
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of a multi-epitope vaccine against SARS-CoV-2: immunoinformatic and computational methods†
Md. Oliullah Rafi af,
Khattab Al-Khafajib,
Md. Takim Sarkera,
Tugba Taskin-Tokcd,
Abdus Samad Ranae and
Md. Shahedur Rahman*af
af,
Khattab Al-Khafajib,
Md. Takim Sarkera,
Tugba Taskin-Tokcd,
Abdus Samad Ranae and
Md. Shahedur Rahman*af
aDepartment of Genetic Engineering and Biotechnology, Jashore University of Science and Technology, Jashore, 7408, Bangladesh. E-mail: ms.rahman@just.edu.bd; rafi.btech.bd@gmail.com; takimsarker27@gmail.com
bDepartment of Medical Laboratory Technology, AL-Nisour University College, Baghdad, Iraq. E-mail: k.a.alkhafaji@gmail.com
cDepartment of Bioinformatics and Computational Biology, Institute of Health Sciences, Gaziantep University, Gaziantep 27310, Turkey. E-mail: ttaskin@gantep.edu.tr
dFaculty of Arts and Sciences, Department of Chemistry, Gaziantep University, Gaziantep, Turkey
eSchool of Biotechnology, Jiangnan University, Wuxi, 214122, PR China. E-mail: ranagebt@gmail.com
fBioinformatics and Microbial Biotechnology Laboratory, Department of Genetic Engineering and Biotechnology, Jashore University of Science and Technology, Jashore 7408, Bangladesh
First published on 2nd February 2022
Abstract
A novel infectious agent, SARS-CoV-2, is responsible for causing the severe respiratory disease COVID-19 and death in humans. Spike glycoprotein plays a key role in viral particles entering host cells, mediating receptor recognition and membrane fusion, and are considered useful targets for antiviral vaccine candidates. Therefore, computational techniques can be used to design a safe, antigenic, immunogenic, and stable vaccine against this pathogen. Drawing upon the structure of the S glycoprotein, we are trying to develop a potent multi-epitope subunit vaccine against SARS-CoV-2. The vaccine was designed based on cytotoxic T-lymphocyte and helper T-lymphocyte epitopes with an N-terminal adjuvant via conducting immune filters and an extensive immunoinformatic investigation. The safety and immunogenicity of the designed vaccine were further evaluated via using various physicochemical, allergenic, and antigenic characteristics. Vaccine-target (toll-like receptors: TLR2 and TLR4) interactions, binding affinities, and dynamical stabilities were inspected through molecular docking and molecular dynamic (MD) simulation methods. Moreover, MD simulations for dimeric TLRs/vaccine in the membrane-aqueous environment were performed to understand the differential domain organization of TLRs/vaccine. Further, dynamical behaviors of vaccine/TLR systems were inspected via identifying the key residues (named HUB nodes) that control interaction stability and provide a clear molecular mechanism. The obtained results from molecular docking and MD simulation revealed a strong and stable interaction between vaccine and TLRs. The vaccine's ability to stimulate the immune response was assessed by using computational immune simulation. This predicted a significant level of cytotoxic T cell and helper T cell activation, as well as IgG, interleukin 2, and interferon-gamma production. This study shows that the designed vaccine is structurally and dynamically stable and can trigger an effective immune response against viral infections.
1. Introduction
Severe acute respiratory syndrome coronavirus 2, at 26–32 kb, is the largest recognized single-stranded RNA enveloped virus. The virus was first detected in late December 2019, with some cases having the symptoms of respiratory diseases like the common cold. Later on, the disease was termed coronavirus disease 2019.1–3 SARS-CoV-2 is a β-coronavirus in the Coronaviridae family. Members of the Coronaviridae family have a wide range of hosts, including birds, mammals, and humans.4 To date, a total of seven coronavirus strains have been identified as being able to infect human hosts, of which two (HCoV-229E and NL63) are of the alpha (α) genera and the other five (SARS-CoV-2, HCoV-HKU1, MERS-CoV, HCoV-OC43, and SARS-CoV) are from the β genera.5,6 To date, only three of the aforementioned stains (MERS-CoV, SARS-CoV-2, and SARS-CoV) have been recognized as dangerous viruses that can cause death. Conversely, the rest of them cause mild symptoms of the common cold.7,8 Researchers consider vaccines to be a compatible and effective alternative that may be capable of generating long-term protection against all virulent strains.9,10 The emergence of the COVID-19 outbreak initially occurred in Wuhan, Hubei Province, central China.11 Investigation suggests that the virus may have spread to humans directly from bats at the Wuhan seafood market and expanded extensively among the human population within a very short period.9,12,13 Health professionals and scientists focused on the mode of transmission as the situation grew worse in other countries, including Brazil, Chile, the United Kingdom, Germany, Russia, India, South Africa, Mexico, Peru, Spain, the United States, and Italy, with substantial fatality caused by community transmission.A study of the SARS-CoV-2 virus particle revealed that it shares structural homology with other beta coronaviruses, maintaining a sequential pattern with a 5′-leader-UTR–replicase–spike–envelope–membrane–nucleocapsid and a 3′ UTR poly(A) tail, including species-specific accessory genes at the 3′ end. The structural proteins consist of (1) the spike (S)/surface glycoprotein, (2) the envelope glycoprotein, and (3) the membrane glycoprotein.14,15 Currently, vaccine development is considered to be the most viable and long-term solution to dealing with this deadly virus. Researchers are particularly targeting some functionally essential viral proteins that are related to viral adhesion and replication processes such as RNA-dependent RNA polymerase (RdRp), involved in RNA catalysis from viral RNA during replication, and main protease (Mpro) that process the synthesized polyproteins derived from translation.16 The viral structural protein, spike glycoprotein, facilitates the viral entry into the host cell. The S glycoprotein is a class 1 single fusion polypeptide chain that contains an S1 subunit, which mediates attachment to the host cell membrane, and an S2 subunit, which fuses to the host membrane. The S1 ectodomain contains 4 beta-rich domains (A, B, C, and D) in its N-terminal region, whereas the C-terminus, containing the S2 ectodomain, is responsible for membrane fusion.4,17,18 It has been suggested that often, fewer CD8+ T cells are activated in response to the natural SARS-CoV-2 infection, although helper T-cell exhibit a robust response in the presence of spike glycoprotein.19 In addition, the S glycoprotein is the leading viral antigen that can neutralize host antibodies during the infection. Therefore, the S glycoprotein is a potential target for vaccine construction.4
Conventional methods to develop vaccines have proven successful; however, they also have some drawbacks. Sometimes generating a vaccine requires sacrificing the entire organism, and in some cases, a heavy load of unwanted antigens can cause an allergic reaction. Therefore, a multi-epitope-based vaccine may be an advantageous approach to reduce the recurrence of infection and generate robust immunity. Epitopes are immune stimulators displayed by the MHC molecules on cells and in turn detected by the T or B cells. Both T and B cells cooperate with various immune cells that secrete cytokines, providing adaptive immunity to the host cells. Records of patients with SARS-CoV-2 suggest that cell-mediated and humoral immune responses contribute equally to the host defense generated by the activation of Th1-skewed and cytotoxic T cells.20 Thus, epitope identification is a focus for researchers working on epitope-based vaccine construction. In addition, it reduces both the time and the expenditures required for this kind of invention.21
This study aims in designing a spike glycoprotein-based multi-epitope vaccine. This could initiate the stimulation of a defensive humoral and cellular immunity to promote recovery from COVID-19. The prediction of epitopes prioritized those that were antigenic, immunogenic, and non-allergenic. The final vaccine was constructed with cytotoxic T-lymphocyte (CTL) and helper T-lymphocyte (HTL) epitopes. Additionally, several characteristics such as physicochemical, immunogenic, and dynamic stability were inspected to understand the safety and effectiveness of the designed vaccine (Fig. 1).
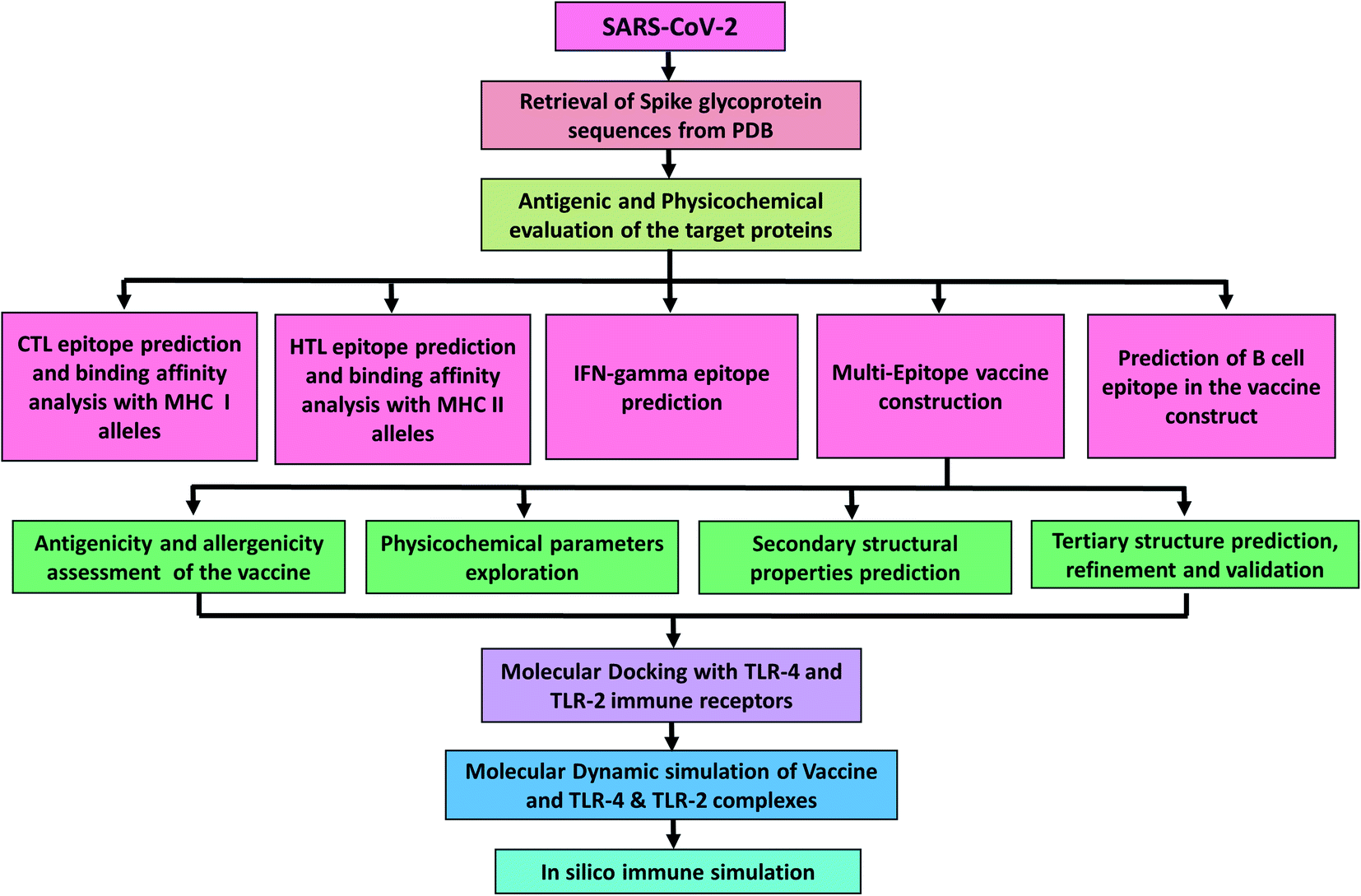 | ||
| Fig. 1 Diagrammatic workflow of the proposed study. The overall methodology is shown in several phases, which include the spike glycoprotein selection from the Protein Data Bank and physicochemical evaluation of the sequences. Epitope prediction from the target sequences (CTL, HTL, interferon-gamma, and B-cell epitopes), vaccine construction, and assessment of its features. Molecular interactions and dynamic stability of the vaccine with toll-like receptors were examined by running molecular docking and MD simulation. While, the ability vaccine to initiate the immune response was also carried out. | ||
2. Materials and methods
2.1. Protein sequence retrieval, phylogenetic tree analysis, and evaluation of antigenic and physiochemical conduct
The complete amino acid sequences of 7 SARS-CoV-2 spike glycoproteins were downloaded from the Protein Data Bank (PDB)22 in fasta format (https://www.rcsb.org/). All sequences were submitted to the VaxiJen v2.0 server,23 to check the antigenicity. The physicochemical properties of the target glycoprotein, including its aliphatic index, extinction co-efficient, gravy, instability index, molecular weight, and theoretical pI were also evaluated using the Expasy ProtParam tool.24 In order to analyze mutation and variation, the sequences were aligned using the MUSCLE tools25 and a phylogenetic tree was constructed through the Maximum likelihood method with 1000 bootstrap replicates by using MEGA-X.262.2. CTL epitopes prediction and assessment of immunogenicity
The NetCTL 1.2 server (http://www.cbs.dtu.dk/services/NetCTL/)27 was used to identify the immunogenic CTL epitope (elicit the cell-mediated immune response) which can generate memory cells. NetCTL 1.2 was built based on a training dataset to predict the CTL epitope from the query sequence. The 9-mer CTL epitopes were predicted that could be recognized by 12 human leukocyte antigen allele class I supertypes of the human population. The thresholds for antigen processing transport efficiency, C-terminal cleavage, and epitope identification were set at 0.05, 0.15, and 0.75 respectively. Additionally, the predicted sequence was subjected to the Immune Epitope Database (IEDB) using a consensus method to identify the binding affinity with HLA class I alleles. The epitopes that displayed a consensus score of less than 2 were considered to be a strong binder to their respective HLA allele.28 To obtain the immunogenic epitope, we used the IEDB class I immunogenicity prediction module.2.3. Prediction of helper T-lymphocyte (HTL) epitope
A fundamental requirement for inducing both humoral and cell-mediated immune responses and accelerating pathogen clearance is the presence of HTLs in association with many cytokines and other immune cells substantial and influential.29 The 15-mer HTL epitope was predicted by utilizing the Net MHC II pan 3.2 server (http://www.cbs.dtu.dk/services/NetMHCIIpan-3.2/) with the commonly occurring MHC class II DEB1 alleles 01:01, 03:01, 04:01, 07:01, 08:01, 08:03, 10:01, 11:01, 12:01, 13:01 13:02, 14:01 and 15:01.30 The MHC class II alleles involved in the investigation are projected to be present in over 95% of the human population in the world.31 The epitopes were divided into the strong binder, intermediate binder, and non-binder with their respective HLA alleles at threshold values of 2%, 10%, and >10%, respectively, in the Net MHC II pan 3.2 servers. Further, to investigate whether the MHC class 2 epitope can activate or not Th1 type immunity followed by interferon-gamma (IFN-γ production), the top-ranked epitope was submitted to the IFN epitope server (http://crdd.osdd.net/raghava/ifnepitope/).32 The server provides a specialized platform for both the development and prediction of IFNγ-induced peptide sequences derived from query sequences. Moreover, it utilizes an enriched dataset for data retrieval containing 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 433 experimentally validated helper T-cell epitopes from the IDEB database.32 It generates overlapping epitopes with IFN-γ inducing from the query protein sequence as presented by a numerical score. An SVM hybrid and motif-based strategy were selected to predict whether the epitope would induce IFN-γ production.
433 experimentally validated helper T-cell epitopes from the IDEB database.32 It generates overlapping epitopes with IFN-γ inducing from the query protein sequence as presented by a numerical score. An SVM hybrid and motif-based strategy were selected to predict whether the epitope would induce IFN-γ production.
2.4. Prediction of B-cell epitope
B-cell plays an essential role in humoral immunity which can secret antibodies for neutralizing antigen. Linear and discontinuous B-cell epitopes were predicted by utilizing the Ellipro web tool (http://tools.iedb.org/ellipro/).33 This method uses three algorithms to perform conformational calculations of proteins as ellipsoids, estimate the protrusion index (PI) of residues, and grouping of adjacent residues based on PI values. Ellipro provides a numerical score for the output of each B-cell epitope which is described as PI values. From ElliPro prediction, 90% of the protein residues involved in an ellipsoid with a PI value of 0.9 and 10% of residues contain without ElliPsoid. Where the most significant predictive tools for all proteins are Ellipsoid and ElliPro.2.5. Prediction of the antigenicity, promiscuity, allergenicity, and overlapping nature of the epitopes
The antigenic, non-toxic, and non-allergenic epitopes were screened out, followed by submitting them to the AllerTop v2.0 (ref. 34) (https://www.ddg-pharmfac.net/AllerTOP/), VaxiJen v.2.0 (ref. 23) (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html), and ToxinPred servers35 (https://webs.iiitd.edu.in/raghava/toxinpred/design.php), respectively. The threshold for antigenicity was set at 0.4 on the VaxiJen v.2.0 server. VaxiJen v2.0 uses open access alignment methods to calculate the antigenicity of a protein sequence based on the auto-covariance and cross-covariance transformation of sequences in uniform vectors with the most important amino acid properties. The accuracy of VaxiJen v2.0 has been assessed at 70–89% for antigen prediction.23 The standard errors of obtained data, precision assessments, were assessed based on the ACC terms and anlyzed them, then we chose the lowest varrient values around the obtained value of VaxiJen v2.0 server. In next step we used descriptive statistics within excel to analysis. Epitopes that have a high molecular interaction with multiple human leukocyte antigen alleles are considered promiscuous epitopes. These sequences are very important in peptide-based vaccine design, as they can generate effective immunity in the host.20 The list of high binding affinity contains promiscuous epitopes that were screened out in this study. The overlapping epitopes can help to activate the CD4 and CD8 T-cells, as they contain both CTL and HTL sequences.20 In the current study, the IEDB server was used to estimate the overlapping capacity of the epitopes.2.6. Epitope conservancy and population coverage calculation
The conservancy of epitopes was analyzed by the Infectious Disease Epidemiology Bureau conservancy tool.36 Whereas the population coverage for each epitope was estimated by the IEDB population coverage web server.372.7. Molecular interaction pattern analysis of epitopes with HLA alleles
The PEPFOLD 3 web tool was applied to generate the 3-D structure of screened-out best epitope sequences.38 The server can analyze molecular interactions between small molecules. The 3-D crystal structure of the most commonly found human leukocyte antigen alleles in the human population, HLA-DRB1-01:01 (MHC class II), HLA-A-02:01 (MHC class I) and were retrieved from RCSB PDB ID 1QEW and 2G9H, respectively. Proteins were prepared before running P–P docking by removing water and co-crystallized ligand. In the next step, we performed energy minimization of each structure. The structure of modeled epitopes docked with respective HLA alleles using PatchDock server39 using default parameters and an RMSD value of 0.4 to analyze the epitope and allele interaction pattern. Further, the complexes with the best docking were refined in the FireDock server40 to find the top 10 complexes of receptor and ligand. Complexes with the lowest global energy (most negative) were nominated for analyzing their molecular interaction patterns.2.8. Vaccine construction
A suitable vaccine sequence must have the capability to generate immune responses by CTLs and HTLs; thus, it should contain appropriate linker-bound HTL and CTL epitopes. Screening of the best promiscuous, antigenic, immunogenic, and non-allergenic CD8+ and CD4+ epitopes was utilized to construct the final multi-epitopic vaccine candidate. Selected CTL epitopes connected via an AAY linker and a GPGPG linker were used to prevent junctional HTL epitope formation. An adjuvant cholera toxin B is linked to the N-terminus of the vaccine sequence via the EAAAK linker.2.9. Study the antigenic and physicochemical properties of vaccines
ANTIGENpro and VaxiJen v2.0 servers were utilized to anticipate the antigenicity of our ultimate multi-epitope-based vaccine. VaxiJen v2.0 is an open-source server for predicting the sequence antigenicity and the prediction was performed on the basis of an auto- and cross-covariance (ACC) transformation method.41 The ANTIGENpro server uses microarray data for antigenic evaluation of a protein and delivers the protein's antigenicity index. While the allergenicity of the designed vaccine was inspected by AllergenFP v1.0 and AllerTOP v2.0 servers.42 AllerTOP v2.0 works to classify the protein allergens based on auto- and cross-covariance (ACC) transformation, the amino acid E-descriptors, and the k nearest neighbor's machine learning methods. Whereas AllergenFP exploits a descriptor-based and alignment-free approach for determining the allergens and non-allergens. Further, various physicochemical parameters of the vaccine (aliphatic index, extinction co-efficient, gravy, instability index, theoretical pI and molecular weight) were investigated by ProtParam server (https://web.expasy.org/protparam/).24 To check for the existence of any signal peptide in the vaccine, we used the SignalP4.1 server.432.10. Structural evaluation of the vaccine
We used the SOPMA web server44 to identify the secondary structure of the vaccine. Whereas the I-TASSER web server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) was used to predict the three-dimensional structure of the vaccine sequence, which is an incorporated platform for predicting both of function and structure of a protein.45 Where this server uses multiple threading alignments and iterative structural assembly simulations to construct the 3-D model of an amino acids sequence.46 The best three-dimensional model found from the I-TASSER server was further refined, first using ModRefiner, then with the GalaxyRefine web tool. ModRefiner refines the protein structure from Cα traces determined by a two-step energy minimization process at the atomic level. It improves global and local structures, with greater accuracy in hydrogen-bonding networks, improved side-chain positions, and fewer atomic overlaps.47 Conversely, the GalaxyRefine web server refinement method is approved by community-wide CASP10 experiments. This server also supports the molecular dynamic simulation in the attainment of the relaxed entire structure and also enhances the quality of local structures according to CASP10 computation techniques.48 The final three dimensional structure of the vaccine was validated by using ProSA-web, which determines the Z-score of global quality for a specific query model.49 The ERRAT server (https://servicesn.mbi.ucla.edu/ERRAT/) was utilized to evaluate non-bonded atom–atom interaction compared to a high-resolution crystallography model.50 The global quality of the vaccine structure was demonstrated through Ramachandran plot analysis followed by evaluation on the ProCheck server (https://servicesn.mbi.ucla.edu/PROCHECK/).512.11. Analysis of molecular docking between the vaccine and toll-like immune receptors
Molecular docking is a computational method that was employed to predict the molecular interaction and binding affinity of receptors and ligands using a scoring function.52,53 Toll-like receptors (TLRs) can recognize viral partial and have a major role in immunity.54,55 To get an insight into the interaction pattern between the vaccine and the TLRs, the crystal structures of TLR2 and TLR4 were obtained from the PDB (ID-2Z7X and 3FXI, respectively). The ClusPro 2.0 server56 (https://cluspro.bu.edu/login.php) was employed separately to analyze the vaccine/TLR2 and vaccine/TLR4 molecular interactions. The server uses three different steps in protein–protein molecular docking including refinement of the structures by energy minimization, lowest energy structure clustering, and rigid body docking.56 The complex having the lowest weighted score was considered to be the best-docked complex and the molecular interaction was visualized using PyMOL.2.12. Molecular dynamic (MD) simulation for the extracellular domain of TLR2 and TLR4/vaccine
MD simulation is an indispensable tool to produce conformational changes in a time scale in which we can examine whether the vaccine–receptor system is stable.57 GROMACS v2018 was implemented to study the query vaccine with both TLR2 and TLR4. First, the vaccine/TLR complex systems were treated with the OPLSS-AA force field and set to the center of the cubic box. The three-point transferable intermolecular potential (TIP3P) solvent was selected for solvation of the vaccine–receptor system58 and neutrality was maintained for the entire system by adding sodium and chloride ions. The steepest descent algorithm was applied for minimizing the energy of the vaccine–receptor complexes. Next, NVT and NPT ensembles were employed for equilibrating the energy-minimized systems 100 ps, as described in our previous work.52 Final MD simulations were run for 50 ns for each of the vaccine/TLR complexes. To maintain the bond constraints and long-range electrostatic interactions, we utilized particle mesh Ewald (PME)59 and the linear constraint solver (LINC)60 algorithm during the employment of NVT, NPT, and MD simulation production. From MD trajectories, we extracted RMSD, RMSF, RG, and SASA using the GROMACS package. Furthermore, the strength of the interactions between vaccine and TLR proteins was assessed through the computation of hydrogen bonding via VMD. In the section that follows, it will provide a detailed description of running MD simulation for molecular features of constructed vaccine with extracellular domain (ECD), transmembrane (TM), and cytoplasmic domains (CD) of TLRs are crucial for innating the immune signaling pathway.2.13. Molecular dynamic (MD) simulation for full domains as dimers of TLR-2 and TLR-4/vaccine
To investigate independently the impact of binding vaccine on the stability of full length of homodimer TLR2 and heterodimer TLR4–MD2, we performed MD simulation. The full length of TLR2 and TLR4 were downloaded from AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/). For simplicity, TLR-2 and TLR-4 contain three main domains extracellular, transmembrane (TM), and cytoplasmic domains. Transmembrane (TM) domains for homodimer TLR-2 (590–610 aa) and heterodimer of MD-TLR-4 (630–650) were placed into a membrane of a single-component 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayer via using a system builder. For TLR2-vaccine complex was placed in the orthorhombic box (buffer of 10 Å SPC) of 4666776 Å3 containing 109394 SPC water molecules, 626 POPC molecules, and 20 Na+ ions, while TLR2/MDM2-vaccine complex was placed in the orthorhombic box (buffer of 10 Å SPC) of 8543937 Å3 containing 218682 SPC water molecules, 922 POPC molecules, and 16 Na+ ions via the aid of system builder tool from Desmond package.61 After then the systems were allowed to relax and equilibrate via implementing the Multisim method.62 The MD simulation calculations were run under the constant pressure of 1.01325 bar and a temperature of 300 K, thermostated and barostated according to the Martyna–Tobias–Klein barostat algorithm with a relaxation time of 2 ps with isotropic coupling style and Nosé–Hoover thermostat algorithm (with a relaxation time of 1 ps) over 50 ns with recording intervals of 1.2 ps for energy and 100 ps for recording interval.63,64 The M-shake algorithm was used to constrain the geometry of water molecules, and heavy atom bond lengths with hydrogen, electrostatic interaction applied using Particle Mesh Ewald (PME) method.65 The van der Waals (vDW) and electrostatic interactions were limited from to 10 Å and the long-term contributions of vDW to energy and pressure were estimated, with a homogeneous distribution of vDW spheres with a dispersion coefficient of 69.5 kcal mol−1 Å−1 was assumed. The optimized potentials for liquid simulations66 (OPLS-2005) forcefield were used. The simulation trajectories were analyzed and visualized by using the simulation interaction diagram tool. The data were plotted as root mean square deviation (RMSD) graphs. Further, the thickness for the POPC membrane was measured, initial frame (59.09 Å) and last frame of MD trajectory (58.93 Å) for POPC with TLR2/vaccine and POPC bilayer thickness is 53.04 Å for the first frame and 55.15 Å for the last frame of MD simulations.
2.14. Protein–protein interactions network analysis
We attempted to take advantage of the Cytoscape plugins (NetworkAnalyzer and RINalyzer) for analyzing the network of interacting residues, where amino acids as nodes and interactions (H-bonding, electrostatic, hydrophobic, and other) as edges. CytoHubba plugin was used to analyze and identify hub residues (the most correlated nodes) of the vaccine/TLR4 or TLR2 systems. The statistics of the generated network were evaluated based on the following parameters: (i) degree of nodes; (ii) betweenness centrality; (iii) eccentricity; and (iv) closeness centrality.672.15. Immune simulation
The immunogenicity of the vaccine structure was computed through the C-ImmSim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/).68 The server is driven by a position-specific scoring matrix (PSSM) and a machine learning approach for predicting the immune interaction and immune epitope. All simulations were carried out at default parameters with time set at 1, 84, 164 (with each time step set at 8 hours).69 In the simulations, 3 injections were administered at 4 weeks apart. In addition, 12 injections of the constructed vaccine were administered four weeks apart to stimulate repeated antigens seen in a typical endemic area. The Simpson index, D, a measure of diversity, was demonstrated from the plot.3. Results
3.1. Sequence retrieval, phylogenetic tree analysis, and evaluation of antigenic and physicochemical properties
To design an antigenic vaccine candidate, the sequences of seven spike glycoproteins were obtained from the PDB (6VSB, 6VXX, 6X2A, 6XR8, 6ZB4, 6ZGE, and 7KDH). The VaxiJen v2.0 server revealed that all sequences are antigenic, with all glycoproteins having antigenicity values between 0.45 and 0.46. Various physicochemical properties related to the S glycoproteins were analyzed using ProtParam, which demonstrated that all of the proteins contained between 1273 and 1370 amino acids and that their molecular weight range was 140.66–144.79 kDa (Table S1†), which indicates a good antigenic nature. The instability index of all proteins was calculated to be below 40, which designates that all sequences are stable; in addition, the negative Gravy values of all proteins imply that the sequences are hydrophilic. Sequences alignment of all spike glycoproteins was performed using the MUSCLE tool which revealed that the sequences have to conserve region although high ranges of point mutation occur in each sequence (Fig. S1†). In order to analyze the variation of the sequences specifically, the phylogenetic tree was constructed and the result displayed that seven S proteins assembled together into multiple clades which specify their significant variation (Fig. S2†). Therefore, a SARS-CoV-2 vaccine that was designed against these strains could potentially be effective against all of the strains.3.2. CTL epitope prediction
Cytotoxic T lymphocytes (CTL) have the ability to destroy cells infected with viruses and bacteria and can promote the development of a robust cellular immune response.70 During infection, whenever CTLs encounter the major histocompatibility complex-I-mounted antigen-specific to their receptor, they go into the cell cycle and undergo several mitotic divisions, followed by differentiation into effector cells.71 In the current work, we predicted immunogenic CTL epitopes using the NetCTL 1.2 server. A total of 231 epitopes 9-mer in length predicted from seven S glycoproteins (Table S2†). Furthermore, the predicted sequences were tested using various immunological filters, including immunogenicity, antigenicity, and hypo allergenicity; finally, 14 CTL immunogenic candidates were selected (Table 1 and Fig. S3†).| PDB ID | Epitopes | Position | Supertype (combined score) | MHC class 1 allele | Binding score | Immunogenicity | Antigenicity |
|---|---|---|---|---|---|---|---|
| 6VSB | WTAGAAAYY | 258 | A1 (3.1128) | HLA-A*01:01 | 0.165 | 0.152 | 0.6306 ± 0.1123 |
| A26 (2.0048) | HLA-B*35:01 | 1.2 | |||||
| B58 (1.2345) | HLA-A*26:01 | 0.11 | |||||
| B62 (1.3574) | HLA-A*30:02 | 0.115 | |||||
| HLA-A*68:01 | 1.185 | ||||||
| HLA-B*15:01 | 1.6 | ||||||
| FTISVTTEI | 718 | A2 (1.1808) | HLA-B*58:01 | 0.4 | 0.044 | 0.8535 ± 0.0479 | |
| A26 (1.0014) | HLA-B*51:01 | 0.7 | |||||
| B58 (0.9262) | HLA-A*02:01 | 0.8 | |||||
| HLA-A*68:02 | 0.2 | ||||||
| HLA-A*26:01 | 0.615 | ||||||
| 6VXX | RLDPPEAEV | 1002 | A2 (0.9436) | HLA-A*02:01 | 0.79 | 0.171 | 0.4496 ± 0.0859 |
| HLA-A*02:11 | 0.395 | ||||||
| HLA-A*02:16 | 0.405 | ||||||
| HLA-A*02:12 | 0.45 | ||||||
| HLA-A*02:19 | 0.45 | ||||||
| VVFLHVTYV | 1079 | A2 (1.0304) | HLA-A*02:01 | 1.2 | 0.127 | 1.5122 ± 0.1999 | |
| HLA-A*02:03 | 0.455 | ||||||
| HLA-A*02:06 | 0.575 | ||||||
| HLA-A*68:02 | 0.6 | ||||||
| HLA-A*69:01 | 0.955 | ||||||
| HLA-A*02:19 | 1.07 | ||||||
| HLA-A*02:11 | 1.19 | ||||||
| HLA-A*02:02 | 1.525 | ||||||
| HLA-A*68:23 | 1.245 | ||||||
| LLMGCVAET | 22 | A2 (1.0322) | HLA-A*02:01 | 0.7 | 0.054 | 0.4298 ± 0.1640 | |
| HLA-A*02:19 | 0.23 | ||||||
| HLA-A*02:03 | 0.585 | ||||||
| HLA-A*02:02 | 0.875 | ||||||
| HLA-A*02:12 | 1.315 | ||||||
| HLA-A*02:11 | 1.975 | ||||||
| HLA-A*02:16 | 1.83 | ||||||
| 6X2A | PYRVVVLSF | 492 | A24 (1.8786) | HLA-A*24:02 | 0.33 | 0.031 | 1.0281 ± 0.0505 |
| HLA-A*23:01 | 0.2 | ||||||
| HLA-A*24:03 | 0.615 | ||||||
| HLA-B*08:03 | 1.895 | ||||||
| IAIPTNFTI | 697 | A24 (1.0788) | HLA-B*51:01 | 0.2 | 0.185 | 0.7052 ± 0.1105 | |
| B58 (1.5865) | HLA-B*53:01 | 0.5 | |||||
| HLA-B*15:17 | 0.72 | ||||||
| HLA-B*57:01 | 0.865 | ||||||
| HLA-A*69:01 | 1.27 | ||||||
| HLA-B*58:01 | 0.3 | ||||||
| HLA-A*24:02 | 1.28 | ||||||
| 6XR8 | FVFLVLLPL | 2 | A26 (1.0076) | HLA-A*02:01 | 0.5 | 0.040 | 0.8601 ± 0.0927 |
| A2 (1.1947) | HLA-B*46:01 | 0.22 | |||||
| B8 (0.8252) | HLA-A*02:06 | 0.36 | |||||
| B62 (0.9849) | HLA-A*25:01 | 0.41 | |||||
| HLA-A*68:02 | 0.6 | ||||||
| HLA-A*02:02 | 0.945 | ||||||
| HLA-A*69:01 | 0.745 | ||||||
| HLA-B*35:03 | 0.565 | ||||||
| HLA-B*39:01 | 0.49 | ||||||
| WTFGAGAAL | 886 | A26 (0.9302) | HLA-B*07:02 | 1.5 | 0.197 | 0.4918 ± 0.1134 | |
| B62 (1.1379) | HLA-B*48:01 | 0.22 | |||||
| HLA-B*39:01 | 0.315 | ||||||
| HLA-B*15:17 | 0.225 | ||||||
| HLA-B*15:02 | 0.285 | ||||||
| HLA-A*68:23 | 0.125 | ||||||
| HLA-B*38:01 | 0.765 | ||||||
| HLA-A*68:02 | 0.7 | ||||||
| 6ZB4 | IPTNFTISV | 714 | B7 (1.5427) | HLA-B*07:02 | 0.7 | 0.172 | 0.8820 ± 0.0649 |
| HLA-B*51:01 | 0.8 | ||||||
| HLA-B*42:01 | 1.535 | ||||||
| HLA-B*83:01 | 1.57 | ||||||
| HLA-B*54:01 | 1.3 | ||||||
| 6ZGE | LPFNDGVYF | 115 | B7 (1.0427) | HLA-B*35:01 | 0.5 | 0.117 | 0.5593 ± 0.0501 |
| HLA-B*53:01 | 0.5 | ||||||
| HLA-B*15:03 | 1.2 | ||||||
| HLA-B*35:03 | 1.81 | ||||||
| HLA-B*42:01 | 1.07 | ||||||
| HLA-B*83:01 | 0.885 | ||||||
| HLA-B*51:01 | 0.9 | ||||||
| 7KDH | HVSGTNGTK | 69 | A3 (1.1575) | HLA-A*03:01 | 1.15 | 0.063 | 1.0956 ± 0.1256 |
| HLA-A*68:01 | 0.905 | ||||||
| HLA-A*11:01 | 1.6 | ||||||
| HLA-A*66:01 | 1.5 | ||||||
| HLA-A*26:03 | 1.265 | ||||||
| TLADAGFIK | 827 | A3 (1.2451) | HLA-A*03:01 | 0.62 | 0.281 | 0.5781 ± 0.0314 | |
| HLA-A*11:01 | 0.44 | ||||||
| HLA-A*68:01 | 0.86 | ||||||
| GVYFASTEK | 89 | A3 (1.4615) | HLA-A*03:01 | 0.19 | 0.090 | 0.7112 ± 0.1176 | |
| HLA-A*66:01 | 0.22 | ||||||
| HLA-A*11:01 | 0.23 | ||||||
| HLA-A*30:01 | 1.1 | ||||||
| HLA-A*68:01 | 1.135 |
3.3. HTL epitope prediction
Helper T-lymphocytes (HTLs) play an essential function in each innate and adaptive immunity.72,73 Hence, a suitable vaccine candidate should contain a receptor-specific HTL epitope. The sequences of all spike glycoproteins were submitted to the NetMHC II pan 3.2 server, and 42 HTL epitopes 15-mer in length were obtained (Table S3†). Top-notch immunogenic epitopes are indicated by a lower percentile rank and IC50 value.74 Based on a cutoff value of below 2 for the lower percentile value, only 11 HTL epitopes were chosen (Table 2). In addition, all selected epitopes displayed an IFN-γ-inducing capacity, as determined by their positive score on the IFN epitope server output32,74 (Table S4†).| PDB ID | Epitopes | Position | Allele | Binding score | Antigenicity |
|---|---|---|---|---|---|
| 6VSB | FQTLLALHRSYLTPG | 238 | DRB1*08:01 | 1.40 | 0.5789 ± 0.0641 |
| DRB1*10:01 | 0.70 | ||||
| DRB1*11:01 | 1.40 | ||||
| DRB1*12:01 | 1.40 | ||||
| DRB1*13:01 | 1.80 | ||||
| DRB1*14:01 | 1.50 | ||||
| DRB1*15:01 | 0.40 | ||||
| QSIIAYTMSLGAENS | 690 | DRB1*01:01 | 1.0 | 0.5728 ± 0.0544 | |
| DRB1*04:01 | 1.0 | ||||
| DRB1*10:01 | 0.30 | ||||
| 6VXX | SIIAYTMSLGAENSV | 710 | DRB1*01:01 | 0.50 | 0.5691 ± 0.0600 |
| DRB1*04:01 | 0.70 | ||||
| DRB1*07:01 | 1.60 | ||||
| DRB1*10:01 | 0.12 | ||||
| IAYTMSLGAENSVAY | 742 | DRB1*01:01 | 0.70 | 0.7072 ± 0.0583 | |
| DRB1*04:01 | 1.30 | ||||
| DRB1*10:01 | 0.17 | ||||
| 6X2A | NFRVQPTESIVRFPN | 302 | DRB1*04:01 | 0.90 | 0.430 ± 0.0211 |
| DRB1*01:01 | 1.90 | ||||
| 6XR8 | IIAYTMSLGAENSVA | 692 | DRB1*01:01 | 0.40 | 0.5426 ± 0.0750 |
| DRB1*04:01 | 0.6 | ||||
| DRB1*07:01 | 2 | ||||
| DRB1*10:01 | 0.80 | ||||
| 6ZB4 | VVLSFELLHAPATVC | 511 | DRB1*01:01 | 0.40 | 0.8618 ± 0.0625 |
| DRB1*10:01 | 0.50 | ||||
| QIPFAMQMAYRFNGI | 895 | DRB1*01:01 | 1.0 | 0.9573 ± 0.1415 | |
| DRB1*10:01 | 0.90 | ||||
| DRB1*14:01 | 0.80 | ||||
| DRB1*15:01 | 1.15 | ||||
| 6ZGE | SQSIIAYTMSLGAEN | 720 | DRB1*10:01 | 1.10 | 0.6141 ± 0.1099 |
| DRB1*07:01 | 2 | ||||
| 7KDH | IRAAEIRASANLAAT | 1013 | DRB1*04:01 | 1.40 | 0.6785 ± 0.0122 |
| DRB1*08:01 | 1.20 | ||||
| DRB1*13:02 | 1.90 | ||||
| RAAEIRASANLAATK | 1031 | DRB1*04:01 | 0.70 | 0.5709 ± 0.0715 | |
| DRB1*08:01 | 0.90 | ||||
| DRB1*13:02 | 1.30 |
3.4. Vaccine construction and structural evaluation
The principles applied for epitopes when designing the final linear vaccine sequence were: (a) must be antigenic, immunogenic, non-allergic, and non-toxic; (b) should have a high binding affinity with multiple MHC alleles; (c) must contain overlapping CTL and HTL epitopes; (d) 100% conserved across S proteins; (e) should be promiscuous; (f) population coverage; and (g) in order to reduce autoimmunity, should not have the capacity to bind with any protein in the human proteome. Based on these properties, a linear vaccine sequence comprising 14 CTL and 11 HTL epitopes was constructed (Tables 1, 2, and S4–S6†). In order to prevent junctional epitope formation, AAY and GPGPG linkers joined the CTL and HTL epitopes, respectively. The cholera toxin B adjuvant is linked to the N-terminus of the vaccine construct by an EAAAK linker (Fig. 2). The sequence of the finished vaccine consists of 494 amino acids and has a molecular weight of 51.51 kDa (ESI SM1†). Epitope sequences included in the vaccine were pictured on the 3-D structure of their respective spike glycoproteins, which revealed that the epitopes retain their original position (Fig. 3).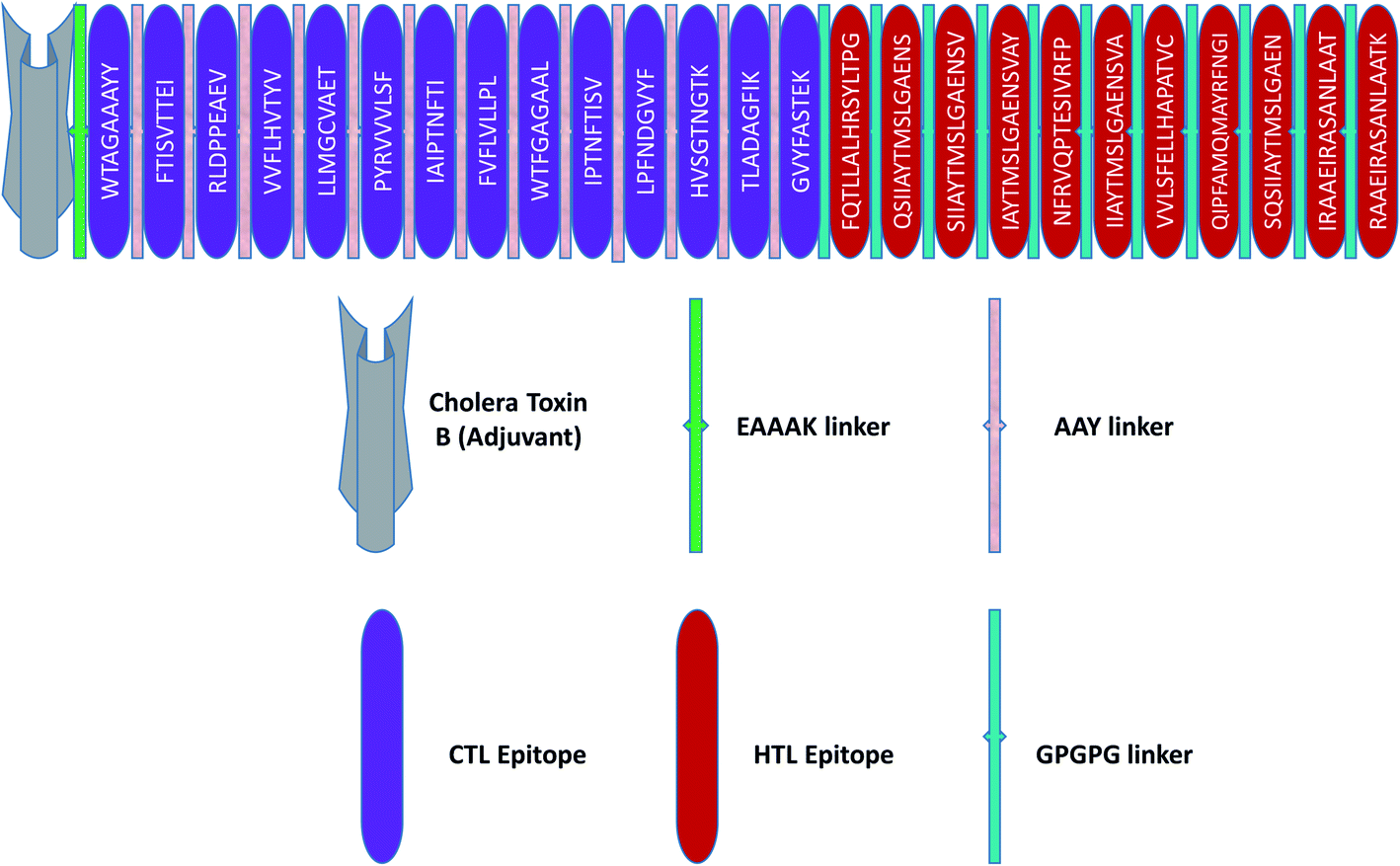 | ||
| Fig. 2 Schematic illustration of the final vaccine design. The HTL and CTL epitopes are shown in red and violet boxes, respectively. A vaccine adjuvant (light grey) is added to the N-terminus of the sequence by an EAAAK linker (green). The CTL and HTL epitopes are joined by AAY (white-gray) and GPGPG (green) linkers, respectively. | ||
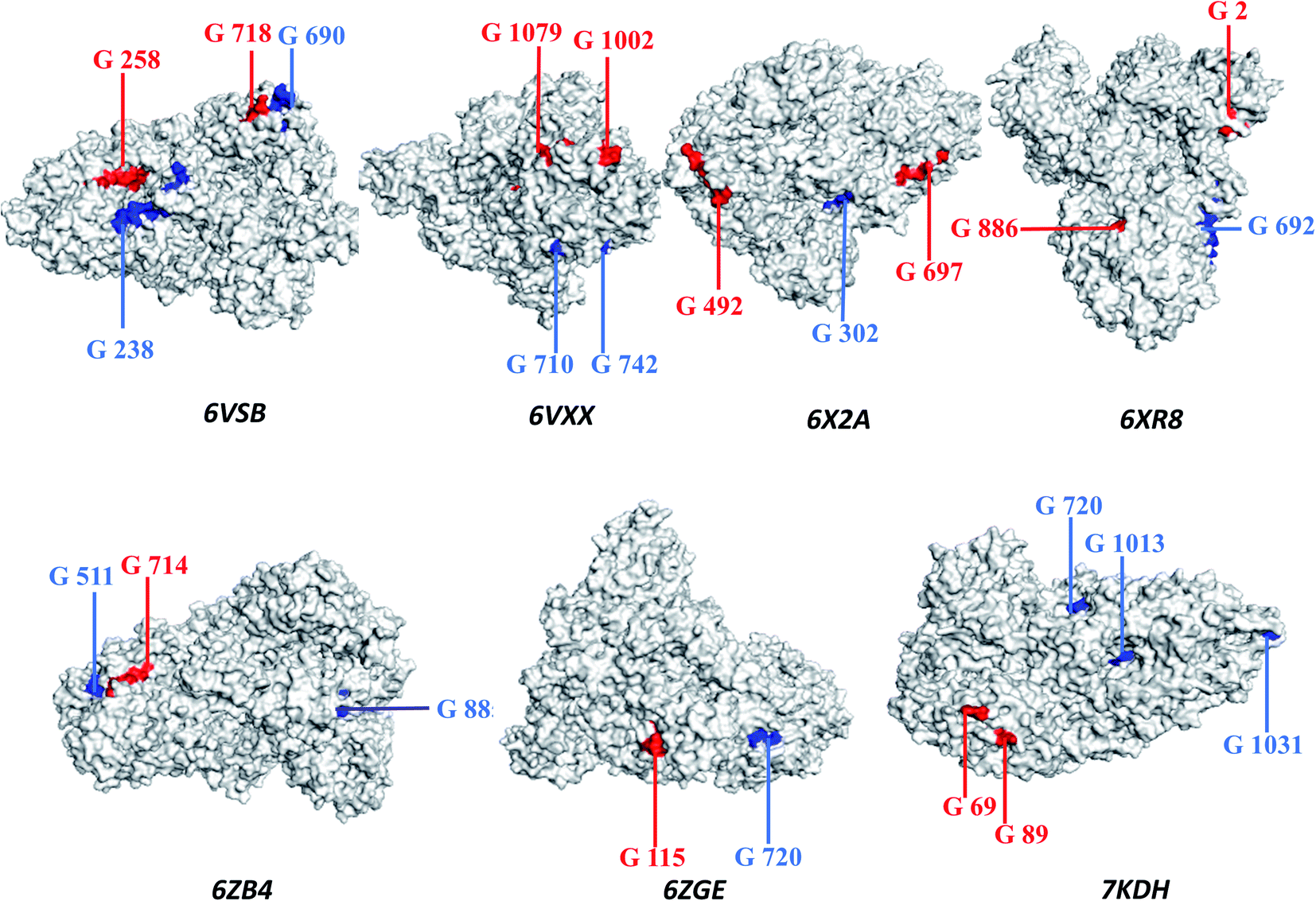 | ||
| Fig. 3 The three-dimensional structure of the spike glycoprotein (Protein Data Bank structure) with cytotoxic T-lymphocyte epitopes marked in red and helper T-lymphocyte epitopes marked in blue. | ||
The predicted secondary structure of the vaccine shows that it contains 5.87% beta turns, 36.3% alpha-helical regions, 32.59% random coils, and 25.51% extended strands (Fig. 4A). The 3D structure of the vaccine was modeled via exploitation of the I-TASSER server, which generated the structures of five vaccines based on 10 threading templates, of which 1ltrA, 4m00A, 3chbD, 5iv5O, and 416t were top-level. The top 10 templates displayed appropriate alignment, as indicated by their Z-scores ranging from 1.74 to 3.03. The C-score values of the five generated models are between −2.97 and −0.30. In the I-TASSER server, model 1 (vaccine structure) showed the highest C-score values and also has a projected TM-score and RMSD of 0.67 ± 0.12 and 8.0 ± 4.4 Å, respectively. The TM-score determines the comparison between two structures.75 A TM score of more than 0.5 means the predicted structure has the correct topology, and a score of less than 0.17 indicates random similarity. Vaccine structure refinement was carried out initially using ModRefiner, then using the GalaxyRefine web server. GalaxyRefine predicted five models of the vaccine structure, with model 1 selected (Fig. 4B and Table S7†) due to its being the best model on the basis of RMSD (0.434), GDT-HA (0.9393), MolProbity (2.537f), and Rama favored (90.7). To evaluate the quality, the structure was subjected to ProSA-web, and its predicted Z-score of −3.08 (Fig. 4D) lay within the score range for the comparable size of the native protein.49 The ERRAT value of the finalized vaccine structure was 79.360 (Fig. 4C), indicating that the percentage of the protein remains below the rejection limit of 95%;50 notably, an ERRAT value higher than 50 suggests a good-quality model.76 Analysis of Ramachandran plot of the finalized vaccine model showed that 86.1% favored, 11.4% additional allowed, 1% allowed, and 1.5% disallowed regions of residues (Fig. 4E and ESI SM2†).
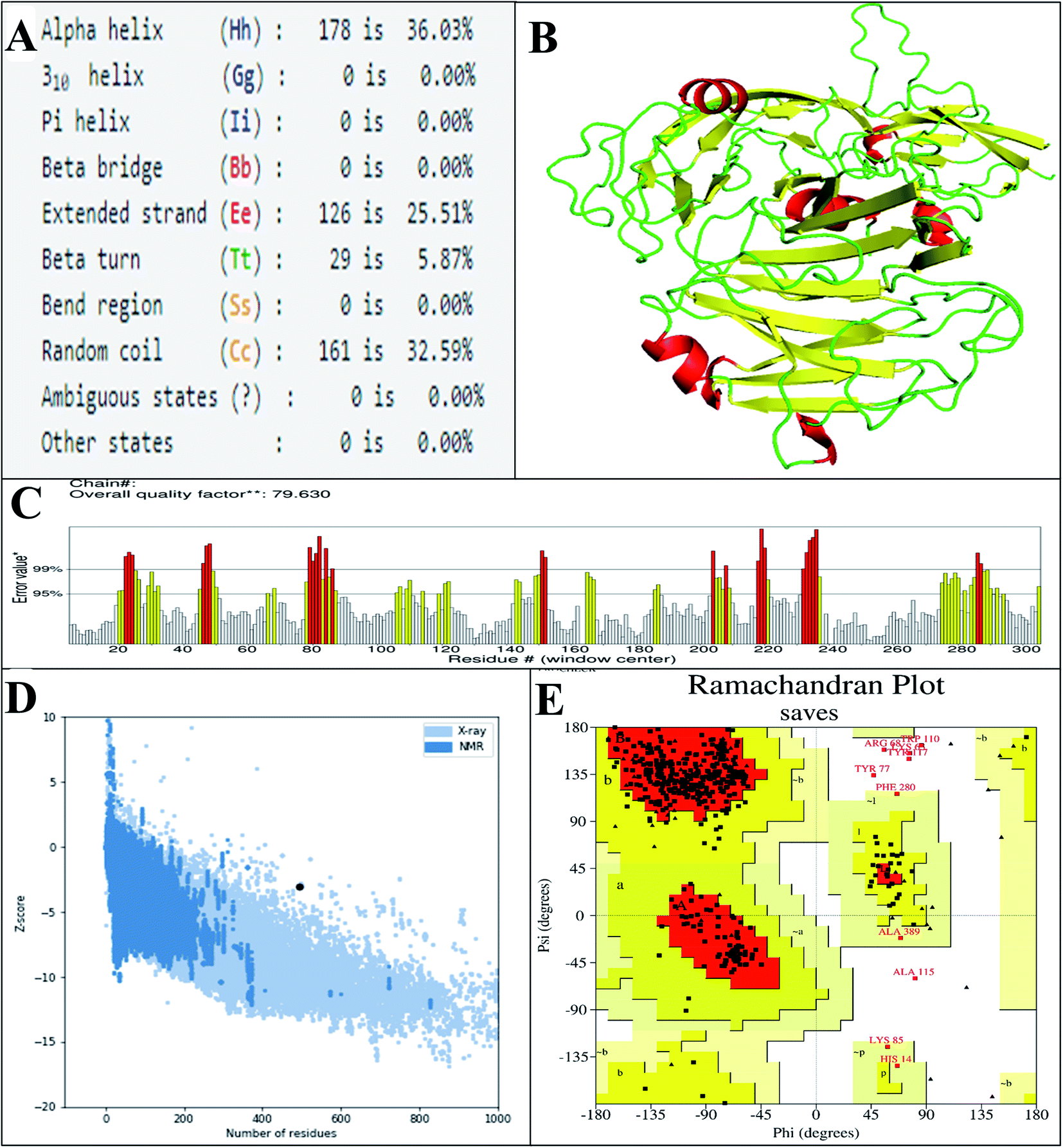 | ||
| Fig. 4 (A) The secondary structural properties of the designed vaccine. (B) 3D model of the vaccine. In this model, the green, yellow, and red sections indicate the loop, sheet, and helix areas, respectively. (C) Evaluation of structure by ERRAT, with a score of 79630. (D) Validation of the 3D structure with a Z-score of −3.08, followed by ProSA. (E) Analysis of the Ramachandran plot using the PROCHECK server shows that 86.1%, 11.4%, 1%, and 1.5% of the residues in preferred regions are additionally allowed, allowed, or not allowed. | ||
3.5. Antigenic, immunogenic, and physicochemical assessment of the vaccine
Immunogenicity is the ability to stimulate adaptive and cell-mediated immune responses, while antigenicity is the capacity to recognize a specific antigenic molecule.77 Hence, a suitable vaccine candidate must have both immunogenic and antigenic characteristics. The immunogenicity score of the final vaccine was found to be 6.23397 by the IDEB class I immunogenicity tool. A higher score indicates a greater ability to stimulate the immune response. The developed multiepitope vaccine has antigenic properties as the candidate's antigenicity score was estimated at 0.5183 by the VaxiJen v2.0 server (a score greater than 0.4 is considered antigenic) and 0.669922 by ANTIGENpro. AllergenFP v.1.0 and AllerTOP v.2.0 servers ensure that the vaccine candidate is not allergic to the human body (Fig. S4†).It is essential to predict several physicochemical characteristics of the vaccine to evaluate its efficacy and safety.78 Several chemical and physical properties have been predicted to be involved in the composition of the ExPASy vaccine, as presented in ESI SM3.† The calculated pI of the vaccine is 6.46 and the vaccine fat index is predicted to be 84.35. This implies that the vaccine would be thermostable in nature, as the higher aliphatic index indicated greater thermostability.24 The predicted half-life of the vaccine is 20 hours in yeast and 30 hours in mammalian reticulocytes, and more than 10 hours in the morning in E. coli. We estimated the instability index of the candidate vaccine at 21.64, which reflects the stable character of the designed vaccine, as an instability index less than 40 (ref. 24) indicates that a protein is stable. In addition, analysis on the Signal P4.1 server suggests that the modeled vaccine candidate does not contain any signal peptide, which indicates prevention of protein localization (Fig. S5†).
3.6. Prediction of B-cell epitope
B cells play an important role in humoral immunity. Epithelial cells can interact with B-cell receptors (BCRs) to generate antibodies and provide long-term immunity.79 To determine if these types of epitopes are present in the structure of the vaccine, the ElliPro tool was applied with the default parameters, and the result indicated 15 linear and 6 discontinuous B-cell epitopes (Tables 3 and 4). The visualization of these epitopes was accomplished with PyMOL (Fig. S6 and S7†).| No. | Start | End | Peptide | No. of residues | Score |
|---|---|---|---|---|---|
| 1 | 398 | 423 | PGVVLSFELLHAPATVCGPGPGQIPF | 26 | 0.817 |
| 2 | 425 | 494 | MQMAYRFNGIGPGPGSQSIIAYTMSLGAENGPGPGIRAAEIRASANLAATGPGPGRAAEIRASANLAATK | 70 | 0.78 |
| 3 | 202 | 208 | LAAYWTF | 7 | 0.776 |
| 4 | 54 | 68 | PGSQHIDSQKKAIER | 15 | 0.762 |
| 5 | 265 | 288 | YGVYFASTEKGPGPGFQTLLALHR | 24 | 0.753 |
| 6 | 134 | 157 | RLDPPEAEVAAYVVFLHVTYVAAY | 24 | 0.745 |
| 7 | 31 | 38 | SLAGKREM | 8 | 0.722 |
| 8 | 102 | 120 | MANEAAAKWTAGAAAYYAA | 19 | 0.705 |
| 9 | 173 | 184 | VVVLSFAAYIAI | 12 | 0.632 |
| 10 | 15 | 19 | NTQIH | 5 | 0.626 |
| 11 | 246 | 258 | TNGTKAAYTLADA | 13 | 0.581 |
| 12 | 164 | 168 | AETAA | 5 | 0.577 |
| 13 | 294 | 301 | GGPGPGQS | 8 | 0.577 |
| 14 | 391 | 395 | NSVAG | 5 | 0.55 |
| 15 | 361 | 380 | FRVQPTESIVRFPNGPGPGI | 20 | 0.501 |
| No. | Epitopes | No. of residues | Score |
|---|---|---|---|
| 1 | A:G388, A:G397, A:P398, A:G399, A:V400, A:V401, A:L402, A:S403, A:F404, A:E405, A:L406, A:L407, A:H408, A:A409, A:P410, A:A411, A:T412, A:V413, A:C414, A:G415, A:P416, A:G417, A:P418, A:G419, A:Q420, A:I421, A:P422, A:M427, A:A428, A:Y429, A:R430, A:F431, A:N432, A:G433, A:I434, A:G435, A:P436, A:G437, A:P438, A:G439, A:S440, A:Q441, A:S442, A:I443, A:I444, A:A445, A:Y446, A:T447, A:M448, A:S449, A:L450, A:G451, A:A452, A:E453, A:N454, A:G455, A:P456, A:G457, A:P458, A:G459, A:I460, A:R461, A:A462, A:A463, A:E464, A:I465, A:R466, A:S468, A:A469, A:N470, A:L471, A:A472, A:A473, A:T474, A:G475, A:P476, A:G477, A:P478, A:G479, A:R480, A:A481, A:A482, A:E483, A:I484, A:R485, A:A486, A:S487, A:A488, A:N489, A:L490, A:A491, A:A492, A:T493, A:K494 | 94 | 0.798 |
| 2 | A:S31, A:L32, A:A33, A:G34, A:K35, A:R36, A:E37, A:M38, A:E52, A:V53, A:P54, A:G55, A:S56, A:Q57, A:H58, A:I59, A:D60, A:S61, A:Q62, A:K63, A:K64, A:A65, A:I66, A:E67, A:R68, A:E84, A:K85, A:M102, A:A103, A:N104, A:E105, A:A106, A:A107, A:A108, A:K109, A:W110, A:T111, A:A112, A:G113, A:A114, A:A115, A:A116, A:Y117, A:Y118, A:A119, A:A120, A:R134, A:L135, A:D136, A:P137, A:P138, A:E139, A:A140, A:E141, A:V142, A:A143, A:A144, A:Y145, A:V146, A:V147, A:F148, A:L149, A:H150, A:V151, A:T152, A:Y153, A:V154, A:A155, A:A156, A:Y157, A:L158, A:G161, A:C162, A:V163, A:A164, A:E165, A:T166, A:A167, A:A168, A:V173, A:V174, A:V175, A:L176, A:S177, A:F178, A:A179, A:A180, A:Y181, A:I182, A:A183, A:I184, A:N187, A:L202, A:A203, A:A204, A:Y205, A:W206, A:T207, A:F208 | 99 | 0.687 |
| 3 | A:N15, A:T16, A:Q17, A:I18, A:T20 | 5 | 0.626 |
| 4 | A:G245, A:T246, A:N247, A:G248, A:T249, A:K250, A:A251, A:A252, A:Y253, A:T254, A:L255, A:A256, A:D257, A:A258, A:I261, A:K262, A:Q281, A:T282, A:L283, A:L284, A:A285, A:L286, A:H287, A:R288 | ||
| 5 | A:L230, A:P231, A:F232, A:N233, A:A264, A:Y265, A:G266, A:V267, A:Y268, A:F269, A:A270, A:S271, A:T272, A:E273, A:K274, A:G275, A:P276, A:G277, A:P278, A:G279, A:F280, A:S289, A:L291, A:G294, A:G295, A:P296, A:G297, A:P298, A:G299, A:Q300, A:S301, A:I302, A:S333, A:V334, A:G335, A:P336, A:G337, A:P338, A:G339, A:T366, A:S368, A:V370, A:F372, A:P373, A:N374, A:G375, A:P376, A:G377, A:P378, A:G379, A:I380 | 24 | 0.6 |
| 6 | A:N391, A:S392, A:V393, A:A394, A:G395 | 51 | 0.597 |
3.7. Population coverage
When developing a vaccine, it is necessary to assess the distribution of human leukocyte antigen alleles in the world population.80 Population coverage of the selected epitopes included in the study was assessed by the Infectious Disease Epidemiology Bureau (IEDB) population coverage tool, which revealed that the epitopes cover 97.31% of the human population (Table 5). The epitopes also displayed 99.09%, 93.24%, 97.66%, 95.45%, and 87.27% coverage in Europe, Oceania, North America, Southeast Asia, and Central Africa, respectively. This result indicates that the vaccine against SARS-CoV-2 would be globally useful.| Population/area | Class combined | ||
|---|---|---|---|
| Coveragea | Average_hitb | pc90c | |
| a Coverage of population on projected.b Population recognized by HLA combinations/epitope hits on the average number.c 90% of the population recognized by HLA combinations/epitope hits on the minimum number. | |||
| Central Africa | 87.27% | 3.96 | 0.79 |
| East Africa | 90.77% | 4.45 | 1.1 |
| East Asia | 97.5% | 5.41 | 2.46 |
| Europe | 99.09% | 6.63 | 3.44 |
| North Africa | 92.35% | 4.9 | 1.35 |
| North America | 97.66% | 6.21 | 2.8 |
| Northeast Asia | 95.41% | 4.63 | 2.02 |
| Oceania | 93.24% | 3.84 | 2.06 |
| South Africa | 91.69% | 4.29 | 1.23 |
| South America | 92.92% | 4.57 | 1.77 |
| South Asia | 94.2% | 4.85 | 2.01 |
| Southeast Asia | 95.45% | 4.61 | 2.14 |
| Southwest Asia | 90.14% | 4.76 | 1.03 |
| West Africa | 91.69% | 4.83 | 1.27 |
| West Indies | 96.58% | 5.87 | 2.43 |
| World | 97.31% | 5.84 | 2.65 |
| Average | 88.96 | 4.69 | 1.8 |
| Standard deviation | 20.2 | 1.36 | 0.81 |
3.8. Molecular docking
Strong interaction between a vaccine and immune cell receptors is an important feature for producing a stable immune response. To analyze such interaction patterns, molecular docking was implemented to analyze the binding affinity pattern of the vaccine with toll-like receptors (TLRs), which can recognize pathogens and play an important role in the cellular immune response.81 TLR4 and TLR2 are involved in the interaction of viral structural proteins leading to the production of inflammatory cytokines.82 Hence, molecular docking of the vaccine with TLR4 and TLR2 was performed using the ClusPro 2.0 online tool, and 30 docked complexes were obtained for each vaccine/TLR4 and vaccine/TLR-2. We selected the top-ranked docked model of vaccine-TLRs from the generated complexes which have the lowest score. In the case of molecular docking of TLR-2 and vaccine, cluster 7 was found to be the best, as it displayed the lowest energy value of −1055.1 (Fig. 5 and Table S8†). Likewise, in the docking study of vaccine with TLR-4, cluster 5 had the lowest energy value (−1010.5) among the complexes (Fig. 6 and Table S9†). Additionally, the selected HTL and CTL epitopes docked individually with human leukocyte antigen alleles HLA-DRB1*01:01 and, HLA-A*02:01 respectively, using the PatchDock server and the best complexes were screened out on the FireDock server. Each docked epitope showed different global energy values with their respective HLA alleles, with value thresholds of global energy −15 or below indicating effective interaction between the epitopes with their corresponding HLA alleles (Table 6).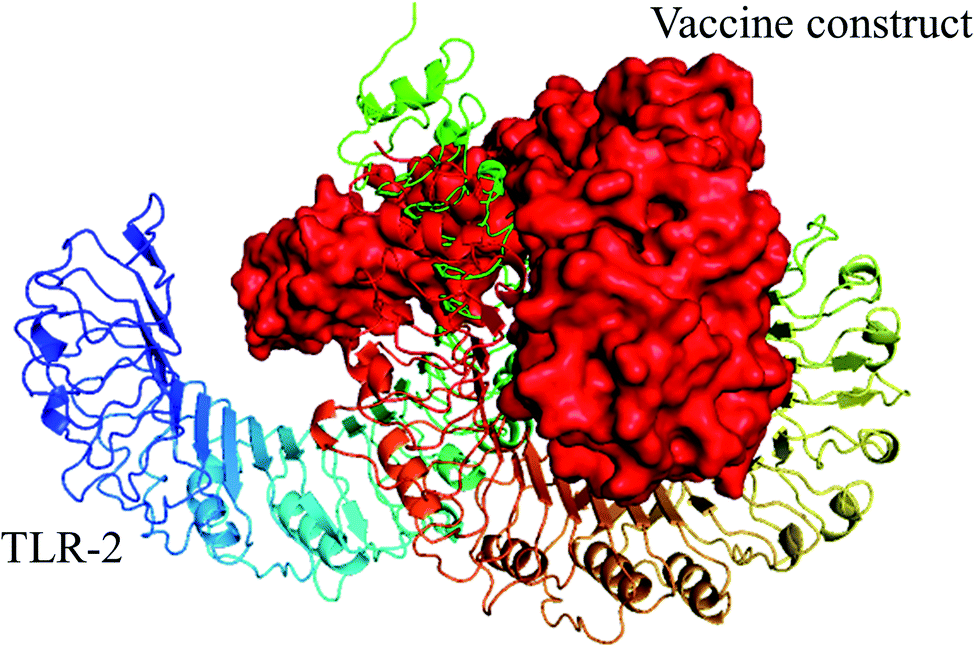 | ||
| Fig. 5 Selected best-docked complex (vaccine/TLR2). The energy value of the complex was predicted to be −1055.1 by the ClusPro web tool. The vaccine construct is shown in red color in the figure. | ||
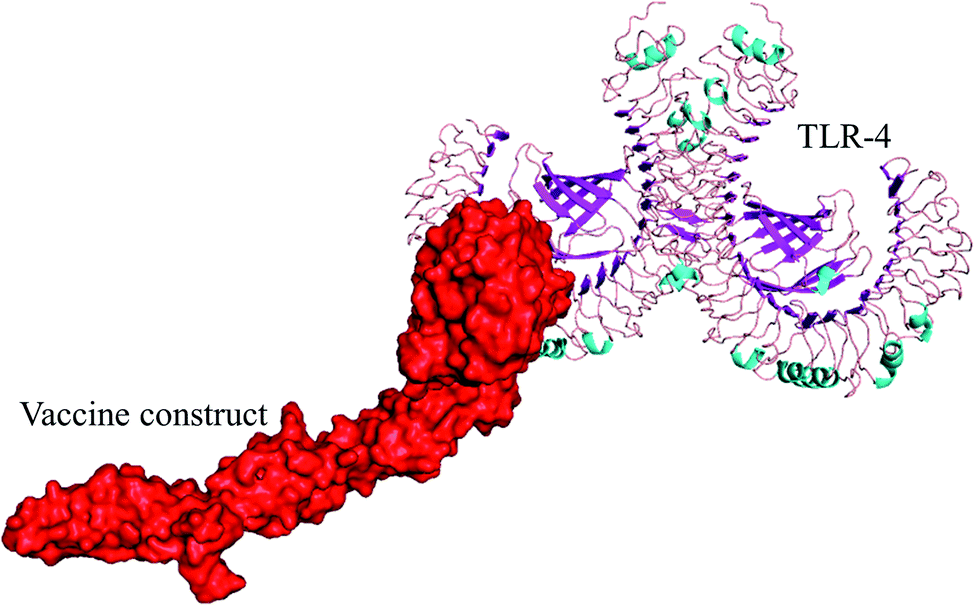 | ||
| Fig. 6 Selected best-docked complex (vaccine/TLR4). The energy value of the complex was found to be −1010.5 by the ClusPro web tool. The vaccine model is shown in red color in the figure. | ||
| CTL epitope | Global energy (HLA-A*02:01) | HTL epitope | Global energy (HLA-DRB1*01:01) |
|---|---|---|---|
| FTISVTTEI | −41.23 | FQTLLALHRSYLTPG | −90.83 |
| FVFLVLLPL | −59.54 | IAYTMSLGAENSVAY | −63.71 |
| GVYFASTEK | −35.36 | IIAYTMSLGAENSVA | −52.15 |
| HVSGTNGTK | −32.85 | IRAAEIRASANLAAT | −33.91 |
| IAIPTNFTI | −35.30 | NFRVQPTESIVRFPN | −16.75 |
| IPTNFTISV | −41.02 | QIPFAMQMAYRFNGI | −48.46 |
| LLMGCVAET | −29.99 | QSIIAYTMSLGAENS | −36.52 |
| LPFNDGVYF | −42.72 | RAAEIRASANLAATK | −33.76 |
| PYRVVVLSF | −37.19 | SIIAYTMSLGAENSV | −54.36 |
| RLDPPEAEV | −17.14 | SQSIIAYTMSLGAEN | −51.83 |
| TLADAGFIK | −33.87 | VVLSFELLHAPATVC | −63.09 |
| VVFLHVTYV | −38.62 | ||
| WTAGAAAYY | −43.64 | ||
| WTFGAGAAL | −38.01 |
3.9. Molecular dynamic simulation
Before studying the structural behaviour of membrane-bound heterodimer TLR4–MD2/vaccine and homodimerTLR2/vaccine systems, we checked the stability of vaccine with the extracellular subunit of TLR2 and TLR4–MD2 as a function of simulation time of 50 ns. Molecular dynamics trajectories of the vaccine-TLR2 and vaccine-TLR4 complexes were used to test for variations in root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and hydrogen bonding. The RMSD plot for the vaccine–TLR-2 and vaccine–TLR-4 systems are presented in Fig. 7, which demonstrates that the RMSD fluctuation of vaccine–TLR-4 is stable and less than 0.9 nm. In contrast, the RMSD value of vaccine–TLR2 showed high fluctuations after 12 ns (Fig. 7), and the RMSD fluctuation reached 2.35 nm. These findings revealed that the docked vaccine has a stabilizing impact on TLR-2 and TLR-4.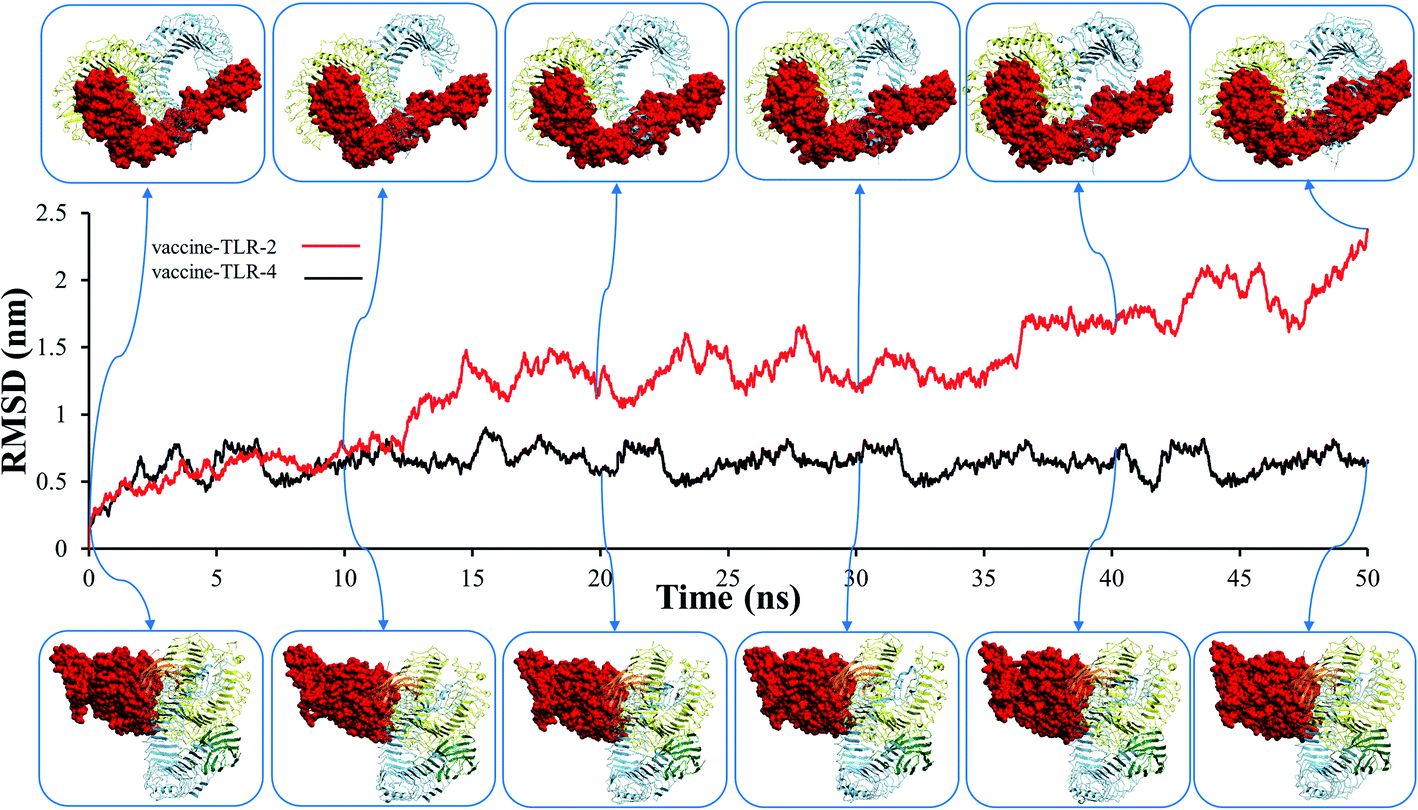 | ||
| Fig. 7 The root mean square deviation (RMSD) plots of vaccine–toll-like immune receptor complex systems. | ||
The RMSF values provide more details about which region in the studied proteins are responsible for causing the RMSD values to fluctuate more in the vaccine/TLR2 system than in the vaccine/TLR4 system. The RMSF for the TLR4 backbone and vaccine backbone is less fluctuated (Fig. 8B). In contrast, the RMSF for the TLR2 backbone revealed that chain B of TLR2 is more dynamic and highly fluctuated, while chain A is conserved (Fig. 8A). Further, the RMSF values for the vaccine backbone are more dynamic when the vaccine is bound to TLR2, and the vaccine is more conserved when it is bound to TL4 (Fig. 8C). The obtained RMSF values of the vaccine (Fig. S8A†) lead to making the TLR2 chain B more dynamic (Fig. S8B†). In addition, computation of the SASA is used to assess the alterations in the accessibility of TLR2 and TLR4 to solvent due to binding to the vaccine. The average SASA value of the vaccine molecule showed the stability and compactness of its structure (Fig. 8E). The radius of gyration, which is a key parameter indicating compactness of structure, also exhibited that the vaccine molecule gained a stable and compact form during MD simulation (Fig. 8D). The stable interactions were assessed through estimation of the hydrogen bonding of the vaccine–TLR-2 and vaccine–TLR-4 systems over 50 ns. The number of hydrogen bonds in the vaccine–TLR-2 complex decreased with time (Fig. 8F), while the hydrogen bonds in the vaccine–TLR-4 were stable over the 50 ns.
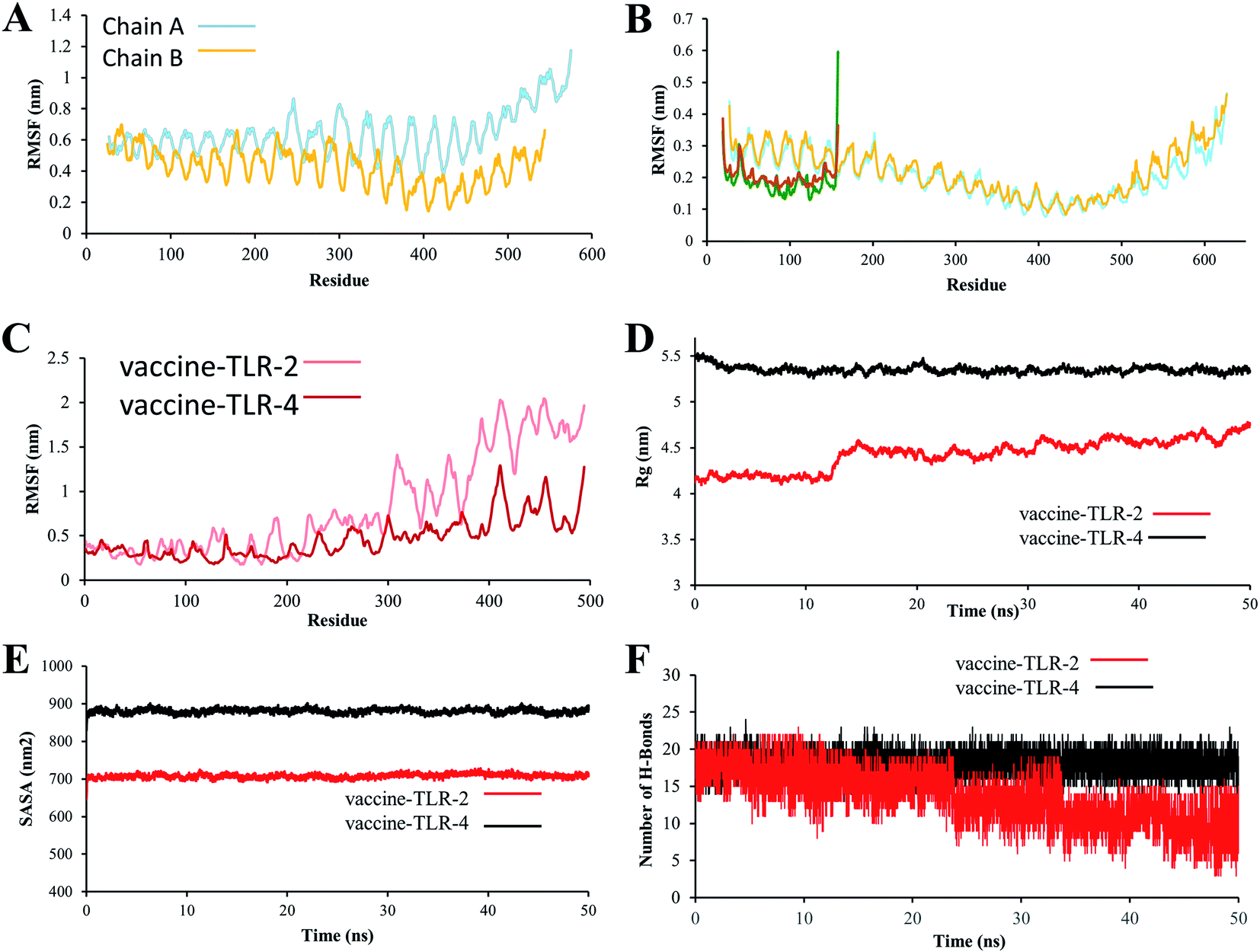 | ||
| Fig. 8 (A) RMSF plot of the TLR2 backbone. (B) RMSF plot of TLR-4 backbone. (C) RMSF plot of vaccine–TLR backbone. (D) Radius of gyration (Rg) plot, (E) Solvent-accessible surface area (SASA). (F) Hydrogen bonds. | ||
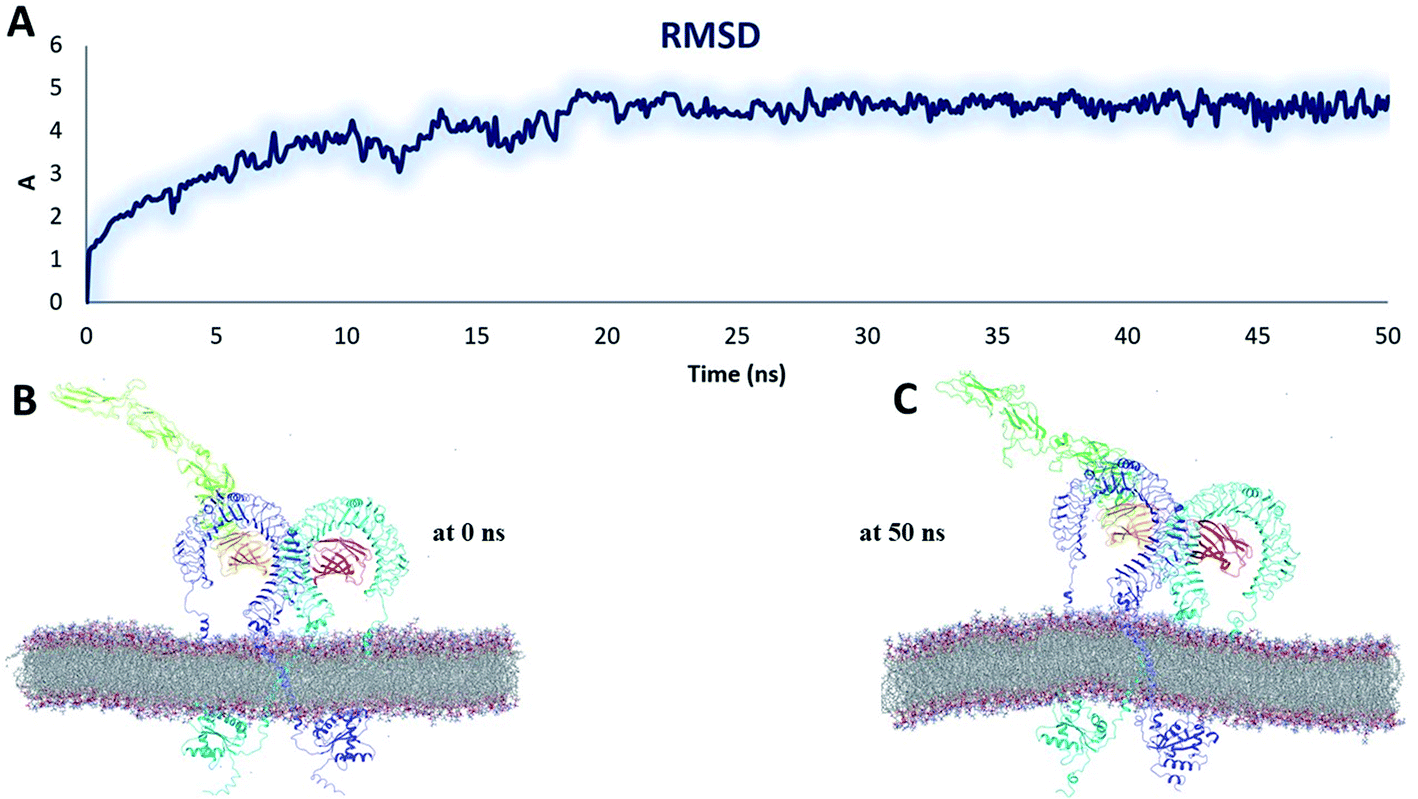 | ||
| Fig. 9 Stability parameters of the heterodimer TLR4–MD2/vaccine complex inside POPC membrane during MD simulation 50 ns (A) root mean square deviation. (B) The initial model of the TLR4–MD2/vaccine complex inside the POPC membrane. (C) Final snapshot of the TLR4–MD2/vaccine complex after 50 ns of MD simulation. | ||
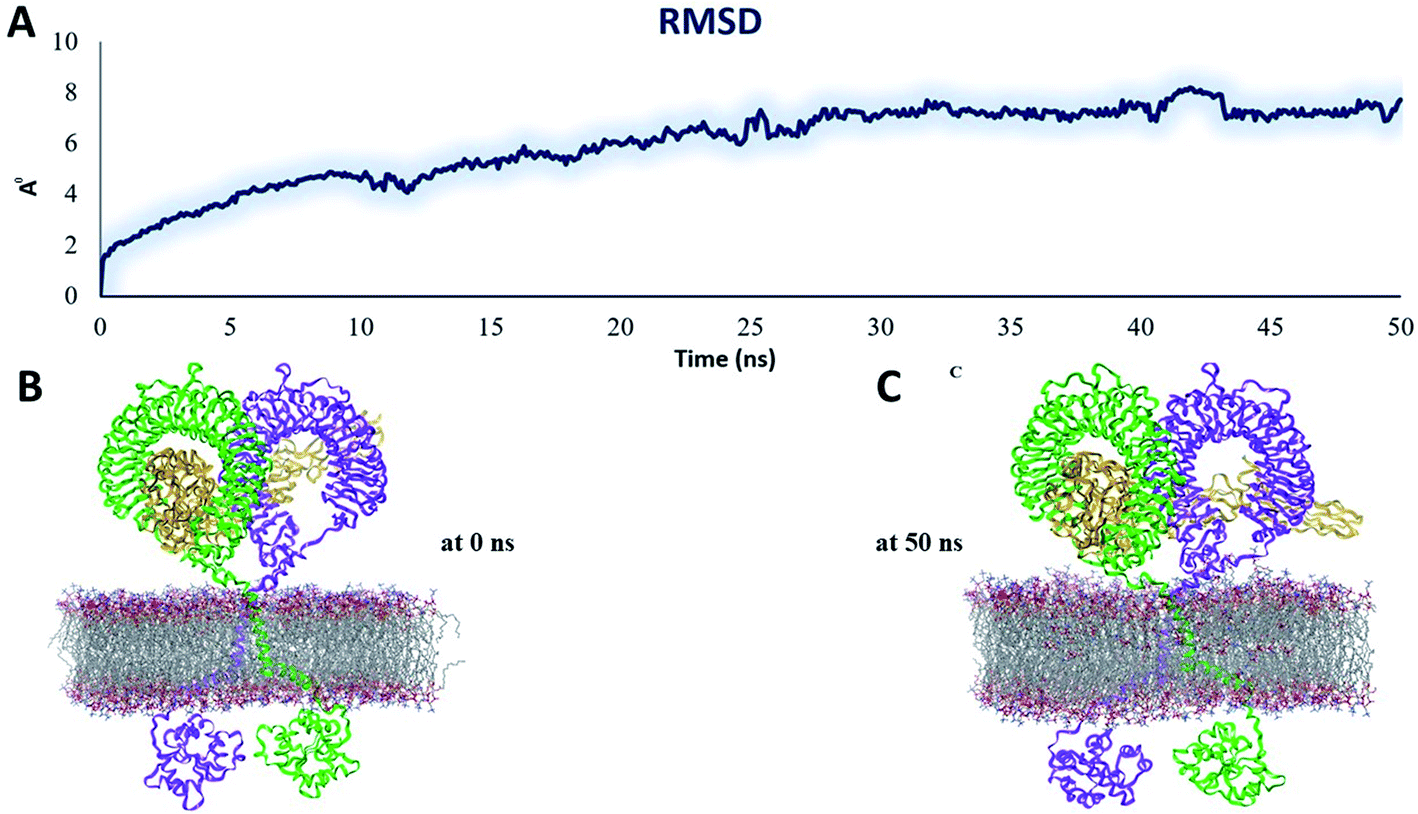 | ||
| Fig. 10 Stability parameters of the homodimer TLR2/vaccine complex inside POPC membrane during MD simulation 50 ns (A) root mean square deviation. (B) The initial model of the TLR2/vaccine complex inside the POPC membrane. (C) Final snapshot of the TLR2/vaccine complex after 50 ns of MD simulation. | ||
3.10. Identification of key residues by residue interaction network analysis during MD simulations
The analysis of residue interaction networks is an important method to identify the residues with the strongest coordination function (i.e. HUB residues) and to obtain information about the transportability or stability of proteins.67 In detail, the analysis of the networks of residual interactions was carried out on the 0 and 50 ns snapshots of the MD simulation time for the heterodimer TLR4–MD2/vaccine and the homodimer TLR2/vaccine by evaluating the following seven topological parameters to identify the HUB nodes: (i) degree of nodes; (ii) betweenness centrality; (iii) eccentricity; and (iv) closeness centrality. The Cytohubba analysis showed that in the MD simulation of snapshots at 0 ns and 50 ns (Fig. 11B and D), E: Lys136, C: Thr166, B: Asn49, E: Gln187, C: Tyr133 were the most conserved HUB residues during the simulation based, suggesting that this residue might have a key structural role heterodimer TLR4–MD2/vaccine during MD simulation. Whereas, for the homodimer TLR2/vaccine complex, C: Glu165, B: Arg155, B: Lys553 were the most vital hub residues in the initial and final snapshots (Fig. 12B and D). Despite the changes in structural behaviors in the TLR2/vaccine complex the Glu165 of the vaccine, and Arg155 and Lys553 of TLR2 were participating in stabilizing the vaccine/TLR2 during the conformational changes.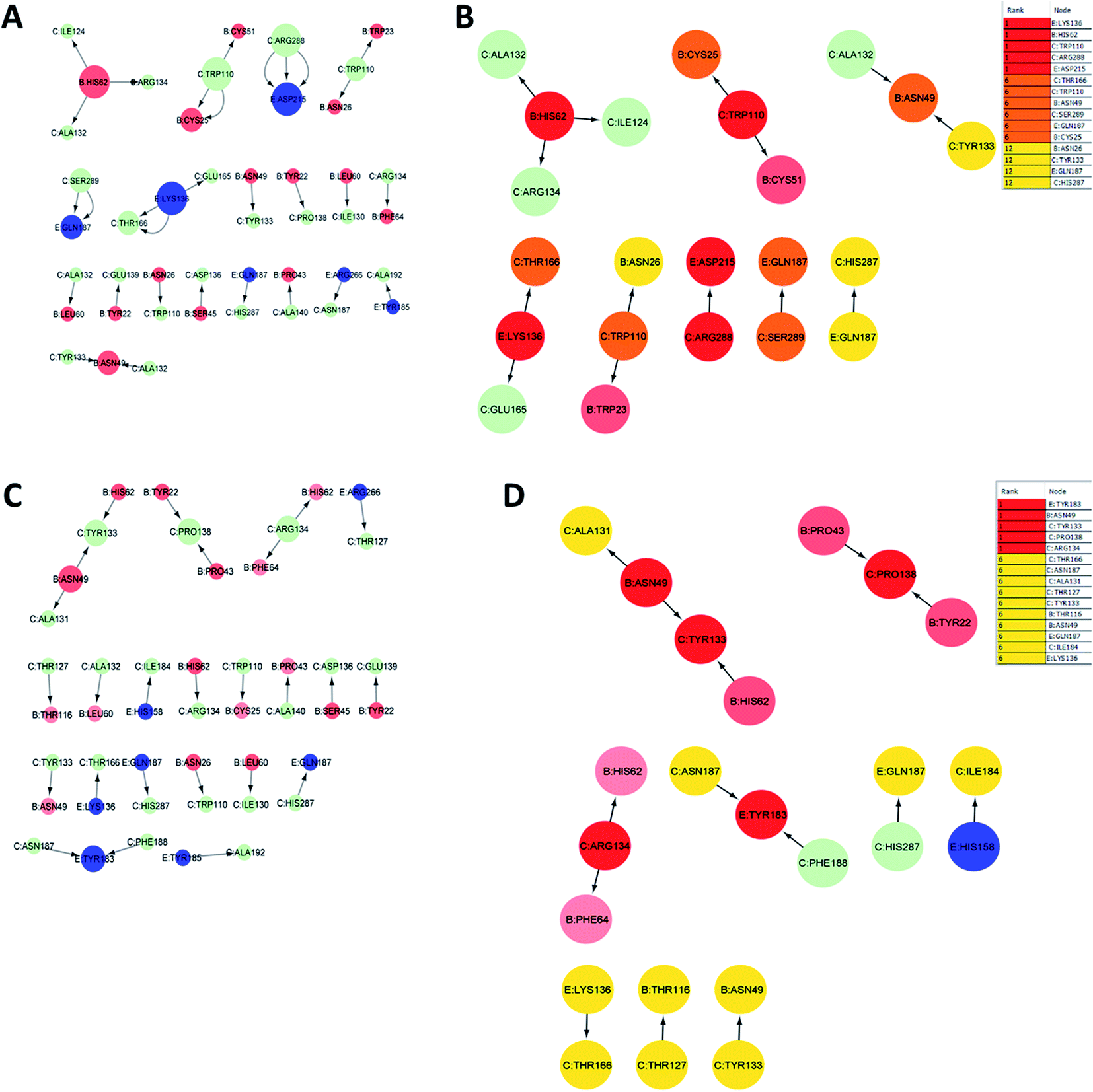 | ||
| Fig. 11 Residue interaction network for TLR4–MD2/vaccine (A) network of interactions at 0 ns (B) hub residues at 0 ns (C) network interactions at 50 ns and (D) hub residues at 50 ns. The vaccine is represented in light green (C chain). TLR4 is represented in blue (E chain) and MD2 is represented in light red (B chain). | ||
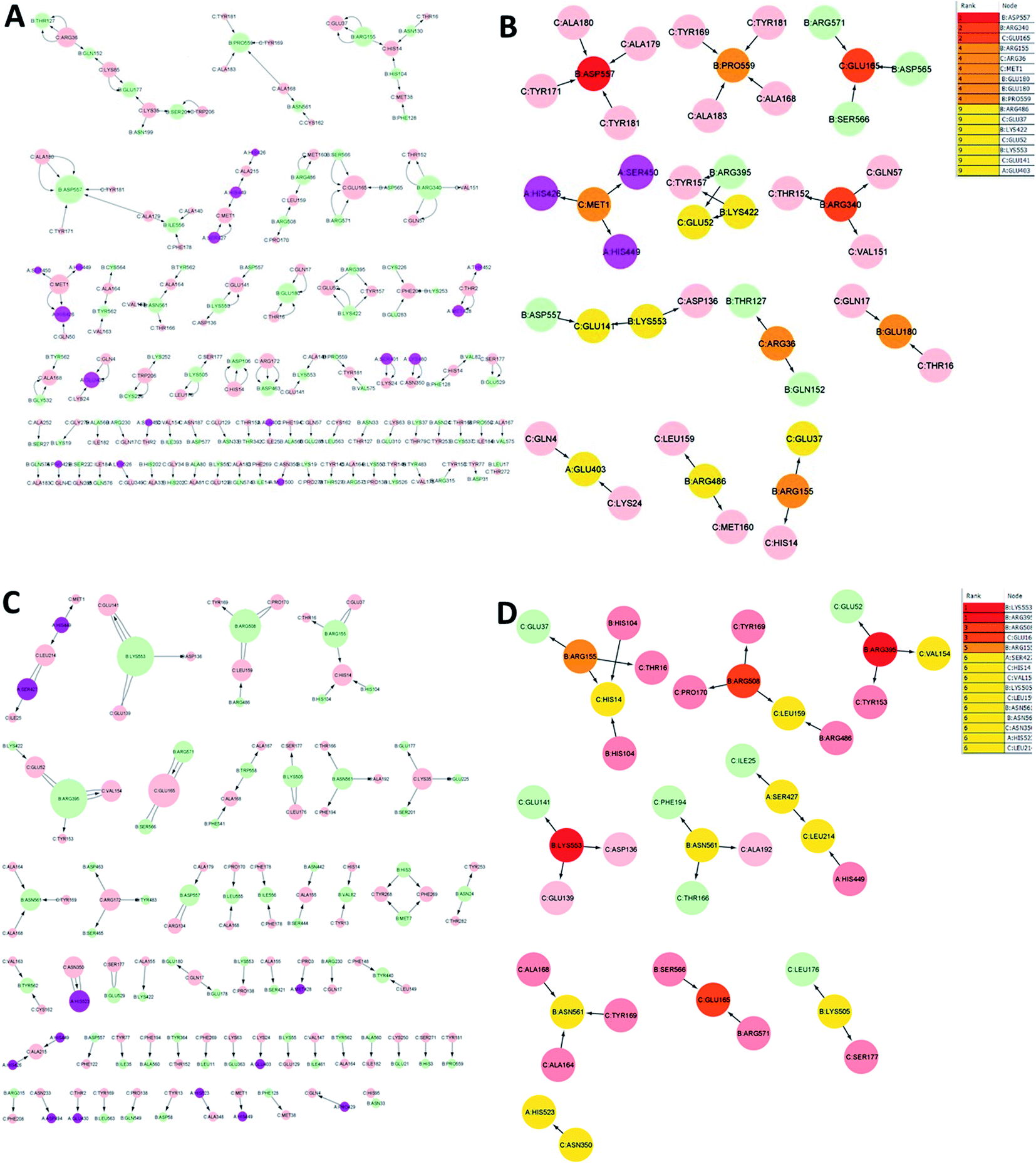 | ||
| Fig. 12 Residue interaction network for TLR2/vaccine (A) network of interactions at 0 ns (B) hub residues at 0 ns (C) network interactions at 50 ns and (D) hub residues at 50 ns. | ||
The vaccine is represented in pink (C chain) and TLR2 is represented in purple (A chain) and light green (B chain).
3.11. Immune simulation
We ran an immune simulation with the C-ImmSim server which indicates that the actual immune response increases with the production of a secondary response.68 The primary response was a high IgM level. The secondary and tertiary reactions showed a significant increase in IgG1 + IgG2, IgG + IgM, and IgM antibodies and the B cell population with a comparable reduction in antigen concentration (Fig. 13A, B, S9A and S9B†). Moreover, the Tc (cytotoxic) and Th (helper) cell populations revealed a high response corresponding with memory cell development (Fig. 13C, D and S9C†). This result also reveals that the evolution of immune memory strengthens upon subsequent exposure to the antigen (Fig. 13D). A high level of interferon-gamma and interleukin-2 was produced after the injection, although the Simpson index is lower (D), it indicates less variety (Fig. 14). The activity of macrophages increased, while the activity of dendritic cells was found to be constant (Fig. S9G and I†). According to the results of the immune simulation, it can be said that the subsequent development of immune memory has increased.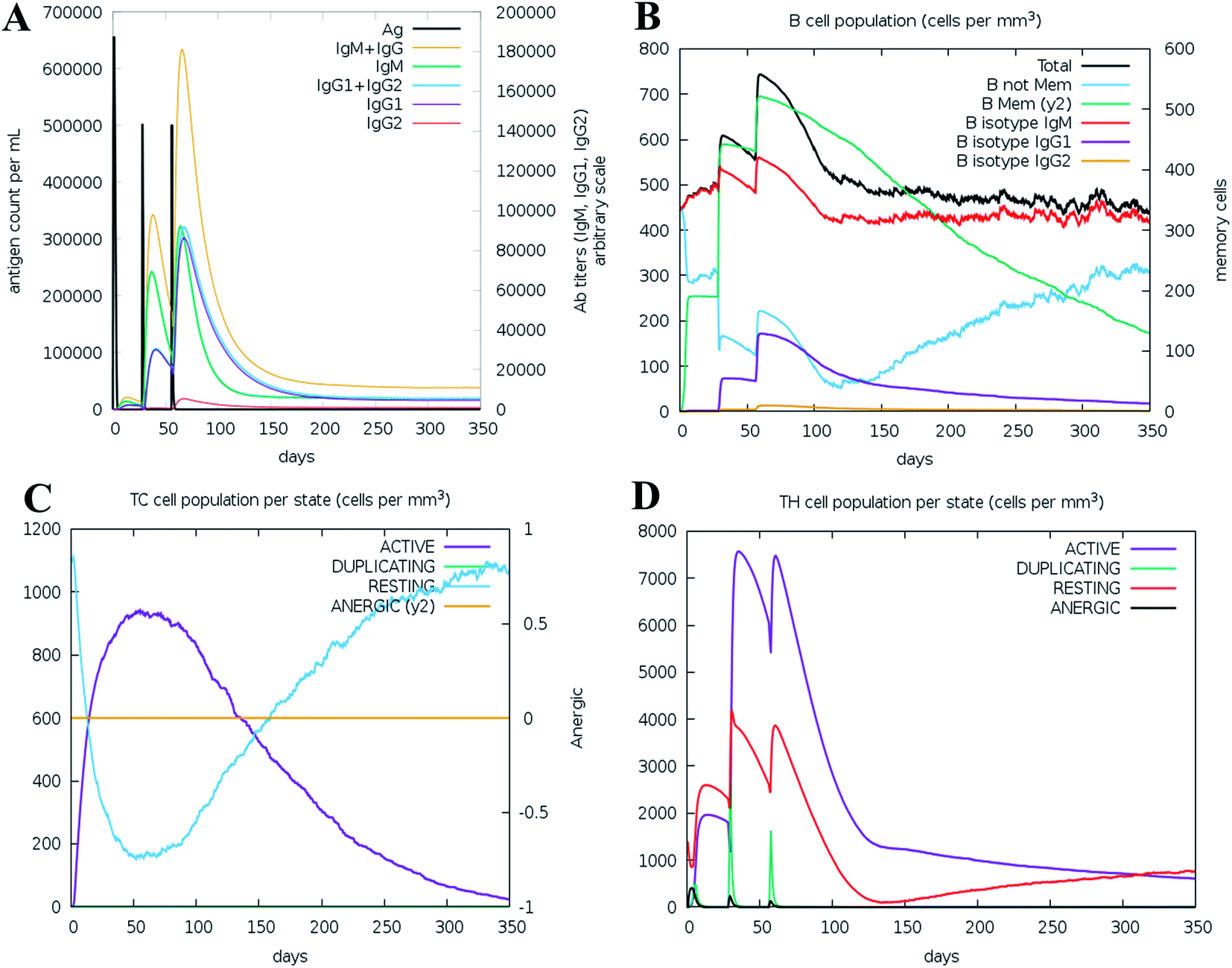 | ||
| Fig. 13 Immune simulation profile of the vaccine construction, measured with the C-ImmSim server. (A) Immunoglobulin production in response to antigen injection (black line); specific subclasses are shown as colored peaks. (B) B cell population after injection. (C) Cytotoxic T-cell population per state after three injections. (D) Helper T-cell population after the injections. | ||
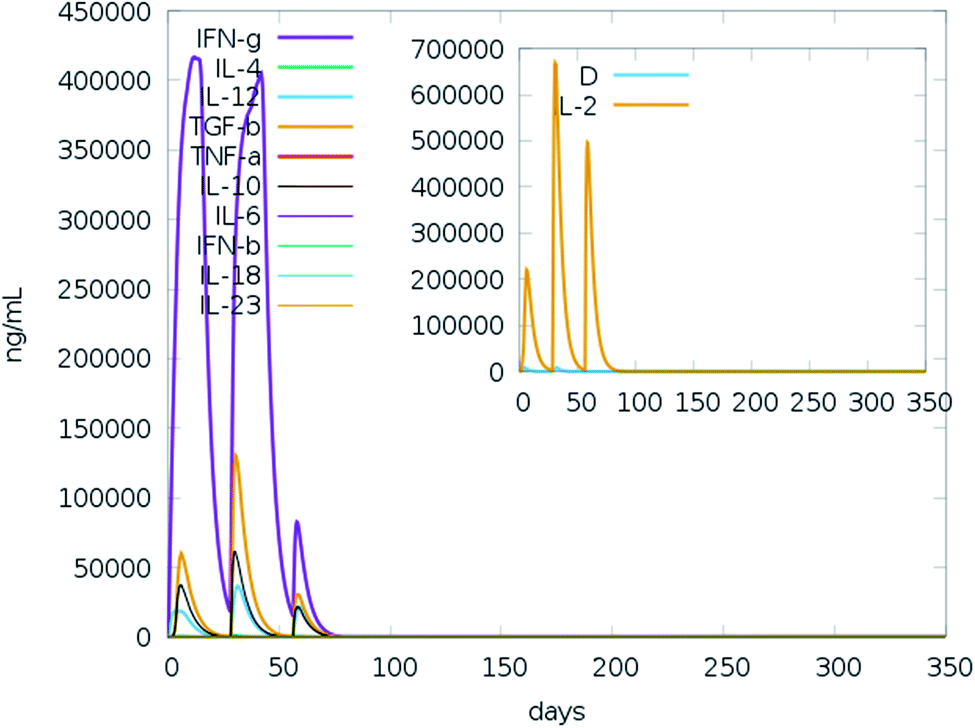 | ||
| Fig. 14 Prediction of cytokine levels instigated by 3 injections given 4 weeks apart. The main plot represents the levels of the cytokine after the injection, and the inset plot represents interleukin 2 (IL-2) levels. | ||
4. Discussion
SARS-CoV-2 infections have spread throughout the world and became a rapidly emerging public health issue affecting whole age groups. It is an emergent demand to produce specific therapeutics, including vaccine and drug candidates, against this disease.83 Immunoinformatics strategies are cost-effective, which assists researchers in predicting antigenic and immunogenic epitopes needed for the construction of epitope-based vaccines against any pathogen.84–86The spike glycoprotein of SARS-CoV-2, an immunogenic protein, has a pivotal role in the virus's attachment to and entrance into the host cells; in addition, it has a high antigenicity and surface contact.87 The aim of this study is to develop a polyepitope-based vaccine that uses various immuno-computational tools that target glycoprotein S to elicit both adaptive and innate immune responses against this deadly virus. The epitope containing vaccine generates immunity based on short immunogenic peptide fragments, whereas the whole genome or a large protein is used for recombinant vaccine technology; hence, the former method cannot produce any allergenic reaction in the host.20,88,89 In addition, our designed vaccine has several advantages compared to single-epitope and conventional vaccines because of the following distinctive characteristics: (a) it comprises multiple MHC epitopes and thus can be recognized by several T-cell receptors, (b) it contains overlapping CTL and HTL epitopes and thus can activate both innate and adaptive immunity, (c) it comprises poly-epitopes from target virulent antigens, and (d) it has an immunostimulator (adjuvant) to stimulate a long-lasting immune response.90–94 Designing a vaccine in this way has already gained importance due to its ability for such vaccines to generate immunity in vivo experiments,95–97 and such vaccines have entered clinical trials.92,98–100
Proper epitope selection is essential for vaccine construction through the in silico biological method.101 In the current study, the epitopes (CTL and HTL) of seven spike glycoprotein sequences were screened out based on several immune filters. The filters applied were that the epitope should be promiscuous, antigenic, and immunogenic; should not have any allergenic properties; should be 100% conserved among target proteins, and should not overlap with any protein in the human proteome. After several immunoassays, a suitable linker-bound epitope-based vaccine was prepared with an adjuvant, cholera toxin B. Cholera B toxin has been shown to act as a potential viral promoter102–104 that is attached to the N-terminus of the vaccine construct through an EAAAK linker. In addition, linkers GPGPG and AAY were added to the sequence, preventing the formation of HTL and CTL binding epitopes, respectively. The use of specialized sequences including linkers can improve vaccine construction. Many previous studies have shown that the addition of GPGPG and AAY linkers between the HTL and CTL epitope sequences, respectively, induces binding immunogenicity, resulting in an impressive variety of vaccines through simplified design and construction.105,106 Arai et al. reported that the EAAAK linker included between epitopes and adjuvants can increase the bioactivity of the fusion protein.107 Bazhan and his colleagues applied similar immunoinformatics strategies to construct a vaccine against Ebola virus. They utilized the IDEB database for predicting potential epitopes and developed a vaccine that was immunogenic when expressed in mice.108 Allergenicity is considered to be a major problem during vaccine development; thus, it is essential to check the allergenic properties of epitopes in the early stage of vaccine development. The epitopes chosen for the vaccine construct were chosen based on their non-allergenic nature; additionally, the AllerTOP v2.0 and AllergenFP v1.0 servers determined that the vaccine sequence designed was inherently non-allergenic.109,110 The antigenic nature of each epitope was measured through the VaxiJen v2.0 tool which has been considered to be a consistent and reliable technique for antigenic evaluation of protein. VaxiJen v2.0 is an independent alignment method used in reverse vaccinology for the prediction of antigen derived from viral, bacterial, and tumor origin. The predictive capacity of this approach was verified by internal training and external test datasets and the accuracy rate of the validation lies in the range of 70% to 89%.23 The combination of negative and positive sets confirmed that this model has remarkable stability for antigen prediction. The spike glycoprotein epitopes of SARS-CoV-2 provoked a robust T-cell immune response. Many studies applied the VaxiJen tool and proposed that the S glycoproteins are the best vaccine candidates.111,112 During the prediction of spike glycoprotein epitopes, the server encompasses biological data of vaccine candidates.111,113 Vary recent study Sami et al.114 used the VaxiJen server for assessing antigenicity of Marburg virus epitopes and they proposed a vaccine candidate that has the ability to generate an immune response against viral pathogenesis. The chemical and physical properties of the vaccine constructs were assessed by ExPASy.24 The predicted vaccine molecular weight and instability index were 51.51 and 21.64 kDa, respectively, classifying the vaccine protein as stable.24 The aliphatic index of the vaccine is 84.35, which indicates the thermostability of the protein.115 The estimated half-life of the vaccine was found to be 20 hours in yeast, 30 hours in mammalian reticulocytes, and greater than 10 hours in Escherichia coli, which determines the time it takes for the protein to reach 50% of its initial concentration after its synthesis in the cell.116 Foroutan and his coworker employed the same types of immunoinformatics approaches for measuring the allergenic and physicochemical parameters of their multi-epitope vaccine designed against Toxoplasma gondii. They conducted an experimental study of their vaccine, the results of which confirmed that a vaccine based on epitopes can elicit strong innate and adaptive immune responses in mice.117 The physicochemical and immunogenic parameters of our developed vaccine candidate, including immunogenicity, aliphatic index, and instability index, were better than those of Foroutan et al. developed and tested vaccine candidates. The secondary structural feature of the vaccine revealed that the protein contains 25.51% extended strands, 36.3% alpha-helical regions, 32.59% random coils, and 5.87% beta turns. The 3D model of the vaccine has been significantly optimized after fine tuning and displaying the appropriate statistics for the Ramachandran batch. An analysis of the vaccine structure by Ramachandran showed that 86.1% of the residual areas were favored, 11.4% were additional allowed, 1% allowed, and 1.5% unauthorized. The ERRAT of the predicted vaccine construct was 79360, and the overall quality assessed by ProSA was Z-score −3.08, indicating that the protein was part of the Z-scores series of previously developed constructs based on experimental X-ray crystallography and nuclear magnetic resonance (NMR) data.49
The spike glycoprotein, a fundamental structural piece of SARS-CoV-2, is recognized by the human TLRs (TLR4 and TLR2), which are expressed in cells' plasma membranes.81,118,119 TLR-4 is expressed in immune cells, including immature dendritic cells, macrophages, monocytes, and granulocytes.120 The direct interaction between TLR4 and cholera toxin B facilitates TLR4 activation.121 Enzyme-linked immunosorbent assays have shown that cholera B toxin can induce NFkB activation in cells expressing TLR4 by direct binding to the receptor.121 In addition, the viral envelope glycoprotein is recognized by TLR2.118 In order to examine the interaction pattern of the vaccine with the immune receptors, molecular docking was carried out with TLR4 and TLR2. The energy values of the docked complexes were found to be −1055.1 and −1010.5 for TLR-2–vaccine, and TLR4/vaccine complexes, respectively, indicating the favourable interaction pattern of the vaccine with the TLRs. Many studies have found that TLR4 and TLR2 in host cells are fundamental to stimulating an active immune response to SARS-CoV. One study by Totura et al. revealed that TLR4 lacking mice are more vulnerable to SARS-CoV infection than wild-type mice.122 Similarly, Hu et al.,123 conducted an investigation in which they have assessed the regulation and expression of TLRs in human monocytic cells upon SARS-CoV infection. Data from that study revealed evidence of upregulated expression of TLR4 and TLR2 24 h after infection with SARS-CoV, indicating its ability to generate an immune response.123 Another study, by Dosch et al., found that TLR-2 expressed by macrophages can trigger IL-8 production by binding to the SARS-CoV spike protein.124 As a result, the cytokine IL-8 is triggered by the activated TLR-2 and helps to generate an innate immune response.124
The RMSD plot showed that the TLR/vaccine complexes were very stable (Fig. 7). Furthermore, as shown in Fig. 8E, the calculated average SASA value for toll-like receptor-2 and toll-like receptor-4 indicated the compactness and stability of their structures. The radius of gyration also showed that the TLR/vaccine complexes had become structurally stable and compact during MD simulation (Fig. 8D). Evaluation of molecular docking between protein and TLR-4 demonstrated that most of the hydrogen bonds between the vaccine and TLR-4 have strong interactions. The consequences of the hydrogen-bond plot suggested reduced flexibility of the vaccine when bound to TLR4, improved binding strength, and increased vaccine–receptor stability (Fig. 8E). Further, proposed the full-length heterodimer TLR4–MD2/vaccine complex was placed in membrane to imitate the dynamic behavior during the MD simulation of the vaccine in biological systems. Lipid–protein, and protein–protein interactions of the ECD, TM, and CD domains were pivotal for determining the biophysical properties behind the signaling competent form of intact TLR4 (Fig. 9) and TLR2 (Fig. 10). Moreover, to understand the possible biological relevance of these systems, we identified hub residues and their role in stabilizing the protein–protein interactions during the MD simulation time. The obtained results revealed that the E: Lys136, C: Thr166, B: Asn49, E: Gln187, C: Tyr133 hub resides are identical in initial and last snapshots among the top 15 hub residues for TLR4–MD2/vaccine system. Whilst network analysis showed that only C: Glu165, B: Arg155, B: Lys553 were identical in the first and final snapshots. This suggests a potential molecular advantage of the HUB residues in stabilizing the interactions of the vaccine with the biological systems.
The results of the immune simulation indicate that the designed vaccine elicits a normal immune response in which iterative exposure to the antigen increases the primary response. The T cells and memory B cells remained durable for several months, with active memory B cells. Helper T-cells were stimulated as well. Similarly, both interferon-gamma and interleukin-2 rose to a high level upon frequent exposure to the antigen and were sustained at the peak. This resulted in a high level of TH cells and subsequent Ig production that helped trigger the humoral immune response. A heterogeneous immune repertoire is suggested by the Simpson index (D), which was used to investigate clonal specificity. This may have been due to the chimeric peptide being constructed from numerous T-cell epitopes. This may occur in response to the stimulation by Th1 and Th2 cells since cytokines released by these cell types (including interleukin-2 and interleukin-4) have been found to be associated with the formation of primary and long-lasting memory CD8+ responses. Furthermore, IFN-γ-dominant Th1-type responses have been found to be increased in immune individuals, as demonstrated by the high levels of neutrophils, cytotoxic T-cells, macrophages, TH1 cells.
5. Conclusions
SARS-CoV-2 has caused the current global COVID-19 pandemic. However, the strategy to deal with COVID-19 rapidly became highly disordered and the situation became unmanageable. An immunoinformatics strategy was employed for designing a vaccine candidate against this lethal virus. The vaccine was constructed based on CTL and HTL epitopes of the viral spike glycoprotein. The vaccine has been found to have both antigenic and immunogenic characteristics. Molecular docking studies have shown that the vaccine has a strong binding affinity for TLRs (TLR2 and TLR4), and MD simulation studies with PPIs network analysis have confirmed a stable interaction between the vaccine and the receptors. Finally, immune simulation has demonstrated the ability of the vaccine to elicit immune responses. Several immunoinformatics approaches have been used sequentially to develop a vaccine candidate that can elicit an effective immune response against viral infection, but the experimental evaluation is necessary to ensure the ability to generate the exact immune response. The laboratory investigations generally involve in vivo and in vitro assays for the vaccine development to reach the experimental standard. In addition, we propose further research including the synthetic process and biological activity of the vaccine.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The datasets supporting the conclusions of this study are included within the article (and its additional files).Author contributions
Conceptualization: Md. Oliullah Rafi, Khattab Al-Khafaji, Md. Shahedur Rahman.Formal analysis: Md. Oliullah Rafi, Khattab Al-Khafaji, Md. Takim Sarker, investigation: Md. Oliullah Rafi, Khattab Al-Khafaji, Md. Takim Sarker, Tugba Taskin-Tok, Abdus Samad Rana, Md. Shahedur Rahman.
Methodology: Md. Oliullah Rafi, Khattab Al-Khafaji, Md. Takim Sarker, Tugba Taskin-Tok.
Project administration: Md. Shahedur Rahman.
Resources: Md. Shahedur Rahman.
Software: Md. Oliullah Rafi, Khattab Al-Khafaji, Md. Takim Sarker.
Supervision: Md. Shahedur Rahman.
Writing – original draft: Md. Oliullah Rafi, Khattab Al-Khafaji, Md. Takim Sarker.
Writing – review & editing: Md. Oliullah Rafi, Tugba Taskin-Tok, Md. Shahedur Rahman.
References
- S. Su, G. Wong, W. Shi, J. Liu, A. C. K. Lai, J. Zhou, W. Liu, Y. Bi and G. F. Gao, Trends Microbiol., 2016, 24, 490–502 CrossRef CAS PubMed.
- J. Ziebuhr, Curr. Top. Microbiol. Immunol., 2005, 287, 57–94 CAS.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS PubMed.
- M. A. Tortorici, A. C. Walls, Y. Lang, C. Wang, Z. Li, D. Koerhuis, G.-J. Boons, B.-J. Bosch, F. A. Rey, R. J. de Groot and D. Veesler, Nat. Struct. Mol. Biol., 2019, 26, 481–489 CrossRef PubMed.
- S. Perlman and J. Netland, Nat. Rev. Microbiol., 2009, 7, 439–450 CrossRef CAS PubMed.
- M. Sarker Takim, M. Rafi Oliullah, R. Capasso and T. Bin Emran, others, Biologics, 2021, 357–383 CrossRef.
- C. Liu, Q. Zhou, Y. Li, L. V. Garner, S. P. Watkins, L. J. Carter, J. Smoot, A. C. Gregg, A. D. Daniels, S. Jervey and D. Albaiu, ACS Cent. Sci., 2020, 6, 315–331 CrossRef CAS PubMed.
- C. Wu, Y. Liu, Y. Yang, P. Zhang, W. Zhong, Y. Wang, Q. Wang, Y. Xu, M. Li, X. Li, M. Zheng, L. Chen and H. Li, Acta Pharm. Sin. B, 2020, 10, 766–788 CrossRef CAS PubMed.
- R. Lu, X. Zhao, J. Li, P. Niu, B. Yang, H. Wu, W. Wang, H. Song, B. Huang, N. Zhu, Y. Bi, X. Ma, F. Zhan, L. Wang, T. Hu, H. Zhou, Z. Hu, W. Zhou, L. Zhao, J. Chen, Y. Meng, J. Wang, Y. Lin, J. Yuan, Z. Xie, J. Ma, W. J. Liu, D. Wang, W. Xu, E. C. Holmes, G. F. Gao, G. Wu, W. Chen, W. Shi and W. Tan, Lancet, 2020, 395, 565–574 CrossRef CAS.
- M. J. Hossain, T. Jannat, S. R. Brishty, U. Roy, S. Mitra, M. O. Rafi, M. R. Islam, M. L. Nesa, M. A. Islam and T. Bin Emran, Biologics, 2021, 1, 252–284 CrossRef.
- Y.-Z. Zhang and E. C. Holmes, Cell, 2020, 181, 223–227 CrossRef CAS PubMed.
- B. L. Haagmans, S. H. S. Al Dhahiry, C. B. E. M. Reusken, V. S. Raj, M. Galiano, R. Myers, G.-J. Godeke, M. Jonges, E. Farag, A. Diab, H. Ghobashy, F. Alhajri, M. Al-Thani, S. A. Al-Marri, H. E. Al Romaihi, A. Al Khal, A. Bermingham, A. D. M. E. Osterhaus, M. M. AlHajri and M. P. G. Koopmans, Lancet Infect. Dis., 2014, 14, 140–145 CrossRef CAS.
- X.-Y. Ge, J.-L. Li, X.-L. Yang, A. A. Chmura, G. Zhu, J. H. Epstein, J. K. Mazet, B. Hu, W. Zhang, C. Peng, Y.-J. Zhang, C.-M. Luo, B. Tan, N. Wang, Y. Zhu, G. Crameri, S.-Y. Zhang, L.-F. Wang, P. Daszak and Z.-L. Shi, Nature, 2013, 503, 535–538 CrossRef CAS PubMed.
- D. Schoeman and B. C. Fielding, Virol. J., 2019, 16, 69 CrossRef PubMed.
- N. Zhu, D. Zhang, W. Wang, X. Li, B. Yang, J. Song, X. Zhao, B. Huang, W. Shi, R. Lu, P. Niu, F. Zhan, X. Ma, D. Wang, W. Xu, G. Wu, G. F. Gao and W. Tan, N. Engl. J. Med., 2020, 382, 727–733 CrossRef CAS PubMed.
- M. O. Rafi, G. Bhattacharje, K. Al-Khafaji, T. Taskin-Tok, M. A. Alfasane, A. K. Das, M. A. K. Parvez and M. S. Rahman, J. Biomol. Struct. Dyn., 2020, 1–20 Search PubMed.
- Z. Li, A. C. Tomlinson, A. H. Wong, D. Zhou, M. Desforges, P. J. Talbot, S. Benlekbir, J. L. Rubinstein and J. M. Rini, eLife, 2019, 8, e51230 CrossRef CAS PubMed.
- N. Rahman, Z. Basharat, M. Yousuf, G. Castaldo, L. Rastrelli and H. Khan, Molecules, 2020, 25, 2271 CrossRef CAS PubMed.
- A. Grifoni, D. Weiskopf, S. I. Ramirez, J. Mateus, J. M. Dan, C. R. Moderbacher, S. A. Rawlings, A. Sutherland, L. Premkumar, R. S. Jadi, D. Marrama, A. M. de Silva, A. Frazier, A. F. Carlin, J. A. Greenbaum, B. Peters, F. Krammer, D. M. Smith, S. Crotty and A. Sette, Cell, 2020, 181, 1489–1501 CrossRef CAS PubMed.
- V. Chauhan, T. Rungta, K. Goyal and M. P. Singh, Sci. Rep., 2019, 9, 1–15 CAS.
- A. C. Walls, M. A. Tortorici, B.-J. Bosch, B. Frenz, P. J. M. Rottier, F. DiMaio, F. A. Rey and D. Veesler, Nature, 2016, 531, 114–117 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- I. A. Doytchinova and D. R. Flower, BMC Bioinf., 2007, 8, 4 CrossRef PubMed.
- J. M. Walker, The Proteomics Protocols Handbook, Springer, 2005 Search PubMed.
- R. C. Edgar, Nucleic Acids Res., 2004, 32, 1792–1797 CrossRef CAS PubMed.
- S. Kumar, G. Stecher, M. Li, C. Knyaz and K. Tamura, Mol. Biol. Evol., 2018, 35, 1547–1549 CrossRef CAS PubMed.
- M. V. Larsen, C. Lundegaard, K. Lamberth, S. Buus, O. Lund and M. Nielsen, BMC Bioinf., 2007, 8, 424 CrossRef PubMed.
- J. J. A. Calis, M. Maybeno, J. A. Greenbaum, D. Weiskopf, A. D. De Silva, A. Sette, C. Keşmir and B. Peters, PLoS Comput. Biol., 2013, 9, e1003266 CrossRef PubMed.
- A. Lerner, T. Matthias and R. Aminov, Front. Immunol., 2017, 8, 1630 CrossRef PubMed.
- K. K. Jensen, M. Andreatta, P. Marcatili, S. Buus, J. A. Greenbaum, Z. Yan, A. Sette, B. Peters and M. Nielsen, Immunology, 2018, 154, 394–406 CrossRef CAS PubMed.
- L. Moise, J. A. McMurry, S. Buus, S. Frey, W. D. Martin and A. S. De Groot, Vaccine, 2009, 27, 6471–6479 CrossRef CAS PubMed.
- S. K. Dhanda, P. Vir and G. P. S. Raghava, Biol. Direct, 2013, 8, 30 CrossRef PubMed.
- J. Ponomarenko, H.-H. Bui, W. Li, N. Fusseder, P. E. Bourne, A. Sette and B. Peters, BMC Bioinf., 2008, 9, 514 CrossRef PubMed.
- I. Dimitrov, D. R. Flower and I. Doytchinova, BMC Bioinf., 2013, 14(suppl 6), S4 CrossRef PubMed.
- S. Gupta, P. Kapoor, K. Chaudhary, A. Gautam, R. Kumar and G. P. S. Raghava, PLoS One, 2013, 8, e73957 CrossRef CAS PubMed.
- H.-H. Bui, J. Sidney, W. Li, N. Fusseder and A. Sette, BMC Bioinf., 2007, 8, 361 CrossRef PubMed.
- H.-H. Bui, J. Sidney, K. Dinh, S. Southwood, M. J. Newman and A. Sette, BMC Bioinf., 2006, 7, 153 CrossRef PubMed.
- A. Lamiable, P. Thévenet, J. Rey, M. Vavrusa, P. Derreumaux and P. Tufféry, Nucleic Acids Res., 2016, 44, W449–W454 CrossRef CAS PubMed.
- D. Schneidman-Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2005, 33, W363–W367 CrossRef CAS PubMed.
- E. Mashiach, D. Schneidman-Duhovny, N. Andrusier, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2008, 36, W229–W232 CrossRef CAS PubMed.
- C. N. Magnan, M. Zeller, M. A. Kayala, A. Vigil, A. Randall, P. L. Felgner and P. Baldi, Bioinformatics, 2010, 26, 2936–2943 CrossRef CAS PubMed.
- I. Dimitrov, L. Naneva, I. Doytchinova and I. Bangov, Bioinformatics, 2013, 30, 846–851 CrossRef PubMed.
- T. N. Petersen, S. Brunak, G. von Heijne and H. Nielsen, Nat. Methods, 2011, 8, 785–786 CrossRef CAS PubMed.
- C. Geourjon and G. Deléage, Bioinformatics, 1995, 11, 681–684 CrossRef CAS PubMed.
- A. Roy, A. Kucukural and Y. Zhang, Nat. Protoc., 2010, 5, 725–738 CrossRef CAS PubMed.
- R. A. Shey, S. M. Ghogomu, K. K. Esoh, N. D. Nebangwa, C. M. Shintouo, N. F. Nongley, B. F. Asa, F. N. Ngale, L. Vanhamme and J. Souopgui, Sci. Rep., 2019, 9, 1–18 CrossRef CAS PubMed.
- D. Xu and Y. Zhang, Biophys. J., 2011, 101, 2525–2534 CrossRef CAS PubMed.
- L. Heo, H. Park and C. Seok, Nucleic Acids Res., 2013, 41, W384–W388 CrossRef PubMed.
- M. Wiederstein and M. J. Sippl, Nucleic Acids Res., 2007, 35, W407–W410 CrossRef PubMed.
- C. Colovos and T. O. Yeates, Protein Sci., 1993, 2, 1511–1519 CrossRef CAS PubMed.
- M. E. Hodsdon, J. W. Ponder and D. P. Cistola, J. Mol. Biol., 1996, 264, 585–602 CrossRef CAS PubMed.
- M. O. Rafi, K. Al-Khafaji, T. T. Tok and M. S. Rahman, J. Biomol. Struct. Dyn., 2020, 1–13 Search PubMed.
- H. R. Abd El-Mageed, D. A. Abdelrheem, M. Rafi, M. Sarker, K. Al-Khafaji, M. Hossain, R. Capasso and T. Bin Emran, others, Biologics, 2021, 1, 416–434 CrossRef.
- S. Mahmud, M. O. Rafi, G. K. Paul, M. M. Promi, M. S. S. Shimu, S. Biswas, T. Bin Emran, K. Dhama, S. A. Alyami, M. A. Moni and M. A. Saleh, Sci. Rep., 2021, 11, 15431 CrossRef CAS PubMed.
- M. C. Patel, K. A. Shirey, L. M. Pletneva, M. S. Boukhvalova, A. Garzino-Demo, S. N. Vogel and J. C. Blanco, Future Virol., 2014, 9, 811–829 CrossRef CAS PubMed.
- D. Kozakov, D. R. Hall, B. Xia, K. A. Porter, D. Padhorny, C. Yueh, D. Beglov and S. Vajda, Nat. Protoc., 2017, 12, 255–278 CrossRef CAS PubMed.
- K. Al-Khafaji and T. T. Tok, Comput. Methods Progr. Biomed., 2020, 195, 105660 CrossRef PubMed.
- K. Al-Khafaji and T. Taskin Tok, J. Biomol. Struct. Dyn., 2021, 39, 1600–1610 CrossRef CAS PubMed.
- K. Al-Khafaji and T. Taskin Tok, J. Biomol. Struct. Dyn., 2021, 39, 1965–1974 CrossRef CAS PubMed.
- D. F. Akın-Balı, K. Al-Khafaji, S. H. Aktas and T. Taskin-Tok, J. Biomol. Struct. Dyn., 2021, 39, 4290–4303 CrossRef PubMed.
- R. Hossain, M. T. Islam, P. Ray, D. Jain, A. S. M. Saikat, L. Nahar, A. Das Talukdar, S. Sarker, S. A. Ayatollahi, M. Martorell, F. Kobarfard, N. Cruz-Martins, K. Al-Khafaji, A. O. Docea, D. Calina and J. Sharifi-Rad, Pharmacogn. Res., 2021, 13, 149–157 CrossRef CAS.
- C. Talarico, S. Gervasoni, C. Manelfi, A. Pedretti, G. Vistoli and A. R. Beccari, Int. J. Mol. Sci., 2020, 21, 2265 CrossRef CAS PubMed.
- S. Saha, E. Panieri, S. Suzen and L. Saso, J. Membr. Biol., 2020, 253, 57–71 CrossRef CAS PubMed.
- G. J. Martyna, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 50, 3234 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110, 1657–1666 CrossRef CAS PubMed.
- C. Nunziata, A. Polo, A. Sorice, F. Capone, M. Accardo, E. Guerriero, F. Z. Marino, M. Orditura, A. Budillon and S. Costantini, Sci. Rep., 2019, 9, 1–14 CAS.
- N. Rapin, O. Lund, M. Bernaschi and F. Castiglione, PLoS One, 2010, 5, 1–14 CrossRef PubMed.
- F. Castiglione, F. Mantile, P. De Berardinis and A. Prisco, Comput. Math. Methods Med., 2012, 2012, 842329 CrossRef CAS PubMed.
- K. A. Jordan and C. A. Hunter, Exp. Parasitol., 2010, 126, 318–325 CrossRef CAS PubMed.
- E. A. Moseman and D. B. McGavern, Immunol. Rev., 2013, 255, 110–124 CrossRef PubMed.
- R. K. Pandey, T. K. Bhatt and V. K. Prajapati, Sci. Rep., 2018, 8, 1125 CrossRef PubMed.
- N. Nezafat, Y. Ghasemi, G. Javadi, M. J. Khoshnoud and E. Omidinia, J. Theor. Biol., 2014, 349, 121–134 CrossRef CAS PubMed.
- M. Ali, R. K. Pandey, N. Khatoon, A. Narula, A. Mishra and V. K. Prajapati, Sci. Rep., 2017, 7, 9232 CrossRef PubMed.
- Y. Zhang and J. Skolnick, Proteins, 2004, 57, 702–710 CrossRef CAS PubMed.
- A. Messaoudi, H. Belguith and J. Ben Hamida, Theor. Biol. Med. Modell., 2013, 10, 22 CrossRef CAS PubMed.
- A. N. Ilinskaya and M. A. Dobrovolskaia, Toxicol. Appl. Pharmacol., 2016, 299, 70–77 CrossRef CAS PubMed.
- A. K. Dey, P. Malyala and M. Singh, Expert Rev. Vaccines, 2014, 13, 671–685 CrossRef CAS PubMed.
- E. D. Getzoff, J. A. Tainer, R. A. Lerner and H. M. Geysen, in Advances in Immunology, ed. F. J. Dixon, Academic Press, 1988, vol. 43, pp. 1-98 Search PubMed.
- Z. Nain, F. Abdullah, M. M. Rahman, M. M. Karim, S. M. R. Rahman, M. S. A. Khan, S. Bin Sayed, M. M. Sheam, Z. Haque and U. K. Adhikari, bioRxiv, 2019, 758219 Search PubMed.
- M. Carty and A. G. Bowie, Clin. Exp. Immunol., 2010, 161, 397–406 CrossRef CAS PubMed.
- S. N. Lester and K. Li, J. Mol. Biol., 2014, 426, 1246–1264 CrossRef CAS PubMed.
- J. S. Morse, T. Lalonde, S. Xu and W. R. Liu, ChemBioChem, 2020, 21, 730–738 CrossRef CAS PubMed.
- S. F. de O. Tosta, M. S. Passos, R. Kato, Á. Salgado, J. Xavier, A. K. Jaiswal, S. C. Soares, V. Azevedo, M. Giovanetti and S. Tiwari, others, J. Biomol. Struct. Dyn., 2020, 1–17 Search PubMed.
- S. Srivastava, M. Kamthania, R. Kumar Pandey, A. Kumar Saxena, V. Saxena, S. Kumar Singh, R. Kumar Sharma and N. Sharma, J. Biomol. Struct. Dyn., 2019, 37, 4345–4360 CrossRef CAS PubMed.
- A. G. Goodman, P. P. Heinen, S. Guerra, A. Vijayan, C. O. S. Sorzano, C. E. Gomez and M. Esteban, PLoS One, 2011, 6, e25938 CrossRef CAS PubMed.
- E. de Wit, N. van Doremalen, D. Falzarano and V. J. Munster, Nat. Rev. Microbiol., 2016, 14, 523–534 CrossRef CAS PubMed.
- H. Sbai, A. Mehta and A. S. DeGroot, Curr. Drug Targets: Infect. Disord., 2001, 1, 303–313 CrossRef CAS PubMed.
- A. Sette and J. Fikes, Curr. Opin. Immunol., 2003, 15, 461–470 CrossRef CAS PubMed.
- C. Lu, S. Meng, Y. Jin, W. Zhang, Z. Li, F. Wang, F. Wang-Johanning, Y. Wei, H. Liu, H. Tu, D. Su, A. He, X. Cao and F. Zhou, Br. J. Haematol., 2017, 178, 413–426 CrossRef CAS PubMed.
- M. Saadi, A. Karkhah and H. R. Nouri, Infect., Genet. Evol., 2017, 51, 227–234 CrossRef CAS PubMed.
- P. Jiang, Y. Cai, J. Chen, X. Ye, S. Mao, S. Zhu, X. Xue, S. Chen and L. Zhang, Vaccine, 2017, 35, 3096–3103 CrossRef CAS PubMed.
- I.-N. Lu, S. Farinelle, A. Sausy and C. P. Muller, Cell. Mol. Immunol., 2017, 14, 511–520 CrossRef CAS PubMed.
- R. He, X. Yang, C. Liu, X. Chen, L. Wang, M. Xiao, J. Ye, Y. Wu and L. Ye, Cell. Mol. Immunol., 2018, 15, 815–826 CrossRef CAS PubMed.
- Y. Cao, D. Li, Y. Fu, Q. Bai, Y. Chen, X. Bai, Z. Jing, P. Sun, H. Bao, P. Li, J. Zhang, X. Ma, Z. Lu and Z. Liu, Antiviral Res., 2017, 140, 133–141 CrossRef CAS PubMed.
- L. Guo, R. Yin, K. Liu, X. Lv, Y. Li, X. Duan, Y. Chu, T. Xi and Y. Xing, Appl. Microbiol. Biotechnol., 2014, 98, 3495–3507 CrossRef CAS PubMed.
- W.-Y. Zhou, Y. Shi, C. Wu, W.-J. Zhang, X.-H. Mao, G. Guo, H.-X. Li and Q.-M. Zou, Vaccine, 2009, 27, 5013–5019 CrossRef CAS PubMed.
- V. Lennerz, S. Gross, E. Gallerani, C. Sessa, N. Mach, S. Boehm, D. Hess, L. von Boehmer, A. Knuth, A. F. Ochsenbein, U. Gnad-Vogt, J. Zieschang, U. Forssmann, T. Woelfel and E. Kaempgen, Cancer Immunol. Immunother., 2014, 63, 381–394 CrossRef CAS PubMed.
- C. L. J. Slingluff, S. Lee, F. Zhao, K. A. Chianese-Bullock, W. C. Olson, L. H. Butterfield, T. L. Whiteside, P. D. Leming and J. M. Kirkwood, Clin. Cancer Res., 2013, 19, 4228–4238 CrossRef CAS PubMed.
- H. Toledo, A. Baly, O. Castro, S. Resik, J. Laferté, F. Rolo, L. Navea, L. Lobaina, O. Cruz, J. Míguez, T. Serrano, B. Sierra, L. Pérez, M. E. Ricardo, M. Dubed, A. L. Lubián, M. Blanco, J. C. Millán, A. Ortega, E. Iglesias, E. Pentón, Z. Martín, J. Pérez, M. Díaz and C. A. Duarte, Vaccine, 2001, 19, 4328–4336 CrossRef CAS PubMed.
- D. Yin, L. Li, X. Song, H. Li, J. Wang, W. Ju, X. Qu, D. Song, Y. Liu, X. Meng, H. Cao, W. Song, R. Meng, J. Liu, J. Li and K. Xu, BMC Infect. Dis., 2016, 16, 219 CrossRef PubMed.
- J. Hou, Y. Liu, J. Hsi, H. Wang, R. Tao and Y. Shao, Hum. Vaccines Immunother., 2014, 10, 1274–1283 CrossRef CAS PubMed.
- H. J. Kim, J.-K. Kim, S. B. Seo, H. J. Lee and H.-J. Kim, Arch. Pharmacal Res., 2007, 30, 366–371 CrossRef CAS PubMed.
- S. Tamura, H. Funato, T. Nagamine, C. Aizawa and T. Kurata, Vaccine, 1989, 7, 503–505 CrossRef CAS PubMed.
- B. Meza, F. Ascencio, A. P. Sierra-Beltrán, J. Torres and C. Angulo, Infect., Genet. Evol., 2017, 49, 309–317 CrossRef CAS PubMed.
- N. Khatoon, R. K. Pandey and V. K. Prajapati, Sci. Rep., 2017, 7, 1–12 CrossRef CAS PubMed.
- R. Arai, H. Ueda, A. Kitayama, N. Kamiya and T. Nagamune, Protein Eng., 2001, 14, 529–532 CrossRef CAS PubMed.
- S. I. Bazhan, D. V. Antonets, L. I. Karpenko, S. F. Oreshkova, O. N. Kaplina, E. V. Starostina, S. G. Dudko, S. A. Fedotova and A. A. Ilyichev, Vaccines, 2019, 7, 34 CrossRef CAS PubMed.
- I. Dimitrov, I. Bangov, D. R. Flower and I. Doytchinova, J. Mol. Model., 2014, 20, 2278 CrossRef PubMed.
- I. Dimitrov, L. Naneva, I. Doytchinova and I. Bangov, Bioinformatics, 2014, 30, 846–851 CrossRef CAS PubMed.
- M. Dalsass, A. Brozzi, D. Medini and R. Rappuoli, Front. Immunol., 2019, 10, 113 CrossRef PubMed.
- E. Ong, M. U. Wong, A. Huffman and Y. He, Front. Immunol., 2020, 11, 1581 CrossRef CAS PubMed.
- A. Rakib, S. A. Sami, N. J. Mimi, M. M. Chowdhury, T. A. Eva, F. Nainu, A. Paul, A. Shahriar, A. M. Tareq, N. U. Emon, S. Chakraborty, S. Shil, S. J. Mily, T. Ben Hadda, F. A. Almalki and T. Bin Emran, Comput. Biol. Med., 2020, 124, 103967 CrossRef CAS PubMed.
- S. A. Sami, K. K. S. Marma, S. Mahmud, M. A. N. Khan, S. Albogami, A. M. El-Shehawi, A. Rakib, A. Chakraborty, M. Mohiuddin, K. Dhama, M. M. N. Uddin, M. K. Hossain, T. E. Tallei and T. Bin Emran, ACS Omega, 2021, 6, 32043–32071 CrossRef CAS PubMed.
- A. Ikai, J. Biochem., 1980, 88, 1895–1898 CAS.
- T. Kar, U. Narsaria, S. Basak, D. Deb, F. Castiglione, D. M. Mueller and A. P. Srivastava, Sci. Rep., 2020, 10, 10895 CrossRef CAS PubMed.
- M. Foroutan, F. Ghaffarifar, Z. Sharifi and A. Dalimi, Comp. Immunol. Microbiol. Infect. Dis., 2020, 69, 101413 CrossRef PubMed.
- K. W. Boehme and T. Compton, J. Virol., 2004, 78, 7867–7873 CrossRef CAS PubMed.
- A. Xagorari and K. Chlichlia, Open Microbiol. J., 2008, 2, 49–59 CrossRef CAS PubMed.
- C. Vaure and Y. Liu, Front. Immunol., 2014, 5, 316 Search PubMed.
- V. Phongsisay, E. Iizasa, H. Hara and H. Yoshida, Mol. Immunol., 2015, 66, 463–471 CrossRef CAS PubMed.
- A. L. Totura, A. Whitmore, S. Agnihothram, A. Schäfer, M. G. Katze, M. T. Heise and R. S. Baric, mBio, 2015, 6, e00638-15 Search PubMed.
- W. Hu, Y.-T. Yen, S. Singh, C.-L. Kao and B. A. Wu-Hsieh, Viral Immunol., 2012, 25, 277–288 CrossRef CAS PubMed.
- S. F. Dosch, S. D. Mahajan and A. R. Collins, Virus Res., 2009, 142, 19–27 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06532g |
| This journal is © The Royal Society of Chemistry 2022 |