
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal- and oxidant-free electrochemically promoted oxidative coupling of amines†
Gang Liua,
Sen Liua,
Zhen Lia,
Hengyu Chena,
Jiashuai Lia,
Yalin Zhanga,
Guodong Shen *a,
Bingchuan Yang*a,
Xiude Hub and
Xianqiang Huang*a
*a,
Bingchuan Yang*a,
Xiude Hub and
Xianqiang Huang*a
aShandong Provincial Key Laboratory of Chemical Energy Storage and Novel Cell Technology, School of Chemistry & Chemical Engineering, Liaocheng University, Liaocheng, Shandong 252059, China. E-mail: hxqqxh2008@163.com
bState Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, Ningxia University, Yinchuan 750021, China
First published on 20th December 2021
Abstract
The selective oxidation of amines into imines is a priority research topic in organic synthesis and has attracted much attention over the past few decades. However, the oxidation of amines generally suffers from the drawback of transition-metal, even noble-metal catalysts. Thus, the strategy of metal- and oxidant-free selective synthesis of imines is highly desirable yet largely unmet. This paper unravels a metal-free and external oxidant-free electrochemical strategy for the oxidative coupling methodology of amines. This general transformation is compatible with various functional amines and led to functionalized imines in moderate to satisfactory yields.
Introduction
Over the past several decades, imine derivatives have been envisaged as a class of important bioactive versatile organic intermediates and these multifunctional imines have been extensively explored as promising feedstocks in the fields of chemistry, biology, materials, drugs, dyes, perfumes and fungicide research.1–3 Thus, tremendous efforts to date have been dedicated to converting amines into imines.4,5Traditionally, imines were usually achieved via the condensation of amines and carbonyl compounds in the presence of Lewis acid catalysts,6,7 however, the homogeneous catalysts generally could not be reutilized in these cases.8 Lately, the oxidative coupling of amines with alcohols or direct self-coupling of amines is one of the promising routes to obtain imines in the presence of transition metal and even noble-metal (Cu, Pd and Au, etc.) catalysts.9,10 For instance, Dong et al. demonstrated that Pd–Au@Mn(II)-MOFs could act as bifunctional heterogeneous catalysts for the efficient tandem synthesis of imines using benzyl alcohols and anilines or benzylamines as the raw materials.11 Recently, the direct aerobic oxidation coupling of amines has attracted considerable attention owing to its atomic economy and environment friendly process.12 Thus far, lots of transition-metal-based catalysts including not only low-cost Cu, Mn and Fe catalysts, but also several noble metal Pd, Au and Pt catalysts have been applied in the coupling reactions of amines and these methods provided sufficient results.13–15 For example, Wang's group reported that functionalized carbon nanotube-supported Au–Pd catalyst showed the efficient heterogeneous catalytic activities in the aerobic oxidation of amines (conv. up to 95%, sele. up to 98%).16 In the presence of Au–Pd@ZrO2 catalyst, dibenzylimine was successfully synthesized via oxidation of benzylamine (95% yield).17 Nevertheless, to the best of our knowledge, these metal-catalyzed strategies are obviously suffered from harsh reaction conditions, higher amount of catalyst, or other additives, etc. Therefore, the alternative efficient strategy of achieving imines without any metal catalysts is highly desirable.
In recent years, electrochemical synthesis is an ideal green sustainable approach to deal with “intractable” synthetic challenges with less waste generation, and it could provide electron as an oxidant and avoid the use of expensive catalysts and toxic oxidants during the reactions, therefore, the efficient and environment friendly electrocatalysis technology has become an emerging field of study.18–20 To our best knowledge, the event of the selective synthesis of imines from amines using electrochemical strategy as a clean and renewable energy resource is still scantly studied. Consequently, the development of mild and efficient approach for targeting imine derivatives using electrocatalysis strategy under exogenous-oxidant-free conditions is highly desirable. Inspired by the above aspects and our continuous interest in green synthetic processes, we present herein an efficient electronic-promoted synthesis of imines and diazenes by the oxidative coupling of amines without metal catalyst and oxidant at room temperature (Scheme 1).
 | ||
| Scheme 1 Representative reactions of oxidation of amines to imines. | ||
Results and discussion
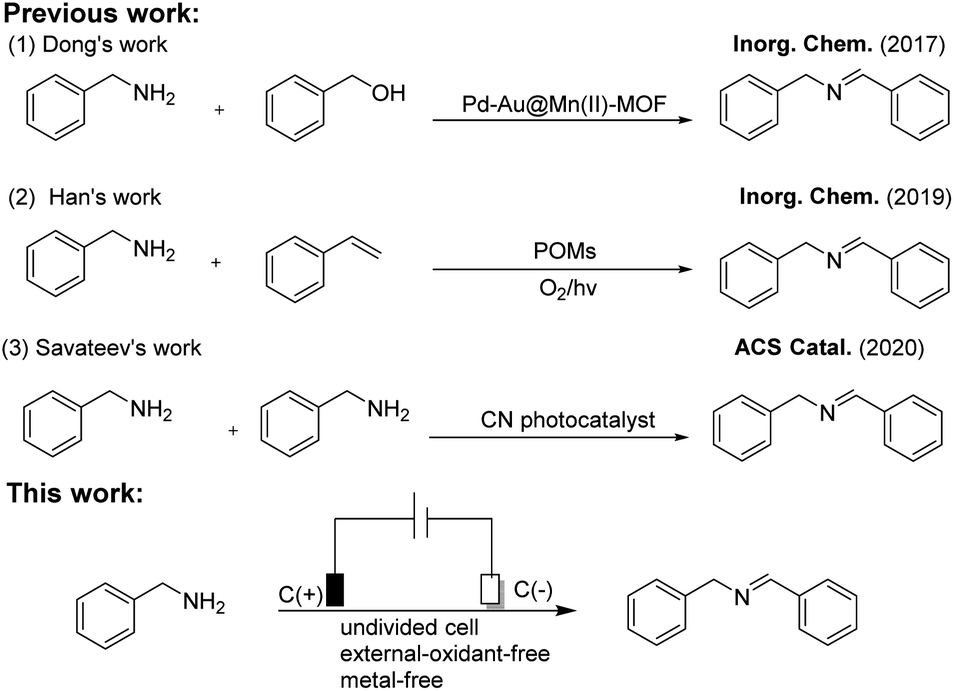
At the outset of the studies, we initially chose benzylamine (1a) as the model substrate to investigate the optimum reaction conditions, and the corresponding results were listed in Table 1. Pleasingly, we found that substrate 1a underwent a self-oxidative coupling reaction in an undivided cell equipped with a carbon anode and a carbon cathode using tetraethyl ammoniumbromide (TBEA) as a electrolyte under constant voltage (5 V) conditions for 10 h, the target product 1b was formed in 96% gas chromatography (GC) yield (Table 1, entry 1). Obviously, both increasing and decreasing the voltage or current have the negative effect on the corresponding product 1b in 88%, 92% and 80% yields, respectively (Table 1, entries 2–4). Notably, the yield decreased sharply to 70% when the reaction time was reduced to 5 h (Table 1, entry 4). Thereafter, a series of solvents (CH3CN, DMF, EtOH and H2O) were successively examined, and none of the other reaction media was superior to CH3CN (Table 1, entries 1, 7–9). Moreover, the conditions of using the 10 mA constant current and platinum plate as the cathode also led to the unsatisfactory results (Table 1, entries 3 and 5). In this regard, we speculated that the surface of the carbon as cathode might be more conducive to electrocatalytic oxidative coupling of benzylamine than other kinds of electrode materials. Also, the reaction yields of imines decreased sharply to 65%, 57% and 41% when TBEA was replaced with tetrabutylammonium tetrafluoroborate (n-Bu4NBF4), LiClO4 and tetrabutyl ammonium iodide (n-Bu4NI), respectively (Table 1, entries 10–12). Expectedly, the reaction did not take place when no electric current passed through the system (Table 1, entry 13).
|
||
|---|---|---|
| Entry | Variation(s) from the standard conditions | Yieldb (%) |
| a Standard conditions: carbon plate (53 mm × 8 mm × 1.5 mm) as anode and cathode, constant voltage = 5 V, benzylamine (0.25 mmol), TBEA (7 mg), CH3CN (3.0 mL), r. t., under air atmosphere, 10 h.b Yields were determined by GC with C6H5Cl as an internal standard and confirmed by GC-MS. | ||
| 1 | None | 96 |
| 2 | 7 V, 12 h | 88 |
| 3 | 3 V | 92 |
| 4 | 10 mA, 10 h | 80 |
| 5 | 5 V, 5 h | 70 |
| 6 | Platinum plate as the cathode | 69 |
| 7 | DMF as solvent | 73 |
| 8 | Ethanol as solvent | 76 |
| 9 | H2O as solvent | NR |
| 10 | n-Bu4NBF4 instead of TBEA | 65 |
| 11 | LiClO4 instead of TBEA | 57 |
| 12 | n-Bu4NI instead of TBEA | 41 |
| 13 | Without electricity | NR |
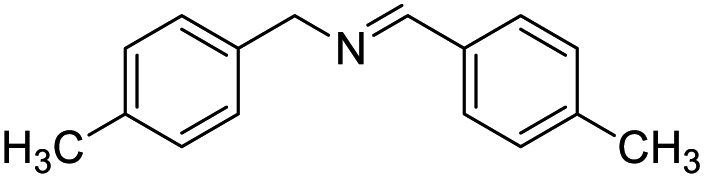
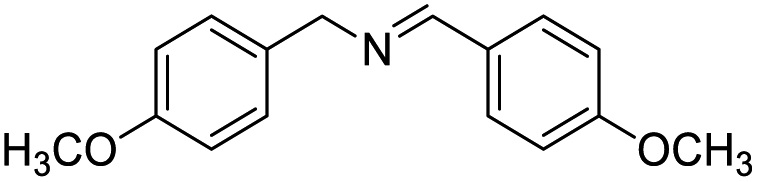
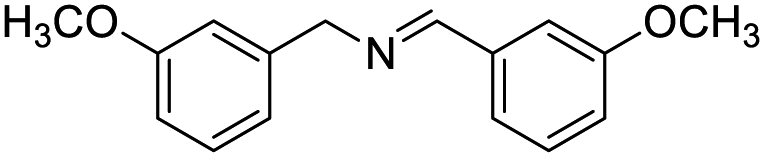
With the optimized conditions established by using electricity as the primary energy input, the scope of reaction substrates with regard to amines were further examined and the results are illustrated in Table 2. First, several para-substituted benzylamines, including –F, –Cl, –Br, –CH3, –OCH3 and t-Bu group, were suitable substrates for this transformation and they could afford the desired products in excellent yields (Table 2, entries 2–8), which was superior to those of the transition-metal catalysts in the oxidation of amines into imines.21–23 In addition, meta-OCH3 group substituted benzylamines also converted smoothly into the corresponding imine in 58% yield (Table 2, entry 7). Thereafter, some secondary aliphatic amines, such as dibenzylamine, N-ethylbenzylamine and N-(tert-butyl)benzylamine, also achieved the corresponding imines with moderate to excellent conversions (Table 2, entries 9–14). Unfortunately, no target products were observed when the aliphatic amines were selected as substrates, respectively (Table 2, entries 15 and 16). Notably, furan-2-ylmethanamine was also well-tolerated and effectively afforded the corresponding product 17b in 90% yield (Table 2, entry 17). Besides, we also found that para-substituted anilines including –CH3CH2, –CH3, –OCH3 and –F groups were suitable for the present process and could survive the optimal conditions to generate the desired 1,2-diphenyldiazene products in moderate yields (Table 3). The electrocatalytic oxidative dehydrogenative coupling of para-methaniline was successfully converted to deliver the self-coupling diazenes compounds in 46% yield (Table 3, entry 1). Moreover, 4-ethylaniline, 4-methoxy-1-aminobenzene successfully underwent this transformation, delivering the corresponding diazenes products in 45% and 41% yields, respectively (Table 3, entry 2). Interestingly, the self-coupling reaction of 4-fluoroaniline gave the corresponding 1,2-di(4-fluoro-phenyl)diazene in 55% yield (Table 3, entry 4), which indicates that substituted anilines with the electron withdrawing group may be more conducive to the reaction.24 However, the 25% and 8% yields of desired diazenes products were detected when 4-chloroaniline, 4-bromoaniline were used instead of 4-fluoroaniline in the reaction, respectively (Table 3, entries 5–6).
|
|||
|---|---|---|---|
| Entry | Amines (a) | Products (b) | Yieldb (%) |
| a Reaction conditions: carbon plate (53 mm × 8 mm × 1.5 mm) as anode and cathode, constant voltage = 5 V, benzylamine (0.25 mmol), TBEA (7 mg), CH3CN (3.0 mL), r. t, under air atmosphere, 10 h.b Yields were determined by GC with C6H5Cl as an internal standard and confirmed by GC-MS. | |||
| 1 |  |
 |
96 |
| 2 |  |
 |
93 |
| 3 |  |
 |
90 |
| 4 |  |
 |
86 |
| 5 |  |
 |
91 |
| 6 |  |
 |
77 |
| 7 | 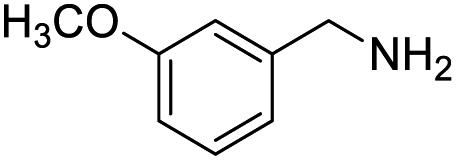 |
 |
58 |
| 8 |  |
 |
77 |
| 9 |  |
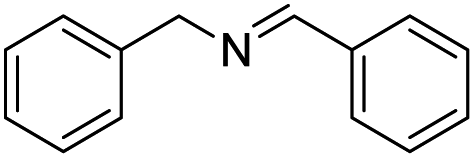 |
97 |
| 10 |  |
 |
26 |
| 11 | 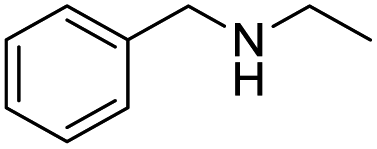 |
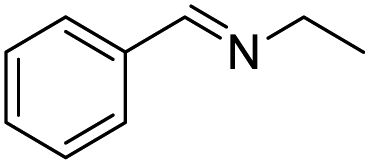 |
56 |
| 12 | 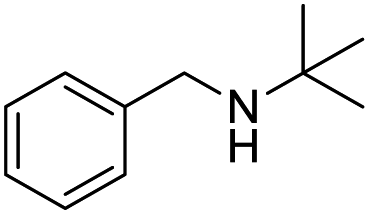 |
 |
96 |
| 13 | 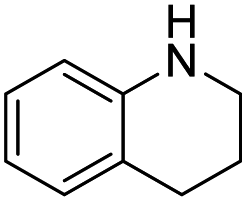 |
 |
85 |
| 14 |  |
 |
86 |
| 15 |  |
 |
NR |
| 16 |  |
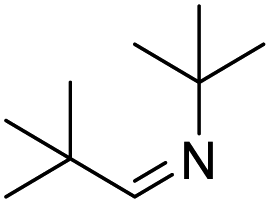 |
NR |
| 17 | 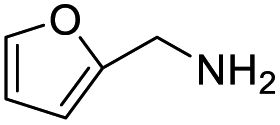 |
 |
90 |

|
|||
|---|---|---|---|
| Entry | Amines | Products | Yieldb (%) |
| a Reaction conditions: carbon plate (53 mm × 8 mm × 1.5 mm) as anode and cathode, constant voltage = 5 V, anilines (0.25 mmol), TBEA (7 mg), CH3CN (3.0 mL), r. t, under air atmosphere, 10 h.b Yields were determined by GC with C6H5Cl as an internal standard and confirmed by GC-MS. | |||
| 1 |  |
 |
46 |
| 2 |  |
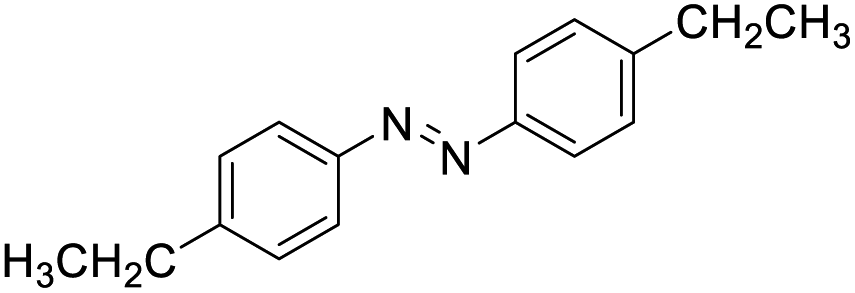 |
45 |
| 3 |  |
 |
41 |
| 4 | 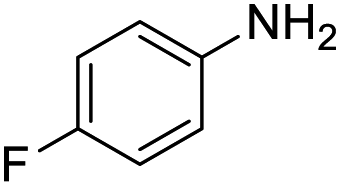 |
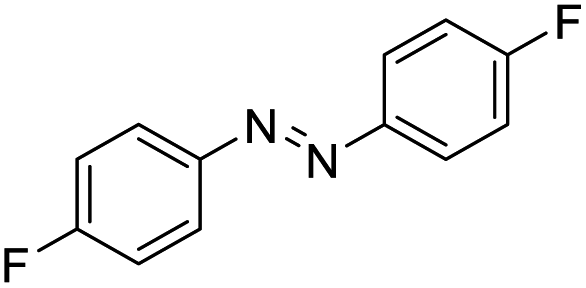 |
55 |
| 5 |  |
 |
25 |
| 6 |  |
 |
8 |

Additionally, to investigate the superiority and practicability of the electrocatalytic reaction of benzylamine, the desired product N-benzyl-1-phenylmethanimine was ease in large-scale production and 0.64 g (64% yield) sample can be readily produced in a batch experiment on a lab scale, holding much promise in the development of the facile and electrochemical method in the production of imine derivatives (Scheme 2). These results indicated that electricity as the ‘reagent’ instead of chemical oxidants or reductants played the key role in the electrochemical catalytic reaction of amines.
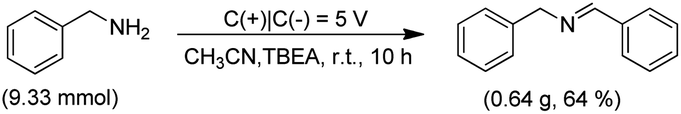 | ||
| Scheme 2 Gram-scale experiments. | ||
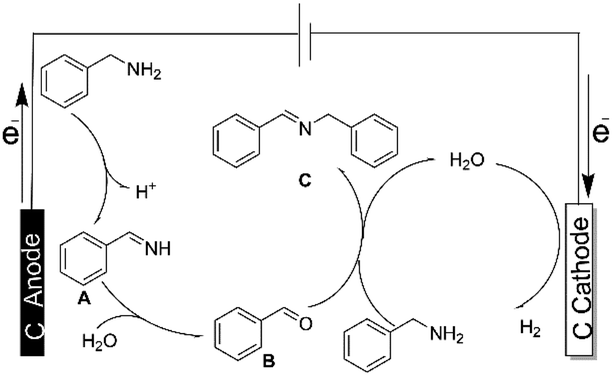
To gain some insights into the mechanism of electrocatalytic oxidative coupling reaction of benzylamine, control experiments were performed (Scheme 3). The yield of imine was trace in the absence of current (Scheme 3a). When the anodic potential was 5 V, benzylamine was easily oxidized to coupling product in 96% yield (Scheme 3b). These results indicated that the control of current and voltage was the key to the electrocatalytic oxidative coupling of benzylamine. In additionally, 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) was added to the electrochemical oxidative reaction under the optimal conditions, the result exhibited TEMPO can't inhibit significantly the formation of the coupled product, which suggested that the reaction didn't involve the radical path (Scheme 3c). Considering the reported proposed mechanism of benzylamine catalytic oxidation,25–28 a possible mechanism was proposed for the electrochemical oxidation of benzylamine is depicted in Scheme 4. Firstly, benzylamine was dehydrogenated to produce PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH (A) as an intermediate, and hydrogen was produced on the cathode.29–31 PhCHNH (A) was further anodized to form benzaldehyde (B) in the presence of water, which has been captured by GC-MS (Fig. S2†), then benzaldehyde coupled with benzylamine to form the final product C.32–34
NH (A) as an intermediate, and hydrogen was produced on the cathode.29–31 PhCHNH (A) was further anodized to form benzaldehyde (B) in the presence of water, which has been captured by GC-MS (Fig. S2†), then benzaldehyde coupled with benzylamine to form the final product C.32–34
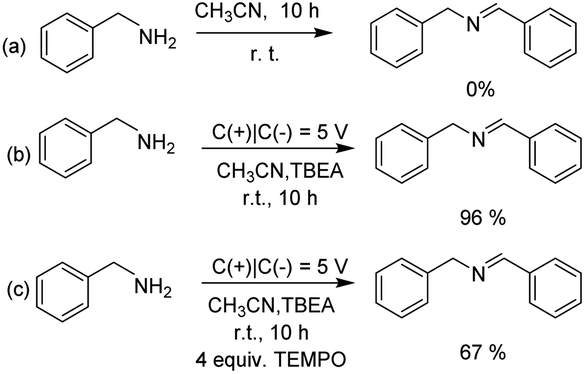 | ||
| Scheme 3 Control experiments. | ||
 | ||
| Scheme 4 Proposed reaction mechanism for formation of N-benzyl-1-phenylmethanimine. | ||
Conclusions
In conclusion, we have developed an efficient electrochemical strategy for synthesis of imines and diazenes derivatives under catalyst- and oxidant-free conditions at room temperature. The process is controlled by fine-tuning of the current and the applied potential to achieve CN bond and NN bond formation. The strategy proceeds smoothly in air at room temperature, providing the corresponding products in moderate to excellent yields. The reaction also features a broad substrate scope, easily scaled-up and simple operation. Thus, this electrochemical process has the potential for industrial application in the future. Further application of electrochemical oxidative coupling of other organic compounds is currently under investigation in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (No. 21871125); the Natural Science Foundation of Shandong Province, China (No. ZR2019MB043 and ZR2019QB022), the Project of State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering (No. 2021-K82) and the Construction Project of Quality Curriculum for Postgraduate Education of Shandong Province (No. SDYKC19057).References
- S. Wang and B. König, Angew. Chem., Int. Ed., 2021, 60, 21624–21634 CrossRef CAS PubMed
.
- C. Wu, C. Zhu, K. Liu, S. Yang, Y. Sun, K. Zhu, Y. Cao, S. Zhang, S. Zhuo, M. Zhang, Q. Zhang and H. Zhang, Appl. Catal., B, 2022, 300, 120288–120298 CrossRef CAS
- D. Uraguchi, Y. Tsuchiya, T. Ohtani, T. Enomoto, S. Masaoka, D. Yokogawa and T. Ooi, Angew. Chem., Int. Ed., 2020, 59, 3665–3670 CrossRef CAS PubMed
- C. Volpe, S. Meninno, A. Roselli, M. Mancinelli, A. Mazzanti and A. Lattanzi, Adv. Synth. Catal., 2020, 12, 5457–5466 CrossRef
- J. Takay, K. Ogaw, R. Nakay and N. Iwasaw, ACS Catal., 2020, 10, 12223–12228 CrossRef
- K. Verma and P. Banerjee, Adv. Synth. Catal., 2017, 359, 3848–3854 CrossRef CAS
- L. K. B. Garve, M. Petzold, P. G. Jones and D. B. Werz, Org. Lett., 2016, 18, 564–567 CrossRef CAS PubMed
- J. T. Reeves, M. D. Visco, M. A. Marsini, N. Grinberg, C. A. Busacca, A. E. Mattson and C. H. Senanayake, Org. Lett., 2015, 17, 2442–2445 CrossRef CAS PubMed
-
(a) B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS
- H. Naeimi, F. Salimi and K. Rabiei, J. Mol. Catal. A: Chem., 2006, 260, 100–104 CrossRef CAS
- G.-J. Chen, H.-C. Ma, W.-L. Xin, X.-B. Li, F.-Z. Jin, J.-S. Wang, M.-Y. Liu and Y.-B. Dong, Inorg. Chem., 2017, 56, 654–660 CrossRef CAS PubMed
- L. Liu, S. Zhang, X. Fu and C.-H. Yan, Chem. Commun., 2011, 47, 10148–10150 RSC
-
(a) R. E. Rodríguez-Lugo, M. A. Chacón-Terán, S. D. León, M. Vogt, A. J. Rosenthal and V. R. Landaeta, Dalton Trans., 2018, 47, 2061–2072 RSC
- H. Huang, J. Huang, Y.-M. Liu, H.-Y. He, Y. Cao and K.-N. Fan, Green Chem., 2012, 14, 930–934 RSC
- R. Kumar, E. H. Gleißner, E. G. V. Tiu and Y. Yamakoshi, Org. Lett., 2016, 18, 184–187 CrossRef CAS PubMed
- W. Deng, J. Chen, J. Kang, Q. Zhang and Y. Wang, Chem. Commun., 2016, 52, 6805–6808 RSC
- S. Sarina, H. Zhu, E. Jaatinen, Q. Xiao, H. Liu, J. Jia, C. Chen and J. Zhao, J. Am. Chem. Soc., 2013, 135, 5793–5801 CrossRef CAS PubMed
- S. Arndt, D. Weis, K. Donsbach and S. R. Waldvogel, Angew. Chem., Int. Ed., 2020, 59, 8036–8041 CrossRef CAS PubMed
- S. P. Blum, L. Schäffer, D. Schollmeyer and S. R. Waldvogel, Chem. Commun., 2021, 57, 4775–4778 RSC
- M. Mehrdadian, S. Khazalpour, A. Amani and M. Jamshidi, Electrochim. Acta, 2021, 381, 138242–138251 CrossRef CAS
-
(a) J. Mondal, P. Borah, S. Sreejith, K. T. Nguyen, X. Han, X. Ma and Y. Zhao, ChemCatChem, 2014, 6, 3518–3529 CrossRef CAS
-
(a) K. Mullick, S. Biswas, A. M. Angeles-Boza and S. L. Suib, Chem. Commun., 2017, 53, 2256–2259 RSC
- P. Sudarsanam, B. Hillary, M. H. Amin, S. B. A. Hamid and S. K. Bhargava, Appl. Catal., B, 2016, 185, 213–224 CrossRef CAS
-
(a) J. Lux and J. Rebek Jr, Chem. Commun., 2013, 49, 2127–2129 RSC
- Y. Xiao, Y. Huang, S. Xue and J. Zhao, Appl. Catal., B, 2020, 265, 118596–118603 CrossRef CAS
- Y. Markushyna, P. Lamagni, J. Catalano, N. Lock, G. Zhang, M. Antonietti and A. Savateev, ACS Catal., 2020, 10, 7336–7342 CrossRef CAS
- K. Wang, P. Jiang, M. Yang, P. Ma, J. Qin, X. Huang, L. Ma and R. Li, Green Chem., 2019, 21, 2448–2461 RSC
- G.-J. Chen, H.-C. Ma, W.-L. Xin, X.-B. Li, F.-Z. Jin, J.-S. Wang, M.-Y. Liu and Y.-B. Dong, Inorg. Chem., 2017, 56, 654–660 CrossRef CAS PubMed
- J. W. Kim, J. He, K. Yamaguchi and N. Mizuno, Chem. Lett., 2009, 38, 920–921 CrossRef CAS
- R. D. Patila and S. Adimurthy, RSC Adv., 2012, 2, 5119–5122 RSC
- L. Al-Hmoud and C. W. Jones, J. Catal., 2013, 301, 116–124 CrossRef CAS
- S. Biswas, B. Dutta, K. Mullick, C.-H. Kuo, A. S. Poyraz and S. L. Suib, ACS Catal., 2015, 5, 4394–4403 CrossRef CAS
- Y. Fu, M. Zheng, Q. Li, L. Zhang, S. Wang, V. V. Kondratievd and B. Jiang, RSC Adv., 2020, 10, 28059–28065 RSC
- B. Venua, V. Shirisha, B. Vishali, G. Naresh, R. Kishore, I. Sreedhar and A. Venugopal, New J. Chem., 2020, 44, 5972–5979 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07263c |
| This journal is © The Royal Society of Chemistry 2022 |