trypsin (Fig. 5a and b). Conversely, the conjugated KT2 peptide remained stable at up to 0.128% trypsin. The semiquantitative image analysis of SDS-PAGE staining, as shown in Fig. 5b, revealed that GNPs could protect the conjugated KT2 peptides and kept them intact against at least 32-fold higher trypsin concentrations than free KT2 peptides. A previous study demonstrated that p53 peptides conjugated on GNPs showed stable up to 0.0156% trypsin before starting to be degraded. Due to higher packing density of KT2 peptides on GNPs, conjugated KT2 peptides showed a greater stability against enzymatic degradation than conjugated p53 peptides.23 In another study about GNPs-peptide stability, GNPs coated with the antimicrobial peptide esculentin-1a(1–21)NH2 (∼16 peptides per GNP) preserved their antibacterial activity in the presence of a proteolytic enzyme.27
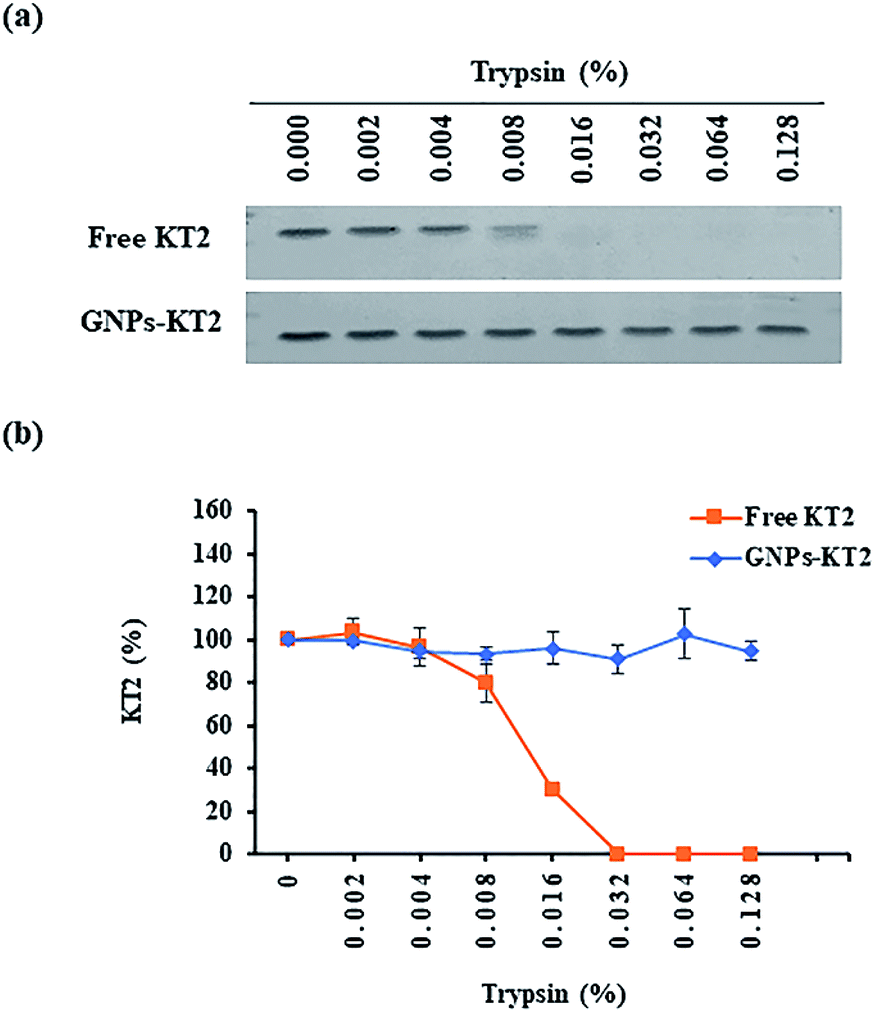 | ||
| Fig. 5 Stability of free and conjugated KT2 peptides under trypsin treatment. (a) Free KT2 peptides and GNPs-KT2 with the same amount of KT2 peptides were exposed to a range of trypsin concentrations, and the amount of undigested KT2 peptides was visualized by SDS-PAGE followed by protein staining. (b) The KT2 peptide band intensity was analyzed using ImageJ software to show the percentage of undigested peptides against the range of trypsin concentrations. | ||
3.3 Intracellular uptake of gold nanoparticles
Cellular uptake of GNPs-KT2-PEG in MDA-MB-231 cells was visualized under dark-field microscopy, where the strong optical scattering of GNPs resulted in bright signals (Fig. 6).28 Here, 23 nm GNPs-KT2-PEG showed a stronger scattering signal than 21 nm GNPs-PEG. The uptake of GNPs of different sizes (2 to 100 nm) conjugated with Herceptin by SK-BR-3 cells was demonstrated to be size-dependent. The highest cellular internalization was found for nanostructures in size ranges of 25–50 nm.29 Previous studies reported that small nanoparticles (<10–15 nm) are related to rapid clearance from the body via renal filtration and urinary excretion.30–32 Therefore, our nanoconstructs showed the optimal size to internalize cells for cancer therapy and were larger than the clearance threshold. However, we observed purple aggregated nanoparticles localized in cells under a light microscope, as viewed at 400× magnification (ESI, Fig. S1†). Therefore, the scattering signal of GNPs could likely come from the aggregates and be detected by darkfield microscopy (Fig. 6). In this imaging, we were not able to distinguish whether the particles were adsorbed or internalized, though it was less consequential since we showed a reduction in cell death, which was ultimately the most important readout (Fig. 7). The cell-penetrating peptide property of KT2 might also enhance the internalization of GNPs into the cells, as in previous reports.33,34 In general, cancer cells present higher net negative charges on their surfaces35,36 that are related to the transformation of normal to cancer cells and from nonmetastatic to metastatic cells.37,38 This unique property of cancer cells allows them to interact preferentially with positive surfaces via electrostatic attraction, including cationic nanoparticles and the positive net charge of GNPs-KT2-PEG. Such an interaction formed the initial step for nanoparticle internalization and cell-targeted cytotoxicity.39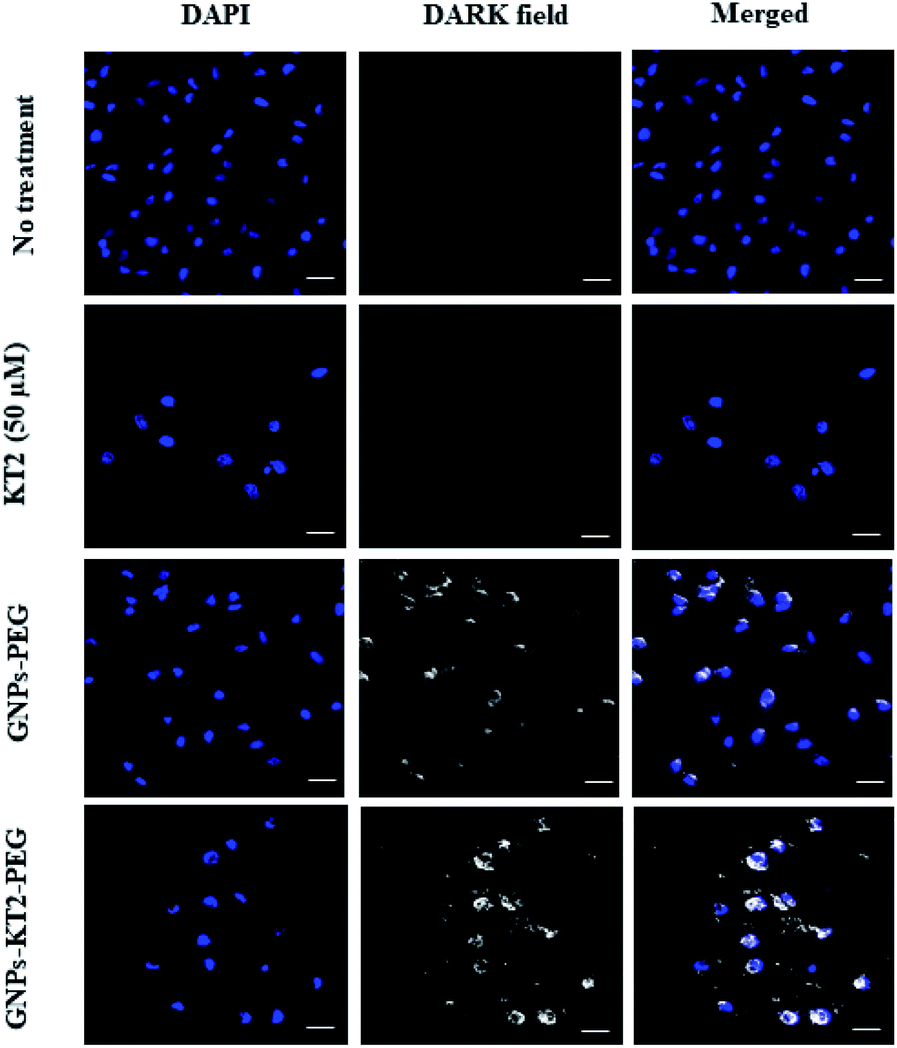 | ||
| Fig. 6 Cellular uptake study. Cellular images were captured after 24 h of incubation, showing the higher localization of GNPs-KT2-PEG than GNPs-PEG in MDA-MB-231 cells. The scale bars are 20 μm. | ||
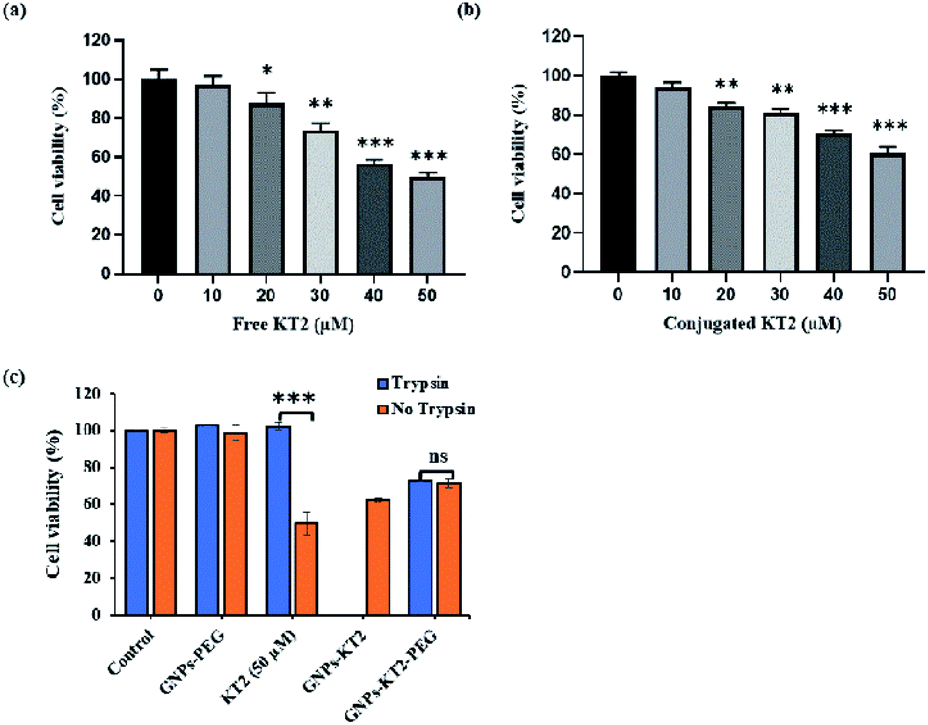 | ||
| Fig. 7 Cell viability after dosing with (a) free KT2 and (b) GNPs-KT2. (c) Cell viability of MDA-MB-231 breast cancer cells after dosing with 33.33 nM GNPs-PEG, GNPs-KT2, GNPs-KT2-PEG, and 50 μM free KT2 peptides in the presence and absence of trypsin pretreatment to demonstrate loss in therapeutic efficacy of KT2 peptides due to proteolytic degradation. The data are shown as the means ± standard deviation (*p < 0.05, **p < 0.01, and ***p < 0.001). | ||
We also observed some bright signals outside MDA-MB-231 cells treated with GNPs-PEG and GNPs-KT2-PEG, which could be due to their decreased colloidal stability and aggregation in the culture medium. The positively charged KT2 peptide on GNPs surface could also exhibit affinity to the coverslip surface. In addition, cellular images showed some overlap of the GNPs signals with the DAPI signals. These overlapping signals resulted from a different imaging plane as the nucleus since optical sectioning could not be performed in the dark field microscopy used in this study.
3.4 Cell cytotoxicity test
MDA-MB-231 breast cancer cells were treated with different concentrations (10–50 μM) of peptides for 24 h to compare efficacy between free KT2 peptide and conjugated KT2 peptide at equal amounts of peptide to induce cytotoxicity in breast cancer cells. Cell viability was measured using a PrestoBlue assay, which determines the metabolic activity of viable cells and emits quantifiable fluorescence. Both free KT2 peptide and GNPs-KT2 showed reduced cell viability with increasing concentrations of KT2 peptide within the range of 20 to 50 μM (Fig. 7a and b). This was in agreement with other previous studies that reported the inhibition of malignant HCT 116 colon cancer cell proliferation and induction of cell apoptosis by KT2 peptides,9,10 likely due to its cationic cell-penetrating property from an amphipathic α-helix structure.8,9,40However, we also observed slightly lower cytotoxicity induced by GNPs-KT2 compared to free KT2 peptide at concentrations of more than 30 μM of KT2 peptide. This could be attributed to the high packing density of peptides on GNPs surfaces, which compromised their functionality or caused a loss in their secondary structure (ESI, Fig. S2†). KT2 is able to penetrate the cell membrane and accumulate in both the cytoplasm and nuclease, and this internalization is involved in its cytotoxicity.9,40 With peptide conjugation, conjugated KT2 peptides formed strong Au–S, which made it difficult for them to release or function freely in cells in a manner similar to unconjugated peptides.
Finally, we investigated the ability of conjugated KT2 to survive proteolytic degradation by 0.032% trypsin to induce cytotoxicity to MDA-MB-231 breast cancer cells (Fig. 7c). This concentration of trypsin was previously shown to completely digest free KT2 but not GNPs-KT2. Here, GNPs-KT2 aggregated after adding 10% FBS-supplemented DMEM to deactivate trypsin; thus, the cell viability of GNPs-KT2 treated with trypsin was not shown. Therefore, we improved the colloidal stability with PEGylation and showed that GNPs-PEG by itself without KT2 peptides was nontoxic to MDA-MB-231 cells in both pretreatment with and without trypsin.
In the absence of conjugation, free KT2 peptides pretreated with trypsin were no longer effective in inducing cell death, as the viability increased from 49.77 ± 2.06% to 100% due to degradation of KT2 peptides by proteolytic enzymes. While GNPs-KT2 killed 39.68 ± 3.53% of MDA-MB-231 cells in the absence of trypsin, its cell viability after treatment with trypsin could not be determined due to aggregation after adding 10% FBS-supplemented DMEM, as mentioned earlier. Nonetheless, we previously observed that the KT2 peptides conjugated on NPs-KT2 remained stable and nondegraded after trypsin treatment. This was likely due to the sufficiently high packing density of peptides on GNPs arising from our conjugation protocol that conferred steric hindrance to trypsin proteins in accessing the cleavage sites and causing trypsin digestion, thus leading to the decreased effectiveness of the enzyme.41
The colloidal stability of GNPs-KT2 was improved with PEGylation, and we observed no aggregation of GNPs-KT2-PEG after treatment with trypsin and subsequent deactivation of trypsin with 10% FBS-supplemented DMEM. While GNPs-KT2-PEG could afford slightly lower levels of cell killing to MDA-MB-231 cells compared to GNPs-KT2 and free KT2 peptides, it could still prevent proteolytic degradation by trypsin with no discernible decrease in cell killing compared to free KT2 peptides.
In fact, free KT2 peptides were susceptible to the proteolytic environment leading to completely lost anticancer activity in the presence of trypsin, while our KT2 peptide-modified GNPs helped improve peptide stability while maintaining cancer cell-killing ability in a hostile proteolytic environment. The stability of KT2 peptides requires less frequent administration to maintain their efficacy in vivo resulting in a lower cost of their therapeutic effect.13
4. Conclusions
We conjugated a cationic anticancer KT2 peptide on GNP surfaces using a facile method. Peptide conjugation was prepared at a low pH below its peptide isoelectric point of 10.0 and using a high peptide-to-GNP ratio to produce colloidally stability of peptide-capped nanoparticles. The conjugated peptides could enhance the internalization of nanoparticles into cells, and the conjugation of unstable peptides with nanoparticles showed improved stability against proteolytic degradation, such that the conjugated peptides could still operate in a hostile proteolytic environment to maintain their functionality in cell killing compared to free peptides, which are quickly degraded and lose their functionality in cell killing.This study has highlighted an interesting strategy that showcases the advantages of nanotechnology to maintain functional forms of biomolecules such as toxicity, biorecognition, and signal transduction for treatment, which would otherwise be susceptible to degradation by digestive enzymes. The same concept of the conjugation of biomolecules to nanoparticles could easily be extended to enhance the stability of other therapeutic biomolecules to serve their function at target sites.
Abbreviations
| GNPs | Gold nanoparticles |
| GNPs-PEG | PEGylated GNPs |
| Au–S | Gold sulfur |
| TEM | Transmission electron microscope |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SPR | Surface plasmon resonance |
Author contributions
Conceptual framework, J. D. and P. M.; experimental design, J. C. Y. K. and P. M.; core facility for materials and analysis tools, J. C. Y. K.; performing experiments, P. M.; data analysis and results interpretation, P. M. and J. D.; drafting manuscript, P. M.; revision of the manuscript, J. C. Y. K. and J. D.; final approval version of the manuscript, J. C. Y. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Nurul 'Ain Azman and Mr Cheah U-Jin Joshua of the Nanomedicine and Nanorobotics lab, Department of Biomedical Engineering, National University of Singapore, for providing lab training and support. This work has received scholarship under the Post-Doctoral Training Program from Khon Kaen University, Thailand (grant no. PD2563-02-30) and the Centre for Research and Development of Medical Diagnostic Laboratories, Faculty of Associated Medical Sciences, Khon Kaen University, Khon Kaen, Thailand. This work was also supported by the Singapore Ministry of Education (MOE) AcRF Tier 1 Grant.References
- F.-M. Li and X.-Q. Wang, Sci. Rep., 2016, 6, 33910 CrossRef CAS PubMed.
- G. Gabernet, A. T. Müller, J. A. Hiss and G. Schneider, MedChemComm, 2016, 7, 2232–2245 RSC.
- W. Chiangjong, S. Chutipongtanate and S. Hongeng, Internet J. Oncol., 2020, 57, 678–696 CrossRef CAS PubMed.
- S. Marqus, E. Pirogova and T. J. Piva, J. Biomed. Sci., 2017, 24, 21 CrossRef PubMed.
- P. Zhang, J. Ma, Y. Yan, B. Chen, B. Liu, C. Jian, B. Zhu, S. Liang, Y. Zeng and Z. Liu, Org. Biomol. Chem., 2017, 15, 9379–9388 RSC.
- E. J. Paredes-Gamero, M. N. C. Martins, F. A. M. Cappabianco, J. S. Ide and A. Miranda, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 1062–1072 CrossRef CAS PubMed.
- S. X. Ren, J. Shen, A. S. L. Cheng, L. Lu, R. L. Y. Chan, Z. J. Li, X. J. Wang, C. C. M. Wong, L. Zhang, S. S. M. Ng, F. L. Chan, F. K. L. Chan, J. Yu, J. J. Y. Sung, W. K. K. Wu and C. H. Cho, PLoS One, 2013, 8, e63641 CrossRef CAS PubMed.
- T. Anunthawan, N. Yaraksa, S. Phosri, T. Theansungnoen, S. Daduang, A. Dhiravisit and S. Thammasirirak, Bioorg. Med. Chem. Lett., 2013, 23, 4657–4662 CrossRef CAS PubMed.
- P. Maraming, S. Klaynongsruang, P. Boonsiri, S. Peng, S. Daduang, C. Leelayuwat, C. Pientong, J. Chung and J. Daduang, J. Cell. Physiol., 2019, 234, 22116–22129 CrossRef CAS PubMed.
- S. Maijaroen, N. Jangpromma, J. Daduang and S. Klaynongsruang, Environ. Toxicol. Pharmacol., 2018, 62, 164–176 CrossRef CAS PubMed.
- P. Maraming, S. Maijaroen, S. Klaynongsruang, P. Boonsiri, S. Daduang, J.-G. Chung and J. Daduang, In Vivo, 2018, 32, 1137–1144 CrossRef CAS PubMed.
- T. Theansungnoen, S. Maijaroen, N. Jangpromma, N. Yaraksa, S. Daduang, T. Temsiripong, J. Daduang and S. Klaynongsruang, Protein J., 2016, 35, 202–211 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- S. K. Golombek, J.-N. May, B. Theek, L. Appold, N. Drude, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2018, 130, 17–38 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- M. Morales-Cruz, Y. Delgado, B. Castillo, C. M. Figueroa, A. Molina, A. Torres, M. Milian and K. Griebenow, Drug Des., Dev. Ther., 2019, 13, 3753–3772 CrossRef CAS PubMed.
- E. Priyadarshini and N. Pradhan, Sens. Actuators, B, 2017, 238, 888–902 CrossRef CAS.
- M. U. Farooq, V. Novosad, E. A. Rozhkova, H. Wali, A. Ali, A. A. Fateh, P. B. Neogi, A. Neogi and Z. Wang, Sci. Rep., 2018, 8, 2907 CrossRef PubMed.
- S. Lee, E.-J. Cha, K. Park, S.-Y. Lee, J.-K. Hong, I.-C. Sun, S. Y. Kim, K. Choi, I. C. Kwon, K. Kim and C.-H. Ahn, Angew. Chem., Int. Ed., 2008, 47, 2804–2807 CrossRef CAS PubMed.
- X. He, H. Liu, Y. Li, S. Wang, Y. Li, N. Wang, J. Xiao, X. Xu and D. Zhu, Adv. Mater., 2005, 17, 2811–2815 CrossRef CAS.
- D. Lee, J. Zhao, H. Yang, S. Xu, H. Kim, S. Pacheco, S. Keshavjee and M. Liu, Nanoscale, 2015, 7, 12356–12360 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- K. P. Chan, S.-H. Chao and J. C. Y. Kah, Bioconjugate Chem., 2019, 30, 920–930 CrossRef CAS PubMed.
- H. Sun, W. Cao, N. Zang, T. D. Clemons, G. M. Scheutz, Z. Hu, M. P. Thompson, Y. Liang, M. Vratsanos, X. Zhou, W. Choi, B. S. Sumerlin, S. I. Stupp and N. C. Gianneschi, Angew. Chem., Int. Ed., 2020, 59, 19136–19142 CrossRef CAS PubMed.
- C. J. Koehler and B. Thiede, JBIC, J. Biol. Inorg. Chem., 2020, 25, 61–66 CrossRef CAS PubMed.
- S. Heissel, S. J. Frederiksen, J. Bunkenborg and P. Højrup, PLoS One, 2019, 14, e0218374 CrossRef CAS PubMed.
- B. Casciaro, M. Moros, S. Rivera-Fernández, A. Bellelli, J. M. de la Fuente and M. L. Mangoni, Acta Biomater., 2017, 47, 170–181 CrossRef CAS PubMed.
- W. Qian, X. Huang, B. Kang and M. A. El-Sayed, J. Biomed. Opt., 2010, 15, 046025 CrossRef PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef PubMed.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344–15349 CrossRef CAS PubMed.
- C. H. J. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656–6661 CrossRef CAS PubMed.
- H. R. Samadikhah, M. Nikkhah and S. Hosseinkhani, Luminescence, 2017, 32, 517–528 CrossRef CAS PubMed.
- S. Khamehchian, M. Nikkhah, R. Madani and S. Hosseinkhani, J. Biomed. Mater. Res., Part A, 2016, 104, 2693–2700 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- B. Chen, W. Le, Y. Wang, Z. Li, D. Wang, L. Ren, L. Lin, S. Cui, J. J. Hu, Y. Hu, P. Yang, R. C. Ewing, D. Shi and Z. Cui, Theranostics, 2016, 6, 1887–1898 CrossRef CAS PubMed.
- R. J. Boohaker, M. W. Lee, P. Vishnubhotla, J. L. M. Perez and A. R. Khaled, Curr. Med. Chem., 2012, 19, 3794–3804 CrossRef CAS PubMed.
- W. Le, B. Chen, Z. Cui, Z. Liu and D. Shi, Biophys. Rep., 2019, 5, 10–18 CrossRef CAS.
- J. Zhao and M. H. Stenzel, Polym. Chem., 2018, 9, 259–272 RSC.
- T. Anunthawan, C. de la Fuente-Núñez, R. E. W. Hancock and S. Klaynongsruang, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1352–1358 CrossRef CAS PubMed.
- P. Wadhwani, N. Heidenreich, B. Podeyn, J. Bürck and A. S. Ulrich, Biomater. Sci., 2017, 5, 817–827 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05980g |
| This journal is © The Royal Society of Chemistry 2022 |