
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and cationic polymerization of halogen bonding vinyl ether monomers†
Yudai Morota,
Takanaga Suzuki and
Kira B. Landenberger *
*
Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo Ward, Kyoto 615-8510, Japan. E-mail: landenberger.kirabeth.2x@kyoto-u.ac.jp
First published on 20th January 2022
Abstract
Halogen bonding is rapidly becoming recognized as a viable and useful intermolecular interaction in supramolecular chemistry. While various monomers amenable to radical polymerization methods containing halogen bonding donors have been developed, this study aims to expand the type of monomers that incorporate this intermolecular interaction to facilitate use of cationic polymerization by developing three novel vinyl ether monomers containing halogen bonding donor moieties: 2,3,5,6-tetrafluoro-4-iodophenoxyethyl vinyl ether (C2I), 2,3,5,6-tetrafluoro-4-iodophenoxybutyl vinyl ether (C4I), and 2-(2,3,5,6-tetrafluoro-4-iodophenoxyethoxy)ethyl vinyl ether (O3I). Well controlled cationic polymerization is achievable through the use of a proton trap, 2,6-di-tert-butylpyridine. The use of SnCl4 as a co-Lewis acid was found to accelerate the reaction. Between the three monomers, the difference in the chain length is shown to influence the reaction rate, with the longest chain demonstrating the fastest polymerization. Initial studies of the halogen bonding ability shows that halogen bonding exists for all three monomers but is most pronounced in C4I. The polymerized vinyl ethers also exhibit halogen bonding. Due to the ease of synthesis and polymerization, these are promising new monomers to increase functionality available for polymers synthesized using cationic polymerization.
Introduction
In the past fifty years, precision synthesis of polymers using a wide variety of polymerization methods has been realized.1,2 With each passing year, an even wider scope of monomers are employed for all polymerization methods, and even previously held conceptions about what monomers are polymerizable or not using various methods are being contested.3 As the toolbox to create polymers becomes more extensive, this enables a wide array of designed and functional self-assembly structures to be readily formed. While intermolecular interactions, such as hydrogen bonding or coordination bonds, can be reliably used to make many intriguing and useful supramolecular materials, these are not the only strong intermolecular interactions available.Halogen bonding has recently been gaining traction as a reliable and useful intermolecular interaction. Halogen-bonding is the electrostatic interaction between the σ-hole of a halogen atom, usually iodine, bromine, or chlorine, although in rare cases fluorine has been shown to exhibit this,4,5 and the lone pair of an electron rich atom such as oxygen, nitrogen, sulfur or even selenium or phosphorous in some cases.6–17 Under appropriate circumstances, even metal centers such as Pd can function as halogen bond acceptors.18 Compared to other intermolecular interactions, halogen bonding has only recently become more recognized as a reliable intermolecular interaction. Indeed, this intermolecular interaction is often overlooked in the discussion of supramolecular materials. Since the halogen bond has the capability to match the bond strength of the more commonly employed hydrogen bonds, it is possible to construct supramolecular materials analogous to those formed via hydrogen bonding.14,19–21
There are several strategies to create halogen-bond donors of sufficient strength. When using halogens in their capacity as halogen bond donors, iodine is the ideal choice since it, being the most polarizable and therefore the largest σ-hole and the most consistent halogen bonding behavior, has reportedly the strongest halogen bonding ability.11,21 For achieving stronger interactions the introduction of positively charged moieties to increase the electron withdrawing character, such as haloimidazolium22,23 or halotriazolium groups24 or electron withdrawing groups, such as nitro groups,25 increases the strength of the resulting halogen bonds. Furthermore, by increasing the number of electron withdrawing groups present, this will also enhance the halogen bonding ability.26 Using fluorinated arenes to achieve an electron withdrawing environment is a relatively common and facile approach to achieving halogen bonding donors6,10,14,26–30 and was used in the design of the vinyl ether monomers presented herein. Indeed, considering the design of monomers for cationic polymerization, avoiding charged species or potentially reactive groups was deemed the wiser approach.9,31
Halogen bonding offers additional qualities that make it attractive as a corollary to hydrogen bonding materials. Firstly, the building block employed almost always exhibits strong hydrophobic characteristics and the donor moiety is aprotic, making halogen bonding materials in general less susceptible to water and pH than hydrogen bonding materials. This means that these materials could be conceivably used in environments that would be unsuitable for hydrogen bonding counterparts.32 Additionally, halogen bonding in general demonstrates much greater specificity in bonding partners than hydrogen bonding. Therefore, the possibility of binding to an unintended target is greatly reduced and a clearer and more designed responses can be obtained.
The bond angles of halogen bonds are also very consistent within a narrow range. In particular, when iodine is used as the halogen bond donor, the bond angle between the covalent bond to the halogen atom and the noncovalent bond formed from the σ-hole to the halogen bond acceptor is almost always very close to 180°, with little variation from this angle in all but the most exceptional circumstances.14,33 This directionality of halogen bonding is one of the key characteristics and is much more pronounced than the range of angles found in hydrogen bonding examples.34 With increasing polarizability in the halogen (commensurate with the size of the halogen), the strong preference for directionality also increases.34,35 Such specificity is also attractive to form supramolecular materials using halogen bonds so that the effect of such bond angle specificity can be investigated.36 In this sense, the increased flexibility of vinyl ether monomers offered in this system could potentially enhance the successful formation of halogen bonds by easing the ability to achieve the desired 180° angle.
While still limited in the reports, the number of polymer systems useful for halogen bonding has been increasing rapidly in recent years.23,34,37–43 Typically radical polymerization methods are used to generate polymers capable of halogen bonding. Most employ acrylic or methacrylic backbones, although there are also examples of styrene based-halogen bonding monomers.23,37–39,41,43 There is a need to expand the type of monomers used to explore the effect that polymer backbones can have on these materials. In this study, cationic polymerization was adopted, which is ideal for polymerizing vinyl ethers. When the acrylates or methacrylates monomers are used, since the side chains are connected via ester groups, there is a weak electrostatic interaction acts between adjacent esters that can increase the rigidity of the main chain. By using vinyl ethers this interaction between adjacent side chains is removed and the main chain can move more flexibly.44 This increase in flexibility could offer a major change in the properties of these materials that would broaden the types of polymer materials available for halogen bonding.
Herein we present, to the best of our knowledge, the first examples of vinyl ether monomers that are capable of serving as halogen bonding donors. These monomers are readily polymerized using cationic polymerization and produce well controlled polymers. An initial study of halogen bonding ability demonstrates that these halogen bonding vinyl ethers are a viable and valuable addition to creating functional materials using cationic polymerization.
Experimental
Monomer synthesis
Monomers C4I, C2I, O3I, C4F, C2F and O3F were synthesized following a similar procedure. Synthesis of C4I is presented below. Additional details for the synthesis of monomers C2I, O3I, C4F, C2F and O3F are provided in the ESI.† All commercially available materials were used as received unless otherwise noted.Isolated yield: 71%; estimated density 1.65 g cm−3; 1H NMR (400 MHz, CDCl3): δ = 6.46 (q, J = 7.07, 1H), 4.28 (t, J = 6.00, 2H), 4.18 (dd, J = 14.4 and 2.00, 1H), 4.00 (dd, J = 6.80 and 2.00, 1H), 3.75 (t, J = 5.80, 2H), 1.88 (m, 4H); 13C NMR (100 MHz, CDCl3): δ = 151.6 (s), 148.4 (m), 146.0 (m), 141.9 (m), 139.4 (m), 138.0 (m), 86.3 (s), 74.8 (s), 67.1 (s), 63.4 (m), 26.6 (s), 25.2 (s); 19F NMR (376 MHz, CDCl3): δ −123.03 (d, J = 17.5 Hz, 2F), −155.87 (d, J = 17.5 Hz, 2F); IR (neat): 3741.2 (w), 2951.5 (w), 2361.4 (m), 1740.4 (m), 1618.9 (s), 1476.2 (s), 1382.7 (s), 1319.1 (s), 1272.8 (s), 1198.5 (s), 1095.4 (s), 971.9 (m), 806.1 (s), 604.6 (s) cm−1; MS (m/z): calcd for C12H11F4IO2, 389.9734; found, 389.9735 M+.
Polymer synthesis
Analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 110000; Mw/Mn = 1.01–1.16) used to estimate the molecular weights and molecular weight distribution. Samples were prepared by dissolving them in THF at approximately 10 mg mL−1.:20:1 ratio (sample:matrix:cationizing agent) mixture.
110000; Mw/Mn = 1.01–1.16) used to estimate the molecular weights and molecular weight distribution. Samples were prepared by dissolving them in THF at approximately 10 mg mL−1.:20:1 ratio (sample:matrix:cationizing agent) mixture.Results and discussion
Monomer synthesis
Multiple strategies were considered for synthesizing vinyl ether monomers containing halogen bonding moieties. While a variety of halogen bonding groups have been reported, halo-perfluorobenzenes were deemed suitable for cationic polymerization since these are the least likely to interfere with the cationic polymerization process. Therefore, methods to synthesize vinyl ethers containing halo-perfluorobenzenes were considered.A common method for synthesizing functional vinyl ether monomers employs an SN2 reaction between a deprotonated alcohol and chloroethyl vinyl ether (CEVE).46 Therefore, as a first approach, synthesis of 2,3,5,6-tetrafluoro-4-iodophenol according to a previously reported method,47 followed by addition of the vinyl ether moiety using CEVE via an SN2 reaction was attempted (Scheme 1).‡ After synthesizing and purifying 2,3,5,6-tetrafluoro-4-iodophenol via column chromatography, the next synthesis step is to deprotonate the phenol; this is expected to proceed readily to due to the strong electron withdrawing nature of the ring. However, the same electron withdrawing effect also results in a very weak nucleophile and it is believed that this is the primary reason that the SN2 reaction did not proceed as anticipated. Instead, the long reaction time (20 h) and relatively high temperatures appear to have allowed a variety of side reactions to occur. Despite attempts to purify the product by column chromatography this method produced so many byproducts, confirmed by TLC, as to make isolation or even an estimation of the crude yield impossible. Considering the low yield, numerous byproducts and intense labor demanded, the synthesis of the desired monomer using this approach, this route was deemed unfeasible and a route from the opposite direction was considered.
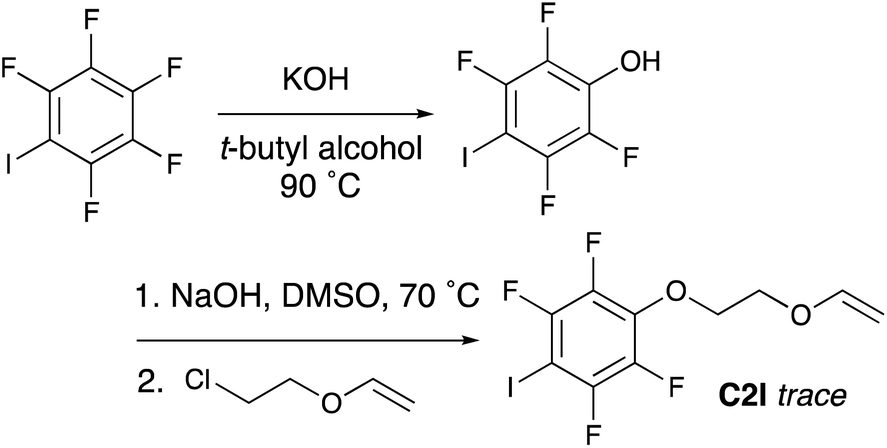 | ||
| Scheme 1 Initial synthesis strategy for C2I that resulted in numerous byproducts and only trace product, making this route unfavorable. | ||
The inverse route employs an SNAr reaction. First a vinyl ether with a terminal hydroxyl group is deprotonated and then this is added to pentafluoroiodobenzene through a nucleophilic aromatic substitution reaction. Initial investigations employed cesium carbonate (Cs2CO3, Wako 1st Grade) as a base and used room temperature conditions. While the desired product was successfully obtained in one step, the yield of the target product was lower than desired (31%), and the presence of byproducts were confirmed by TLC. To suppress the side reactions, lower reaction temperature (−10 °C) synthesis conditions were explored. Moreover, the base was changed to sodium hydride (NaH) to improve the efficiency of the removal of the hydrogen on the hydroxyl group in the first step.
Thus, in the final synthesis method (Scheme 2) used to synthesize C2I, NaH was added to ethylene glycol monovinyl ether (EGVE) to deprotonate the terminal alcohol. Pentafluoroiodobenzene was then slowly added at low temperature (−10 °C), after which the temperature was allowed to gradually rise to approximately 20 °C. The reaction was stirred overnight and the products were diluted with diethyl ether at the end of the reaction and washed with water and base (10% NaOH(aq)) to remove any salts or unreacted alcohols. After drying, the resulting residue was purified via column chromatography. This resulted in an 81% yield for C2I.
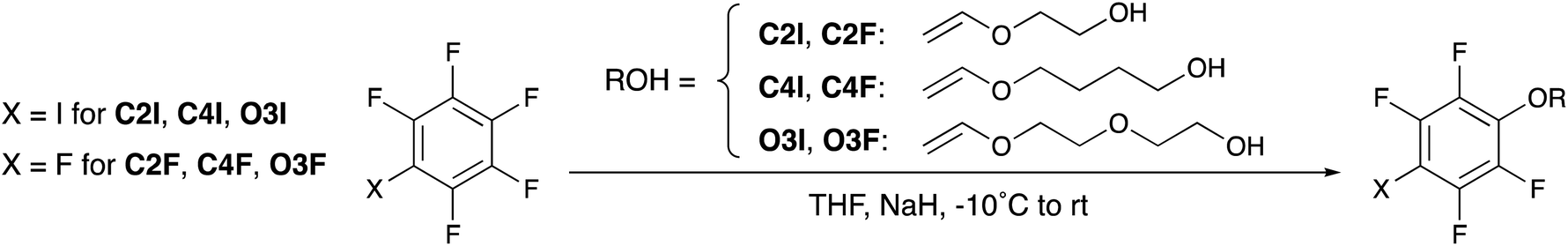 | ||
| Scheme 2 Final synthetic strategy for monomers C2I, C4I, O3I, C2F, C4F, and O3F. Alcohols were deprotonated by NaH first, followed by addition of the perfluorinated arene. | ||
Considering the significantly higher yield and reduction in impurities, this general synthesis method was used to synthesize the other halogen bonding monomers. For synthesis of C4I and O3I tetramethylene glycol monovinyl ether (TGVE) and diethylene glycol monovinyl ether (DGVE) were used respectively (see ESI†). The yields were comparable to C2I (Table 1).
| Monomer | Reaction concentration (mM) | Reaction time (h) | Yield % |
|---|---|---|---|
| C2I | 75 | 20 | 81 |
| C4I | 75 | 20 | 71 |
| O3I | 250 | 20 | 74 |
| C2F | 40 | 2 | 49 |
| C4F | 250 | 23 | 62 |
| O3F | 40 | 2 | 40 |
Since the planned future work with these halogen bonding monomers (C2I, C4I and O3I) entails polymerization and investigation of the formation of various supramolecular structures based upon their ability to halogen bond, a way to accurately assess the extent to which halogen bonding is responsible for interactions between polymers of C2I, C4I and O3I and various donors is necessary. Therefore, to compare the behavior of polymers of C2I, C4I and O3I, monomers that are similar in structure but lack the iodine which allows for halogen bonding are ideal. Consequently, monomers C2F, C4F and O3F were synthesized in order to provide a control case to confirm the presence of halogen bonding. Monomers C2F, C4F and O3F correlate with monomers C2I, C4I and O3I respectively, but with a fluorine group replacing the iodine. Synthesis of monomer C2F had already been reported using comparable conditions,48 but monomers C2F, C4F and O3F for this study were synthesized following the general method used for monomers C2I, C4I and O3I. Compared to their iodine-containing counterparts, the completely fluorinated monomers exhibited a somewhat lower yield.
Polymer synthesis
As the roots of living cationic polymerization start in the use of a hydroiodic acid initiating system with elemental iodine as the stabilizer for the cationic end,49,50 and more recent uses of iodine containing catalysts for achieving living cationic polymerization have also been reported,51–54 the idea of including a monomer that contains highly polarized iodine is potentially complicating. If the iodine on the monomer acts as an added base or adds to the propagating cation chain end in an unwanted fashion this could preclude the use of this functional group, iodo-perfluorobenzenes, in cationic polymerization. Before attempting to synthesize the halogen bonding vinyl ethers, the effect of the presence of the iodo-perfluorobenzenes in a cationic polymerization system first needed to be considered. To investigate the influence of iodine-containing perfluorobenzenes, the polymerization of isobutyl vinyl ether (IBVE), a common and reliable monomer in cationic polymerization, was conducted in the presence of pentafluoroiodobenzene. The polymerization of IBVE proceeded with a comparable speed and produced controlled product consistent in reaction conditions without the addition of pentafluoroiodobenzene, suggesting that the mere presence of iodine in a benzene ring, or a strongly electron withdrawing group within the system would not hinder the progress of cationic polymerization. Based on this study alone, however, the potential for the iodine on the iodoperfluorobenzene to interact with the propagating end cannot be eliminated. It is at least clear that cationic polymerization of monomers containing this functional group is not deterred by the presence of this group in the reaction mixture.Initial studies on polymerization of the novel vinyl ether monomers containing iodoperfluorobenzene began with C2I. Using Et1.5AlCl1.5 as the Lewis acid, 1-(isobutoxy)ethyl acetate (IBEA) as the preformed initiating species, and 1,4-dioxane as the added base, the polymerization proceeded in a toluene solution at 0 °C. However, as is readily apparent from the SEC charts (Fig. 1a), while the polymerization did indeed proceed, it exhibited a lack of control and produced polymers with relatively broad polymer weight distributions from 1.4–1.8. Furthermore, the SEC chart shows the clear formation of oligomer species and remaining monomer despite relatively long reaction times. While these conditions prove that polymerization is possible, the investigation continued to determine conditions which would allow for a more controlled polymerization.§
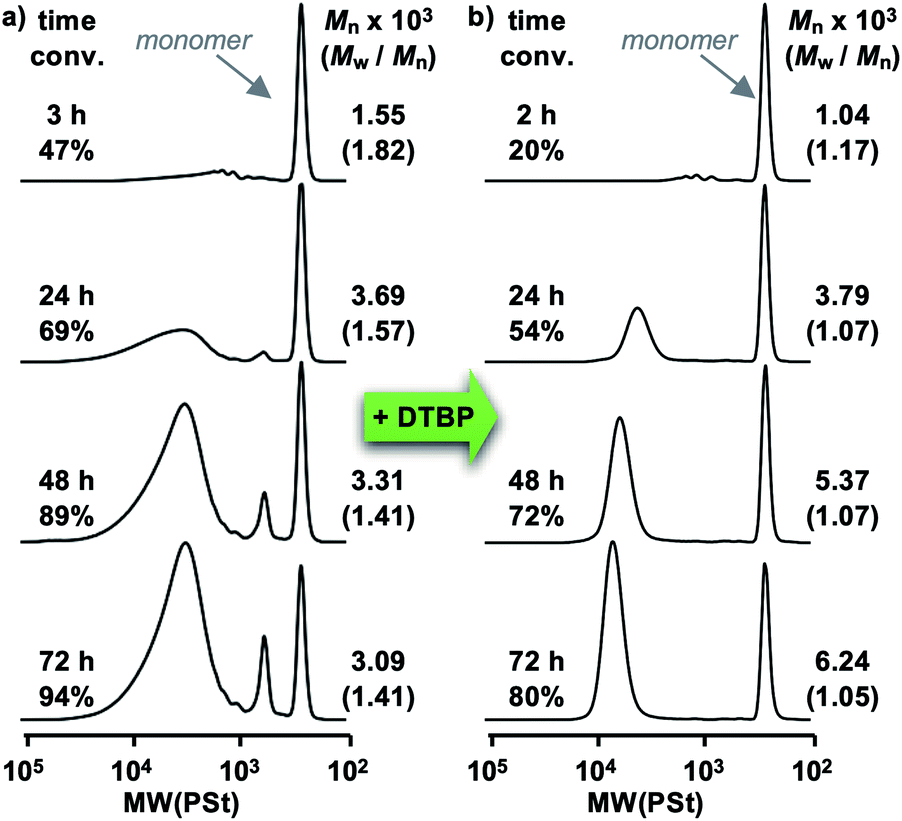 | ||
| Fig. 1 (a) SEC charts of the initial polymerization of C2I using the following conditions: [C2I]0 = 160 mM, [IBEA]0 = 4 mM, [Et1.5AlCl1.5]0 = 20 mM, [DO]0 = 1.2 M, 0 °C in toluene. (b) SEC charts of the precision polymerization of C2I using 2,6-di-tert-butylpyridine (DTBP) using the following conditions: [C2I]0 = 160 mM, [IBEA]0 = 4 mM, [Et1.5AlCl1.5]0 = 20 mM, [DO]0 = 1.2 M, [DTBP]0 = 4 mM, 0 °C in toluene. | ||
Since even small amounts of adventitious protons in cationic polymerization can impede the living nature of this system, it was determined that a proton trap, 2,6-di-tert-butylpyridine (DTBP) could prove beneficial.55 The addition of DTBP had a remarkable effect on the polymerization of C2I (Fig. 1b) and the resulting polymers became much more controlled as evidenced by the narrow molecular weight distribution as the polymer near completion (1.05). There was also a reduction in side reactions compared to the polymerizations where DTBP is absent. In particular, peaks hinting at the formation of oligomers in the low molecular weight region were not observed, indicating the effectiveness in DTBP in suppressing unwanted side reactions.
Due to the presence of electron withdrawing fluorine groups in all of the monomers in this study, there is an inherently strong electron withdrawing nature present that deactivates the propagating cation, resulting in noticeably slower reaction speeds. For example, the non-fluorinated analog of monomer C2I, phenoxy ethyl vinyl ether (PhOVE) when polymerized in toluene at 0 °C using Et1.5AlCl1.5 as the Lewis acid and dioxane as the added based generally nears completion around 4 hours,56 while in the polymerization reaction of C2I using the same conditions fails to reach completion even at 72 hours. To increase the polymerization rate, therefore, the addition of an accelerator, SnCl4, was considered. Previous studies reported the use of EtAlCl2 and SnCl4 as a mixed catalyst system to successfully accelerate the polymerization rate for the living cationic polymerization of IBVE.57,58 Through the addition of SnCl4 as an accelerator to the system, and keeping all other factors consistent, the time required for the polymerization of C2I was significantly reduced (Fig. 2). The polymerization reaction which was estimated to take more than 3 days to complete using the initial conditions, could instead reach completion closer to 20 minutes.
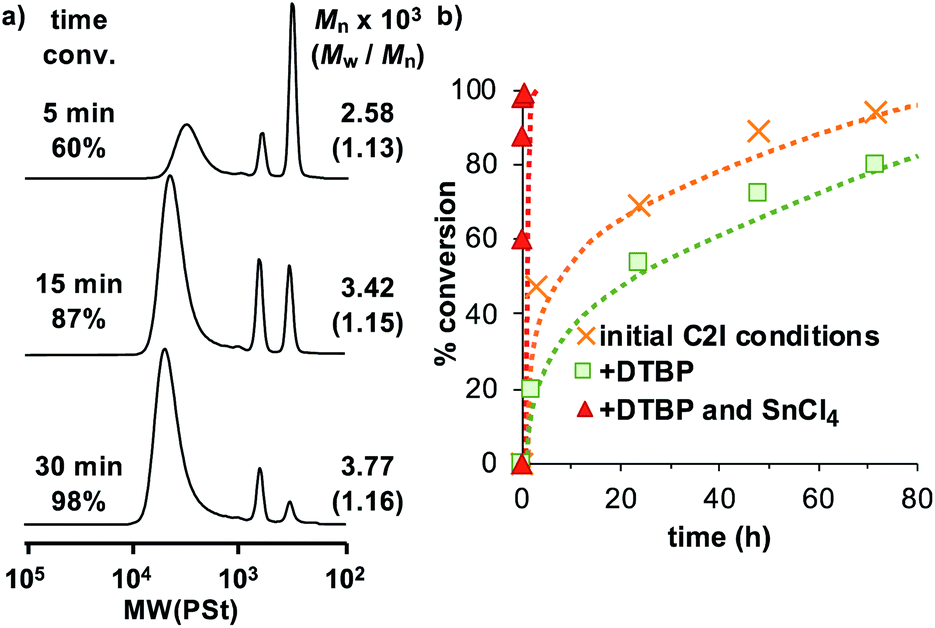 | ||
| Fig. 2 (a) SEC charts of the accelerated polymerization of C2I using the following conditions: [C2I]0 = 160 mM, [IBEA]0 = 4 mM, [Et1.5AlCl1.5]0 = 20 mM, [DO]0 = 1.2 M, [DTBP]0 = 4 mM, [SnCl4]0 = 10 mM, 0 °C in toluene. SnCl4 functioned as the accelerator. (b) A comparison of the conversion vs. the reaction time. While DTBP does slow down the reaction rate somewhat, the accelerator increases the rate quite significantly. | ||
Since it was determined that polymerization was possible for C2I, and that polymers incorporating DTBP and SnCl4 usually resulted in polymers with sufficiently narrow molecular weight distributions, cationic polymerization of monomers C4I, O3I, C2F, C4F, and O3F were conducted using conditions found to be amenable for C2I (Et1.5AlCl1.5 as the Lewis acid, DTBP as a proton trap and SnCl4 as an accelerator). The difference in chain length was found to have rather significant influence over the polymerization rate for monomers C2I, C4I, and O3I, with the polymerization rate varying in the order of O3I > C4I > C2I, where O3I is the fastest. Indeed, using these conditions, O3I reached completion in less than 1 minute while C4I took approximately 5 minutes to near completion. These are notably faster than C2I at approximately 20 minutes (Fig. 3).
 | ||
| Fig. 3 A comparison the rate of polymerization of monomers C2I, C4I and O3I using the following conditions: [monomer]0 = 160 mM, [IBEA]0 = 4 mM, [DO]0 = 1.2 M, [Et1.5AlCl1.5]0 = 10 mM, [DTBP]0 = 4 mM, [SnCl4]0 = 4 mM, 0 °C in toluene. | ||
The synthesis of the fluorine-only monomer analogues (C2F, C4F and O3F) was also performed using the same reaction conditions to both verify the effect of the spacer between the electron withdrawing group as well as test for any potential influence from the inclusion of the iodine moiety. The reaction rate for these fluorine-only monomer analogues were very similar to their iodinated counterparts, C2I, C4I and O3I (Fig. 4). For O3F the reaction also reaches completion the fastest, with at least 94% completion in 1 minute. The second fastest is still the second longest chain, C4F, reaching completion within 10 minutes. And the slowest reaction rate is still the shortest chain monomer of C2F. While quite similar in reaction rate, it is at least noteworthy that all of the fluorine-only monomers have slightly slower reaction rates than their iodine counterparts. This is thought to be due to the more significant electron withdrawing effects of fluorine as compared to iodine.
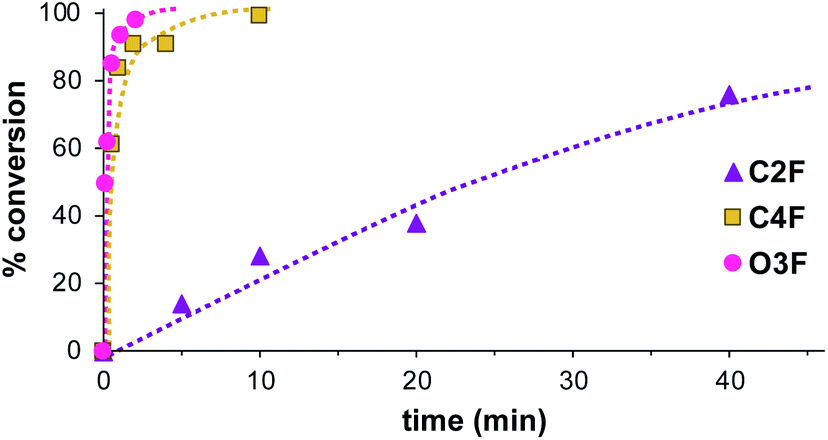 | ||
| Fig. 4 A comparison the rate of polymerization of monomers C2F, C4F and O3F using the following conditions: [monomer]0 = 160 mM, [IBEA]0 = 4 mM, [DO]0 = 1.2 M, [Et1.5AlCl1.5]0 = 10 mM, [DTBP]0 = 4 mM, [SnCl4]0 = 4 mM, 0 °C in toluene. | ||
Nevertheless, the reaction rate results for both the iodine-monomer series (C2I, C4I and O3I) and the fluorine-only monomer series (C2F, C4F and O3F) seem to indicate that the main contributing factor to the difference in reaction rate is the distance of the propagating vinyl ether end from the electron withdrawing perfluorobenzene group. During propagation for cationic polymerization, having an electron withdrawing group near the cationic species at the growth terminal destabilizes this intermediate and biases the reaction to the dormant species, reducing the polymerization rate. Considering the inductive effect, the closer the electron withdrawing group is to the oxygen which assists in stabilizing the cationic species during the propagation reaction the more significant the effect. The monomers C2I and C2F, have the shortest spacer and longest reaction times. With progressively longer chain length in C4I and C4F a faster reaction rate is observed. With the longest chain length O3I and O3F show the fastest reaction rate. The electron withdrawing effects can be observed in part in the peak shifts in the 1H NMR of the hydrogens on the non-vinylic carbons in the α position to the oxygen of the vinyl ether. The peak shift is around 4.02 ppm in C2I and 4.01 ppm in C2F, while the same carbon in C4I and C4F show 1H NMR peak shifts at 3.75 ppm for both monomers. O3I and O3F have peak shifts at 3.81 ppm and 3.83 ppm respectively, however, in the case of O3I and O3F, an extra oxygen atom is present increasing the electron rich nature of monomer and therefore adding a factor beyond mere chain length.
Monomer C4I was used for a further investigation into optimal polymerization conditions. The polymerization results are shown in Fig. 5. While Et1.5AlCl1.5 was initially used as the Lewis acid it is entirely possible that this is not the ideal Lewis acid. Therefore, a similar but distinct Lewis acid, EtAlCl2 was also investigated. Additionally, while SnCl4 was initially used as an accelerator, the softer SnBr4 was also investigated.
 | ||
| Fig. 5 SEC spectra of the polymerization of C4I investigating the two different Lewis acids (EtAlCl2 and Et1.5AlCl1.5) and accelerators (SnCl4 and SnBr4) to investigate optimal polymer synthesis conditions. Oligomer peaks fall in the range of approximately 400, 800, 1200 and 1600 g mol−1; [C4I]0 = 160 mM, [IBEA]0 = 4 mM, [DO]0 = 1.2 M, [Et1.5AlCl1.5 or EtAlCl2]0 = 10 mM, [DTBP]0 = 4 mM, [SnCl4 or SnBr4]0 = 4 mM, 0 °C in toluene. | ||
In the case of samples where EtAlCl2 was used as the Lewis acid, there is a broadening in the SEC spectra as well as the appearance of many peaks indicating the presence of oligomers and potentially other side products. Even though analysis by NMR indicates complete conversion (particularly EtAlCl2 with SnBr4), the SEC spectra clearly shows that this is not entirely due to formation of a polymer. The SEC charts suggest the formation of dimers and oligomers. Notably, this increases with time suggesting degradation of the polymer to some extent when too much time elapses after completion of the polymerization. A fact that is also corroborated by the decrease in molecular weight at the longest time point. In contrast, the use of Et1.5AlCl1.5 as the Lewis acid appears to produce more well-controlled polymers in the case of both SnCl4 and SnBr4. There is less evidence of byproducts and a reduction in the PDI over time. When using Et1.5AlCl1.5 as the Lewis acid, SnCl4 appears to be a more favorable choice as an accelerator when compared to SnBr4. A comparison of the conversion of the polymers synthesized with either SnCl4 or SnBr4 also suggests that SnBr4 accelerates the reaction far more significantly (reaction times were kept consistent). The samples polymerized using SnBr4 finished faster, but generally resulted in broader molecular weight distributions. While the spectra of polymer samples synthesized using Et1.5AlCl1.5 as the Lewis acid do not show as pure a sample as would be desired, the overall decrease in side reactions still makes this the more attractive condition to use.
Analysis of halogen bonding
The halogen bonding capability of both the monomers and the resulting polymers were investigated using primarily 19F NMR analysis. 19F NMR is very sensitive to changes in the electronic environment and can therefore be used to correlate the degree of interaction between molecular species, in other words, the degree of halogen bonding present in the system.11 Though a variety of halogen bonding acceptors have been reported in the past, most strong examples contain an exposed oxygen (ethers, alcohols, etc.), nitrogen atom (pyridine, amine, etc.) or sulfur, all of which can function as Lewis bases.15,16As an initial study, the halogen bonding ability of iodo-perfluorobenzenes and C4I were tested with both nitrogen and oxygen containing acceptors. Potential halogen bond acceptors included: triethylamine, 1,4-dioxane and tetrahydropyran. In all cases there was a clear peak shift (see ESI†), but since nitrogen-containing acceptors appeared to demonstrate more significant halogen bonding ability with the donors tested, triethylamine was the primary acceptor employed for investigating the halogen bonding of the monomers and polymers for this study.
The halogen bonding ability of monomers C2I, C4I, O3I and C2F were examined. As is shown in Fig. 6, monomer C4I shows more halogen bonding ability than either C2I or O3I. An apparent association constant (Ka,app) can be calculated for the halogen bonding by assuming a 1:1 binding and fitting a linear curve to the samples.38,41 While these are not the true binding constants, they do assist in quantifying the difference in the halogen bonding ability. Based on this a Ka,app of 0.28 M−1 can be estimated for C2I, 0.49 M−1 for C4I, and 0.25 M−1 for O3I. Even though the iodo-perfluorobenzene group is identical for these monomers, the slight changes in the distance between the chain between the vinyl ether and halo-perfluorobenzene as well as the number of oxygens present clearly has an impact. Indeed, it cannot be overlooked that the vinyl ether monomers contain a readily available oxygen for halogen bonding. While nitrogen containing halogen bond acceptors appear to be more effective as a halogen bonding acceptor for these monomers (see ESI†), halogen bonding does still exist to some extent.
 | ||
| Fig. 6 Comparison of the halogen bonding abilities of monomers C2I, C4I, O3I and C2F as measured using 19F NMR peak shifts. Monomers were dissolved in toluene at 100 mM. Triethylamine was used at the acceptor. | ||
Notably, the fluorine-only monomer, C2F, shows no significant peak shift, clearly showing that the peak shifts are due to the presence of the iodine, the halogen bonding group in these samples. Monomers C2F, C4F and O3F are approximately the same size as C2I, C4I and O3I, with a similar strong electron withdrawing capability, and in the polymer form similar chain entanglement effects. In this way any changes in the materials due to the halogen bonding influence become clear. As the monomer C2F containing only fluorine showed almost no shift in the 19F NMR spectra even with the addition of significant amounts of acceptor molecules, this strongly suggests a lack of halogen bonding. In contrast to this, compounds containing the iodine all showed some degree of peak shift with a linear relationship to the amount of acceptor added.
The halogen ability of the polymerized halogen bonding vinyl ethers (approximately 40 monomer units) was also measured using triethylamine as the acceptor. As can be seen in Fig. 7, the polymer samples appears to exhibit less halogen bonding ability than the monomer form using the same condition. An estimation of the Ka,app, shows values of 0.38 M−1, 0.32 M−1 and 0.40 M−1 for polymers of C2I, C4I and O3I respectively. These binding constants are not significantly different from the value for the monomer forms. This is a somewhat unexpected result based on previous literature,38,39,41 but considering the expected 1:1 binding for this system and the close proximity of the halogen bonding units to one another in the polymer samples, it is possible that binding is discouraged compared to the monomer form.
 | ||
| Fig. 7 A comparison of the halogen bonding ability of polymers (DP ≈ 40) of C2I, C4I, and O3I as estimated using peaks shifts in 19F NMR. All polymer samples were dissolved in toluene at 100 mM. Triethylamine was used as the acceptor. | ||
Conclusions
In conclusion, novel vinyl ether monomers capable of halogen bonding suitable for living cationic polymerization were successfully synthesized and polymerized. These monomers make it possible to easily introduce halogen bonding into a wide variety of precisely controlled polymer structures synthesized using cationic polymerization. Monomer synthesis employing vinyl ethers that had been deprotonated with NaH that could affect a SNAr reaction was found to be the most effective means for synthesizing the monomers. The polymerization of the monomers revealed that use of a proton trap, DTBP, and an accelerator, SnCl4, was an effective way to produce controlled polymers quickly. The chain length influences the reaction rate, with the longest chain exhibiting the fastest reaction. All of the monomers containing the halogen bonding moiety exhibited halogen bonding as evidence by peaks shifts in the 19F NMR spectra.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the Society of Iodine Science as well as the Grants-in-Aid for Early-Career Scientists (19K15635) from the Ministry of Education, Culture, Sports, Science and Technology in Japan for their generous support of this research. The authors also thank Dr Duncan Brown for insightful advice and discussion.Notes and references
- M. Kamigaito and M. Sawamoto, Macromolecules, 2020, 53, 6749–6753 CrossRef CAS.
- R. B. Grubbs and R. H. Grubbs, Macromolecules, 2017, 50, 6979–6997 CrossRef CAS.
- S. Sugihara, A. Yoshida, T. Kono, T. Takayama and Y. Maeda, J. Am. Chem. Soc., 2019, 141, 13954–13961 CrossRef CAS PubMed.
- V. Elakkat, C. C. Chang, J. Y. Chen, Y. C. Fang, C. R. Shen, L. K. Liu and N. Lu, Chem. Commun., 2019, 55, 14259–14262 RSC.
- P. Metrangolo, J. S. Murray, T. Pilati, P. Politzer, G. Resnati and G. Terraneo, Cryst. Growth Des., 2011, 11, 4238–4246 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS PubMed.
- G. Berger, J. Soubhye and F. Meyer, Polym. Chem., 2015, 6, 3559–3580 RSC.
- F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058–3071 RSC.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC.
- M. Fourmigue, Curr. Opin. Solid State Mater. Sci., 2009, 13, 36–45 CrossRef CAS.
- P. Politzer, P. Lane, M. C. Concha, Y. G. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- Y. Le Gal, A. Colas, F. Barrière, V. Dorcet, T. Roisnel and D. Lorcy, CrystEngComm, 2019, 21, 1934–1939 RSC.
- D. Cinčić, T. Friščić and W. Jones, CrystEngComm, 2011, 13, 3224–3231 RSC.
- A. M. Siegfried, H. D. Arman, K. Kobra, K. H. Liu, A. J. Peloquin, C. D. McMillen, T. Hanks and W. T. Pennington, Cryst. Growth Des., 2020, 20, 7460–7469 CrossRef CAS.
- E. A. Katlenok, A. V. Rozhkov, O. V. Levin, M. Haukka, M. L. Kuznetsov and V. Y. Kukushkin, Cryst. Growth Des., 2021, 21, 1159–1177 CrossRef CAS.
- C. F. Li, Y. Y. Chai, X. L. Zhou, Z. Shen, B. B. Ma, B. J. Chen, R. D. Huang, H. Chen, W. M. Li and Y. He, CrystEngComm, 2018, 20, 3006–3010 RSC.
- C. B. Aakeröy, T. K. Wijethunga, M. A. Haj, J. Desper and C. Moore, CrystEngComm, 2014, 16, 7218–7225 RSC.
- A. M. Abeysekera, V. W. Day, A. S. Sinha and C. B. Aakeroy, Cryst. Growth Des., 2020, 20, 7399–7410 CrossRef CAS.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- R. Tepper, S. Bode, R. Geitner, M. Jäger, H. Görls, J. Vitz, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 4047–4051 CrossRef CAS PubMed.
- F. Zapata, L. Gonzalez, A. Bastida, D. Bautista and A. Caballero, Chem. Commun., 2020, 56, 7084–7087 RSC.
- K. B. Landenberger, O. Bolton and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 5074–5079 CrossRef CAS PubMed.
- M. Saccone, M. Spengler, M. Pfletscher, K. Kuntze, M. Virkki, C. Wölper, R. Gehrke, G. Jansen, P. Metrangolo, A. Priimagi and M. Giese, Chem. Mater., 2019, 31, 462–470 CrossRef CAS.
- A. S. Novikov, D. M. Ivanov, Z. M. Bikbaeva, N. A. Bokach and V. Y. Kukushkin, Cryst. Growth Des., 2018, 18, 7641–7654 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem.–Eur. J., 2013, 19, 16240–16247 CrossRef PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- Y. J. Zhao, Y. Cotelle, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 4270–4277 CrossRef CAS PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- V. Stilinović, G. Horvat, T. Hrenar, V. Nemec and D. Cinčić, Chem.–Eur. J., 2017, 23, 5244–5257 CrossRef PubMed.
- A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn-Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Adv. Funct. Mater., 2012, 22, 2572–2579 CrossRef CAS.
- M. G. Chudzinski, C. A. McClary and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 10559–10567 CrossRef CAS PubMed.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. M. Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- S. Välimäki, L. Gustavsson, N. K. Beyeh, V. Linko and M. A. Kostiainen, Macromol. Rapid Commun., 2019, 40, 1900158 CrossRef PubMed.
- A. Vanderkooy, P. Pfefferkorn and M. S. Taylor, Macromolecules, 2017, 50, 3807–3817 CrossRef CAS.
- A. Vanderkooy and M. S. Taylor, Faraday Discuss., 2017, 203, 285–299 RSC.
- R. Milani, N. Houbenov, F. Fernandez-Palacio, G. Cavallo, A. Luzio, J. Haataja, G. Giancane, M. Saccone, A. Priimagi, P. Metrangolo and O. Ikkala, Chem, 2017, 2, 417–426 CAS.
- A. Vanderkooy and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 5080–5086 CrossRef CAS PubMed.
- N. Houbenov, R. Milani, M. Poutanen, J. Haataja, V. Dichiarante, J. Sainio, J. Ruokolainen, G. Resnati, P. Metrangolo and O. Ikkala, Nat. Commun., 2014, 5, 4043 CrossRef PubMed.
- F. Wang, N. Ma, Q. X. Chen, W. B. Wang and L. Y. Wang, Langmuir, 2007, 23, 9540–9542 CrossRef CAS PubMed.
- M. Naya, K. Kokado, K. B. Landenberger, S. Kanaoka, S. Aoshima and K. Sada, Macromol. Chem. Phys., 2020, 221, 1900455 CrossRef CAS.
- S. Aoshima and T. Higashimura, Macromolecules, 1989, 22, 1009–1013 CrossRef CAS.
- M. Sawamoto, S. Aoshima and T. Higashimura, Makromol. Chem., Macromol. Symp., 1988, 13–4, 513–526 CrossRef.
- J. X. Wen, H. B. Yu and Q. Chen, J. Mater. Chem., 1994, 4, 1715–1717 RSC.
- W. Li, P. Feng, Y. Q. Zou and B. Hai, J. Appl. Polym. Sci., 2014, 131, 41019 Search PubMed.
- M. Miyamoto, M. Sawamoto and T. Higashimura, Macromolecules, 1984, 17, 265–268 CrossRef CAS.
- M. Miyamoto, M. Sawamoto and T. Higashimura, Macromolecules, 1984, 17, 2228–2230 CrossRef CAS.
- K. Takagi and K. Okamura, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2436–2441 CrossRef CAS.
- K. Takagi, K. Yamauchi and H. Murakata, Chem.–Eur. J., 2017, 23, 9495–9500 CrossRef CAS PubMed.
- R. Haraguchi, T. Nishikawa, A. Kanazawa and S. Aoshima, Macromolecules, 2020, 53, 4185–4192 CrossRef CAS.
- K. Takagi, H. Murakata, K. Yamauchi and K. Hashimoto, Polym. Chem., 2020, 11, 6739–6744 RSC.
- A. Kanazawa, S. Shibutani, N. Yoshinari, T. Konno, S. Kanaoka and S. Aoshima, Macromolecules, 2012, 45, 7749–7757 CrossRef CAS.
- Y. Shinke, A. Kanazawa, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5041–5048 CrossRef CAS.
- T. Yoshida, T. Tsujino, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 468–472 CrossRef CAS.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, discussion of molecular weight estimation supplementary halogen bonding data and IR spectra. See DOI: 10.1039/d1ra06957h |
| ‡ This was reacted by deprotonating with NaOH by heating for 2 hours at 70 °C, then slowly adding 2-chloroethyl vinyl ether (CEVE, TCI, >97.0%) dropwise for 25 minutes at room temperature, followed by heating at 70 °C for an additional 20 hours. |
| § The molecular weight estimates from SEC are often smaller than anticipated based on the theoretical mass from the conversion calculated (see ESI Fig. S28†). It was also smaller than the weights estimated when the samples were investigated using MALDI-TOF MS. The underestimation of the molecular weight when using SEC is due to the large deviation in the structure of these heavily halogenated monomers as compared to the polystyrene standard used in the SEC. The presence of halogens on the benzene instead of hydrogen atoms ring greatly increase the density and therefore there is a underestimation of the molecular weight for all samples in this study using this method. To be consistent, however, molecular weights as determined by SEC will be presented in this paper. |
| This journal is © The Royal Society of Chemistry 2022 |