
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering bacteria to control electron transport altering the synthesis of non-native polymer†
Mechelle R. Bennetta,
Akhil Jain ab,
Katalin Kovacsb,
Phil J. Hillc,
Cameron Alexanderd and
Frankie J. Rawson*a
ab,
Katalin Kovacsb,
Phil J. Hillc,
Cameron Alexanderd and
Frankie J. Rawson*a
aDivision of Regenerative Medicine and Cellular Therapies, Biodiscovery Institute, School of Pharmacy, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Frankie.Rawson@nottingham.ac.uk
bSynthetic Biology Research Centre, School of Life Sciences, University of Nottingham, University Park,, Nottingham, NG7 2RD, UK
cDivision of Microbiology, Brewing and Biotechnology, School of Bioscience, University of Nottingham, Sutton Bonington Campus, Nottingham LE15 5RD, UK
dDivision of Molecular Therapeutics and Formulation, Boots Science Building, School of Pharmacy, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 21st December 2021
Abstract
The use of bacteria as catalysts for radical polymerisations of synthetic monomers has recently been established. However, the role of trans Plasma Membrane Electron Transport (tPMET) in modulating these processes is not well understood. We sort to study this by genetic engineering a part of the tPMET system NapC in E. coli. We show that this engineering altered the rate of extracellular electron transfer coincided with an effect on cell-mediated polymerisation using a model monomer. A plasmid with arabinose inducible PBAD promoters were shown to upregulate NapC protein upon induction at total arabinose concentrations of 0.0018% and 0.18%. These clones (E. coli(IP_0.0018%) and E. coli(IP_0.18%), respectively) were used in iron-mediated atom transfer radical polymerisation (Fe ATRP), affecting the nature of the polymerisation, than cultures containing suppressed or empty plasmids (E. coli(IP_S) and E. coli(E), respectively). These results lead to the hypothesis that EET (Extracellular Electron Transfer) in part modulates cell instructed polymerisations.
1. Introduction
Electron transfer plays a fundamental role in modulating biology, enabling almost all cellular functions.1 Transplasma membrane electron transport systems [tPMETs] are essential from a biological energetic perspective and for mediation of cell redox and signaling.2,3 It has recently been shown that electron transfer via tPMETs can be used to synthesise biopolymers (defined as polymers synthesised by cells) and may have a role in radical polymerisations of synthetic monomers.4–8 This was achieved by tPMET modulating the redox state of metal ions facilitating atom transfer radical polymerisation or redox-mediated oxidative polymerisation events. Importantly, the fabrication of in situ generated biopolymers via microbes, opens new strategies for interfacing cells with new materials and may find applications controlling cell behaviour and connecting cellular bioelectrical relays and improving application in microbial fuel cells.8–12 To date however, the underlying cellular redox systems that modulate polymerisation have not been studied in depth. One area that is expected to modulate the synthesis of polymers is bacterial electron transfer, however, there is a lack of understanding of how biologically derived electron transfer affects polymer synthesis.One approach to controlling bacterial electron transfer is to adopt synthetic biology protocols to tune bacterial behaviour.13 Consequently, we were inspired to explore a synthetic biology approach to tune electron transfer from bacteria for Fe ATRP under aqueous conditions. It has been previously shown that electrogenic Cupriavidus metallidurans (C. met)14–17 and Escherichia coli (E. coli) are able to start Fe ATRP reactions.4 Metal ion reduction takes place via microbial external electron transfer (EET) involving membrane bound C type cytochrome (C-Cyt) proteins, possibly in combination with synergistic Fe homeostasis efflux pumps.9 To further understand the role of microbial EET in Fe catalysis and resulting polymerisation, C-Cyt (NapC of E. coli) was our engineering target. C-Cyt was chosen as a target to mediate cell instructed ATRP as cytochromes are known to play a substantial role in bacterial EET that is required for energy production, particularly involving the reduction of metal ions.18–21 The electron transport chain of S. oneidensis MR1 is well researched and the corresponding C-Cyts can contribute to metal ion reduction,22,23 with protein CymA integral to the reduction of iron.24 NapC is also a member of the NapC/NirT family of C-Cyt, existing in the periplasmic membrane of E. coli.25 NapC showed ferric reductase activity like its homologue, CymA, with the ability to substitute for CymA when inserted into Shewanella strains.26 As E. coli K12 has been previously shown to initiate ATRP with Cu-,7 and Fe-catalysts,4 it was sought as a vehicle to investigate the effect of EET on polymerisation by altering NapC levels. To study the resulting change in EET we used an electrochemical method that we have recently reported.2,27 Herein we find that overexpression of the NapC protein in E. coli alters EET and this changes the kinetics of the reported cell instructed bio-mediated polymerisations.
2. Methods
2.1 Materials
All chemicals were purchased from the supplier and used without further purification unless stated. Iron(II) chloride hydrate FeCl2·4H2O and L-arabinose were purchased from Sigma Aldrich. Iron(III) chloride hexahydrate (FeCl3·6H2O) ≥98% was purchased from Scientific Laboratory supplies. Ascorbic acid (AscA) >99% was purchased from Alfa Aesar. For bacteria growth lysogeny broth (LB) was used. GenElute™ bacterial genomic DNA kit was purchased from Sigma-Aldrich. Monarch® plasmid miniprep kit, Monarch® DNA gel extraction kit and Monarch® PCR & DNA cleanup kit was purchased from New England Biolabs (NEB). Pierce™ BCA protein assay kit was purchased from Thermo Fisher Scientific. Polyethylene glycol methacrylate (PEGMA) and 2-hydroxyethyl 2-bromoisobutyrate (ATRP inhibitor; HEBIB) were purchased from Sigma, UK.napC gene sequence
ATGGGAAATTCTGACCGTAAGCCTGGTCTGATTAAGCGCCTGTGGAAATGGTGGCGTACCCCCAGCCGTCTGGCGCTGGGGACGCTGCTGTTGATCGGTTTTGTTGGCGGCATCGTCTTCTGGGGTGGCTTTAACACCGGGATGGAAAAAGCCAATACCGAAGAGTTCTGCATTAGCTGCCACGAAATGCGCAACACGGTGTATCAGGAATACATGGATTCCGTGCACTACAACAACCGTAGCGGCGTCCGTGCGACCTGTCCGGATTGTCACGTTCCGCACGAGTTTGTGCCGAAGATGATACGCAAGCTCAAAGCAAGTAAAGAGCTGTATGGTAAAATTTTTGGCGTTATTGACACGCCGCAGAAATTTGAAGCTCATCGTCTGACGATGGCACAGAATGAGTGGCGGCGCATGAAGGACAATAACTCGCAGGAGTGCCGTAACTGTCACAACTTCGAGTATATGGATACAACCGCC CAGAAATCGGTTGCCGCGAAGATGCATGACCAGGCGGTGAAAGATGGGCAAAC CTGTATTGATTGCCATAAAGGGATAGCGCACAAGCTGCCCGATATGCGTGAAGTCGAGCCAGGTTTTTAA (sourced using Kegg genome database for Escherichia coli K-12 MG1655: b2202, https://www.genome.jp/dbget-bin/www_bget?eco:b2202).
2.2 Bacteria, plasmid and primers
Bacterial strains, plasmids and primers used in this work are listed in Tables S1 and S2,† respectively.2.3 Storage, growth conditions and transformation
Bacterial cultures were stored at – 80 °C on beads from Microbank™ long term bacterial storage system (Prolabs Diagnostics). For recovery of cultures, E. coli top 10 wild type were grown from beads at 37 °C overnight (at a fixed 18 hours for all cultures) in Lysogeny Broth (LB; 5 mL) with agitation. E. coli harbouring pMTL8000 series plasmids with Cm resistance gene (catP) were grown from beads at 30 °C overnight (18 hours) in LB (5 mL) and 2.5 μL Cm stock solution with agitation. The chemical transformation of complete plasmids into competent cells was carried out according to the NEB chemical transformation protocol. Chemically competent cells were thawed for 10 minutes on ice and then 15 μL of plasmid DNA was added. The mixture was incubated on ice for 30 minutes before being heat shocked at 42 °C for exactly 30 seconds. Afterwards, they were placed on ice for another 5 minutes and later 500 μL SOC (Super Optimal broth with Catabolite repression) media (NEB) was added before incubating at 37 °C for 60 minutes. After this, the cells were spread on agar plates (with chloramphenicol, Cm) and left to grow at 24 °C for 4 days.2.4 Construction of inducible promoter vector containing for NapC overexpression
For overexpression of NapC, pMTL83153_PBAD_NapC vector containing an inducible promoter PBAD upstream of napC was generated. Firstly, napC region was isolated from E. coli gDNA and amplified through PCR. During the next step, an arabinose inducible promoter (PBAD_araC) region was obtained through PCR amplification from the plasmid pMTL71101_PBAD_araC using PBAD_araC_fwd and PBAD_araC_rev primers. Next, the plasmid pMTL83153 was digested with restriction enzymes Not1 and Sal1 to remove the Pfdx constitutive promoter. Finally, Hifi ligation assembly was carried out to insert the PBAD_araC and napC region into the pMTL83153_(-Pfdx) vector to obtain pMTL83153_PBAD_NapC inducible promoter vector.2.5 Colony PCR and sequencing
Colony PCR (primers: ColE1+tra_F2 and pCB102_R1) was conducted to determine napC insertion into pMTL83153_PBAD_NapC inducible promoter vector. Colony PCR amplified DNA regions were sequenced using sanger sequencing (Source Bioscience service) and results analysed using Benchling sequence alignment tool.2.6 NapC expression and quantification
Clones containing the PBAD promoter were induced using L-arabinose according to manufacturer protocol in manual ‘pBAD/His A, B, and C pBAD/Myc-His A, B, and C’ (Invitrogen™). Briefly, a culture of the clone was grown in 5 mL LB with 2.5 μL Cm stock at 30 °C overnight. A total of 3 tubes containing 10 mL LB were labelled and 5 μL culture was added to achieve OD600 nm 0.05. These were grown to OD600 nm 0.5 and arabinose (0.1 mL, Table S3†) was added to each. After 8 hours (or stationary phase achieved), 2 mL of each culture was harvested by centrifugation (8000 rpm, 10 minutes, 4 °C) and stored at −20 °C overnight. The total protein content was evaluated using Pierce™ BCA protein assay kit (Thermo Scientific™) according to the manufacturer's instructions. A BSA standard sample was analysed for protein content using the BCA assay at different concentrations to create a standard curve (Fig. S2†). A standard curve was used to compare the absorbance of protein from cell lysates to calculate total protein concentration in each sample. SDS-PAGE was performed to confirm the expression of NapC.2.7 Arabinose toxicity
Cultures were grown overnight in 5 mL of LB with 2.5 μL Cm. These were adjusted to 0.05 OD in 2 mL LB (+Cm) and grown to OD600 0.4. Finally, the cultures were induced with arabinose at a predetermined concentration as shown in Table S3.†2.8 Fe atom transfer radical polymerisation (ATRP)
E. coli(IP) cultures were grown and induced according to protocol mentioned in earlier section (NapC expression and quantification). The induced clones were grown to OD600 ∼1.1 (20 mL LB + 10 μL Cm), centrifuged (6000 rpm, 20 min), washed using PBS and then resuspended in degassed PBS (1.5 mL, approximately 1.35 × 1010 CFU mL−1 in final reaction volume (4 mL)) before using in the Fe ATRP reactions. For each reaction an appropriate volume of FeCl3·6H2O and Me6TREN (Table S5†) were added to a 5 mL Eppendorf in PBS under stirring. Alongside in separate tubes, polyethylene glycol methacrylate (PEGMA) and 2-hydroxyethyl 2-bromoisobutyrate (ATRP inhibitor; HEBIB) were dissolved in PBS. The above mixture was placed in the anaerobic cabinet for >1 hour to allow degassing. After degassing, bacteria were added to FeCl3/Me6TREN mix and pre-mixed for 15 minutes before adding a monomer/initiator mixture. The reaction was left overnight and terminated by exposure to air. The kinetics of each reaction were monitored by 1H NMR and the resulting polymer were analysed with Size Exclusion Chromatography (SEC).2.9 Electrochemical studies
Linear Sweep Voltammetry (LSV) analyses were carried out using a 3-electrode system: carbon fibre micro-disk working electrode (33 μm), Ag/AgCl reference electrode and platinum wire counter electrode from ALS Co. Ltd, Japan. Experiments were conducted using potentiostat ‘Autolab PGStat302A’ with low current module (EDC). Measurements and analysis were carried out using NOVA 2.1 software. All electrochemical experiments were carried out in 1× phosphate buffer saline solution (PBS) supporting electrolyte, at current range 100 pA, scan rate 100 mV s−1 from 1.25 V to −0.25 V at room temperature.2.10 Calibration curve for Fe reduction
For the calibration curve, LSV of potassium ferricyanide and ferrocyanide (1 mM) mixed in different ratios (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 8:2, 5:5, 2:8, 0:10) was performed in PBS (10 mM). A sample of PBS was applied to all measurements as a control baseline subtraction. A PK-3 electrode polishing kit (ALS Co. Ltd) was used to polish the working electrode between measurements. The first derivative functions of the calibration voltammograms were taken to create a cross-referencing tool to determine steady state reduction (Iss,red), steady state oxidation (Iss,ox) peaks and extract the rate of change (d(Iss)/dt). The concentration of the redox analyte was calculated using Randles–Sevcik equation. The steady state current (Iss) is measured:
:0, 8:2, 5:5, 2:8, 0:10) was performed in PBS (10 mM). A sample of PBS was applied to all measurements as a control baseline subtraction. A PK-3 electrode polishing kit (ALS Co. Ltd) was used to polish the working electrode between measurements. The first derivative functions of the calibration voltammograms were taken to create a cross-referencing tool to determine steady state reduction (Iss,red), steady state oxidation (Iss,ox) peaks and extract the rate of change (d(Iss)/dt). The concentration of the redox analyte was calculated using Randles–Sevcik equation. The steady state current (Iss) is measured:| Iss = 4nFDCr |
Finally, the calibration graph was created by plotting the d(Iss)/dt peak values against ferricyanide or ferrocyanide concentrations. The lines of best fit could then be compared to first derivative values of LSV graphs for samples reduced by bacterial.
2.11 1H NMR
1H NMR spectra were recorded at room temperature on a 400 MHz (Bruker DPX400 Ultrashield) using deuterated solvents (D2O). NMR spectra were analysed using MestReNova 11.0.0-17609 2016 Mestrelab Research S.L.2.12 Size exclusion chromatography (SEC)
THF SEC was performed Shimadzu prominence LC-20AD system equipped with 2× Agilent PLgel Mixed-D columns heated to 40 °C. Samples were eluted with THF + 2% TEA + 0.01% BHT (flow rate 1 mL min−1) over 30 min. Injection vol. was set to 50 μL. Mn and Đ were calculated using PMMA standards (InfinityLab EasiVial) with MWs ranging from 600–1.5 million g per mol.2.13 Electrochemical determination of Fe3+
Bacteria were induced/grown overnight (10 mL, LB + Cm) and pellets were resuspended in potassium ferricyanide (5 mL, 1 mM) for 1 hour at 37 °C. Bacteria were removed by centrifugation (6000 rpm, 10 minutes) and the supernatant was analysed by LSV. Prior to incubation with ferricyanide clones were either (i) suppressed by addition of glucose E. coli(IP_S), (ii) activated by 0.0018% total arabinose concentration E. coli(IP_0.0018%) or (ii) activated by 0.18% total arabinose concentration E. coli(IP_0.18%). Three aliquots of each supernatant sample were scanned (n = 3) using LSV. The whole experiment was repeated twice with new biological samples. The first derivative of the voltammograms was taken and Fe concentrations were calculated using the calibration graph.3. Results and discussion
We initially took a synthetic biology approach to rewire tPMET behaviour. We began by cloning napC gene isolated from E. coli into the plasmid pMTL83153 (details are given in Table S1†), immediately downstream from the non-constitutive inducible promoter (PBAD). We chose PBAD because high quantities of transcribed protein (NapC) were desired to facilitate investigations into its effect on iron reduction which is indicative of EET. PBAD would also allow us to control any potential toxicity and NapC expression by optimising the concentration of inducer (arabinose). To achieve this, a pMTL8000 modular plasmid collection was designed for ease of component selection during cloning. This can also be useful for tuning via plasmid replicons (controls replication efficiency), markers (antibiotic resistance selection), promoters (drives the transcription of the target gene), and multiple cloning sites (MCSs) (containing restriction sites for restriction enzyme cloning).28 Although these were created to aid cloning in Clostridium cultures, they are hosted in E. coli and so were convenient in the cloning of napC into E. coli. The plasmid constructed, based on pMTL83153 (Fig. 1a), confers resistance to chloramphenicol29 and napC under the control of PBAD.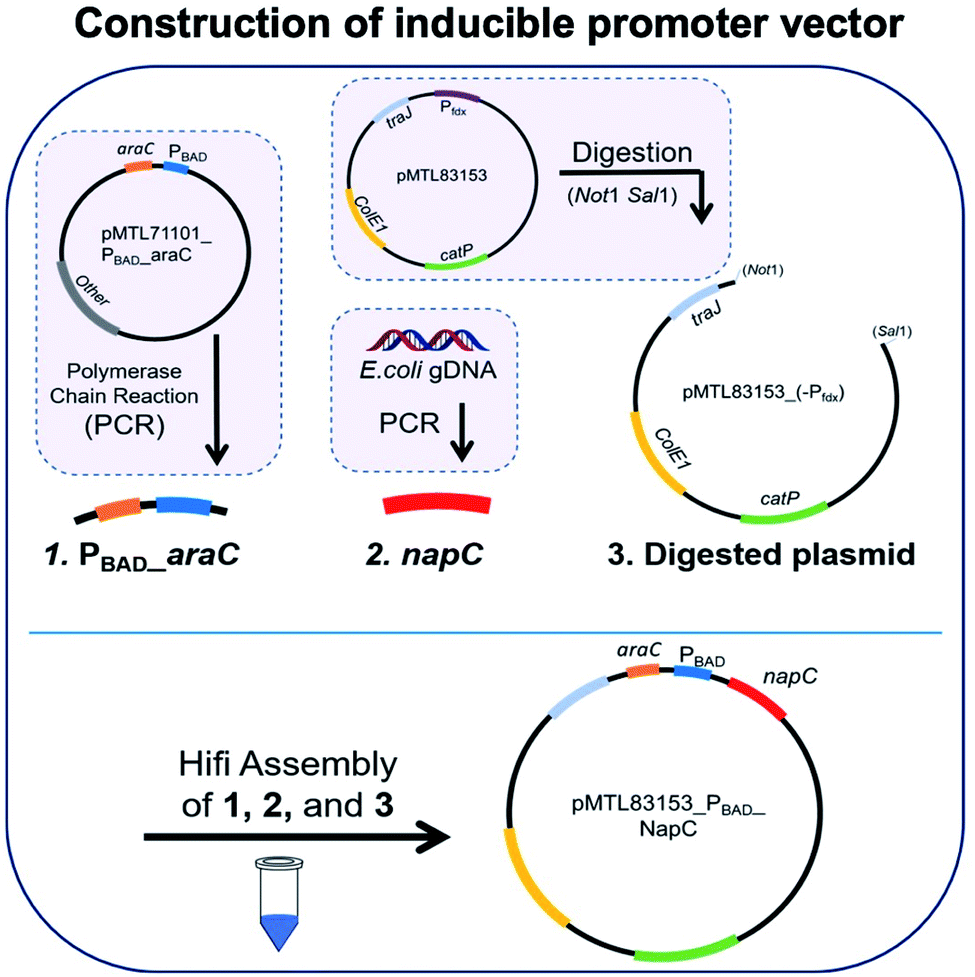 | ||
| Fig. 1 Assembly of inducible promoter vector containing promoter PBAD for NapC overexpression and control. PCR was carried out with specific primers to extract and amplify regions (1) PBAD_araC and (2) napC. Region 1 was obtained from the plasmid pMTL71101_PBAD_araC and region 2 was obtained from E. coli gDNA. The plasmid pMTL83153 was digested with REs NotI and SalI to remove the Pfdx promoter, resulting in region 3, digested plasmid. The three regions were ligated together with Hifi assembly to create the completed vector. Gel electrophoresis of colony PCR products for DNA regions making up the inducible promoter vector, against 1 KB Plus DNA ladder PBAD_araC DNA region (1278 bps) and napC DNA region (633 bps) are as expected. Sanger sequencing diagrams for inducible promoter vector showing matching DNA regions of sequencing with forward and reverse primers compared to a model sequence. | ||
Plasmid pMTL83153 was assembled using the napC gene and PBAD promoter region, amplified from E. coli and plasmid pMTL71101_PBAD_araC, respectively, amplified and purified. These napC and PBAD regions were inserted in Not and SalI digested pMTL83153. The transformation of the cloned vectors led to the growth of colonies that were selected on Cm plates and screened by colony PCR (primers: ColE1+tra_F2 and pCB102_R1; Table S2†). Gel electrophoresis was used to analyse the resultant amplicons and confirm the successful insertion of PBAD_araC DNA region (1278 bps) and napC DNA region (633 bps) (Fig. 1b). Finally, the authenticity of the cloned pMTL83153_PBAD_NapC inducible promoter vector colonies (E. coli(IP)) was confirmed using Sanger sequencing of the amplified DNA fragments (Fig. 1c) and designated pBADNAP.
After the successful cloning of napC, we examined the resulting NapC protein expression by E. coli. alongside clones harbouring a control plasmid, pMTL83151, referred to as the ‘empty plasmid’ (E. coli(E)). NapC expression in bacteria containing pBADNAP (E. coli(BADNAP)) was induced by the addition of arabinose, which binds to AraC, activating the PBAD promoter and initiates transcription of the protein. This expression was tuned by optimising the arabinose concentration (Table S3†) and its effect on NapC content was examined by performing SDS-PAGE (Fig. S1†). E. coli(BADNAP) were exposed to a final arabinose concentration of 0% (band I0), 0.000018% (band I1), 0.0018% (band I2), and 0.18% (band I3). As a control measure, arabinose was also added to E. coli(E) (band E), permitting a consistent comparison between protein expression levels. The total protein quantification was analysed using bicinchoninic acid (BCA) protein assay (Fig. S2 and Table S4†). A certain band of interest was identified in the resulting protein gel (Fig. S1 and S3,† circled green area).25 From the BCA assay the maximum expression was found when at 0.00018% inducer was used.
As shown previously, Fe ATRP can be activated by bacteria including E. coli.4 To investigate the effects of changing NapC expression on Fe reduction rates, E. coli(BADNAP) were used in Fe ATRP reactions (Table S5†) alongside E. coli(E). An iron catalysed ATRP of the water-soluble monomer poly(ethylene glycol)methyl ether methacrylate (PEGMA, Mn = 300 gmol @ 1) was carried out at 37 °C with tris(2-dimethylaminoethyl)amine (Me6TREN) for Fe3+ reduction. The kinetics of each reaction was monitored by 1H NMR (Fig. 2a) and the resulting polymers were analysed by SEC (Fig. S4† and Table 1). A larger polymer yield for E. coli(BADNAP_0.0018%) and E. coli(BADNAP_0.18%) activated reactions was observed, compared to those activated by E. coli(E) or E. coli(BADNAP_S) cultures. Importantly, all cell polymerisation studies were performed when cells had reached an OD of 500 and therefore observed effects were not due to differing growth rates. This suggested that the upregulation of the NapC protein had some effect on the rates of Fe3+ reduction to Fe2+. As this difference is small, it suggests that there could be other factors contributing to EET from the bacteria to the Fe catalyst. These might include electron transfer (ET) across the periplasm via other cascade proteins in the electron transport chain, or EET between shuttle molecules and the Fe catalyst (Fig. 2b).
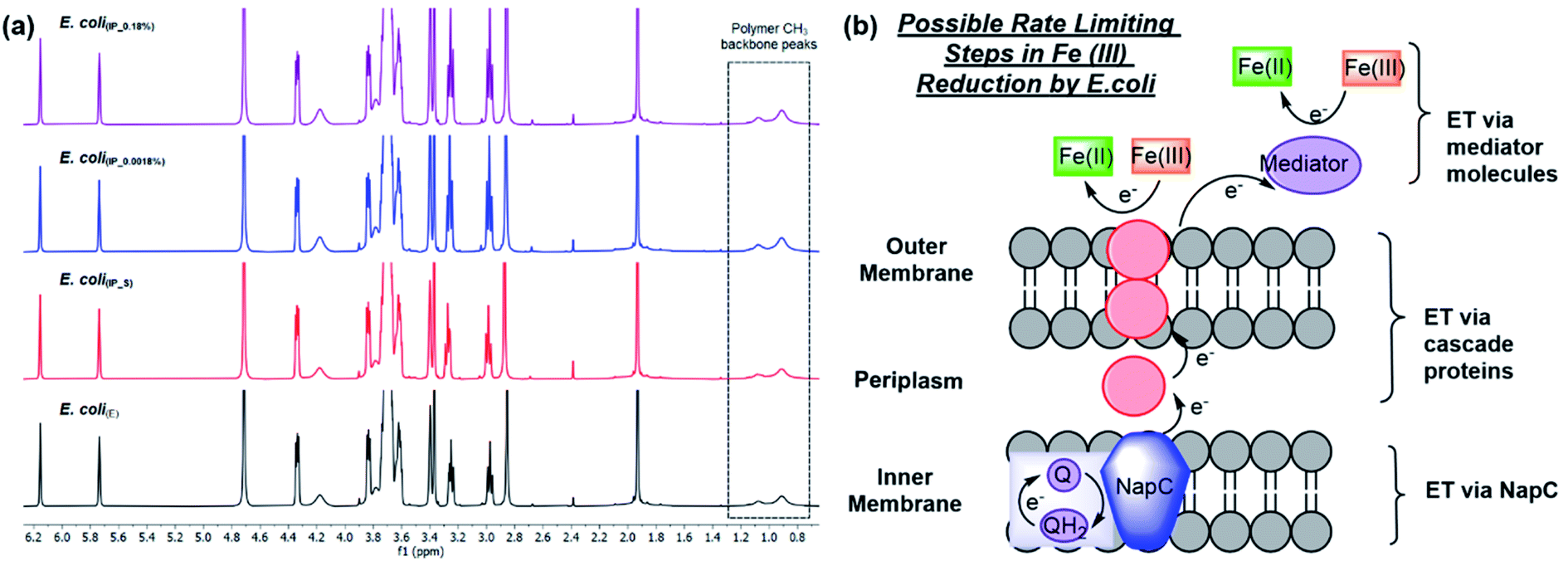 | ||
| Fig. 2 Fe ATRP polymerisations activated by E. coli cultures harbouring different plasmids to compare the effects of NapC protein upregulation. (a) 1H NMR spectra of Fe ATRP activated by E. coli(E) (black), or inducible promoter plasmids, E. coli(IP) either (i) suppressed by addition of glucose E. coli(IP_S) (red), (ii) activated by 0.0018% total arabinose concentration E. coli(IP_0.0018%) (blue) or (ii) activated by 0.18% total arabinose concentration E. coli(IP_0.18%) (purple). (b) Possible rate limiting steps in Fe(III) reduction including electron transfer (ET) via NapC, ET via cascade proteins and ET via mediator molecules. | ||
| Culture | Conversion | Mnth (kDa) | MnSEC (kDa) | Đ[c] |
|---|---|---|---|---|
| E. coli(E) | 38.4% | 23.3 | 251.9 | 3 |
| E. coli(IP_S) | 38.8% | 23.5 | 237.4 | 2.9 |
| E. coli(IP_0.0018%) | 46.5% | 28.1 | 228 | 2.7 |
| E. coli(IP_0.18%) | 45.6% | 27.6 | 230 | 3 |
To note the Đ[c] values suggest the polymerisation is not very well controlled. However, we do show bioelectrical activity is one factor affecting the mechanism of polymerisation and will be discussed in more detail in the proceeding experiments. However, more research is required to deconstruct the other biological controlling factors.
We next wanted to study the effect on electron transfer when altering the NapC expression levels of the tPMET. This was achieved using linear sweep voltammetry (LSV), an electrochemical technique that reports on the concentration of ferricyanide and ferrocyanide (Fe reduction), which is indicative of the electron transfer rate when measured over time. We have previously reported this LSV based method to detect concentration changes corresponding to EET in cells.27,30,31 In brief, the first derivative function of the LS voltammograms (Fig. S5†) obtained from known concentrations of ferricyanide/ferrocyanide redox couple was used to calculate the rate of change in steady-state current (d(Iss)/dt) (Fig. 3a). A calibration curve of concentration vs. (d(Iss)/dt) (Fig. S6†) was generated to determine whether E. coli(IP) clones with upregulated NapC protein could reduce more Fe3+ in the form of ferricyanide. Next, the samples of E. coli; E. coli(E), E. coli(IP_S) (suppressed by addition of glucose to inhibit PBAD activation of NapC expression),32 E. coli(IP_0.0018%), and E. coli(IP_0.18%) were incubated with ferricyanide for 1 hour at 37 °C, after which the bacteria were removed by centrifugation and the supernatant was taken and LSV was performed on the supernatant. The average current was taken for each sample and the first derivative graph was plotted to determine d(Iss)/dt values. Finally, the d(Iss)/dt values of each sample were compared to the calibration graph and the concentration of ferrocyanide (Fe2+) in solution was plotted as a percentage of total Fe concentration.
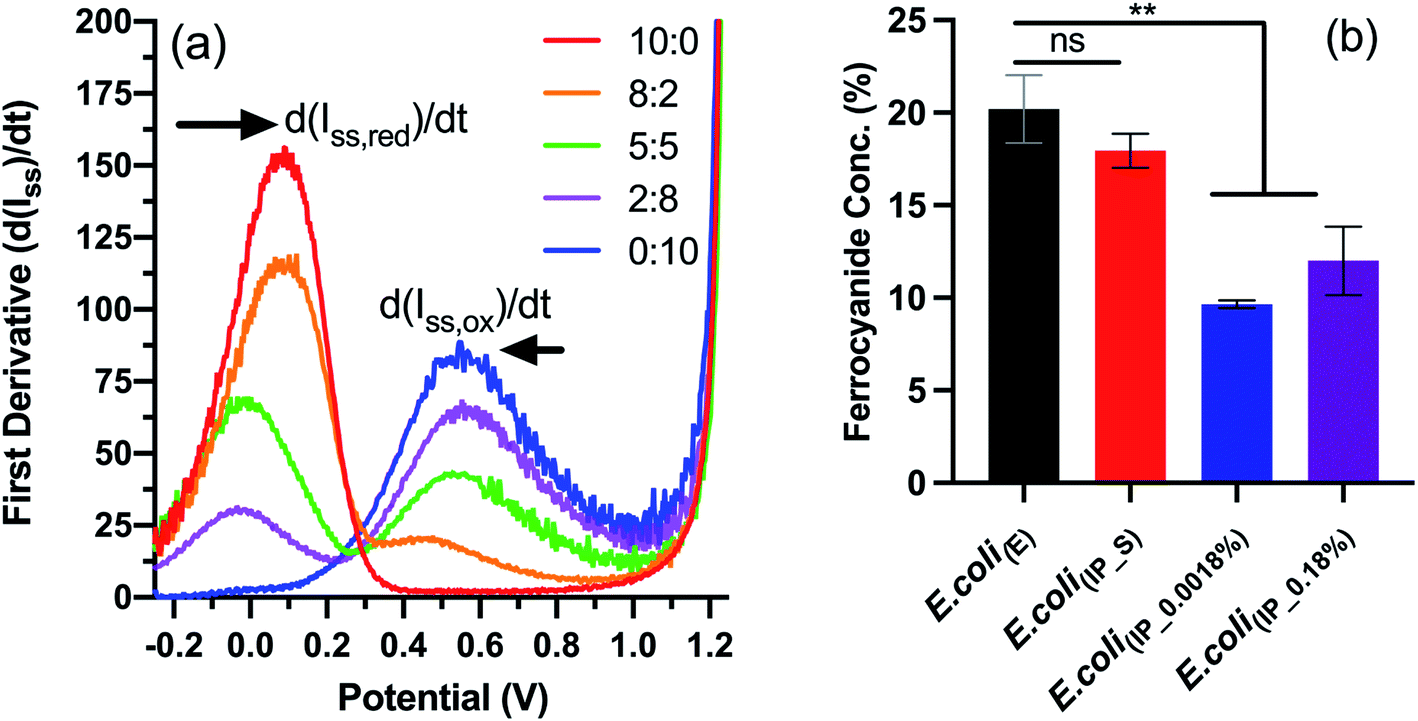 | ||
| Fig. 3 Electrochemical detection of ferrocyanide using linear sweep voltammetry. (a) First derivative function applied to linear sweep voltammogram of ferricyanide/ferrocyanide redox couple (Fig. S6†) to determine d(Iss)/dt values. (b) Concentrations (%) of ferrocyanide detected in the supernatant of samples incubated for 1 hour with E. coli(IP) or E. coli(E). Results are expressed as mean ± S.D. ** P < 0.005 vs. E. coli(E), obtained using 1-way ANOVA with a Dunnett's post-test. LSV was used to analyse the supernatant of incubated samples (N = 2, n = 6) and the first derivative function was applied to resulting voltammograms. The concentrations were determined using the calibration graph showed in Fig. S7.† | ||
The data indicate that E. coli(E) and E. coli(IP_S) cultures reduced more ferricyanide to ferrocyanide than the E. coli(IP) clones (Fig. 3b). This is of note because higher ferricyanide reduction was expected from E. coli clones that were induced with arabinose as indicated by the western blot which indicated increased NapC expression when compared to the cells modified with the control plasmid (Fig. S3†). We attribute this to a shift in the bioenergetics of the bacteria, that could have been triggered in E. coli(IP_0.0018%) and E. coli(IP_0.18%) cultures, whereby EET reduction pathways were stunted due to the use of excess energy required for the over-production of the NapC protein. Interestingly, there was a slight increase in the ferricyanide reduction with E. coli(IP_0.18%) cultures compared to E. coli(IP_0.0018%) cultures. We show arabinose induction as 0.18% slowed the growth of E. coli(IP) (Fig. S7†). This increased stress in E. coli(IP_0.18%) cultures could have triggered EET systems (such as NapC) increasing ferricyanide reduction. Stress induced tPMET upregulation pathways were also observed in analogous experiments with other cell types,2 suggesting that cells utilise EET systems to balance bioenergetic requirements. Although the bioenergetics of Fe metabolism in living organisms remains challenging to study,33 further studies are underway to gain more insights through monitoring Fe reducing behaviour in stress-induced environments, such as temperature, pH and chemical treatments.
4. Conclusions
In summary, the cloning of the napC gene into E. coli was achieved and controlled with the PBAD promoter. We established that there were differences in electron transfer rates dependent on the arabinose concentration. The nature of polymerisation for ATRP catalysed by E. coli(IP_0.0018%) and E. coli(IP_0.0018%) were different, suggesting that NapC regulation has some effect on the Fe3+ reduction system. This subsequently affects the nature of the polymerisation. On the modification of NapC levels, there appear to be alterations in extracellular electron transfer which coincides with variations in differences in polymerisation kinetics.Author contributions
Conceptualization: M. R. B., F. J. R., C. A. P. J. H., data curation: M. R. B., F. J. R., C. A. and K. K. Formal analysis: M. R. B., F. J. R., and K. K. Funding acquisition: F. J. R. Investigation: M. R. B. Methodology: M. R. B., F. J. R. and C. A. Project administration: F. J. R. Resources: F. J. R., C. A., P. J. H., K. K. Supervision: C. A., P. J. H., F. J. R. Validation: M. R. B., A. J., C. A., F. J. R. Visualization: F. J. R., M. R. B. and C. A. Writing: A. J., M. R. B. and F. J. R. Writing – review & editing: A. J., M. R. B., F. J. R., C. A., P. J. H., K. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (grant numbers EP/R004072/1, EP/N03371X/1), the Biotechnology and Biological Sciences Research Council (grant number BB/L013940/1), and the Royal Society (Wolfson Research Merit Award WM150086 to C. A.). We thank Christian Arenas Lopez and Michaella Whittle for assistance with bacterial cell culture.Notes and references
- J. Blumberger, Recent advances in the theory and molecular simulation of biological electron transfer reactions, Chem. Rev., 2015, 115(20), 11191–11238 CrossRef CAS PubMed.
- H. G. Sherman, C. Jovanovic, A. Abuawad, D.-H. Kim, H. Collins, J. E. Dixon, R. Cavanagh, R. Markus, S. Stolnik and F. J. Rawson, Mechanistic insight into heterogeneity of trans-plasma membrane electron transport in cancer cell types, Biochim. Biophys. Acta, Bioenerg., 2019, 1860(8), 628–639 CrossRef CAS PubMed.
- A. J. Robinson, A. Jain, H. G. Sherman, R. J. Hague, R. Rahman, P. Sanjuan-Alberte and F. J. Rawson, Toward Hijacking Bioelectricity in Cancer to Develop New Bioelectronic Medicine, Adv. Ther., 2021, 4(3), 2000248 CrossRef CAS.
- M. R. Bennett, P. Gurnani, P. J. Hill, C. Alexander and F. J. Rawson, Iron-Catalysed Radical Polymerisation by Living Bacteria, Angew. Chem., 2020, 132(12), 4780–4785 CrossRef.
- G. Fan, C. M. Dundas, A. J. Graham, N. A. Lynd and B. K. Keitz, Shewanella oneidensis as a living electrode for controlled radical polymerisation, Proc. Natl. Acad. Sci., 2018, 115(18), 4559–4564 CrossRef CAS PubMed.
- G. Fan, A. J. Graham, J. Kolli, N. A. Lynd and B. K. Keitz, Aerobic radical polymerisation mediated by microbial metabolism, Nat. Chem., 2020, 12(7), 638–646 CrossRef CAS PubMed.
- E. P. Magennis, F. Fernandez-Trillo, C. Sui, S. G. Spain, D. J. Bradshaw, D. Churchley, G. Mantovani, K. Winzer and C. Alexander, Bacteria-instructed synthesis of polymers for self-selective microbial binding and labelling, Nat. Mater., 2014, 13(7), 748–755 CrossRef CAS PubMed.
- A. Ramanavicius, E. Andriukonis, A. Stirke, L. Mikoliunaite, Z. Balevicius and A. Ramanaviciene, Synthesis of polypyrrole within the cell wall of yeast by redox-cycling of [Fe(CN)6]3−/[Fe(CN)6]4−, Enzyme Microb. Technol., 2016, 83, 40–47 CrossRef CAS PubMed.
- M. Ishikawa, K. Kawai, M. Kaneko, K. Tanaka, S. Nakanishi and K. Hori, Extracellular electron transfer mediated by a cytocompatible redox polymer to study the crosstalk among the mammalian circadian clock, cellular metabolism, and cellular redox state, RSC Adv., 2020, 10(3), 1648–1657 RSC.
- M. Kaneko, K. Ishihara and S. Nakanishi, Redox-Active Polymers Connecting Living Microbial Cells to an Extracellular Electrical Circuit, Small, 2020, 16(34), 2001849 CrossRef CAS PubMed.
- S. Ramanavicius and A. Ramanavicius, Conducting Polymers in the Design of Biosensors and Biofuel Cells, Polymers, 2021, 13(1), 49 CrossRef CAS PubMed.
- R.-M. Apetrei, G. Carac, A. Ramanaviciene, G. Bahrim, C. Tanase and A. Ramanavicius, Cell-assisted synthesis of conducting polymer – polypyrrole – for the improvement of electric charge transfer through fungal cell wall, Colloids Surf., B, 2019, 175, 671–679 CrossRef CAS PubMed.
- K. Stephens and W. E. Bentley, Synthetic biology for manipulating quorum sensing in microbial consortia, Trends Microbiol., 2020, 633–643 CrossRef CAS PubMed.
- M. J. Colombo, J. Ha, J. R. Reinfelder, T. Barkay and N. Yee, Oxidation of Hg(0) to Hg(II) by diverse anaerobic bacteria, Chem. Geol., 2014, 363, 334–340 CrossRef CAS.
- A. Espinoza Tofalos, M. Daghio, M. González, M. Papacchini, A. Franzetti and M. Seeger, Toluene degradation by Cupriavidus metallidurans CH34 in nitrate-reducing conditions and in bioelectrochemical systems, FEMS Microbiol. Lett., 2018, 365(12), fny119 Search PubMed.
- F. Reith, B. Etschmann, C. Grosse, H. Moors, M. A. Benotmane, P. Monsieurs, G. Grass, C. Doonan, S. Vogt and B. Lai, Mechanisms of gold biomineralization in the bacterium Cupriavidus metallidurans, Proc. Natl. Acad. Sci., 2009, 106(42), 17757–17762 CrossRef CAS PubMed.
- N. Wiesemann, L. Bütof, M. Herzberg, G. Hause, L. Berthold, B. Etschmann, J. Brugger, G. Martinez-Criado, D. Dobritzsch and S. Baginsky, Synergistic toxicity of copper and gold compounds in Cupriavidus metallidurans, Appl. Environ. Microbiol., 2017, 83(23), 1–17 CrossRef CAS PubMed.
- M. J. Edwards, G. F. White, C. W. Lockwood, M. C. Lawes, A. Martel, G. Harris, D. J. Scott, D. J. Richardson, J. N. Butt and T. A. Clarke, Structural modeling of an outer membrane electron conduit from a metal-reducing bacterium suggests electron transfer via periplasmic redox partners, J. Biol. Chem., 2018, 293(21), 8103–8112 CrossRef CAS PubMed.
- H. M. Jensen, M. A. TerAvest, M. G. Kokish and C. M. Ajo-Franklin, CymA and exogenous flavins improve extracellular electron transfer and couple it to cell growth in Mtr-expressing Escherichia coli, ACS Synth. Biol., 2016, 5(7), 679–688 CrossRef CAS PubMed.
- K. Tanaka, S. Yokoe, K. Igarashi, M. Takashino, M. Ishikawa, K. Hori, S. Nakanishi and S. Kato, Extracellular electron transfer via outer membrane cytochromes in a methanotrophic bacterium Methylococcus capsulatus (Bath), Front. Microbiol., 2018, 9, 2905 CrossRef PubMed.
- G. F. White, M. J. Edwards, L. Gomez-Perez, D. J. Richardson, J. N. Butt and T. A. Clarke, Mechanisms of bacterial extracellular electron exchange, Adv. Microb. Physiol., 2016, 68, 87–138 CrossRef CAS PubMed.
- H. M. Jensen, A. E. Albers, K. R. Malley, Y. Y. Londer, B. E. Cohen, B. A. Helms, P. Weigele, J. T. Groves and C. M. Ajo-Franklin, Engineering of a synthetic electron conduit in living cells, Proc. Natl. Acad. Sci., 2010, 107(45), 19213–19218 CrossRef CAS PubMed.
- M. Shi, Y. Jiang and L. Shi, Electromicrobiology and biotechnological applications of the exoelectrogens Geobacter and Shewanella spp, Sci. China: Technol. Sci., 2019, 62(10), 1670–1678 CrossRef.
- C. R. Myers and J. M. Myers, Cloning and sequence of cymA, a gene encoding a tetraheme cytochrome c required for reduction of iron (III), fumarate, and nitrate by Shewanella putrefaciens MR-1, J. Bacteriol., 1997, 179(4), 1143–1152 CrossRef CAS PubMed.
- M. L. Cartron, M. D. Roldán, S. J. Ferguson, B. C. Berks and D. J. Richardson, Identification of two domains and distal histidine ligands to the four haems in the bacterial c-type cytochrome NapC; the prototype connector between quinol/quinone and periplasmic oxido-reductases, Biochem. J., 2002, 368(2), 425–432 CrossRef CAS PubMed.
- J. S. Gescher, C. D. Cordova and A. M. Spormann, Dissimilatory iron reduction in Escherichia coli: identification of CymA of Shewanella oneidensis and NapC of E. coli as ferric reductases, Mol. Microbiol., 2008, 68(3), 706–719 CrossRef CAS PubMed.
- H. G. Sherman, C. Jovanovic, S. Stolnik and F. J. Rawson, Electrochemical system for the study of trans-plasma membrane electron transport in whole eukaryotic cells, Anal. Chem., 2018, 90(4), 2780–2786 CrossRef CAS PubMed.
- J. T. Heap, O. J. Pennington, S. T. Cartman and N. P. Minton, A modular system for Clostridium shuttle plasmids, J. Microbiol. Methods, 2009, 78(1), 79–85 CrossRef CAS PubMed.
- V. Hershfield, H. W. Boyer, C. Yanofsky, M. A. Lovett and D. R. Helinski, Plasmid ColE1 as a molecular vehicle for cloning and amplification of DNA, Proc. Natl. Acad. Sci., 1974, 71(9), 3455–3459 CrossRef CAS PubMed.
- F. J. Rawson, A. J. Downard and K. H. Baronian, Electrochemical detection of intracellular and cell membrane redox systems in Saccharomyces cerevisiae, Sci. Rep., 2014, 4(1), 1–9 Search PubMed.
- F. J. Rawson, A. J. Gross, D. J. Garrett, A. J. Downard and K. H. Baronian, Mediated electrochemical detection of electron transfer from the outer surface of the cell wall of Saccharomyces cerevisiae, Electrochem. Commun., 2012, 15(1), 85–87 CrossRef CAS.
- A. Roux, C. Beloin and J.-M. Ghigo, Combined inactivation and expression strategy to study gene function under physiological conditions: application to identification of new Escherichia coli adhesins, J. Bacteriol., 2005, 187(3), 1001–1013 CrossRef CAS PubMed.
- L. J. Bird, V. Bonnefoy and D. K. Newman, Bioenergetic challenges of microbial iron metabolisms, Trends Microbiol., 2011, 19(7), 330–340 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06403g |
| This journal is © The Royal Society of Chemistry 2022 |