
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable production of curable maltodextrin-based electrospun microfibers†
Claudio Cecone *a,
Gjylije Hotia,
Marco Zanettiabc,
Francesco Trottaa and
Pierangiola Braccoa
*a,
Gjylije Hotia,
Marco Zanettiabc,
Francesco Trottaa and
Pierangiola Braccoa
aDepartment of Chemistry, Nis Interdepartmental Centre, University of Turin, Via P. Giuria 7, Turin, 10125, Italy. E-mail: claudio.cecone@unito.it
bInstm Reference Centre, University of Turin, Via G. Quarello 15A, Turin, 10135, Italy
cICxT Interdepartmental Centre, University of Turin, Via Lungo Dora Siena 100, Turin, 10153, Italy
First published on 24th December 2021
Abstract
Maltodextrins are inexpensive, water-soluble starch hydrolysis products composed of high molecular weight polysaccharide molecules. This feature allows their water solutions to be processed by electrospinning to produce bio-based microfibrous mats. Also, the presence of hydroxyl functions along the maltodextrin's backbone enables cross-linking reactions to be performed, necessary to overcome the intrinsic solubility of the starting material, by exploiting suitable functional molecules. In this work, the electrospinning process to obtain fibre deposition from water solutions of five different commercial maltodextrins was firstly optimized. Well-defined fibres with diameters ranging between 1.1 μm and 1.5 μm were successfully obtained using water as the unique solvent. Subsequently, the same maltodextrin-containing water solutions with citric acid added were then processed again. The presence of citric acid did not hinder the spinnability of the studied systems, while the possibility to achieve a one-step thermal curing of the obtained fibres was proved via solubility tests, TGA, and FTIR-ATR analyses. Eventually, bio-based cross-linked mats with fibre diameters ranging from 0.7 μm to 1.4 μm were obtained from the electrospinning of commercial maltodextrins and citric acid, employing water as the unique solvent and environmentally friendly curing processes. This approach enables the reported mats to be further studied for environmental, pharmaceutical, and medical applications.
Introduction
The plastic waste accumulation observed during the past decades has generated severe environmental complications and concerns of the public.1 Currently, about 99% of plastics are produced through petrochemicals, and their manufacture requires approximately 80% of the whole non-fuel chemicals production.2 In this frame, the increasing demand of such materials, for which degradation time is estimated to be higher than one hundred years, emphasizes the need to reduce the production of single-use goods and to replace the non-degradable polymers with sustainably-produced and degradable choices.3 In this context, bioplastics are being studied and developed as sustainable alternatives, aiming for a transition towards a circular economy.1 Generally speaking, bioplastics are defined as polymers which are bio-based and/or biodegradable. For this reason, they can be broadly divided into three groups: those that are both bio-based and biodegradable, those that are only bio-based, and those that are only biodegradable.4 Among the first group, polysaccharides such as starch and cellulose but also lignocellulosic fibres, have attracted increasing scientific and industrial interest.5,6 They have been applied in industrial processes such as packaging and bio-composites production,7–11 and exploited in food, environmental, pharmaceutical, and medical applications.12–20 Maltodextrins are water-soluble starch hydrolysis products, hence considered D-glucose polymers characterised by α-(1,4) and α-(1,6) glycosidic linkages, employed mostly as food and pharmaceutical additives.21 In their definition, maltodextrins are characterised by a dextrose equivalent (DE) < 20, a value which represents the reducing equivalent of a given carbohydrate against the same mass of glucose.22 Also, due to their low-price and bio-derived nature, maltodextrins have been exploited as building-block for the production of bio-based cross-linked polymers. Suitable cross-linkers such as epichlorohydrin, di-glycidyl ethers, isocyanates, poly-amines, carbodiimide, glutaraldehyde, and poly-carboxylic acids have been reported for this purpose.23–30Being a well-established technique, electrospinning is a facile, cost effective, and flexible approach which allows the production of fibres at micron, submicron and nano-scale, from an electrically charged jet of polymer solutions or melts.31 The past decades have witnessed a tremendous development of this technique which now has consolidated its role in the processing of a rich variety of synthetic and natural polymers.32 Filtration, biosensors, drug delivery, tissue engineering, and wound dressings are just few of the applications in which the electrospinning has shown evident advantages, mainly related to the hight surface-to-volume ratio of the mats produced.33 However, in most cases, a limitation is represented by the need of toxic or flammable organic solvents, which can limit industrial productions due to environmental and safety regulations.34–39 In this regard, developing processes aimed to reduce or eliminate the use of hazardous substances represents one of the key points of the Green Chemistry, and for this reason an active field of research.40,41
Starch derivatives, such as maltodextrins and cyclodextrins have been extensively explored in multiple applications recently and, for example, have proved to be powerful tools for the encapsulation and slow release of therapeutic agents, flavours, and food preservatives.42–45 However, the search for new solutions to maximize the versatility of applications is still active. In particular, while the spray-drying technique has already proved to be a valid preparation method to maximize the surface area of the products, the potential of electrospinning on these matrices has not yet been adequately explored.
With the above in mind, the possibility to obtain fibrous mats via electrospinning from the aforementioned polysaccharides, being maltodextrins water-soluble, allows to avoid the use of any organic solvent, synthetic polymer, or toxic compound, describing a sustainable process.46–48 However, the high solubility in aqueous environment of these biobased precursors, which is one of the strengths for a green processing, often turns into a limit for a subsequent application in any hydrophilic environment. Different approaches have been developed to make the obtained fibres water insoluble and characterized by higher mechanical properties. Vapor cross-linking with glutaraldehyde or in moist state with 1-ethyl-3-dimethyl-aminopropyl carbodiimide hydrochloride, N-hydroxyl succinimide, and glucose, have been reported in combination with e.g. polyvinyl alcohol, chitosan, and gelatin.49–57 In addition, to overcome the intrinsic toxicity imparted by glutaraldehyde, also citric acid was employed to obtain polyvinyl alcohol-based cross-linked fibres.58–60 In this context, citric acid has been also reported as suitable linking molecule to achieve the synthesis of both hyper-branched and cross-linked beta cyclodextrin-based polymers, exploiting sustainable conditions such as absence of organic solvents or the use of natural deep eutectic solvents.61–63 Besides, the residual carboxyl functions observed after the synthesis of a water-soluble pyromellitic dianhydride/beta cyclodextrin-based polymer, have been exploited to achieve a thermal cross-linking of the resulting mats, obtained through the electrospinning of the aforementioned polymer. However, even if the deposition was carried out using only water solutions, the synthetic approach employed beta cyclodextrin as building-block, which are more expensive if compared to maltodextrins, organic solvents and hazardous reactants.64 Yet, to the best of our knowledge, there are no studies reported in literature related to the use of citric acid in combination with maltodextrins to obtain cross-linked fibres.
The aim of this study is therefore to preliminarily demonstrate the facile processability via electrospinning of inexpensive, water soluble, matrices such as maltodextrins and to explore a sustainable method to overcome the disadvantage of the solubility in aqueous environment of the mats obtained, with the purpose of paving the way for broad spectrum applications, for example in the field of encapsulation of food additives or therapeutics.
Experimental
Materials
Glucidex 2® (GLU2; DE value of 2), Glucidex 6® (GLU6; DE value of 6), Glucidex 9® (GLU9; DE value of 9), Glucidex 12® (GLU12; DE value of 12), and Glucidex 19® (GLU19; DE value of 19), were provided by Roquette Freres (Lestrem, France). Maltodextrins can give yellowing phenomena if kept at 100 °C for a longer period, for this reason the latter were dried in oven at 75 °C up to constant weight before use. Citric acid (99%) was purchased from Sigma-Aldrich (Darmstadt, Germany).Sample preparation
In order to identify the optimal conditions of spinnability, series of solutions with different concentration were first prepared for each sample. All maltodextrins were solubilized in deionized water at 70 °C, in sealed 20 mL glass vials, under stirring. Homogeneous solutions were obtained after approximately 30 minutes. Afterwards, the samples were degassed in a heated ultrasound bath for approximately 15 minutes at 70 °C, and subsequently let to cool down to room temperature. Otherwise, when the addition of citric acid was required with the purpose to obtain curable fibres, the latter was solubilized before the maltodextrin and then the same procedure was followed. The list of investigated samples is shown in Table 1.| Maltodextrin | Optimal spin conc. (wt%) | wt% citric acid (in respect of the maltodextrin) | ||||
|---|---|---|---|---|---|---|
| 0 | 8.3 | 16.6 | 25.0 | 33.3 | ||
| GLU2 | 50.0 | GLU2_0 | GLU2_8 | GLU2_16 | GLU2_25 | GLU2_33 |
| GLU6 | 55.0 | GLU6_0 | GLU6_8 | GLU6_16 | GLU6_25 | GLU6_33 |
| GLU9 | 57.5 | GLU9_0 | GLU9_8 | GLU9_16 | GLU9_25 | GLU9_33 |
| GLU12 | 60.0 | GLU12_0 | GLU12_8 | GLU12_16 | GLU12_25 | GLU12_33 |
| GLU19 | 62.5 | GLU19_0 | GLU19_8 | GLU19_16 | GLU19_25 | GLU19_33 |
Processing and curing
A self-made electrospinning apparatus composed of a 3 mL plastic syringe coupled with a volumetric pump, and a power supply needed to generate a potential between the syringe's nozzle (electrode) and the collector (ground), was used to process all the maltodextrin solutions. The deposition was carried out at room temperature and relative humidity comprised between 30% and 45%, by setting a working distance of 15 cm, 30 kV field strength and 1.2 mL h−1 flow. A Linari NanoTech Easy Drum (Pisa, Italy) rotary system equipped with an aluminium cylinder was used as the collector, with rotation speed of 75 rpm; a gauge 18 nozzle was employed. The thermal curing was performed by treating the samples in oven at 180 °C in air, for 30 minutes.Solubility test
For each sample, 50 mg of polymer mat were placed in a 5 mL Eppendorf tube, and then 5 mL of deionized water were added. After 15 hours at room temperature the solution was separated from the mat via centrifugation, and the latter was dried in oven at 60 °C up to constant weight. Each test was carried out in triplicate. The soluble fraction was calculated as follows,
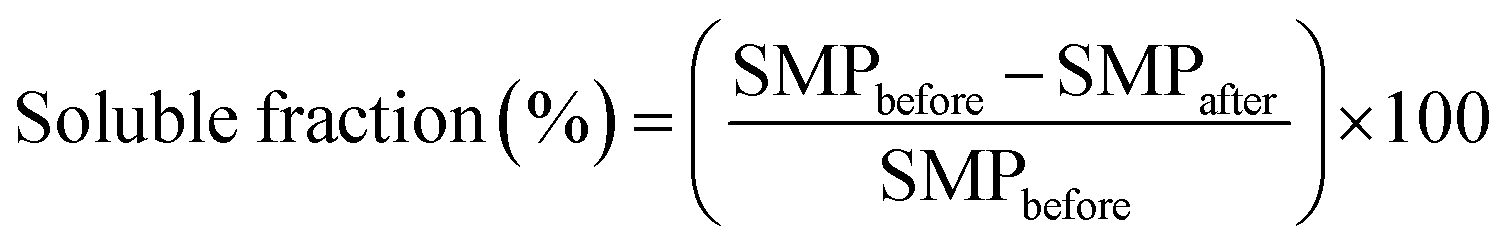 | (1) |
Thermogravimetric analysis (TGA)
TGA were carried out with a TA Instruments Q500 TGA (New Castle, DE, USA), from 50 °C to 700 °C with a heating ramp of 10 °C min−1, under 100 mL min−1 nitrogen flow, using approximately 10 mg of sample.Attenuated total reflectance Fourier transform infrared (FTIR-ATR) spectroscopy
A PerkinElmer Spectrum 100 FT-IR Spectrometer (Waltham, MA, USA) equipped with a Universal ATR Sampling Accessory was used for the FTIR-ATR characterisation. All spectra were collected in the wavenumber range of 650–4000 cm−1, at room temperature, with a resolution of 4 cm−1 and 8 scans per spectrum.Scanning electron microscopy (SEM)
The morphology of the samples was studied using scanning electron microscopy. The images were acquired with a Tescan VEGA 3 SEM (Brno, Czech Republic) using secondary electrons and 3 kV accelerating voltage. The samples were analysed without any previous coating, while the diameter distribution was evaluated manually using ImageJ software. To generate reliable statistics, 100 measurements were acquired for each SEM image analysed. In addition, the fibers lying outside the focal plane were not evaluated.Size exclusion chromatography (SEC)
SEC analyses were performed using a PL aquagel–OH MIXED-M 8 μm (Agilent, CA, USA) column installed on a PerkinElmer Flexar chromatograph. The mobile phase was a 0.1 M sodium nitrate water solution at a flow rate of 0.3 mL min−1. Refractive index was used as detector, while the duration of a single run was set at 15 minutes. A calibration curve was constructed before the sample analysis using dextran standards with specific average molecular weights ranging from 6 kDa to 500 kDa, dissolved in 0.1 M sodium nitrate water solution.Rheological measurements
The rheological characterisation of the solutions was performed using a TA Instruments Discovery HR 1 Rheometer (New Castle, DE, USA). The shear stress (τ) as a function of shear rate (γ′) was recorded with Trios software coupled with the rheometer. The viscosity of the solutions was measured at 25 °C as a function of shear rate (0.1–120 s−1), using a 40 mm 0.9939° cone-plate geometry with a 0.028 mm gap size. At first, the sample was placed between the upper parallel plate and the stationary surface. Afterwards, the gap was closed, and the sample was carefully trimmed with a spatula. Measurements were carried out in steady flow using a three steps protocol: (1) conditioning, the sample was equilibrated for 2 minutes before the experiments; (2) flow-ramp, the shear rate was increased linearly from 0.1 to 100 s−1 in 2 minutes; and (3) flow-peak hold, a constant shear rate of 100 s−1 was maintained for 2 minutes. All measurements were performed in triplicate and the data obtained were expressed as mean values.Results and discussion
Processing optimization
Electrospinning offers unique possibilities for developing and producing fibres and mats with controllable structures and morphologies. However, to obtain fibres without defects, an optimization of the processing parameters, such as the concentration of the starting solution, is required. Further, an appropriate viscosity is also necessary to obtain a uniform jet and to hamper effects of surface tension, such as the beads.33 The solution viscosity reflects the number of entanglements generated between the polymer chains and is proportional to the molecular weight of the latter. In this regard, the DE value of a maltodextrin which represents the amount of reducing sugar ends, is also inversely correlated to the molecular mass of the oligosaccharide: higher DE values indicate lower molecular mass molecules.48,65 With the above in mind, the first step of the work was screening the molecular weight of each maltodextrin via SEC technique. As a result (Fig. 1), all the samples appeared to be composed of both high molecular weight fractions, related to the maltodextrin chains, and of lower molecular weight fractions in the range between hundreds to a few thousand Daltons. This second fraction was related to hydrolysis by-products and glucose oligomers still present in the products after the production process, as stated by the manufacturer and confirmed by Castro et al.66 Nevertheless, by considering only the higher molecular weight fraction, an average molecular weight of 272 ± 12 kDa for GLU2, 46 ± 1 kDa for GLU6, 32 ± 3 kDa for GLU9, 27 ± 3 kDa for GLU12, and 20 ± 1 kDa for GLU19 was estimated.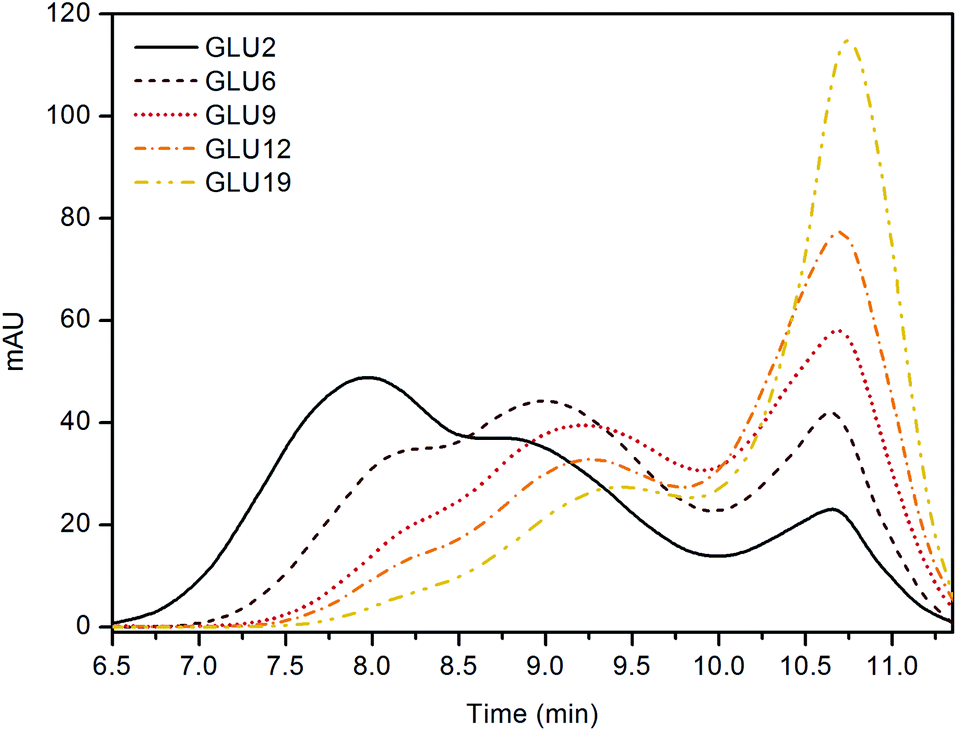 | ||
| Fig. 1 SEC analyses. | ||
Afterwards, the electro-spinnability, thus the possibility to spin the chosen solution into dry, homogeneous fibres, was evaluated for each maltodextrin processed without additives, using water as unique solvent. To screen for the optimum concentration, different amounts of each maltodextrin were solubilized in deionized water at room temperature with the purpose to obtain distinct solutions with concentration ranging from 40 wt% to 70 wt% at increasing amounts of 2.5 wt% one to the next. After being spun, the optimum condition for each system was identified as lack of defects on the obtained fibrous mats and absence of difficulties, such as clogging of the nozzle, during the processing step. From this first screening, all the chosen maltodextrins resulted suitable to be processed via electrospinning technique. As reported in Table 1 and Fig. 2, an optimum concentration of 50.0 wt% for GLU2 (Fig. 2A, GLU2_0), 55.0 wt% for GLU6 (Fig. 2F, GLU6_0), 57.5 wt% for GLU9 (Fig. 2K, GLU9_0), 60.0 wt% for GLU12 (Fig. 2P, GLU12_0), and 62.5 wt% for GLU19 (Fig. 2U, GLU19_0) was determined. The increasing concentration needed for higher DE values is consistent with the decreasing molecular weight of the maltodextrin, thus related to the need of higher chain entanglements to obtain defined fibres. However, despite the higher concentration values reported for higher DE maltodextrins, the results obtained from the viscosity measurements followed the opposite trend (Fig. 3), due to the decreasing molecular weight of the polysaccharides. In this context, the highest viscosity value was observed for GLU2_0 and was equal to 6.89 ± 1.79 Pa s, while the lowest was related to GLU19_0 and equal to 1.39 ± 0.07 Pa s. As a consequence, GLU2_0 and GLU6_0 solutions gave the mats characterised by the highest fibre diameters, of 1.38 ± 0.21 μm and 1.49 ± 0.25 μm, respectively, whereas from GLU9_0, GLU12_0, and GLU19_0 a mean diameter of 1.10 ± 0.21 μm, 1.25 ± 0.20 μm, and 1.12 ± 0.29 μm was observed. The results obtained are consistent with the work of Kutzli et al.67 in which 1.61 ± 0.44 μm was observed as mean diameter of fibres obtained from the electrospinning of maltodextrins displaying DE value of 2.
 | ||
| Fig. 2 SEM images of spun maltodextrin solutions. Scale bars: 20 μm. | ||
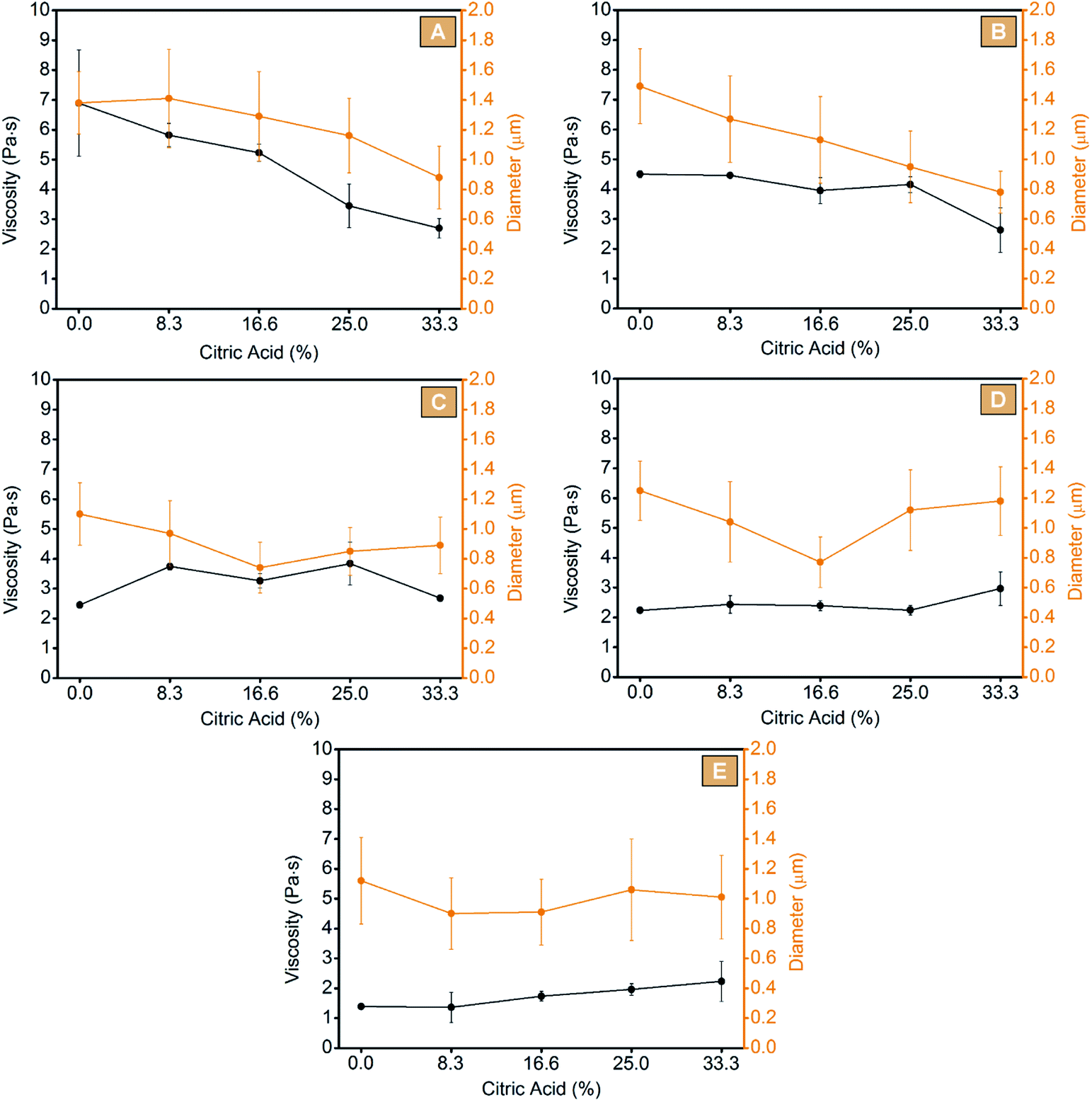 | ||
| Fig. 3 Viscosity and spun fiber diameters obtained from GLU2 (A), GLU6 (B), GLU9 (C), GLU12 (D), and GLU19 (E) systems. | ||
As mentioned, a strong limitation of producing electrospun fibres from water solutions of maltodextrins, is that the resulting mats are still water soluble, and thus not suitable to be used in presence of water or moisture. The second step of the work was then focused on the study of the electro-spinnability of water solutions of maltodextrins containing increasing amounts (8.3 wt%, 16.6 wt%, 25 wt%, and 33.3 wt% in respect of the maltodextrin) of citric acid, added as cross-linking agent. At first, the effects of citric acid on the viscosity of the solution were evaluated. As reported in Fig. 3, a decrease of the viscosity proportional to the amount of citric acid was observed for GLU2 and GLU6 systems. On the contrary, for GLU9 no clear trend was obtained, while for GLU12 and GLU19 a slight increase of the viscosity was detected. Citric acid appeared to act like a plasticizer for higher molecular weight maltodextrins, probably by hampering the strong hydrogen interactions occurring between the polysaccharide chains. For lower molecular weight structures, on the other hand, the polar interactions generated between citric acid and the polysaccharide's chains, were hypothesized to increase the overall viscosity of the solution. Nevertheless, citric acid appeared to affect the solutions of high molecular weight maltodextrins, more than that bearing shorter polysaccharide molecules. Afterward, each system was electrospun and the resulting mats are shown in Fig. 2. Clearly, citric acid did not hinder the electro-spinnability of the solutions, since it was still possible to obtain defined fibres from each maltodextrin added with different amounts of citric acid. However, by including changes in the viscosity of the solutions, citric acid also influenced the fibre diameters. In the case of GLU2 and GLU6, the decrease of the mean fibre diameters was proportional to the citric acid content, thus consistent with the viscosity measurements previously observed. Considering GLU2_33 and GLU6_33, the fibre diameters were approximately 36% and 48% lower, if compared to GLU2_0 an GLU6_0, respectively. However, in the case of GLU9, GLU12, and GLU19 an increasing amount of citric acid did not produce a proportional decrease of mean diameters, whereas the presence of a minimum centred at citric acid content of 16.6 wt% was observed instead. A decrease of 32% for GLU9_16, 38% for GLU12_16, and 19% for GLU19_16 was observed if compared to GLU9_0, GLU12_0, and GLU19_0, respectively.
Cross-linking procedure
After demonstrating the possibility to obtain fibres from the electrospinning of water solution containing maltodextrins and citric acid, the study was dedicated to assessing the effectiveness of the curing treatment aimed at cross-linking the fibres and give them water stability. Kutzli et al. reported how the glycation of proteins via Maillard reaction can be exploited for improving the water stability of electrospun fibres of maltodextrins.68 Differently, in this work the approach chosen was to exploit the carboxylic functions of the citric acid molecules for binding together the maltodextrin chains via ester formation (Fig. 4A). To this end, an oven thermal treatment was identified as the best procedure. In order to choose the optimal temperature to carry out the curing, the samples were firstly characterised via TGA. For the clarity of the description, being the observed profiles similar, only the results obtained from GLU2_16 will be reported and discussed (Fig. 4). Fig. 4B shows that the fibres resulted thermally stable up to approximately 200 °C, while the degradation took place in a two-step process roughly between 200 °C and 400 °C, giving a stable carbon residue at 500 °C, corresponding approximately to the 14% of the initial weight. However, the derivative curves evidenced a weight loss phenomenon centred approximately at 180 °C, occurring only in the analysis carried out on the sample before the thermal treatment (Fig. 4B, dot line). This weight loss was attributed to the evaporation of water, as a product of the esterification reactions between citric acid and the maltodextrin molecules. For this reason, 180 °C was chosen as temperature to perform the cross-linking process. As a confirmation, the FTIR-ATR spectra reported in Fig. 4C, showed a shift of the peak associated to carbonyl stretching from 1716 cm−1 to 1726 cm−1. This behaviour was associated to the different chemical surrounding generated after the thermal treatment, caused by the condensation of carboxylic acids and hydroxyl groups resulting in esters formation. Subsequently, SEM images were acquired to determine if the thermal treatment had any influence on the morphology of the spun mats. As reported in Fig. 5, all the mats obtained from GLU2 solutions did not display any change in the fibrous morphology (Fig. 5A–D). Conversely, some of the samples obtained from GLU6, GLU9, GLU12, and GLU19 solutions showed alterations after curing. In these cases, the fibres were no longer well-defined, but coalescence phenomena occurred, resulting in the fibres attached to each other. Furthermore, after a careful observation, the degree of morphology loss resulted correlated to both the amount of citric acid present in the spun mat and the DE value of the starting maltodextrin. As a proof, in GLU6 and GLU9 systems, only in the case of GLU6_33 (Fig. 5H) and GLU9_33 (Fig. 5L) a coalescence of the fibres was observed. For GLU12 samples, the alteration is visible in both GLU12_16 and GLU12_33, reported in Fig. 5O and P, respectively. In GLU19, only GLU19_8 (Fig. 5Q) retains the starting morphology, whereas deformations are detected in GLU19_16 (Fig. 5R) and GLU19_25 (Fig. 5S) samples; finally, for GLU19_33 (Fig. 5T) the alteration was so severe that the fibres appear to lack continuity. This phenomenon was attributed to the presence of water within the spun mats before curing, thus other from the water generated as a product of the condensation reactions taking place during cross-linking. Being citric acid hygroscopic,69,70 the hypothesis was that even if the reagent employed for the study was anhydrous, it got hydrated during the preparation of the solution. The subsequent spinning step was sufficient to evaporate the unbound solvent, as demonstrated by the well-defined morphology of the collected mats. However, being the fibres deposition carried out at room temperature, this step was not sufficient to restore the citric acid to its anhydrous form, and thus the latter was present within the mat as monohydrated. Subsequently, during the curing step as the temperature increased, the hydration water was also released from the polymer, together with the water generated as product of the condensation reactions. The early stages of this release, in particular when the fraction of citric acid is high, might have caused a local dissolution of the polymer, resulting in a deformation of the fibre, giving coalescence phenomena and in the most severe cases, loss of the sample morphology. This phenomenon resulted more pronounced for higher DE maltodextrins, because of the lower molecular weight of the polysaccharide, thus more prone to get involved in dissolution phenomena. Nevertheless, in those cases in which the thermal treatment did not cause any damage to the fibrous morphology, the mean diameter values appeared to be not affected. This observation differs from what reported by Kutzli et al.,71 where the presence of whey and soy proteins as cross-linking agents led to fibres characterized by diameters approximately 30% to 300% higher if compared to the fibres obtained without the addition of protein (1.61 ± 0.44 μm).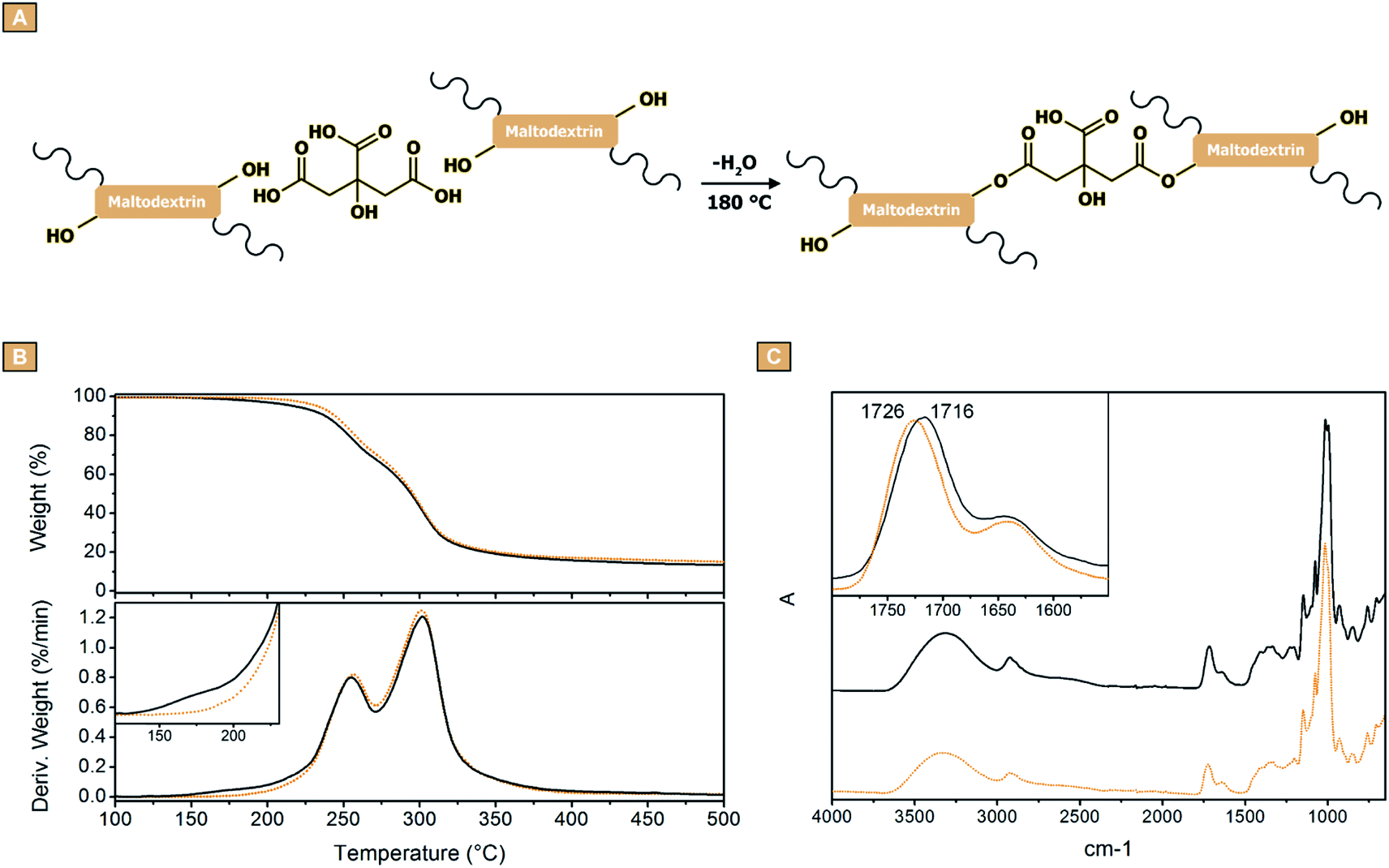 | ||
| Fig. 4 (A) Reactions taking place during the cross-linking treatment. TGA analyses (B), and (C) FTIR-ATR measurements of GLU2_16 before cross-linking (solid) and after cross-linking (dot) process. | ||
 | ||
| Fig. 5 SEM images of spun maltodextrin solutions after thermal treatment at 180 °C. Scale bars: 20 μm. | ||
Eventually, water solubility tests were carried out as a further proof of the thermal curing previously described. The mats were dipped in deionized water at room temperature and recovered via centrifugation after 15 hours. After been dried up to constant weight, the soluble fraction was calculated according to eqn (1) and reported in Table 2. At increasing amounts of citric acid, the soluble fraction decreased accordingly. As mentioned, the absence of citric acid within the formulation hindered the possibility to achieve a thermal cross-linking, thus resulting in fibres that are still water soluble. The lowest soluble fraction was detected on sample GLU2_33 and was equal to 12.7 ± 3.4%; this value was probably still affected by the presence of low molecular weight fractions composing the starting maltodextrin (see Fig. 1), less prone to be bound to the polymer network during the curing. Overall, maltodextrins displaying lower DE values, thus higher average molecular weight, displayed lower soluble fractions, probably due to the possibility to obtain an insoluble network with a lower number of cross-links. On the contrary, higher amounts of citric acid were required to turn the mats obtained from maltodextrin with higher DE insoluble. In this frame, the samples in which coalescence phenomena resulted more pronounced (GLU12_33, GLU19_25, and GLU19_33) were not further characterised, due to difficulties observed during the recovery of the sample from the collector, and for this reason they are marked as “—” in Table 2. Probably, the coalescence taking place during curing also caused the mats to get stick to the collecting substrate, hence hindering their recovery.
| wt% citric acid | Maltodextrin (soluble fraction, %) | ||||
|---|---|---|---|---|---|
| GLU2 | GLU6 | GLU9 | GLU12 | GLU19 | |
| 0 | Fully soluble | Fully soluble | Fully soluble | Fully soluble | Fully soluble |
| 8.3 | 28.3 ± 3.2 | Fully soluble | 47.4 ± 6.1 | Fully soluble | Fully soluble |
| 16.6 | 14.0 ± 0.7 | 31.1 ± 1.9 | 21.2 ± 2.1 | 22.4 ± 2.1 | 32.1 ± 0.7 |
| 25.0 | 14.9 ± 1.4 | 13.8 ± 1.9 | 14.5 ± 2.2 | 25.2 ± 0.1 | — |
| 33.3 | 12.7 ± 3.4 | 13.9 ± 2.1 | 13.6 ± 3.8 | — | — |
Circular specimens obtained from GLU2_16 electrospun mat are reported in Fig. 6. As reported in Fig. 6A, the mat appears white before the thermal treatment, while the cross-linked one (Fig. 6B) is characterized by a slight yellow colour, as a consequence of the curing process.
 | ||
| Fig. 6 GLU2_16 spun mat (A) before and (B) after the thermal treatment. | ||
Conclusions
Commercial maltodextrins Glucidex 2®, Glucidex 6®, Glucidex 9®, Glucidex 12®, and Glucidex 19® demonstrated to be suitable choices to obtain bio-based fibres deposition via electrospinning technique, without the use of any synthetic carrier polymer. Further, thanks to the intrinsic water solubility of the chosen maltodextrins it was possible to process the latter using water as unique solvent, thus exploiting a sustainable approach. For all maltodextrins, it was possible to identify an optimum condition to obtain the deposition of well-defined fibres, with diameters ranging from roughly 1.1 μm to 1.5 μm. Subsequently, to overcome the solubility of the obtained mats, the maltodextrins solutions were added with increasing amounts of citric acid, and the spinnability of the so-prepared solutions was evaluated. The presence of citric acid did not hinder the overall spinnability of the systems since well-defined fibres with diameters ranging from approximately 0.7 μm to 1.4 μm were observed. Moreover, citric acid allowed to obtain a one-step thermal cross-linking of the spun mats via ester formation; a thermal curing carried out at 180 °C was demonstrated to trigger condensations reactions between the citric acid molecules and the maltodextrins chains, giving cross-linked, water insoluble networks as the product. However, the retention of the fibrous morphology of the cured mat resulted to be affected by increasing amounts of citric acid and increasing DE value characterising the maltodextrins. In this context, when the morphology was retained, also the diameter's distribution did not vary, while the loss of morphology was accompanied by coalescence phenomena generated from local dissolution mechanism taking place during the curing step.This study demonstrated the possibility to obtain polysaccharide-based fibrous mats via electrospinning of suitable maltodextrins through an eco-friendly approach. Also, the addition of citric acid allowed to turn the obtained fibres from water soluble to water insoluble via sustainable thermal treatment and to avoid the use or generation of toxic by-products. As a prospective, the introduction of a purification step carried out with e.g. ultrafiltration technique, can be exploited to separate the low molecular weight fractions from the maltodextrin samples. This approach can be useful to reduce the drawbacks observed in this study, such as the coalescence phenomena and the presence of soluble fractions in all the cured mats. Nevertheless, the processing and curing chosen for this work constitute an overall green process, suitable to obtain bio-based cross-linked mats which can be further studied for environmental, pharmaceutical, and medical applications.
Author contributions
Claudio Cecone: conceptualization, methodology, investigation, validation, formal analysis, visualization, writing – original draft, writing – review & editing. Gjylije Hoti: methodology, investigation, validation, writing – review & editing. Marco Zanetti: supervision, methodology, writing – review & editing. Francesco Trotta: supervision, project administration, writing – review & editing. Pierangiola Bracco: supervision, methodology, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Roquette Frères (Lestrem, France) for providing the maltodextrins used for this work.References
- G. Bishop, D. Styles and P. N. L. Lens, Resour., Conserv. Recycl., 2021, 168, 105451 CrossRef
.
- H. Karan, C. Funk, M. Grabert, M. Oey and B. Hankamer, Trends Plant Sci., 2019, 24, 237–249 CrossRef CAS PubMed
- S. Walker and R. Rothman, J. Cleaner Prod., 2020, 261, 121158 CrossRef CAS
- A. Di Bartolo, G. Infurna and N. T. Dintcheva, Polymers, 2021, 13, 1229 CrossRef CAS PubMed
- Y. Yu, M. Shen, Q. Song and J. Xie, Carbohydr. Polym., 2018, 183, 91–101 CrossRef CAS PubMed
- X. Qi, L. Wu, T. Su, J. Zhang and W. Dong, Colloids Surf., B, 2018, 170, 364–372 CrossRef CAS PubMed
- A. Soroudi and I. Jakubowicz, Eur. Polym. J., 2013, 49, 2839–2858 CrossRef CAS
- F. Xie, E. Pollet, P. J. Halley and L. Avérous, Prog. Polym. Sci., 2013, 38, 1590–1628 CrossRef CAS
- J. Yang, Y. C. Ching and C. H. Chuah, Polymers, 2019, 11, 1–27 Search PubMed
- M. Brodin, M. Vallejos, M. T. Opedal, M. C. Area and G. Chinga-Carrasco, J. Cleaner Prod., 2017, 162, 646–664 CrossRef CAS
- S. Ochi, Composites, Part A, 2006, 37, 1879–1883 CrossRef
- H. J. Prado and M. C. Matulewicz, Eur. Polym. J., 2014, 52, 53–75 CrossRef CAS
- M. R. Kweon, F. W. Sosulski and P. R. Bhirud, Starch/Staerke, 1997, 49, 59–66 CrossRef CAS
- R. S. Blackburn, Environ. Sci. Technol., 2004, 38, 4905–4909 CrossRef CAS PubMed
- A. Mignon, N. De Belie, P. Dubruel and S. Van Vlierberghe, Eur. Polym. J., 2019, 117, 165–178 CrossRef CAS
- J. Desbrières and E. Guibal, Polym. Int., 2018, 67, 7–14 CrossRef
- L. Liu, Z. Y. Gao, X. P. Su, X. Chen, L. Jiang and J. M. Yao, ACS Sustainable Chem. Eng., 2015, 3, 432–442 CrossRef CAS
- A. Ali and S. Ahmed, Int. J. Biol. Macromol., 2018, 109, 273–286 CrossRef CAS PubMed
- J. M. Dang and K. W. Leong, Adv. Drug Delivery Rev., 2006, 58, 487–499 CrossRef CAS PubMed
- S. Bratskaya, S. Schwarz, J. Laube, T. Liebert, T. Heinze, O. Krentz, C. Lohmann and W. M. Kulicke, Macromol. Mater. Eng., 2005, 290, 778–785 CrossRef CAS
- L. Dokic-Baucal, P. Dokic and J. Jakovljevic, Food Hydrocolloids, 2004, 18, 233–239 CrossRef CAS
- X. Qi and R. F. Tester, Starch/Staerke, 2018, 70, 10–14 Search PubMed
- N. Reddy, R. Reddy and Q. Jiang, Trends Biotechnol., 2015, 33, 362–369 CrossRef CAS PubMed
- N. A. O'Connor, A. Abugharbieh, F. Yasmeen, E. Buabeng, S. Mathew, D. Samaroo and H.-P. Cheng, Int. J. Biol. Macromol., 2015, 72, 88–93 CrossRef PubMed
- M. Castro-Cabado, F. J. Parra-Ruiz, A. L. Casado and J. San Román, Polym. Polym. Compos., 2016, 24, 643–654 CAS
- D. Gyawali, P. Nair, Y. Zhang, R. T. Tran, C. Zhang, M. Samchukov, M. Makarov, H. K. W. Kim and J. Yang, Biomaterials, 2010, 31, 9092–9105 CrossRef CAS PubMed
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef
- L. Hovgaard and H. Brøndsted, J. Controlled Release, 1995, 36, 159–166 CrossRef CAS
- A. J. Kuijpers, P. B. Van Wachem, M. J. A. Van Luyn, G. H. M. Engbers, J. Krijgsveld, S. A. J. Zaat, J. Dankert and J. Feijen, J. Controlled Release, 2000, 67, 323–336 CrossRef CAS PubMed
- C. Cecone, G. Costamagna, M. Ginepro and F. Trotta, RSC Adv., 2021, 11, 7653–7662 RSC
- J. Anu Bhushani and C. Anandharamakrishnan, Trends Food Sci. Technol., 2014, 38, 21–33 CrossRef CAS
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS
- N. Bhardwaj and S. C. Kundu, Biotechnol. Adv., 2010, 28, 325–347 CrossRef CAS PubMed
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson, U. Von Gunten and B. Wehrli, Science, 2006, 313, 1072–1077 CrossRef CAS PubMed
- P. E. Stackelberg, E. T. Furlong, M. T. Meyer, S. D. Zaugg, A. K. Henderson and D. B. Reissman, Sci. Total Environ., 2004, 329, 99–113 CrossRef CAS PubMed
- A. J. Watkinson, E. J. Murby, D. W. Kolpin and S. D. Costanzo, Sci. Total Environ., 2009, 407, 2711–2723 CrossRef CAS PubMed
- R. Loos, R. Carvalho, D. C. António, S. Comero, G. Locoro, S. Tavazzi, B. Paracchini, M. Ghiani, T. Lettieri, L. Blaha, B. Jarosova, S. Voorspoels, K. Servaes, P. Haglund, J. Fick, R. H. Lindberg, D. Schwesig and B. M. Gawlik, Water Res., 2013, 47, 6475–6487 CrossRef CAS PubMed
- E. M. Golet, I. Xifra, H. Siegrist, A. C. Alder and W. Giger, Environ. Sci. Technol., 2003, 37, 3243–3249 CrossRef CAS PubMed
- E. Gonzalez, A. Barquero, B. Muñoz-sanchez, M. Paulis and J. R. Leiza, Nanomaterials, 2021, 11, 1–14 Search PubMed
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed
- P. T. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC
- T. C. Kha, M. H. Nguyen and P. D. Roach, J. Food Eng., 2010, 98, 385–392 CrossRef CAS
- P. Robert, T. Gorena, N. Romero, E. Sepulveda, J. Chavez and C. Saenz, Int. J. Food Sci. Technol., 2010, 45, 1386–1394 CrossRef CAS
- F. Sansone, T. Mencherini, P. Picerno, M. D'Amore, R. P. Aquino and M. R. Lauro, J. Food Eng., 2011, 105, 468–476 CrossRef CAS
- M. Ahmed, M. S. Akter, J. C. Lee and J. B. Eun, LWT--Food Sci. Technol., 2010, 43, 1307–1312 CrossRef CAS
- A. C. Stijnman, I. Bodnar and R. Hans Tromp, Food Hydrocolloids, 2011, 25, 1393–1398 CrossRef CAS
- I. Kutzli, D. Griener, M. Gibis, C. Schmid, C. Dawid, S. K. Baier, T. Hofmann and J. Weiss, Food Hydrocolloids, 2020, 101, 105535 CrossRef CAS
- I. Kutzli, D. Beljo, M. Gibis, S. K. Baier and J. Weiss, Food Biophys., 2020, 15, 206–215 CrossRef
- Y. Z. Zhang, J. Venugopal, Z. M. Huang, C. T. Lim and S. Ramakrishna, Polymer, 2006, 47, 2911–2917 CrossRef CAS
- J. D. Schiffman and C. L. Schauer, Biomacromolecules, 2007, 8, 594–601 CrossRef CAS PubMed
- A. G. Destaye, C. K. Lin and C. K. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 4745–4752 CrossRef CAS PubMed
- M. Hulupi and H. Haryadi, Mater. Today: Proc., 2019, 13, 199–204 CAS
- W. Lu, M. Ma, H. Xu, B. Zhang, X. Cao and Y. Guo, Mater. Lett., 2015, 140, 1–4 CrossRef CAS
- X. Xu, J. F. Zhang and Y. Fan, Biomacromolecules, 2010, 11, 2283–2289 CrossRef CAS PubMed
- S. Zhang, Y. Huang, X. Yang, F. Mei, Q. Ma, G. Chen, S. Ryu and X. Deng, J. Biomed. Mater. Res., Part A, 2009, 90, 671–679 CrossRef PubMed
- K. Siimon, H. Siimon and M. Järvekülg, J. Mater. Sci.: Mater. Med., 2015, 26, 1–9 CrossRef CAS PubMed
- C. Tang, C. D. Saquing, J. R. Harding and S. A. Khan, Macromolecules, 2010, 43, 630–637 CrossRef CAS
- J. Shi and E. Yang, J. Nano Res., 2015, 32, 32–42 CAS
- D. Yu, Y. Y. Feng, J. X. Xu, B. H. Kong, Q. Liu and H. Wang, Packag. Technol. Sci., 2021, 34, 361–370 CrossRef CAS
- D. Nataraj, R. Reddy and N. Reddy, Eur. Polym. J., 2020, 124, 109484 CrossRef
- S. Bednarz, M. Lukasiewicz, W. Mazela, M. Pajda and W. Kasprzyk, J. Appl. Polym. Sci., 2010, 119, 3511–3520 CrossRef
- B. Martel, D. Ruffin, M. Weltrowski, Y. Lekchiri and M. Morcellet, J. Appl. Polym. Sci., 2005, 97, 433–442 CrossRef CAS
- C. Cecone, G. Hoti, I. Krabicova, S. L. Appleton, F. Caldera, P. Bracco, M. Zanetti and F. Trotta, Green Chem., 2020, 22, 5806–5814 RSC
- C. Cecone, F. Caldera, A. Anceschi, D. Scalarone, F. Trotta, P. Bracco and M. Zanetti, J. Appl. Polym. Sci., 2018, 135, 1–6 CrossRef
- Y. Rong, M. Sillick and C. M. Gregson, J. Food Sci., 2009, 74(1), C33–C40 CrossRef CAS PubMed
- N. Castro, V. Durrieu, C. Raynaud and A. Rouilly, Carbohydr. Polym., 2016, 144, 464–473 CrossRef CAS PubMed
- I. Kutzli, M. Gibis, S. K. Baier and J. Weiss, J. Appl. Polym. Sci., 2018, 135, 46328 CrossRef
- I. Kutzli, M. Gibis, S. K. Baier and J. Weiss, J. Agric. Food Chem., 2018, 66, 10283–10291 CrossRef CAS PubMed
- C. Peng, M. N. Chan and C. K. Chan, Environ. Sci. Technol., 2001, 35, 4495–4501 CrossRef CAS PubMed
- C. Peng, C. K. Chan and A. H. L. Chow, Aerosol Sci. Technol., 2001, 35, 753–758 CrossRef CAS
- I. Kutzli, M. Gibis, S. K. Baier and J. Weiss, Food Hydrocolloids, 2019, 93, 206–214 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06785k |
| This journal is © The Royal Society of Chemistry 2022 |