DOI:
10.1039/D1RA05062A
(Review Article)
RSC Adv., 2022,
12, 1287-1309
The inchoate horizon of electrolyzer designs, membranes and catalysts towards highly efficient electrochemical reduction of CO2 to formic acid
Received
30th June 2021
, Accepted 30th October 2021
First published on 6th January 2022
Abstract
The economic viability of CO2 reactors is contingent on the selectivity of the CO2 reduction reaction and the rate of product formation. For this, the rational design of electrolyzers also has a substantial impact on the figures of merit (current density, faradaic efficiency, cell durability). Thus, herein we portray a short review on the shortcomings, challenges and the recent developments on different reactor configurations, components and membrane structures for the efficient electrochemical CO2 reduction (CO2R) into HCOO−/HCOOH. Despite their low CO2 solubility and poor mass transport, H-type electrolyzers are commercialized due to their screening of a vast number of catalysts. In contrast, membrane-based gas and liquid phase flow reactors break the barriers faced by H-types through the incorporation of gas diffusion electrodes (GDEs) and the membrane electrode assembly (MEA). As the GDE forms the gas–liquid–solid interface, it allows the electrolyzers to generate current densities at the industrial level (200 mA cm−2). Intriguingly, a continuous liquid fed intermittent flow electrolyzer can control the electrolyte flow at a desired frequency and allow sufficient time for CO2 gas molecules to effectively reduce into HCOOH. Therefore, a high and stable faradaic efficiency (95%) is achieved in 4 h for HCOOH (576.98 mg) using the boron-doped diamond catalyst. Very recently, a novel strategy to enhance the CO2R to HCOO−/HCOOH has been adopted via the recirculation of by-products to the liquid phase MEA flow reactors, which substantially improves HCOO− selectivity, lowers material costs, and promotes CO2 mass transfer. In the end, the zero-gap electrolyzer has newly emerged and affords reduced ohmic losses, leading to a straight-forward implementation of industrial systems for CO2R to value-added products in the future. Besides, the efficiency of HCOO−/HCOOH production is also explored against proton exchange, anion exchange and bipolar membranes, and the pH of the electrolyte plays a dominant role in deciding the stability and characteristics of the membranes. It is also depicted that the product selectivity depends on different electrolyzer configurations. Recently, bimetallic alloys (Bi–Sn, Bi–In) and 2D layered composites (SnO2/rGO/CNT) have proven to be potential electrocatalysts (faradaic efficiency > 95%, highly selective and durable) assigned to the abundant active sites for CO2R. Based on the recent findings and future research directions, we draw reader's attention to construct economic, scalable and energy-efficient CO2R electrolyzers to realize the techno-economic predictions.
| P. Senthilkumar P. Senthilkumar presently is a senior project associate at Council of Scientific & Industrial Research-Institute of Minerals and Materials Technology (CSIR-IMMT), Odhisa. He received his Ph.D. in Physics in 2020 under the mentorship of Prof. S. Dhanuskodi (Retd.) at Bharathidasan University, Tamil Nadu. His research interests across multidisciplinary areas in energy conversion such as photo/photocatalytic/electrochemical CO2 reduction, water splitting, electrolyzer designs, and density functional theory calculations. |
| Mamata Mohapatra Dr Mamata Mohapatra received her Ph.D. from Utkal University, Bhubaneswar. She got her post-doctoral experience from University of Waterloo, Canada by availing BOYSCAST fellowship. She joined at Hydro & Electrometallurgy department, CSIR-IMMT in 2006 (October) as Jr Scientist and has been undertaking research independently on the interdisciplinary field of research in the aqueous processing of ores and secondaries, synthesis and surface modification of transition metal oxide/oxalate and carbonate materials for environment and energy application. Currently, she is engaged in energy material development through the urban mining of sent batteries. She has many contributory well-cited publications in international peer review journals (about 85) in collaboration with IIT, Kanpur, Utkal University (India), KIIT, Murdoch University (Australia), and WATLab (Canada), University of Southern Denmark. Her work has received 3485 citations with an h index of 27. She has successfully supervised 4 PhD students, 14 M.Sc students, 2 M.Tech students. |
| Suddhasatwa Basu Prof. Suddhasatwa Basu completed his M.S. and Ph.D. from IISc, Bangalore prior to completing B.Tech. in Chemical Engineering from Calcutta University. He is Professor (on lien) at the Chemical Engineering Department, IIT Delhi. At present, he is the Director of CSIR-Institute of Minerals & Materials Technology (IMMT), Bhubaneswar. He was earlier Associate Dean (R&D), IIT Delhi and Head of Chemical Engineering Department, IIT Delhi. He has received Herdillia Award for Excellence in Basic Research in Chemical Engineering and A V Rama Rao Research Excellence Award and Indus Foundation award. He is a Fellow of The National Academy of Sciences of India (NASI), Indian Chemical Society. International Association of Advanced Materials, Sweden, Indian National Academy of Engineering, and Royal Society of Chemistry, UK. His current broad area of research consists of electrochemical, photoelectrochemical water splitting, high and low temperature fuel cells, electrolyzer, electrochemical sensor, micro fuel cell, artificial photosynthesis-CO2 reduction, batteries and supercapacitors. He has published 250 high impact journals articles with H-index 39. He has authored 10 books and book chapters and patented 5 technological innovations. He is the editor and editorial board member of many journals published by ACS, Springer-Nature and Taylor and Francis. |
1. Introduction
A drastic rise in global population (∼7.9 billion as of 2021) and industrialization have brought a serious impact on humanity and the environment in the form of energy demand, global warming, and climate change. The heavy dependence of humankind on fossil fuels has pushed the energy consumption rate to 19.0 TW in 2019 and it will steadily increase in the coming century as the average energy consumption per person increases with the rise in the number of vehicles.1 On the other hand, a large part of global warming and climate change originates from the burning of carbon-based fossil fuels, releasing carbon dioxide (CO2) into the atmosphere (411.85 ppm in 2019).2 According to the International Energy Agency,3,4 CO2 emission should be curtailed by at least 50% to keep the global temperature rise <2 °C by 2050. To mitigate the environmental footprint of current technologies and sort out the constraints of fossil fuels, solutions towards the projected growth of the energy crisis and environment pollution should adopt carbon neutral energy production and carrier systems.5 As a potential solution to these issues, CO2 capture and sequestration (CCS) and CO2 utilization (CCU) are frequently employed approaches to address the release of CO2 to the atmosphere.6 In CCS, the storage of CO2 is done with geological sub-surfaces/oceans; however, there is still the possibility of leakage into the atmosphere, and thus, CCU has become a prominent approach to overcome the above problems. Therefore, tremendous efforts are being made to recycle CO2 as renewable fuels and chemicals by the approach called as electrochemical reduction (ER), which decreases the CO2 emissions and also reduces the energy crisis (Fig. 1).
 |
| | Fig. 1 Schematic representation of the electrochemical reduction of emitted CO2 to value-added products and fuels curtailing the energy crisis and environmental pollution. | |
Three decades ago, Hori et al., studied electrochemical CO2 reduction (CO2R) on metal catalysts7,8 where the reaction took place at the electrode and electrolyte interface. The CO2R process involves the absorption of CO2 on the catalysts surface, transformation of protons or electrons to dissociate the C–H bonds, and desorption of the final products from the electrode. Despite CO2R becoming an interdisciplinary research area between materials science, chemistry, and physics, it is still facing a series of technical challenges (enormous potential, low energy efficiency, etc.) before reaching commerciality. Unfortunately, the dissociation energy of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond is quite large (750 kJ mol−1) compared to C–H (411 kJ mol−1), C–C (336 kJ mol−1), and C–O (327 kJ mol−1), making them energetically unfavorable in the absence of external catalysts. Without the electrocatalyst, the formation of the anion *CO2˙− radical from CO2 via the first electron transfer requires a more negative cathodic potential of −1.9 V vs. standard hydrogen electrode (SHE); hence, it is very much important to establish electrocatalysts to overcome the activation barrier and promote the reduction kinetics.9 Therefore, the efficiency and selectivity of the CO2R process depend on the electrocatalysts, local electrode potential, and the composition of the electrolytes. Although CO2R is observed with complex intermediates during diverse steps including electron and proton transfer, it is frequently accomplished with high overpotential (more negative potential must be biased than the theoretical potential to reduce CO2). This not only required additional energy but also gave undesirable competitive products (H2), causing a decrease in both the efficiency and selectivity. Consequently, the reduction of the overpotential remains a major concern in the overall CO2R performance.
O double bond is quite large (750 kJ mol−1) compared to C–H (411 kJ mol−1), C–C (336 kJ mol−1), and C–O (327 kJ mol−1), making them energetically unfavorable in the absence of external catalysts. Without the electrocatalyst, the formation of the anion *CO2˙− radical from CO2 via the first electron transfer requires a more negative cathodic potential of −1.9 V vs. standard hydrogen electrode (SHE); hence, it is very much important to establish electrocatalysts to overcome the activation barrier and promote the reduction kinetics.9 Therefore, the efficiency and selectivity of the CO2R process depend on the electrocatalysts, local electrode potential, and the composition of the electrolytes. Although CO2R is observed with complex intermediates during diverse steps including electron and proton transfer, it is frequently accomplished with high overpotential (more negative potential must be biased than the theoretical potential to reduce CO2). This not only required additional energy but also gave undesirable competitive products (H2), causing a decrease in both the efficiency and selectivity. Consequently, the reduction of the overpotential remains a major concern in the overall CO2R performance.
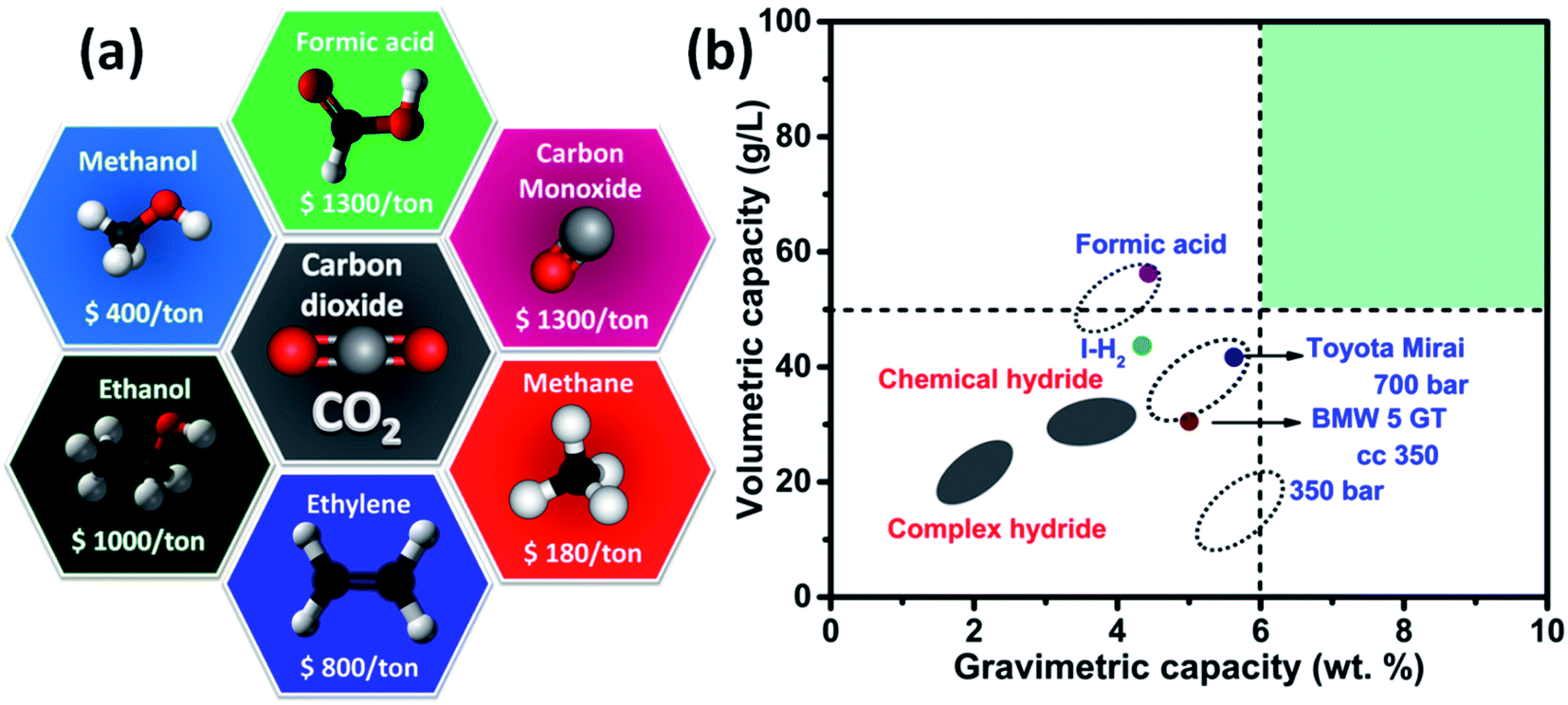
CO2R generates a variety of products due to the number of electrons transferred to the CO2 molecule and they are classified into one (oxalic acid/H2C2O4 or oxalate/C2O42− (ref. 1)), two (carbon monoxide/CO,7 formic acid/HCOOH or formate/HCOO− (ref. 1 and 5)), four (formaldehyde/CH2O4), six (ethylene/CH2CH2,8 methanol/CH3OH,10 ethanol/CH3CH2OH11), and eight electron (methane/CH4 (ref. 8)) pathways, and the corresponding thermodynamic half-reactions are illustrated in Table 1.12 The economic viability of the aforementioned products has been assessed via the gross-margin model by Verma et al.13 including the cost of goods sold and revenue; thereby, the figures of merit such as maximum potential, minimum current density, faradaic efficiency (FE), and electrocatalyst stability have been explored. Based on this model, it is predicted that the production of HCOO− or HCOOH and CO with an enriched operation voltage could be commercialized in the future over other products (CH4, C2H4, CH3OH, and C2H5OH) due to the larger variation in the market size. Thus, HCOOH and CO derived from electrochemical CO2 reduction have more economic profitability, in which HCOOH in the context of fuel cell and energy carrier applications dominate CO because of its toxicity and environmental hazard. Formic acid is one of the foremost value-added products due to a number of factors. A large-scale ER of CO2 to formic acid is demonstrated to be feasible (Fig. 2a) as the price is about 1300 USD per ton, which is similar to that of carbon monoxide but much higher compared to methanol and ethylene. It is a simple carboxylic acid existing naturally and the global market price is estimated to hit 620 million dollars.14 Formic acid is utilized in agriculture as a preservative agent due to the antibacterial property and also in fermentation processes, production of leather, and textile dyeing though it evaporates without leaving any surplus, unlike mineral acids. More importantly, formic acid can be used as a promising hydrogen carrier because of its 4.4 wt% of H2, high density (1.22 g cm−3), and volumetric capacity (53 g H2 per L). The energy density of formic acid (1.77 kW h L−1) exceeds the commercial 70 MPa hydrogen pressure tanks (1.4 kW h L−1 for Toyota Mirai) so that it may find potential applications in automotive and mobile sectors (Fig. 2b).5 According to the techno-economic prediction by Spurgeon and Kumar,17 the deep capital cost inspection on the simple C1 chemicals (HCOO− or HCOOH and CO) could be profitable when the current density is well above 200 mA cm−2 and stable operation duration should be in the range of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h. However, the production of HCOOH and CO will be commercialized in the near future; this review deliberately focuses on the electrochemical reduction of CO2 to HCOOH and how the current density, faradaic efficiency, and stability are tuned by extracting the optimistic performance of various electrolyzers.
000 h. However, the production of HCOOH and CO will be commercialized in the near future; this review deliberately focuses on the electrochemical reduction of CO2 to HCOOH and how the current density, faradaic efficiency, and stability are tuned by extracting the optimistic performance of various electrolyzers.
Table 1 Illustration of thermodynamic potential (E) of CO2R reactions against the aqueous solution yielding different products at pH = 7 and 25 °C [ref. 12]
| Thermodynamic half reactions for CO2 reduction |
E (V vs. SHE) |
| CO2 (g) + 2H2O (l) + 2e− = HCOOH (l) + 2OH− |
−0.610 |
| CO2 (g) + 5H2O (l) + 6e− = CH3OH (l) + 6OH− |
−0.380 |
| CO2 (g) + 6H2O (l) + 8e− = CH4 (g) + 8OH− |
−0.240 |
| 2CO2 (g) + 10H2O (l) + 14e− = C2H6 (g) + 14OH− |
−0.270 |
| 2CO2 (g) + 9H2O (l) + 12e− = C2H5OH (l) + 12OH− |
−0.33 |
| 2CO2 (g) + 8H2O (l) + 12e− = C2H4 (g) + 12OH− |
−0.34 |
| 3CO2 (g) + 13H2O (l) + 18e− = C3H7OH (l) + 18OH− |
−0.320 |
| CO2 (g) + 3H2O (l) + 4e− = CH2O (l) + 4OH− |
−0.480 |
| CO2 (g) + H2O (l) + 2e− = CO (g) + 2OH− |
−0.520 |
 |
| | Fig. 2 (a) Global market price for CO2 reduced valuable commodity chemicals [ref. 12]. (b) FA as a hydrogen carrier overtakes other reversible hydrogen storage options [reproduced with permission from ref. 5, 15 and 16]. | |
In recent years, extensive research has been conducted for the development of CO2R performance while taking care of the activity, energy efficiency, and long term stability but indeed noteworthy progress has been acquired on HCOOH production. For example, the grain boundary-enriched Bi catalyst showed a high current density (450 mA cm−2), FE (97%), and stability (100 h) toward ultra-high concentration of HCOOH (up to 100 wt%).18 Han et al.19 observed 100% FE at −1.3 V vs. SCE for Bi nanosheet electrodes, whereas bismuth oxyiodides prepared by Wang et al.,20 have shown high current density (40 mA cm−2) at −0.9 V vs. RHE for HCOO− production. Besides, many continuous flow reactors deliver current density over 100 mA cm−2 and lifetime (h to a several months) with a high selectivity of HCOO−/HCOOH products. Despite these efforts, CO2R is still lacking in the scale up and commercialization process owing to the slow kinetics of CO2 reduction, large overpotential, and weak product selectivity as result of competitive hydrogen evolution reaction and the degradation of reactor performance over time. To take forward CO2R in a practically viable and efficient manner, the present technology needs a sudden migration from the half-cell or batch scale to continuous flow CO2 reactors, which overcome the existing issues such as poor mass transfer, solubility, low operation current density, and selectivity toward products. To address these issues, immense research interest has been dedicated to fundamental aspects of electrocatalyst fabrication, electrolyte composition, pH, pressure, and temperature utilizing the lab scale reactors;10,21–23 however, there is a lack of attention on reactor engineering, its components, membrane separators, and reaction mechanisms.24,25 Hence, one should explore CO2R reaction kinetics with respect to the electrolyzer configurations as they govern the local reaction environment near the electrode. Although there are few review and perspective articles exploring the CO2R of value-added products, the conversion of CO2 to HCOOH/HCOO− is still at an initial stage accompanied with key challenges, which have to be addressed toward the implementation of the commercial devices. Herein, we depart from the literature by exploring the recent advances in the reactor configurations, operating principles, and suitability of membranes and electrocatalysts to the scientific community.
2. CO2R reactor configurations
In order to enhance the figures of merit (current density, FE, and stability) and to address the major shortcomings (ohmic loss, mass transport, and solubility), the CO2R reactor engineering is an indispensible strategy since the reaction takes place between the open circuit potential of the cell and the applied external voltage. Recently, the extensive research has enabled the different geometries of electrolyzers (H-type, flow, and microfluidic reactors) and major components (gas diffusion electrode and membrane electrode assembly). In the present section, the current research updates and existing challenges of various electrolyzers toward CO2R to HCOO−/HCOOH have been emphasized.
2.1 H-type reactors
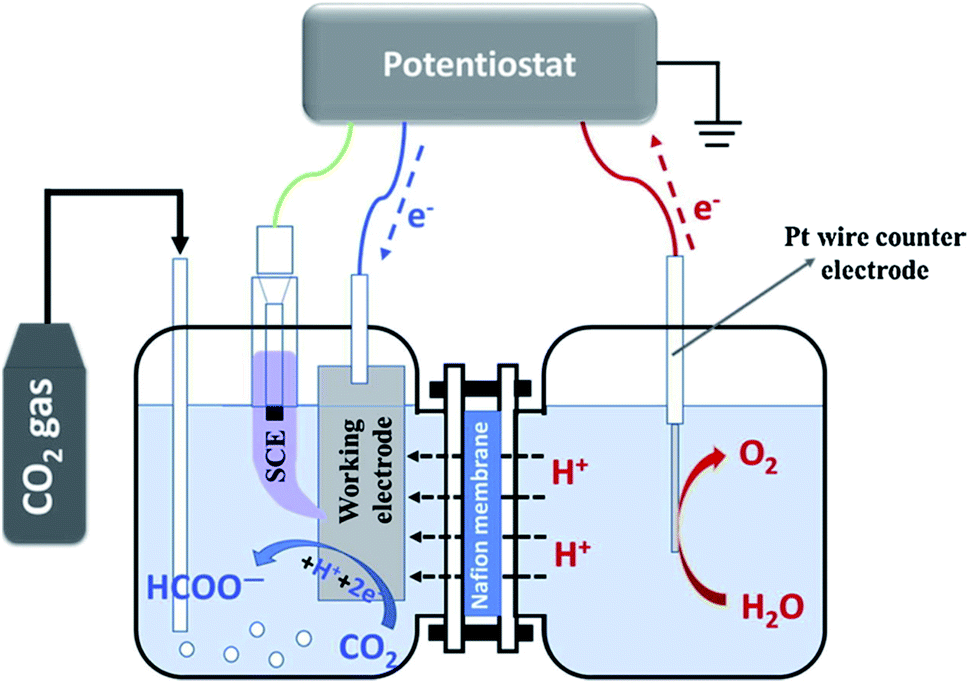
However, H-type reactors (Fig. 3) are commercially available lab scale reactors for CO2R due to its adaptability with different electrodes and geometries, and ease of operation, leading to rapid and economic assessment. The cell consists of two compartments prefilled with the electrolyte in which the working and reference electrodes occupy the cathodic compartment, whereas the counter electrode lies at the anodic compartment. The ion exchange membrane connects these compartments via the channel, exposing the H-form; thereby, it prevents the reduced products from further oxidation by limiting the fuel crossover. CO2 gas is fed into the cathodic chamber wherein gas and liquid products can be collected after the reduction reaction. On the other hand, oxidation (oxygen evolution reaction with water) happens at the anodic chamber to maintain the charge balance by closing the circuit. H-type electrolyzers are capable of screening a number of catalysts in a short span of time, making it easy to operate and cost-effective. Recently, Zhang et al.26 have employed an air tight H-type electrolyzer with Nafion 117 membrane and porous SnO2 catalysts (Fig. 4a–c) have been loaded on carbon paper by drop casting. The optimal catalyst loading delivered the increased partial current density (∼11.5 mA cm−2), a high faradaic efficiency (94.5%), and excellent long-term stability (100 h) for the reduction of CO2 to HCOOH + CO. Li's group27 investigated both electrocatalytic (EC) and photoelectrocatalytic (PEC) CO2 reduction using TiO2 nanotube arrays as the photoanode and electrodeposited CoOx@Cu foam as the cathode. The product yield of HCOOH at the potential from 0.5 to 0.9 V vs. Ag/AgCl is 2 times higher for the photoanode-driven PEC system compared to the EC system at 0.6 V vs. Ag/AgCl. The optimal performance for CO2R to HCOOH is obtained for the as-prepared CoOx cathode with an FE of 60.9% at −0.85 V vs. Ag/AgCl. Likewise, Sn catalysts on Cu film prepared by Wang et al.,28 showed an initial current density of 0.9–1.4 mA cm−2 along with the maximum FE 91.7% at −1.4 V vs. SCE in CO2 saturated KHCO3. When the reduction reaction time is increased, the FE declines to 81.6% for 2 h and further drops to 69.6% for 3 h; therein, the current density declined to 0.7 mA cm−2. This is attributed to the formation of the alkali metal intermetallic compound during CO2R; meanwhile, the degradation of the Sn electrode commenced. Very recently, the ER of a CO2 electrolyzer bearing the boron-doped diamond (BDD) as the cathode has been used to achieve the best HCOOH selectivity. In this two-electrode CO2 electrolyzer, the reference electrodes are inserted on both the cathode and the anode compartments to monitor the actual potentials; therefore, the energy distribution in each part could be analyzed. Under the optimistic conditions, a remarkable FE (96%) at 3.5 V and the highest electrical-to-chemical energy (ECE) conversion efficiency (43%) at 3 V were observed for FA, and the ECE further increased by 45% if H2 and CO products are included.29 One of the important targets in CO2R is to attain a single product with 100%, which has not been frequently achieved; thus, H-type electrolyzers are still active for screening the numerous electrocatalysts. Despite this significant research progress, a vast amount of metal electrodes suffer from a lack of surface area, huge interelectrode distance, and poor reactor performance. Besides, a thick diffusion layer (>50 μM) and low solubility (0.034 M) of CO2 in aqueous electrolyte (dissolved CO2 rapidly forms neutral pH carbonate mixture in the alkaline electrolyte) lead to poor mass transport and selectivity. In order to increase the CO2 solubility, Li et al.30 fabricated the high-pressure H-type electrolyzer working at 60 atm of CO2 partial pressure, wherein the HER activity is almost quenched. The selectivity of CO2R is changed from complex C1–C3 products (1 atm) to single C1 (HCOO−) product while exhibiting FE > 98% at 45 atm. These drawbacks limit the current density of the H-type electrolyzer below 100 mA cm−2, which hinders the scaling up to the practical devices.
 |
| | Fig. 3 Schematic illustration of a conventional H-type cell for CO2 conversion to formic acid/formate [ref. 28, Copyright © 2019 Elsevier]. | |
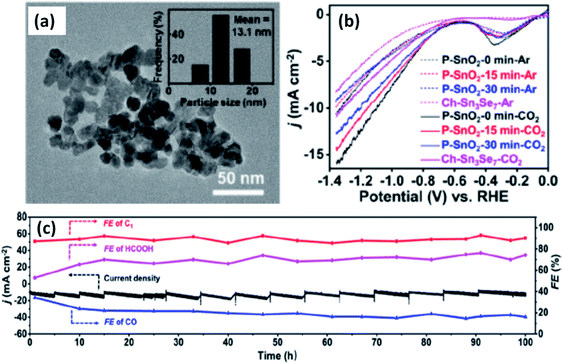 |
| | Fig. 4 (a) TEM picture of SnO2 particles in P-SnO2-0 min, (b) LSV curves measured for P-SnO2 samples for CO2-saturated 0.1 M KHCO3, (c) long-term catalytic test (100 h) showing FE and the current density for P-SnO2-0 min at −1.06 V vs. RHE [ref. 26, Copyright © 2021, American Chemical Society]. | |
2.2 Flow cell reactors
To circumvent the shortcomings of the H-cell, flow cell reactors are developed in three different configurations (Fig. 5a–c), (i) gas phase, (ii) solid phase, and (iii) liquid phase reactors.31 From continuous flow reactors, the limitations experienced in H-type cells prevailed, indicating a more realistic CO2R and ability to provide a current density more than 100 mA cm−2. To begin, gas phase reactors employ the membrane electrode assembly (MEA) structure (a detailed explanation can be seen in the upcoming Section 2.4), which is made up of a catalyst coated on the gas diffusion electrode (GDE) as the cathode layer attached on an ion exchange membrane. Here, the solid polymer electrolyte is used as the membrane, which transports the ionic charges, enabling a continuous and a stable charge transfer in the circuit. CO2R takes place when humidified CO2 is supplied to the cathode and the source of water to the electrocatalyst is availed from the aqueous anolyte next to the MEA. Without the catholyte, there are some experimental results showing increased partial current density (PCD), higher stability, and better control over the liquid product toward formate production.32 The use of solid electrolytes in a gas phase flow reactor does not require the additional steps for final product separation as in the case of a liquid phase where the products are mixed with the solute in the electrolyte. Nevertheless, the solid ion conductors transport the generated cations/anions and prevent mixing with the solute, forming the pure products, thereby shortening the separation process. Xia et al.33 developed the four-chamber solid electrocatalytic cell with a solid electrolyte to produce the proton (H+), hydroxyl (OH−/KOH), and HCOOH. A peak PCD of 150 mA cm−2 has been achieved at 3.36 V for HCOOH in addition to a pure KOH (concentration up to 0.66 M) with the flow of de-ionized water. The use of such water decreased the product concentration and thus necessitated the liquid electrolyte for oxidation at the anode. Gas phase reactors are not much explored owing to the presence of contaminants, which obviously depend on the cell architecture and need to be studied more. With a solid phase reactor, CO2R is performed at the high temperature from 300 to 600 °C, where the cathode or anode is in an oxide form and the electrolyte is either the oxygen ion or proton conductive. The oxygen ion conductive electrolyte transports the reduced oxygen ions at the cathode to the anode to form the oxygen gas by oxidation. On the other hand, CO2 gas molecules at the cathode reduced to form hydrogen. In the proton-conductive electrolyte, the water molecules oxidized at the anode, leaving oxygen gas and protons, in which the latter flows through the proton conductive layer to the cathode reducing the CO2 molecules. Therefore, the major advantage of this reactor is the high current density at high temperature resulting from the enhanced kinetics and mass transport. At high temperature, the difficulty in sealing and CO as the only reduced carbon product are the key drawbacks of the solid phase reactors. Hence, it is out of the scope of this review as it ultimately focuses on HCOO−/HCOOH products.
 |
| | Fig. 5 (a) Gas phase reactor, (b) solid phase reactor, and (c) liquid phase reactor [reproduced from ref. 31, no permission is required]. | |
The ER of CO2 in the liquid phase electrolyzer (Fig. 5c) is carried out in the presence of the liquid electrolyte, the gas diffusion layer (GDL) on both the anodic and the cathodic compartments and ion exchange membrane at the electrolyte bulk. Depending on the membranes (cationic, anionic, bipolar), the charges flow via the electrolyte to the anode/cathode and thereby completes the circuit. Unlike gas-phase reactors, CO2 inlet in the liquid phase does not need to be humidified; rather, it can be maintained in a pressurized mode.34 The key component of gas and liquid phase electrolyzers is the GDE (more discussion is available in the upcoming Section 2.3), forming gas–liquid–solid interfaces and permit CO2 to react in the gas form with the catalysts before the aqueous solutions. The GDE component fails to carry metal/foil catalysts but, fortunately, it often measures the wide variety of powder catalysts, leading to the operation of the reactors at industrial current densities.35 CO2 conversion to HCOO− using the cation exchange membranes has been studied as a function of pressure (from 10 to 50 bar), anolyte (0.5 M H2SO4), and catholyte (1 M KHCO3). It is found that the current density, FE, and HCOO− concentration increase with CO2 pressure. A maximum of ∼90% FE is observed for 40 bar but it is decreased thereafter owing to HCOO− crossover via the cation exchange membrane, and a pH drop due to CO2 dissolution.36 Wu et al.37 discussed that the electrochemical flow cell circulating a liquid-phase electrolyte significantly enhanced the formation of HCOO−, with a FEmax >90% at −1.7 V vs. SHE and the PCD of 9 mA cm−2 while the anode is fed into H2. However, FEmax gain is reduced to 85% at −2.0 V with a PCD of 6 mA cm−2. This is due to the fact that buffer circulation has enabled the conversion of formate at η of 200 mV even though there are gaseous or aqueous reactants at the anode. In one of the recent outstanding work done by Yasuaki Einaga group,38 the mass transport and CO2 reduction parameters are optimized with a continuous liquid fed intermittent flow cell (Fig. 6a) controlled using a synchronized dual-phase double-action cylindrical pump. The cell is operated by stopping the electrolyte flow at a desired frequency and allowed a sufficient time for CO2 gas molecules to effectively reduce into HCOOH. The synchronized cylindrical pump creates the stop-start motion (0.02 s) such that a high and a stable FE (95%) is achieved (for BDD catalyst) in 4 h for HCOOH (576.98 mg). Further, electrochemical CO2 reduction is conducted with and without air chambers to counter and minimize the pressure lost when the pump stops the electrolyte flow. When the air chamber flow is regular as in the continuous flow system, the FE decreased <10% after 2 h. This conveys that controlling the stop-start motion of the electrolyte (Fig. 6b) flow is also a crucial factor behind the HCOOH production. Moreover, the observed result is 10 times higher than the conventional flow cell system39 and this type of CO2 electrolyzer system seek industrial-scale applications. The discrepancy between liquid and gas phase electrolyzers is the introduction of a liquid electrolyte layer among the cathode and the ion exchange membrane. Besides, the production of HCOOH by these electrolyzers showed notable differences if GDEs are used. When switching from the liquid-phase electrolysis to the gas-phase, CO2R for HCOO− has been promoted and the figures of merits are increased such as the average overpotential (0.91 to 1.09 V), the average FE (40.5 to 50.6%), and the average current density (9.2 to 28.2 mA cm−2).32 It is also important to notify the challenges in the flow cells such as the crystallization of salts by GDL, high ohmic losses due to the aqueous electrolytes, the water management issues, and sensitivity to contaminants. At the gas-liquid-solid three-phase interfaces in GDE, the carbonate/bicarbonate salts have lower solubility assigned to the reaction of CO2 with the alkaline electrolyte, which makes the GDE more hydrophilic and flooded over time.40 In addition, the absorption of CO2 into the alkaline catholyte leads to carbonates, which influences the system stability and increases the capital costs. These discussions show that it is necessary to jump from the liquid-phase to the gas-phase systems as they have the ability to raise the current densities well above the base level of the techno-economic analysis. However, a further analysis would be required, though the economic feasibility parameters of liquid and gas phase processes are dissimilar.
 |
| | Fig. 6 (a) Pictorial representation of the intermittent flow cell and (b) demonstration of the stop-start motion on the flow conditions (i) before the piston pump closes the pipe (as in the continuous cell), (ii) when the piston pump closes the pipe, and (iii) after the piston pump opens the pipe [ref. 38, Copyright © 2021, American Chemical Society]. | |
2.3 Gas diffusion electrode (GDE)
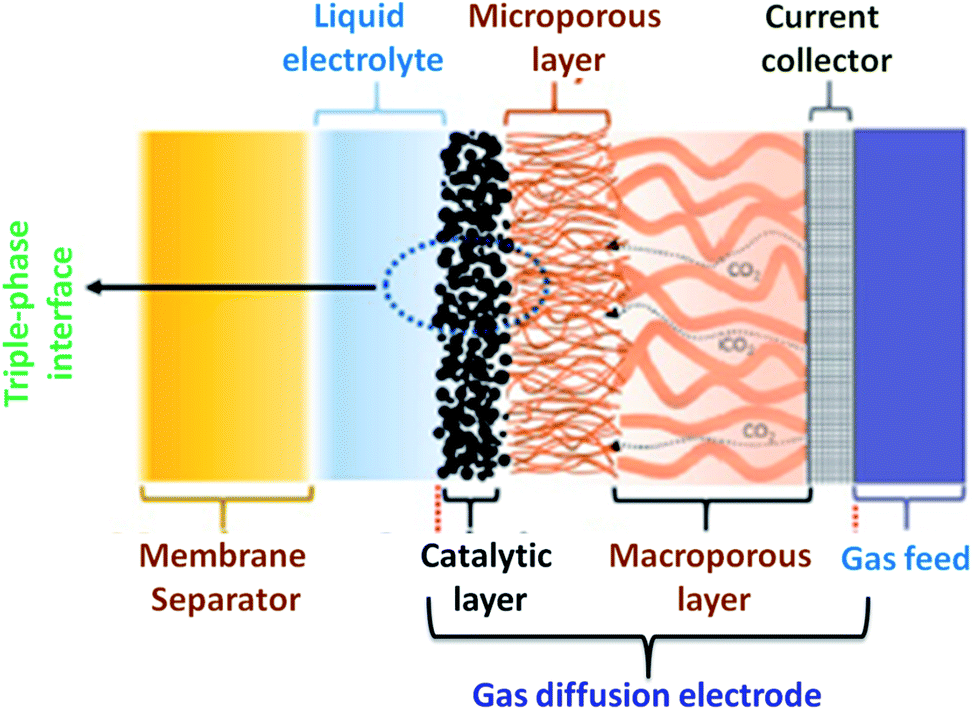
Fortunately, the mass transport limitation from two zone reactors could be sorted out by the three zone gas diffusion electrodes (Fig. 7), which operate the catalysts in the powder form on gas diffusion layers (GDL). CO2 is supplied to the catalyst by diffusion through the GDL in contact with the electrolyte flow. CO2R commences at the gas and the liquid electrolyte interface thereby enhances the reaction kinetics, causing efficient mass transport. In H-type cells, CO2 is to be saturated with the bulk electrolyte. When CO2R happens, the entire products move from the surface in the opposite direction to the incoming CO2. In contrast, GDE with GDL carries the liquid products in the aqueous state because of the hydrophilicity and the gas products migrate from the gas side. The advantage of using GDE over H-type cells, GDL is linked to the good mass transfer characteristics and the short diffusion length. It is understood that the diffusion path is 50 μm for dissolved CO2 from the bulk to the non-permeable catalyst, whereas from gaseous CO2 to the liquid surrounded catalyst is at the level of only about 50 nm.41,42 This shorter diffusion path length enables the CO2-saturated electrolyte layer on the catalyst, preventing the hydrogen evolution reaction.42
 |
| | Fig. 7 Schematic of a gas diffusion electrode (GDE) with the membrane separator in the liquid phase CO2 electrolyzer showing three-phase interfaces as the liquid electrolyte, catalytic layer and the CO2 gas molecules [ref. 9, Copyright © 2020, Elsevier Ltd]. | |
Further, CO2 gas approaches the active sites faster before it reacts with OH− ions in the electrolyte, enabling the use of alkaline electrolytes. Another distinct feature of GDL is the reaction direction of CO2, whereas in the case of H-cells, it approaches the catalyst surface from the bulk. Meanwhile, in GDL, the reactant CO2 gas is purged via the hydrophobic layer, which lacks the agglomeration of gaseous products and blocks the catalyst surface, facilitating the adsorption of CO2 molecules. Thus, the use of GDL simultaneously overcomes the challenges of mass transfer and the availability of CO2 at the liquid/catalyst interface, resulting in a significant activity and lower cell potential so that GDE-based electrolyzers have a great potential for scaling up the devices. However, under this circumstance, it is prone to show product crossflow via the GDL due to the pressure difference and CO2 bubbles in the catholyte leads to a fluctuation in the cell voltage upon time.43
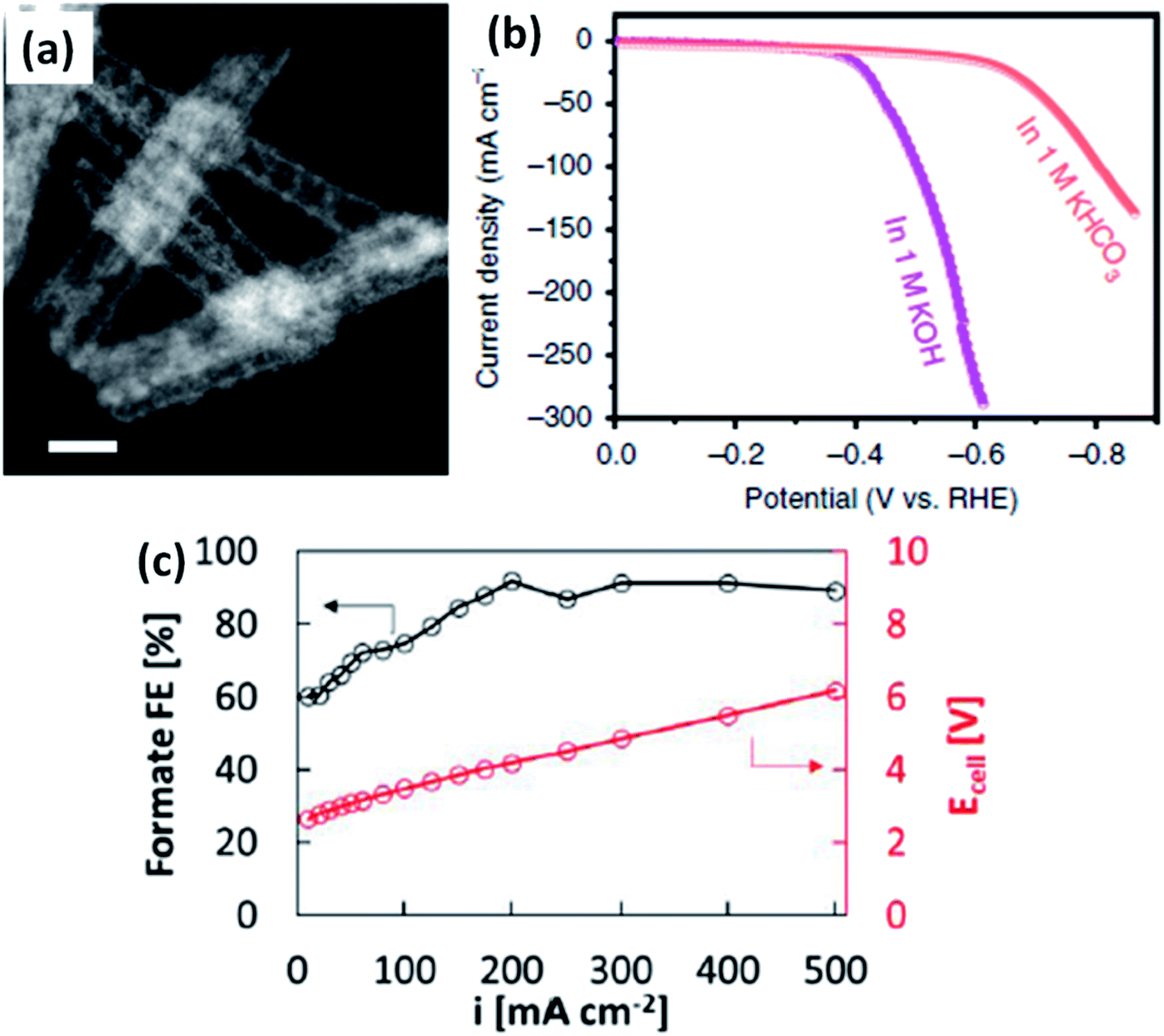
In this configuration, extensive studies have been proven to be efficient over conventional H-type cells. For example, Bi2O3 nanotube catalyst showed 100% selectivity at −1.05 V vs. RHE, a high current density (60 mA cmgeo−2), and a long-term catalytic stability >48 h in 0.5 M KHCO3 under a standard H-type cell.44 When the reactor is subjected to GDL, the nanotube-derived Bi delivered a larger current density of 288 mA cm−2 for −0.61 V in 1 M KOH (Fig. 8a and b). In the other study,45 CO2R to HCOO− at the gas diffusion electrode (25 cm2) with a carbon-supported SnO2 electrocatalyst produced 90% FE at 500 mA cm−2 while retaining a total cell voltage ≤ 6 V, as shown in Fig. 8c. Besides the several improvements, GDL undergoes the crystallization of the hydroxide and bicarbonates salts on the porous layer because of the richness of the hydroxide ions at the cathode. Moreover, the local pH increases ≥ 12 for the current densities more than 50 mA cm−2, leading to a different local environment for CO2R to occur. In the context of the operation conditions, the higher current gives rise to a notable ohmic loss iRs and then the pH of the electrolyte, temperature of the electrodes, and the charge conductivity are varied over time due to the concentration polarization between the electrodes. Due to the hydrophobic nature of the GDL nanostructure, the contaminants in the electrolyte are harmful to the active sites at the catalyst surface. In order to resolve these ambiguities, Dinh et al.46 framed a robust configuration, where the electrocatalyst was sputter-coated on a porous layer of PTFE and the nanoparticle carbon is spray-deposited, thereby stabilizing the copper catalytic layer in KOH solution. Such an architecture is extended to the CO2 gas diffusion zone with the catalyst surface and thus promoted more CO2 conversion and higher current density. Wang's group33 reported that the continuous CO2R utilizing solid electrolytes generated cations (H+) and anions (HCOO−) that combined to form pure HCOOH solution. With the Bi catalyst, the cathode produced a pure HCOOH concentration up to 12 M (FE > 90%) and a stable generation of 0.1 M HCOOH for 100 h; besides, a negligible degradation is seen in selectivity and activity. Further, in an interesting research article by Delafontaine et al.,47 the concentration of CO2 molecules in aqueous solution with H-type electrolyzer is found to be 0.038 M and the corresponding diffusion coefficient is 0.0016 mm2 s−1 in CO2-saturated 0.1 M KHCO3. When the humidified CO2 is supplied to the flow cell with GDE, the concentration of CO2 is increased up to 0.041 M and 10000-fold enriched diffusion coefficient (16 mm2 s−1) is obtained. A more gaseous CO2 has been reached at the catalyst, resulting in a large CO2 availability and a consequent raise in the CO2R PCD.24,47 Therefore, the usage of GDEs is the prominent strategy to increase the HCOO− PCD but it is very much challenging to avail a quick HCOO− production rate and a low overpotential with a high FEHCOOH.
 |
| | Fig. 8 (a) STEM-HAADF images of Bi2O3 nanotubes, (b) polarization profiles of nanotube-derived Bi in 1 M KHCO3 and KOH [ref. 44, Copyright © 2019, Springer Nature] and (c) the formate FE and polarization curve [ref. 45, Copyright © 2020, American Chemical Society]. | |
2.4 Membrane electrode assembly (MEA)
To overcome the drawbacks in the H-type cell, gas and liquid phase flow reactors equipped MEA have been developed, in which the cathodic layer of GDE has made direct contact with the polymer electrolyte membrane (PEM) in a zero-gap configuration (Fig. 9). MEA recovers from the ohmic losses and the contamination of the cathode catalyst in the catholyte by the removal of the catholyte between the membrane and GDE, which brings it higher efficiency and stability. Thus, MEA consists of an anode compartment with a liquid anolyte and the cathode chamber with only a gas phase. Therefore, water management on the cathode catalyst surface became one of the major problems in MEA. By giving humidified CO2 as the feed, the above problem can be solved but still the membrane turned dry even under humidification; hence, the anolyte is necessitated to supply water on the cathode catalyst. Since MEA does not enclose the reference electrode, CO2R is performed at the cell voltage or the current compared to the working electrode potential. However, CO is a targeted product in MEA reactors and a remarkable current density of 0.5 A cm−2 with more than 85% of FE was achieved.48 However, CO2R on the liquid products such as alcohols are scarce due to MEA swelling, affecting the durability too. But, fortunately, MEAs are capable of generating high purity products; in particular, HCOO− production has been vigorously studied.49 Lee et al.32 investigated the catholyte-free CO2R to avoid the solubility limitation by commercial Sn nanoparticles as the cathode catalyst. Despite a decrease in CO2 solubility, the PCD of HCOO− enhanced two times with 52.9 mA cm−2 when the reaction temperature increased from 303 to 363 K. Also, an appreciably high HCOO− concentration of 41.5 g L−1 has been observed with a significant PCD (51.7 mA cm−2) and high FE (93.3%) at 2.2 V. An improved electrochemical reduction is demonstrated (Fig. 10a and b) for Sustainion™ anion exchange membrane with a nanoparticle Sn GDE carrying an imidazole ionomer, which displayed a stable electrochemical cell performance for 500 h at 140 mA cm−2 and at a cell voltage of only 3.5 V.50 More recently, Thijs et al.51 adopted a novel strategy to enhance the CO2R to HCOO− or HCOOH by the recirculation of the by-products to the reactor along with unconverted CO2. It has been elaborated that different MEA-type gas and liquid phase reactors (Fig. 11a) have garnered a predominant role in the electrochemical reduction of CO2 to HCOO− or HCOOH. With the gas phase reactor using type I configuration, FE toward HCOOH is limited in the range from 5 to 12.5% while the H2 product is predominant when H2 is employed as the source of protons at the anode to the cathode via the Nafion membrane.52 In another study, the alkaline anion exchange membrane displayed promoted selectivity for CO2R in contrast to the acidic proton exchange membrane owing to the inaccessibility of the protons. Such type of MEA-based reactors are adopted for CO rather than HCOO− or HCOOH and because the HCOO− crossover to anode is a serious issue and more HCOOH acidification is required to maintain the proton balance.50,53 Therefore, a center flow compartment was integrated to the MEA, which drives the water liquid flow between the anodic proton exchange membrane and the cathodic anion exchange membrane to acidify the HCOO− and discard the HCOOH from the reactor (type II).50 On the other hand, the type III configuration studied by Park's group32 showed FE against HCOO− up to 93% with liquid KOH as the anolyte, highly humidified CO2, and increased temperature. Further, type IV54 is developed for the entire gas phase CO2R where MEA consists of an alkaline ionomer layer at the cathode, attached with a membrane, and separates the anode from the cathode. This configuration demonstrates excellent results; however, it would be further explored for HCOO− or HCOOH. Among these four configurations, H2 evolves as the main by-products and the aqueous environment is needed for the proton source against HCOOH formation. In order to avoid the formation of H2 by-product, a new strategy has been developed by Thijs et al.51 based on type III configuration. A typical gas phase CO2R reactor with recirculation has been proposed in Fig. 11b and herein, water vapour is supplied. The created protons due to the water oxidation transport via the membrane from the anode to the cathode and further the water at the cathode is originated by an electro-osmotic drag. The cathodic compartment composes by-products such as HCOOH, H2O, unconverted CO2, H2, CO, and some higher order products. Therefore, the cathode outlet gas stream has been directed to the condenser wherein HCOOH and H2O are discharged by condensation. Unconverted CO2 and by-products are re-circulated to the cathode and amalgamated with the CO2 inlet. At the end, the pressure is tuned by a valve P, evacuating the unconverted CO2 and by-products to keep away from excessive build up. For the initial accumulation of the by-products (H2), the electrochemical formation potential of the undesired by-products in the reactor is moved to higher values and favored the formation of HCOOH, for which the potential is unaffected. Besides, mathematical simulation has found that the re-circulation of the by-products necessitated substantially enhanced HCOO− selectivity at low pressure. Further, experimental validation with an MEA reactor validated as the decrease in the H2 selectivity for Sn-nanoparticle on the carbon cathode is about 40% (in gas mixtures 0.07 MPa of H2 and 0.03 MPa of CO2) as opposed to pure CO2. This has led to a notable increase in the HCOO− selectivity and energy efficiency. In conclusion, CO2R from the MEA-based reactor excels in the view point of lowered material costs, reduced cell potential promoted CO2 mass transfer, and selectivity. Water management at the cathode, liquid product collection difficulty, and crossover of liquid products are the challenges in the MEA setup. To better understand, more investigations will be needed on the mechanism of electrocatalytic reactions, catalyst, and the ion–exchange interface for the selective formation of products.55
 |
| | Fig. 9 Membrane electrode assembly with GDE [reproduced from ref. 25, Copyright © 2016 Elsevier]. | |
 |
| | Fig. 10 (a) Formic acid cell performance using graphite flow field, nanoparticle Sn@GDE, Nafion® 212 membrane, and IrO2 coated GDE and (b) cell voltage polarization plot [ref. 50, Copyrights © 2017 Elsevier Ltd]. | |
 |
| | Fig. 11 (a) MEA-type reactor configurations for CO2R. Type I and IV operated in the vapor phase, type II carries a water liquid flow in the center compartment and type III holds a liquid anolyte flow. (b) MEA based gas phase CO2R reactor with the recirculation of by-products and maximized HCOOH production. The effect of recirculation on the reaction selectivity has been simulated by calculating the variation of partial pressures of the gases at four steps. At the inlet (A) and outlet (B), at the outlet of a condenser differentiating HCOOH and H2O from the gas stream (C), and behind a pressure valve (P) allowing a part of the gas (D) [reproduced from ref. 51, no permission is required]. | |
2.5 Microfluidic reactor
Kenis' group56 proposed the microfluidic reactor (MR), also called the microfluidic flow reactor (Fig. 12), where the liquid electrolyte stream separating the anodes and the cathodes is passed through a thin channel (<1 mm) in a laminar flow instead of an ion exchange membrane (membrane less configuration). The product formation in MR happens with the redox reaction via diffusion rather than by the membrane. This excludes the challenges associated with mass transfer and neutralization by the alkaline anolyte and acidic catholyte attributed to the crossover. As a consequence, it reduces the concentration of the OH− and H+ ions for the enrichment of the current density. The uniqueness of the membraneless configuration enables the compositional tunability of the electrolytes unlike membrane-based CO2R reactors (one of the electrodes is in an acidic environment and other is in an alkaline environment). This results in the optimization of the kinetics and thermodynamics at both the electrodes, thereby enhancing the overall performance of the cell. To increase the current density, MR with GDE contains a very thin diffusion layer (50 nm) composed of three components such as a cathodic layer, GDL, and a gas flow field. Its porous structure gives rise to the physical support and the gas transport tunnels to the cathodic layer. Moreover, the hydrophobic property of GDL is treated by hydrophobic additives such as polytetrafluoroethylene, which safeguards the blocking of the pores while facilitating the CO2R transport to the cathodic layer. Although a high current density more than 400 mA cm−2 can be achieved by MR, it undergoes product crossover and re-oxidation (cathodic products to the anode and the reduction of oxygen to the cathode), thus decreasing the productivity and efficiency. In this regard, an attempt has been made with a dual electrolyte system to produce HCOOH under the optimized anolyte and catholyte pH conditions.57 The current density has been increased from 10 to 60 mA cm−2 at 2.8 V (Fig. 13a–c) upon anolyte optimization (pH = 14) along with FE (81.6 to 95.6%) and the peak energy efficiency (41.7 to 48.5%). Further, Whipple et al.56 reported efficient CO2R to HCOOH using Sn-coated GDE (pH values from 4–10), which worked as an effective MR with a notable FE (89%), energy efficiency (45%), and PCD (100 mA cm−2) at pH = 4. At lower pH, Sn dissolved in a highly acidic medium. It is worthwhile to mention that the comparative study between the H-type electrolyzer and MR has been demonstrated on BiOBr-deposited GDE carbon electrode.58 Even though the H-type liquid electrochemical cell had a mass transport limitation, it delivered a current density of 80 mA cm−2 and stable operation for 65 h toward HCOO−.
 |
| | Fig. 12 Microfluidic reactor setup [reproduced from ref. 25, Copyright © 2016 Elsevier]. | |
 |
| | Fig. 13 (a) Current density at 2.8 V, (b) cathode potential at 100 mA cm−2 under catholyte pH of 7, 1 and 0; anode potentials at 150 mA cm−2 under anolyte pH of 7, 13 and 14, (c) peak faradaic and energy efficiencies before and after electrolyte optimization [ref. 57 Copyright © 2016 Elsevier] and (d) FE of 90% up to 200 mA cm−2 for BiOBr deposited on the GDE carbon electrode [ref. 58 Copyright © 2018 Wiley]. | |
On the other hand, it further shows a high HCOO− selectivity (over 90%) with a PCD (200 mA cm−2) for MR configuration (Fig. 13d). This comparison dictates that the gas phase MR reactors have the potential for high productivity compared to the liquid phase electrolyzers. Castillo et al.59 investigated the influence of Sn catalyst loading and size on carbon paper and superior performance was identified for the loading (0.1–0.75 mg cm−2), smallest particle size (150 nm), FE (70%), and the current density (90 mA cm−2). These investigations show that the advantages and importance of MR may eventually find large-scale applications.
2.6 Zero-gap electrolyzer
In the direction of large-scale applications, the zero-gap electrolyzer has newly emerged and the absence of a cathode flow channel results in a difference when compared to the flow reactor. This enables the conversion of CO2 into desired products even without the liquid catholyte and its circulation loop is also not needed, resulting in a very low cell resistance. This configuration not only affords reduced ohmic losses but also attenuates the problems associated with poor membrane hydration and electrode flooding at higher current densities. Further, this could minimize any losses attributed to the dissolution of CO2 in the catholyte, which in turn decreases the power required to produce the same quantity of CO by 50% with a similar electrocatalyst as in the flow cell. In this context, the zero-gap electrolyzer is the promising configuration for CO2R to value-added products in the future. Fig. 14 shows two types of zero-gap electrolyzers based on the parallel and the interdigitated flow field patterns, and their electrochemical CO2R performances are compared with a well-known catholyte flow by the electrolyzer studied by Mot et al.60 For the CO2 flow rate (200 mL min−1), water injection rate (0.3 mL min−1) and at 100 mA cm−2 for 1 h, the FE of HCOO−, H2, and CO against the cathode flow by parallel and interdigitated patterns are obtained to be 67, 43, and 81%, respectively. Unfortunately, the parallel pattern zero-gap electrolyzer is less efficient than the catholyte flow but the interdigitated pattern zero-gap electrolyzer has shown better performance than the former. Because the interdigitated flow field patterns have forced the CO2 into GDE, hence, efficient mass transfer occurred toward the catalyst surface as a result of diffusion and convection. Furthermore, the rich water availability at GDE provides more humidification of the MEA.60 Recently, the CO2R to HCOO− was investigated by the gas-fed zero-gap and H-type electrolyzers in alkaline and almost basic electrolytes. It is found that the FE and PCD at −1.87 V are prominent as 20.1% and 148 mA cm−2, respectively, when the hydroxide solution is used in the gas-fed zero-gap electrolyzers. Conversely, the H-type cell generated an FE of 2.6% and a PCD of 7.5 mA cm−2 for the optimized potential. This observation underpins that the knowledge garnered by batch type experiments does not lead to practical approaches aimed at industrial CO2R. Therefore, many more experiments should be done beyond the catalyst development-oriented approach such as rational electrolyzer design, engineering, and process optimization to pave strong and stable gas–liquid interfaces for achieving high current density and durability toward large-scale applications.
 |
| | Fig. 14 Schematic representation of the two types of zero-gap electrolyzers with (a) the parallel flow field patterns (mass transfer due to diffusion) and (b) the interdigitated patterns (mass transfer due to both diffusion and convection) [ref. 60, Copyright © 2020, Wiley-VCH]. | |
Besides, Table 2 demonstrates the figures of merit (potential, current density, and FE) of HCOOH/HCOO− by the CO2R process for different reactor designs. Based on the above discussions, it is understood that the engineering of the reactor design for CO2 electrochemical reduction to formic acid has gained tremendous attention in the past few decades for fuel cell and energy storage applications. Nevertheless, the practical application is hampered by several limitations as elucidated in this perspective; hence, the CO2R is still being processed at the laboratory scale. Currently, H-type cells are commercialized despite their limitations in CO2 solubility and mass transport, causing low energy efficiency. However, the flow type reactor could be a potential candidate in the future to attain the base line current density and long-term stability for industrial applications. The prime motivation of the review is to bring readers' attention toward the construction of economic, scalable, and energy-efficient CO2R electrolyzers to produce value-added formic acid. MEA-based and MR configurations the enhanced energy efficiency due to the zero-gap mode and membraneless diffusion mechanism, respectively, lacking the ohmic losses and facilitating the mass transport. In particular, MEA-based reactors lead to the pure form of the liquid products, enabling the choice for future large-scale applications.
Table 2 Performances and results of CO2R to formic acid/formate (HCOOH/HCOO−) by CO2 reactors with different designs
| Reactor design |
Electrocatalysts |
Electrolyte |
Potential (V vs. RHE) |
Current density (mA cm−2) |
FE (%) |
Ref. |
| H-cell |
Electrodeposited Bi dendrites |
0.5 M KHCO3 |
−1.0 |
38.1 |
92 |
61 |
| Sn/SnOx |
0.1 M KHCO3 |
−1.2 |
11.2 |
89.6 |
62 |
| Sulfur-doped indium |
0.5 M KHCO3 |
−0.98 |
∼60 |
93 |
63 |
| PdPt/C |
0.1 M K2HPO4/0.1 M KH2PO4 |
−0.4 |
5 |
88 |
64 |
| Bi–PMo nanosheets |
0.5 M NaHCO3 |
−0.86 |
30 |
93 |
65 |
| Flow cell |
In/Pb |
1 M NaHCO3 |
|
40 |
80 |
66 |
| Chain-like mesoporous SnO2 |
0.1 M KHCO3 |
−1.06 |
13.6 |
95 |
67 |
| Sn nanoparticles (commercial) |
1 M KOH anolyte |
−0.2 |
51.7 |
93.3 |
31 |
| Sn/C-GDE |
DI water |
3.3 |
140 |
94 |
50 |
| 2D-Bi GDE |
0.25 M K2SO4 |
3 |
30 |
80 |
34 |
| GDE |
SnOx–CNT |
0.5 M KHCO3 |
−1.25 |
10 |
77 |
68 |
| Ru, Pd alloy particles |
0.5 M KHCO3 |
−1.1 V vs. NHE |
80 |
90 |
69 |
| CuSn alloy particles |
1 M KOH |
−1.0 |
148 |
82 |
70 |
| Sn particles/C-GDE |
0.45 mol L−1 KHCO3 + 0.5 mol L−1 KCl |
−1.3 |
90 |
70 |
71 |
| SnO2/C-GDE |
0.1 M KHCO3 |
2.5 |
133 |
81 |
72 |
| MEA |
Commercial Sn with a GDE |
0.5 M KHCO3 |
−1.3 |
150 |
60 |
73 |
| Sn powder |
0.1 M KHCO3 |
−1.7 |
9 |
90 |
52 |
| Sn nanostructure |
— |
−0.7 |
5 |
12.5 |
37 |
| SnO2 catalyst carbon black |
1.0 M KOH |
−1.43 |
251 |
80 |
74 |
| MR |
RuPd/Sn |
0.5 M KCl + 1 M HCl |
−0.55 |
100 |
89 |
56 |
| Sn nanocatalyst/graphite rod |
0.5 M KHCO3 |
2.5 |
— |
84.25 |
75 |
| SnO2 nanoparticles |
1 M KOH |
0.95 |
147 |
97 |
76 |
3. Membrane separator
The membrane is one of the crucial parts of electrochemical reactors, which contributes a crucial role in the current density and FE of the CO2R process by preventing product crossover between the electrodes. Ion exchange membranes are often employed due to their selectively permeable characteristic for desired type of ions while holding other ions or neutral molecules. There are three different types, such as monopolar exchange membranes (cation exchange membrane and anion exchange membrane) and the bipolar membrane (BPM) as both the cationic and anionic exchangeable characteristics.77–79 This governs the ion transport pathway between the anode and cathode depending on the pH of the reaction environment, thereby affecting the kinetics of the CO2R reactions. The current perspective in the upcoming sections discusses the conductivity issues and challenges on CO2R parameters (faradaic efficiency and current density) with the introduction of different ionic exchange membranes for HCOOH/HCOO− production.
3.1 Cation exchange membrane
The cation exchange membrane, also known as the proton exchange membrane (PEM), permits the transit of cations while keeping the anions and has potential applications in water electrolyzers and fuel cells. A well-studied PEM consists of the perfluorosulfonic acid derivatives with sulfonic acid groups on the polymer backbone, which is obviously named as Nafion. It has been widely used in the PEM flow cell and H-type CO2R reactors because of its outstanding proton transfer property. Though the ion transport depends on the pH of the reaction medium, proton conduction by PEM will take place thorough sulfonic acid groups (R-SO3Na) in the presence of alkaline solution (e.g., NaOH). It is well known that in the lower pH condition, the transport of anions such as SO42−, Cl−, and NO3− are available as the dissociated states by the occurrence of protons. Such anions are blocked owing to the Donnan exclusion effect and therefore the permeability via PEM is very feeble. Conversely, weak acid anions (F− and NO2−) in their state diminish the Donnan exclusion effect and thus increase the permeation rate.80 Wu et al.37 introduced the buffer layer of KHCO3 between the cathode Sn catalyst and the PEM (Nafion 212) in the PEM flow cell for the production of HCOO−. It is found that CO2R has led to the FE of 70% by the buffer layer while suppressing the H2 evolution reaction. This dictates that ion management depends on the selection of the membrane, which is indispensable for the flow cell. In another study, CO2R in a zero gap flow cell equipped with Nafion by the In–Pb bimetallic catalyst produced 80% FE, which further reduced after 1 h as a result of diffusion barriers occurring at the interface of the membrane and the electrode.81 A comparative investigation between PEM and BPM has been reported by Ramdin et al.36 under a pressure of 50 bar in the flow cell. The generation of HCOO− is assessed by supplying a high-pressure CO2 gas to the cathodic compartment and 90% of FE is observed for the pressure of 40 bar. As the reduction reaction keeps on increasing, HCOO− crossover is seen via PEM in the form of variation in the pH between the compartments and this limitation is addressed with BPM. In general, the membrane thickness also plays a significant role in the product formation, as described by Li and Oloman et al.82 In most of the cases, Nafion 117 with a thickness 180 μm has been adopted as the electrolyte; however, only a few articles have dealt with the thinner Nafion 115 having 127 μm thickness, leading to the loss of the catholyte owing to product crossover. It is worth noting that even Nafion 117 also undergoes a problem associated with HCOO− crossover with catholyte feeding. Hence, it is not only mandatory to study the membrane thickness but also other properties such as the membrane surface, water uptake, and the ionic conductivity, which need to be thoroughly assessed for an efficient CO2R process.
3.2 Anion exchange membrane
Anion exchange membrane (AEM) allows transiting the anions while holding the cations and is also known as the hydroxide conducting membrane due to the conduction of hydroxide (OH−). One of the properties of AEM is the reduction of crossover of the gas molecules (hydrogen and oxygen) in the CO2R electrolyzers. The AEM originates from polymer backbones such as poly(arylene ethers) derivatives (polysulfones, poly(ether ketones)) functionalized with quaternary ammonium anion exchange sites.83 The CO2R reactor equipped with AEM consists of a strong anolyte at the anode compartment, which facilitates the conductivity and oxygen evolution reaction. The conducting species such as OH−, HCO3−, and CO32− ions are formed at the cathode during product formation and this transfer via the AEM separator to anode compartment. The major issue with AEM is CO2 pumping to the anode in the presence of the anion electrolyte. In order to understand the apparent consumption of product formation and CO2 pumping, Ma et al.84 carried out a systematic study on the CO2 utilization in AEM type CO2 reactors. Unfortunately, 70% of consumed CO2 got pumped out to the anodic compartment and 30% remains for product formation. The three-compartment reactor using Sn catalyst on GDE with proprietary Sustanion AEM imidazole has delivered a current density 140 mA cm−2 and FE of 94% over 500 h toward formic acid.50 In addition, Kaczur et al.85 reported a remarkable CO2 toward HCOO− conversion efficiency of 99.2% with a current density of 100 mA cm−2 using Sn as the cathode electrocatalyst and Sustainion X37-50 as the membrane separator. It is important to emphasize that CO2R to HCOOH/HCOO− utilizing AEM is scarce due to the increased ohmic losses, instability in strong alkaline solutions, and product crossover issues due to the pH variation among the respective compartments. Hence, the proper selection of AEM derivatives has an indispensable role in the mass transfer of CO2 within the compartments and the kinetics of the CO2R process.
3.3 Bipolar membrane
BPM is manufactured by the stacking of cation and anion exchange layers forming the interface and it can be operated either in forward bias or reverse bias to drive the electrochemical reactions. When the reaction is performed under forward bias, the cation or proton exchange layer (PEL) is kept toward the anodic compartment while the anion exchange layer is directed to the cathodic compartment (vice versa for reverse bias). On the one hand, the oxygen evolution reaction commences at the anode by proton formation as it requires a stable noble metal oxide electrocatalysts such as IrO2. On the other hand, CO2 reduction will occur at the cathode side resulting from the creation of OH− ions and bicarbonate species. In fact, AEM shows good conductivity toward OH− and HCO3− when the Donnan effect co-ion exclusion has no influence. Importantly, the recombination of H+ and OH− ions at the interface of PEM/AEM forms water and the bicarbonate species react with H+ to yield CO2 as this is a pumping issue in the case of AEM. For the efficient CO2R process, the operation of BPM under reverse bias is more feasible as compared to forward bias because the water splitting reaction is encountered at the PEM/AEM interface. Though the reverse bias enables AEM toward the anode with the flow of alkaline electrolyte and the CEM at the cathode accepts the CO2 feed, the water splitting reaction leads to the accumulation of H+ ions at the acidic CEM side and OH− ions at the AEM side.86 The underlying mechanism behind water splitting at the BPM interface is caused by proton transfer between the water and the fixed groups and the enriched electric field behavior described by second Wien's consequence from the Onsager's theory.87,88 As discussed earlier, H+ and OH− ions are formed during reverse bias and are transferred from the BPM interface to the cathode and anode. As a result, this balances the overall H+ and OH− ions at the reduction process, which in turn maintains the pH. Compared to monopolar membranes, BPM excels in pH stabilization for the better design and optimization of the economic CO2R reactors, as discussed in the literature.79,89,90 When CO2R is carried out for HCO3− salts under reverse bias, H+ ions accumulate at the cathode. The acid–base equilibrium among H+ ions and HCO3− creates CO2 and water (eqn (1) and (2)) near the membrane solution interface, which have a profound impact on the efficiency of the CO2R reaction at the cathode. Besides, the migration of HCO3− to the anode is highly hampered owing to the Donnan exclusion effect and the transfer of H+ is promoted at the CEM in such a way that the CO2 pumping issue experienced in AEM| | |
HCO3− + H+ → H2O + CO2
| (1) |
| | |
CO32− + 2H+ → H2O + CO2
| (2) |
by HCO3− and CO32− at the anodic compartment is remarkably curtailed.78 Consequently, CO2R reactors functioning with BPM makes HCO3− a major thread for the enhancement of local CO2 concentration in the cathode rather than the solubility limit of CO2 in the aqueous media. This in turn provides a novel way to facilitate the current density of the liquid phase CO2R. At the end, the BPM in the context of water splitting at monopolar junctions displays the minimization of product crossover over CEM and AEM as they possess a low capacity for stabilizing the pH. For example, Zhou et al.90 used the conventional H-type cell to study the CO2R performance of the cathode electrocatalyst as the Pd/C nanoparticle coated Ti with the bipolar membrane (Fumasep FBM-PK). A maximal current density of 8.5 mA cm−2 and the 94% of FE for CO2 reduction to formate in 2.8 M KHCO3 have been obtained. Even though BPM shows several advantages over CEM and AEM, the research investigations on CO2R for HCOOH/HCOO− are not well explored; rather, notable reports are available on the gaseous products such as CO.86,91 A large amount of potential develops at BPM under reverse bias, thereby increasing the required voltage for CO2R, declining the efficiency. This intensified the scientific community to further take forward the research directions on advanced BPM-based CO2R reactors for large scale applications. Table 3 demonstrates that progress has been made on CO2R toward HCOOH/HCOO− using different ion exchange membranes.
Table 3 The comparison of CO2R performances to HCOOH/HCOO− by different ion exchange membranes (CEM, AEM and BPM)
| Type |
Membranes |
Electro- catalysts |
Electrolyte |
J (mA cm−2) |
FE (%) |
Ref. |
| CEM |
Nafion 211 |
Sn |
0.1 M KHCO3 |
15 |
91 |
28 |
| Fumasep FKB-PK |
Sn |
1 M KHCO3 |
30 |
90 |
36 |
| Nafion 212 |
Sn |
0.1 M KHCO3 |
−9 |
90 |
37 |
| Nafion 117 |
SnO2 |
0.5 M KHCO3 |
12.5 |
62 |
92 |
| Nafion 117 |
Cu |
0.5 M KHCO3 |
— |
60 |
93 |
| AEM |
Sustainion X37-50 |
Sn |
Humidified CO2 |
100 |
99.2 |
86 |
| PSMIM AEM |
2D-Bi |
Humidified CO2 |
30 |
90 |
33 |
| Sustainion X37-50 |
Sn/PTFE |
CO2 |
140 |
94 |
50 |
| AMI-7001S |
In–Pb/C |
1 M NaHCO3 |
40 |
80 |
83 |
| BPM |
Fumasep FBM-PK |
Sn |
0.5 M KHCO3 |
90 |
30 |
36 |
| Fumasep FBMPK |
Pd/C |
2.8 M KHCO3 |
8.5 |
∼100 |
91 |
In this section, it is elucidated that the membrane is one of the principle factors to tailor the electrochemical CO2 reduction for the desired product. Therefore, the polymer electrolyte membranes used in the PEM fuel cells can also be subjected to CO2 electrolyzers to attain the performance metrics necessitated for commercial implementation. It is described as the optimal performance of the CEM and AEM should possess a good swelling property, selectivity, and ionic conductivity. To date, Nafion CEM is a well-developed membrane and considered as an efficient component for CO2 reduction into HCOOH/HCOO−. Further, the increased selectivity and swelling characteristics of BPM are essential for efficient water dissociation at the interface, which in turn decreases the membrane voltage and increases the energy efficiency.
4. Selectivity of CO2 electrolyzers
The selectivity of CO2R products is not only governed by the nature of the catalyst, morphology, temperature, pressure, potential, current density, pH, electrolyte media, aqueous or non-aqueous solvents, and membranes but also by the electrochemical reactor configurations.36 Successful attempts have been made with continuous-flow electrolyzers of reasonable size (300 cm2), delivering the CD (over 100 mA cm−2), durability (h to several months), and high product selectivity (mostly HCOOH/HCOO− and CO). For example, one of the unique CO2 electrolyzers was developed to produce pure HCOOH based on three-compartment design. The generation of water electrolysis has been done using mixed metal oxide-coated Ti anode and AEM was deployed to prevent the transfer of HCOO− to the anode. This peculiar configuration has makes contact as the zero-gap between the anodic compartment and the anode membrane and the optimal FE of 85–95% for HCOOH (5 to 30 wt%) was produced with a large CD of 140–200 mA cm−2.94 In a microfluidic cell configuration, a very narrow (∼1 mm thick) dual channel electrolyte arrangement enabled a high product selectivity of 95% for HCOOH and CD > 300 mA cm−2. The effect of micro-channel thickness was found to be one of the crucial factors for the desired products.95 Interestingly, the production of HCOOH acquired a notable difference in the current density, overpotential, and FE or selectivity when the CO2R reaction was changed from the liquid phase to gas phase electrolysis.96 Based on the above discussions and prior observations,97 it is understood that the product selectivity can be highly tailored by the partial pressure of CO2 and components (GDE, MEA, membrane, etc.) of the electrolyzer. Ultimately, such factors vary for different configurations and hence the selectivity of CO2R also varies.
5. Fabrication of CO2 electrolyzers and membranes
5.1 CO2 electrolyzers
The CO2 electrolyzer is often fabricated with either in two or three compartments wherein the anodic and cathodic counterparts are separated by an oxidant-resistant permeable ion exchange membrane. These compartments are machined using thick acrylic plastic or polytetrafluoroethylene with the required dimensions. The anodic compartment was packed with a thick titanium anode back plate on one side bearing gold coating (2.54 microns) and a long titanium post on the other side. The conductor posts were tightened via holes from the outside of the anodic compartment. In order to prevent the fluid from flowing away from the anodic back plate, the gold-coated titanium plate was glued and sealed in inside the compartment with a silicone adhesive. On the other hand, the cathodic compartment was packed with perforated stainless-steel plate with a similar dimension as that of the anodic compartment. The ionic exchange membrane was sandwiched between the anodic and cathodic compartments; thereby, the cell assembly was completed using rubber gasket glued to each compartment's surface. Finally, the CO2 electrolyzer was compressed and sealed with nuts and bolts and allowed for DC power supply. However, the cell fabrication material was suggested to be corrosion-resistant as acrylic and other materials such as titanium, tantalum, zirconium, polyethylene, and polystyrene can also be utilized. Depending upon the requirements and modifications, three compartments and stacked electrolyzers can also be fabricated to promote the CO2R reaction and figure of merits.98
5.2 Membranes
The frequently-used cationic exchange membrane Nafion is made up of perfluorosulfonic acid (PFSA) groups consisting of perfluorocarbon backbones with side chains terminated with sulfonated groups. The precursor PFSA is usually converted into resin (H+ type) using 6 M NaOH and 2 M H2SO4 aqueous solutions under stirring for a long time. Thereafter, the PFSA resin was dissolved in N,N-dimethylformamide (DMF) in an autoclave for 230 °C to attain 5 wt% solution. Then, the solution was condensed to 13.6 wt% and was casted onto a glass plate with the help of a stainless-steel scraper. Followed by this, it was again kept in an oven at 180 °C for 4 h. After the oven temperature was cooled down to the room temperature, the PFSA membrane with a desired thickness (in terms of microns) was peeled off from the glass plate. Further, it was soaked in 1 M KOH solution for 24 h to activate the ion exchange and its capacity was finally determined by the titration method.99,100 Similarly, the anion exchange membranes bearing a variety of precursors, e.g., poly(arylene ethers), polysulfones, and poly(ether ketones), are dissolved in dimethyl sulfoxide (DMSO) with a content of 5 wt%. Then, the solution was filtered and cast onto a glass plate. The resultant glass plate was heated at 80 °C for 12 h to yield the membrane. Then, the above membrane on a glass plate was soaked in water until it was automatically peeled off from the plate. At last, the membrane was submerged in 1 mol L−1 NaOH solution for 48 h and finally washed with deionized water.101 The bipolar membrane can be prepared by choosing proper cationic (e.g., Nafion NR-211) and anionic membrane layers (e.g., hexamethyl-p-terphenyl poly(benzimidazolium), known as PBI). To begin, PBI in the iodide form was changed to the OH− form by immersing the membrane layer in 1 M KOH for 48 h at room temperature. Further, the membrane was washed several times with deionized water, followed by soaking for few days prior to usage. On the other side, Nafion was transferred to the H+ form by soaking in H2SO4 for a day. After several washes with deionized water, the membrane was kept in the same for a longer duration. Then, BPM was fabricated with or without the interlayer (e.g., polyethylene glycol) by laminating with the help of post-treatment processes such as a hot melt or solvent fumigation. Due to the electrostatic force of attraction, the Nafion and PBI membranes were adhered together when they were placed one over another. According to the procedure followed for AEM or CEM, BPM can also be prepared step-by-step by taking the initial precursors of the desired membranes with or without the interlayer.102,103
6. Electrocatalysts
To track the targeted chemicals/fuels along with the enriched current density, feeble overpotential, and long-term stability, the proper choice of electrocatalysts is essential so as to make the device operational cost lesser and increase the energy efficiency. The electrocatalysts subjected to the CO2R process can be classified into homogeneous and heterogeneous, where the former could be homogeneously dispersed in the electrolyte to activate a large contact area, leading to high utilization efficiency. However, heterogeneous electrocatalysts have been reserved for CO2R because of the easy separation of the products from the electrolyte and catalyst recyclability. The product selectivity is primarily tackled by exploring the relationship between the binding affinities and strength of the intermediates and the electrocatalyst surface. The plausible reaction intermediates are *OCHO, *COOH, *CO, and *H (for H2), in which the adsorption of *OCHO and *COOH is via the endothermic reaction. Formic acid or formate is produced by the catalyst surface, which stabilizes the *OCHO rather than *COOH, thus favoring carbon monoxide.104,105 Based on the above predictions, CO2R to HCOOH/HCOO− conversion is performed so far with metal-based (group II metals) and metal-free (carbon, N (B)-doped carbon materials) electrocatalysts. Due to the poor electronic conductivity and selectivity, the metal free electrocatalysts hinder their application in CO2R toward HCOOH/HCOO−. Thus, metals such as Pb, Pd, Sn, Bi, In, Hg, Cd, Sb, and Co have been well established as electrocatalysts for the CO2R toward HCOOH/HCOO−; however, recently, Pb, Sn, Bi, and In have become popular electrocatalysts when alloying, doping, and compositing for efficient CO2R because doping and alloying are the most predominant ways to develop new electrocatalysts, thereby affecting the electronic structure and their association with selectivity and activity of CO2R toward HCOOH/HCOO−. Therefore, the current section particularly aids very recent developments on Pb, Sn, Bi, and In catalysts to the readers as there are already significant progresses made on the same catalysts.106
Despite the intrinsic toxicity and environmental unfriendliness of Pb, it is of fundamental interest to assess CO2R owing to its excellent selectivity toward HCOOH/HCOO−, which shows the similarities and discrepancies compared to main group electrocatalysts. Pb electrodes in the form of flat plates, nanowires, and granules have been demonstrated to be the catalyst for CO2R.107,108 For instance, PbS nanocrystals prepared under in situ electrochemical CO2R conditions reveal CO2 to HCOO− conversion >97.6 ± 5.3% of FE at −1.2 V and a maximal mass activity of 74.9 ± 4.7% mA mgPb−1 at 1.4 V. Besides, in situ synchrotron X-ray diffraction unraveled the structural evolution of PbS via PbCO3–Pb as a result of increased concentration of the CO2/HCO3/CO32− species.109 In another study, 3D layered porous CNT aerogel hybridized with Pb nanoparticles showed stable CO2 reduction performance for 10 h with a FE of 84.6% and a current density of 28 mA cm−2 for the production of HCOO−. This excellent activity is attributed to the typical 3D porous structure, large specific surface area, and favorable conductive network, leading to promoted selectivity.110 One of the interesting approaches by Widiatmoko et al. has reported alloy fabrication using the Pb–Sn combination for the separation of HCOOH. It is found that the productivity and selectivity were higher in KHCO3 when compared to NaHCO3. The maximum amount of HCOOH is observed to be 9400 μmol with an FE of 67.19% and the electrode is stable over a period of 7 h of electrolysis.111 Therefore, a high FE can be obtained against HCOO− using Pb electrodes even though a higher overpotential is required to drive the CO2R reaction compared to Sn, Bi, and In. Sn-based electrocatalysts display excellent environmental and economic benefits such as abundance, low toxicity, and high selectivity toward HCOO− but with a large overpotential and low current density. Fortunately, Sn catalysts were not only restricted to HCOO− but the selectivity of the product can also be tuned (CH4 and CO) by making composites with oxides and zeolites.112 It is prone to oxidize under air and its surface is normally covered by oxide layers. These surface oxide layers reduce before taking part in the CO2R reaction depending on the standard redox potential. Hence, a large overpotential is necessary for commencing the CO2R due to a strong kinetic barrier for electron transfer from Sn to CO2 to create the initial CO2˙− intermediate. Nevertheless, long-term stability is a degrading factor in Sn electrodes, which arises from cathodic deterioration of the Sn surface, formation of non-catalytic species from intermediates, and contaminants from electrolyte. Very recently, Tsujiguchi et al.113 investigated the electrochemical CO2 reduction of Sn particles decorated on reduced graphene oxide (rGO) and observed an FE of 98 ± 0.7% at −0.82 V (Fig. 15a–d). To support this, density functional theory (DFT) calculation predicted the formation of various oxidized functional groups by rGO adjacent to the Sn surface and thus the production of HCOO− occurred via the *COOH intermediate rather than *CO. Similarly, Wen et al.114 fabricated a highly selective Sn nanosheets decorated with Bi nanoparticles, which were further reduced into Sn nanosheets to form a Bi–Sn bimetallic catalyst. Owing to the orbital interaction of the Bi–Sn and Sn–O interface, the Bi–Sn bimetallic catalyst delivered a remarkable FE (96%) and productive rate (0.07 mmol h−1 cm−2) at −1.1 V vs. RHE. Beneficially, the catalyst sustained efficiency over a large period of electrolysis time (100 h). Moreover, DFT calculations elucidated that the loading of Bi nanoparticles lifted the electronic states of Sn just away from the Fermi level, allowing the HCOO* intermediate to adsorb easily at the Bi–Sn interface. This facilitated the migration of electrons for the selective and durable conversion of CO2 into HCOO−. Moreover, with the in situ formation of the SnO2/Sn heterostructure, an efficient CO2R activity was noted against HCOO−, possessing an FE of 93% and a partial current density of 28.7 mA cm−2 at −1 V vs. RHE. It was found that Mott–Schottky heterojunctions were formed at SnO2/Sn, which favored the HCOO− productivity while suppressing H2.115 Another pioneering work was carried out by Ito et al.116 for the phase-dependent CO2R performance of NiSn bimetallic alloys, where one of the phases stimulated the primary reaction and other induced the secondary side reaction. NiSn alloys in the forms of Ni3Sn4, Ni3Sn, and Ni3Sn2 were fabricated and the synergetic impact was identified for Ni3Sn4; herein, the Sn concentration is richer, creating more Ni–Sn bonds instead of Ni–Ni bonds. Thus, the FE of HCOO− generation of 79.1 ± 1.5% was achieved at −0.8 V vs. RHE for Ni3Sn4. This insightful investigation leads to a better understanding of CO2R and provides a novel strategy to design bimetallic alloys when mixed with other metals such as Bi and In.
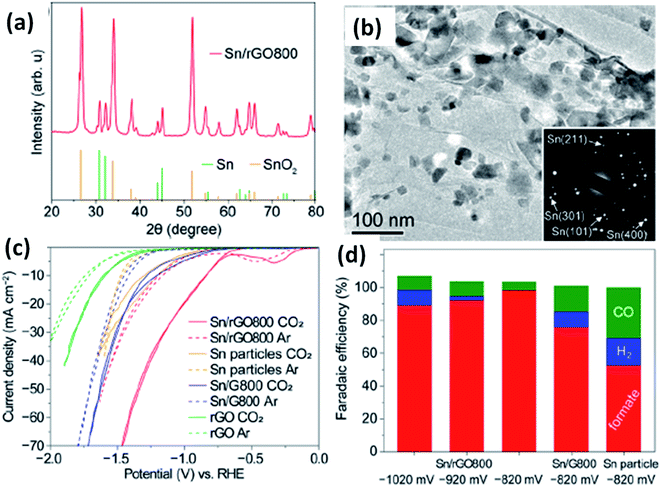 |
| | Fig. 15 (a) XRD pattern, (b) TEM micrograph of Sn/rGO composites, (c) hydrodynamic voltammograms of different cathodes with and without rGO, and (d) FE of HCOO−, H2, and CO for Sn/rGO800, Sn/G800, and Sn particles [ref. 113, Copyright © 2021, American Chemical Society]. | |
Unlike Pb and Sn, Bi is well-known for its low toxicity and environmental benignity. After the first observation of CO2R by Komatsu et al.,117 it has been developed as a catalyst for CO generation in ionic liquids. However, in recent days, Bi is one of the leading electrocatalysts for the selective production of HCOO− in aqueous solution and can be simply fabricated by electrodeposition while carefully controlling the deposition potential, time, and base substrate. Importantly, the product formation happened through *OCHO intermediate and ultimately resulted in almost 100% FE of HCOO−. To start with, the in situ electrochemical transformation of BiVO4 flakes to bismuthene nanosheets demonstrated a large FE toward HCOO− (97.4%) and a very huge current density −105.4 mA cm−2 at −1 V vs. RHE. Both the experimental and theoretical backgrounds show that the outstanding CO2R reaction of the ultrathin bismuthene nanosheets is assigned to the faster kinetics with regards to the HCOO* intermediate in comparison with *COOH and H*.118 Though most of the research efforts have been dedicated to form HCOO− depending upon the catalyst, a novel Bi–Sn based bimetallic catalyst has also proven to be efficient for HCOOH.119 In this case, FE for the production of HCOOH gradually increased with moles of Bi in the BixSny catalysts on Cu and Bi/Cu is shown to have promoted the values of current (30 mA) and FE (90.4%) at −0.84 V vs. RHE. In addition to this, Wu et al.120 prepared a non-precious bimetallic aerogel of Bi–Sn with 3D interconnected channels, abundant interfaces, and hydrophilic surfaces. Unlike pure Bi or Sn, Bi–Sn aerogel exhibited more active sites, endowing a high FE (93.9%) and 90% against HCOOH after 10 h in the flow battery (Fig. 16a–g). This investigation paves a novel way to design non-precious metal aerogels having numerous reactive sites for the selective detection of products. Finally, In-based electrocatalysts almost resemble Bi and Sn except its mild toxicity and 10 times higher cost, which become major obstacles in the practical applications. It is a highly oxophilic metal and so, normally, the oxide layer is wrapped on its surface, resulting in poor thermal stability. Meanwhile, the oxide layers still actively take part in the CO2R reactions. Hou et al.121 realized the efficient CO2R on InN–C catalysts in membrane electrode assembly systems. When the reaction is done in aqueous phase, the maximum FE for HCOO− is found to be 92.2% at 0.9 V vs. RHE and it is slightly enhanced to 92.5% at 2.96 V with a partial current density of 60.1 mA cm−2 by membrane electrode assembly electrolysis. Likewise, In-coated gas diffusion electrode enhanced the selectivity of HCOOH with a notable FE (38%) at −1.65 V vs. Ag/AgCl in comparison with In foil (11%).122 Most recently, In/ZnO@C hollow nanocubes have proven to be a promising electrocatalyst for CO2R to HCOOH via HCOO* intermediates (Fig. 17a–d). The unique nanocubes exhibited superior catalytic activity and selectivity as the PCD of 23.5 mA cm−2 with a FE of 90% at −1.2 V vs. RHE.123 Since the CO2 reduction activity relies on the nature of the active sites and the surface hydroxyl groups of the electrocatalyst, Li et al.124 investigated such parameters on electrodeposited Bi, Sn, and In by in situ XRD and DFT calculations. The active sites such as Bi0, SnO, and In2O3 with affinities declared that there are two pathways for the production of HCOO−: (i) interaction of CO2 with hydroxyl ions to form bicarbonate species, (ii) hydroxyl free sites of the *OCHO intermediate. The role of hydroxyl species in CO2R gives the order Bi < Sn < In for the affinity of these species.
 |
| | Fig. 16 (a) Representation of the synthesis of Bi–Sn aerogel, (b) TEM image of Bi–Sn, (c) LSV curves of Sn, Bi, Bi–Sn and bulk Bi–Sn, (d) partial CD for HCOOH of the prepared electrocatalysts, (e) FEs of various products of Bi–Sn aerogels, (f) comparison of HCOOH FEs of the synthesized Bi, Sn, Bi–Sn and bulk Bi–Sn at different applied potentials and (g) long term durability study of Bi–Sn for 10 h [ref. 120, Copyright © 2021, Wiley-VCH GmbH]. | |
 |
| | Fig. 17 (a) SEM micrograph, (b) TEM of In/ZnO hollow nanocubes@C, (c) LSV sweeps of In/ZnO@C, ZnO@C and In2O3 nanoparticles in N2− and CO2− saturated 0.5 M KHCO3 solutions and (d) variation of FEHCOO−, FECO and FEH2 for In/ZnO@C as a function of the applied potential [ref. 123, no permission is required]. | |
7. Economic feasibility and future research directions
Despite a sufficient number of lab scale experiments that have been performed on CO2R to produce several liquid and gas fuels, there have been less attempts so far to establish the foundation of electrolyzer systems at the pilot scale. The current benchmark of CO2R in the pilot scale was developed for CO production by Dioxide Materials using the ionic liquid-assisted MEA and was obtained as the current density (400 mA cm−2) for 4500 h.125 Mantra Venture Group Ltd. (Canada)126 deployed a pilot plant for capturing CO2 from the Lafarge plant that emits 100 kg daily and the same was designated for the electrochemical conversion to HCOO− salts that sell for $1500 per ton. Similarly, a plant for HCOOH production with a capacity of 10 kg of CO2 per day was developed by Techwin Co., Ltd. (South Korea).127 To further scale up CO2R, the Det Norske Veritas (DNV, Norway) employed a semi-pilot size reactor (surface area of 600 cm2) for the electrochemical reduction of CO2 to HCOO−/HCOOH with a reducing capacity of ∼1 kg of CO2 per day.128 In 2019, the techno-economic analysis of CO2R for the production of HCOOH was analyzed by De Luna et al.129 and the fossil fuel-derived electrocatalyst has become cost-effective with electricity costs of 4 cents per kW h, 90% FE, and 70% energy conversion efficiency compared with that of the biocatalytic process. This brief survey conveys that the production of HCOO−/HCOOH has an indispensable scope for commercialization such as CO and, currently, the research on this domain has expanded globally. Thus, electrolyzer design and membrane are the focus of the present review, which could also determine the efficiency of the figures of merits in the electrochemical CO2 reduction, as highlighted in this perspective. Therefore, achieving a high performance in CO2 electrolyzers not only demands promoted electrokinetics but also requires precise device engineering.130,131 To meet a high reaction rate, CO2R must be carried out in continuous flow reactors encompassing MEA with GDE, leading to a thin hydrodynamic boundary layer, which allows the strong diffusion of CO2 to the catalyst, resulting in the high current density. In this aspect, flow reactors would be a major development direction in the future and the gas-phase CO2 stream, and relevant polymer electrolyte membrane and gas diffusion electrodes are carefully considered for further industrial application. In addition, the channel length and the flow rate of the CO2 stream associated with CO2 concentration on the cathode surface must also be optimized. Moreover, the highly alkaline environment in MR reveals a current density greater than 1 A cm−2; however, now the production is efficient for CO rather than HCOOH, necessitating further research advancements. To date, the use of zero-gap electrolyzers offers the straight-forward implementation of industrial systems to generate high value-added products with a large energy efficiency. The ultimate goal of CCU will be realized when the electrolyzer design is sufficiently advanced and compatible with the techno-economic requirements. Thus, the industrial processing architecture could be established, safeguarding the world by renewable technology.
8. Summary and outlook
Energy crisis and global warming are the most challenging issues existing in the world and the electrochemical CO2 reduction provides unprecedented opportunities to address the above obstacles by closing the carbon cycle. The electrochemical reduction of CO2 to formic acid has great scope as a hydrogen energy carrier, mobility households, and in different industrial sectors. There has been a significant effort made to get the base line current density with FE; however, those are yet to be assessed by the electrocatalysts, electrode modification, and stabilizing the electrolyte. CO2R not only depends on such factors but also relies on the reactor design and the membrane structure. Therefore, a series of configurations (H-type, flow cells, microfluidic, and zero-gap reactors), its components (gas diffusion electrodes, membrane electrode assembly), the different types of membranes (proton exchange membranes, anion exchange membranes, and bipolar membranes), and the recent advancement on the catalyst have been reviewed in this perspective to explore scientific insights for efficient CO2R.
Yet, the conventional H-type reactor is the universal architecture for the CO2R screening of a vast amount of catalysts; however, still ER is gaining more attention to observe the enhanced and accurate performance by modifying the position of the reference electrodes and the inside pressure of the reactor. The flow reactors are becoming alternatives to H-type cells, conquering the mass transfer and low CO2 solubility wherein reactants and products are subsequently transferred among the electrodes. Unlike the liquid phase flow reactors, the use of solid electrolytes in a gas phase flow reactor does not require additional steps the final product separation, nominating as an emerging one. Interestingly, it is proved that controlling the stop-start motion of the electrolytes flow is also a crucial factor behind HCOOH production, allowing a sufficient time for CO2 gas molecules to effectively reduce. It is also realized as switching from the liquid-phase electrolysis to the gas-phase is desired for industrial-scale applications. Besides, the different MEA type gas and liquid phase reactors have emerged for the electrochemical reduction of CO2 into HCOO− or HCOOH and the recirculation of by-products has been introduced to improve the efficiency of HCOOH. The membraneless MR is formed with the accommodation of gas products; however, this is also prone to suffer from product crossover and the re-oxidation of products. Membrane-based CO2R electrolyzers are configured to minimize the distance between the electrodes, obviously called ‘zero-gap’, which in turn decrease the internal ohmic resistance and enhance the efficiency as well. Further, the gas fed zero-gap electrolyzer has been newly existing in the direction of large-scale applications but the CO2R toward HCOOH is at an initial stage. Despite the several improvements, all the reactors still show drawbacks that have to be addressed with further investigations. In particular, CO2R to HCOO−/HCOOH by MEA based flow reactors and gas fed zero-gap configurations have potential in the future to boost the current density and FE for the commercial commodity. All the reactors govern a similar electrochemical principle with CO2 reducing cathode, materials stream, and oxygen-generating anode. In these, the cathode holds special attention, carrying the electrocatalyst for the production of value-added chemicals (e.g., formic acid). The current scientific research also lacks in the anode design, which remains one of the biggest aspects to conquer the full cell overpotential. Moreover, the key points (pH of the electrolyte, ionic conductivity, selectivity, and swelling property) of different ion exchange membranes have been taken into the consideration and a very few reports are available on HCOOH by the CO2R process using BPM as compared to CEM and AEM. While screening the performance of different electrocatalysts Pb, Sn, Bi, In, novel alloy combinations (Pb–Sn, Bi–Sn, Bi–Sn–In) have delivered promising results due to the unique morphologies and the aerogel designs. Importantly, aerogels have garnered numerous reactive sites (3D interconnected channels, abundant interfaces, and hydrophilic surfaces) for the selective detection of desired products. Hence, the futuristic improvements should also rely on the proper choice of the electrolyzers, membranes, and electrocatalysts to meet the target proposed by the techno-economic analyses (current density of 200 mA cm−2 and the reactor duration 20000 h). As our ultimate motive of the global is freedom from energy shortage and pollution, it is also better to transition from electrochemical to photoelectrochemical CO2 reduction utilizing the abundant solar energy, which is clean, green, and renewable.
Conflicts of interest
Authors have not any conflict of interest.
Acknowledgements
Authors acknowledge GAP-308, funded by Ministry of Steel, Government of India.
References
- S. Chatterjee, I. Dutta, Y. Lum, Z. Lai and K. W. Huang, Energy Environ. Sci., 2021, 14, 1194 RSC.
- NOAA, Earth's CO2 home page, accessed on January 2, 2020 Search PubMed.
- I. E. Agency, Energy Technology Perspectives 2014, Paris Cedex, France, 2014 Search PubMed.
- Y. Y. Birdja, Electrocatalytic CO2 reduction toward liquid fuels: on heterogeneous electrocatalysts and heterogenized molecular catalysts, PhD thesis, Leiden Institute of Chemistry (LIC), Science, Leiden University, 2018.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188 CrossRef CAS.
- Niharika, Y. S. Chaudhary and S. Basu, CO2 Conversion into Chemicals and Fuel: India's Perspective, in Climate Change and Green Chemistry of CO2 Sequestration. Green Energy and Technol., ed. M. Goel, T. Satyanarayana, M. Sudhakar and D. P. Agrawal, Springer, 2021, ch. 8, pp. 105–122 Search PubMed.
- H. Yoshio, K. Katsuhei and S. Shin, Chem. Lett., 1985, 14, 1695 CrossRef.
- H. Yoshio, K. Katsuhei, M. Akira and S. Shin, Chem. Lett., 1986, 15, 897 CrossRef.
- K. Malik, S. Singh, A. Verma and S. Basu, Wiley Interdiscip. Rev.: Energy Environ., 2016, 6(4), 1 Search PubMed.
- G. Gag and S. Basu, Electrochim. Acta, 2015, 177, 359 CrossRef.
- D. Chanda, R. A. Tufa, D. Aili and S. Basu, Nanotechnology, 2021, 33, 055403 CrossRef PubMed.
- R. A. Tufa, D. Chanda, M. Ma, D. Aili, T. B. Demissie, J. Vaes, Q. Li, S. Liu and D. Pant, Appl. Energy, 2020, 277, 115557 CrossRef CAS.
- S. Verma, B. Kim, H. R. Jhong, S. Ma and P. J. Kenis, ChemSusChem, 2016, 9, 1972 CrossRef CAS PubMed.
- D. Du, R. Lan, J. Humphreys and S. Tao, J. Appl. Electrochem., 2017, 47, 661 CrossRef CAS.
- V. P. Roan, et al., Review of the research program of the freedom CAR and fuel partnership, 3rd report, National Academy of Sciences, The National Academies Press, Washington, DC, 2010 Search PubMed.
- J. Alazemi and J. Andrews, Renewable Sustainable Energy Rev., 2015, 48, 483 CrossRef CAS.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536 RSC.
- L. Fan, C. Xia, P. Zhu, Y. Lu and H. Wang, Nat. Commun., 2020, 11, 3633 CrossRef CAS PubMed.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef PubMed.
- Q. Wang, M. Ma, S. Zhang, K. Lu, L. Fu, X. Liu and Y. Chen, ChemPlusChem, 2020, 85, 672 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 1 Search PubMed.
- N. Kumari, A. Haider, M. Agarwal, N. Sinha and S. Basu, J. Phys. Chem. C, 2016, 120(30), 16626 CrossRef CAS.
- M. Ma, K. Liu, J. Shen, R. Kas and W. A. Smith, ACS Energy Lett., 2018, 3, 1301 CrossRef CAS PubMed.
- N. Kumari, N. Sinha, A. Haider and S. Basu, Electrochim. Acta, 2015, 177, 21 CrossRef CAS.
- S. Liang, N. Altaf, L. Huang, Y. Gao and Q. Wang, J. CO2 Util., 2020, 35, 90 CrossRef CAS.
- S. Zhang, M. Sun, K. y. Wang, L. Cheng, S. Zhang and C. Wang, ACS Sustainable Chem. Eng., 2021, 9, 2358 CrossRef CAS.
- D. Pan, X. Ye, Y. Cao, S. Zhu, X. Chen, M. Chen, D. Zhang and G. Li, Appl. Surf. Sci., 2020, 511, 145497 CrossRef CAS.
- C. Zhao and J. Wang, Chem. Eng. J., 2016, 293, 161 CrossRef CAS.
- J. Du, A. Fiorani and Y. Einaga, Sustainable Energy Fuels, 2021, 5, 2590 RSC.
- J. C. Li, Y. Kuang, Y. T. Meng, X. Tian, W. H. Hung, X. Zhang, A. W. Li, M. Q. Xu, W. Zhou, C. S. Ku, C. Y. Chiang, G. Zhu, J. Guo, X. Sun and H. Dai, J. Am. Chem. Soc., 2020, 142, 7276 CrossRef CAS PubMed.
- R. Lin, J. Guo, X. Li, P. Patel and A. Seifitokaldani, Catalysts, 2020, 10(473), 1 Search PubMed.
- W. Lee, Y. E. Kim, M. H. Youn, S. K. Jeong and K. T. Park, Angew. Chem., Int. Ed., 2018, 57, 6883 CrossRef CAS PubMed.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitsk, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776 CrossRef CAS.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C. T. Dinh, T. Burdyny, M. G. Kibria, C. P. O'Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442 RSC.
- M. Ramdin, A. R. T. Morrison, M. Groen, R. Haperen, R. Kler, L. J. P. Broeke, J. P. M. Trusler, W. Jong and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 1834 CrossRef CAS PubMed.
- J. J. Wu, F. G. Risalvato, P. P. Sharma, P. J. Pellechia, F. S. Ke and X. D. Zhou, J. Electrochem. Soc., 2013, 160, 953 CrossRef.
- Irkham, S. Nagashima, M. Tomisaki and Y. Einaga, ACS Sustainable Chem. Eng., 2021, 9, 5298 CrossRef CAS.
- K. Natsui, H. Iwakawa, N. Ikemiya, K. Nakata and Y. Einaga, Angew. Chem., Int. Ed., 2018, 57, 2639 CrossRef CAS PubMed.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, ChemSusChem, 2020, 13, 400–401 CrossRef CAS PubMed.
- L. C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973 RSC.
- A. Seifitokaldani, C. M. Gabardo, T. Burdyny, C. T. Dinh, J. P. Edwards, M. G. Kibria, O. S. Bushuyev, S. O. Kelley, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 3833 CrossRef CAS PubMed.
- C. Lee, B. Zhao, J. K. Lee, K. F. Fahy, K. Krause and A. Bazylak, iScience, 2020, 23, 101094 CrossRef CAS PubMed.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Nat. Commun., 2019, 10, 2807 CrossRef PubMed.
- Y. Chen, A. Vise, W. E. Klein, F. C. Cetinbas, D. J. Myers, W. A. Smith, T. G. Deutsch and K. C. Neyerlin, ACS Energy Lett., 2020, 5, 1825 CrossRef CAS.
- C. T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835 CrossRef CAS.
- L. Delafontaine, T. Asset and P. Atanassov, ChemSusChem, 2020, 13, 1688 CrossRef CAS PubMed.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455 RSC.
- G. Díaz-Sainz, M. Alvarez-Guerra, J. Solla-Gullón, L. García-Cruz, V. Montiel and A. Irabien, Catal. Today, 2020, 346, 58 CrossRef.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2017, 20, 208 CrossRef CAS.
- B. Thijs, J. Rongé and J. A. Martens, Sustainable Energy Fuels, 2021, 5, 1867 RSC.
- S. Lee, H. Ju, R. Machunda, S. Uhm, J. K. Lee, H. J. Lee and J. Lee, J. Mater. Chem. A, 2015, 3, 3029 RSC.
- G. O. Larrazábal, P. Strøm-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen, I. Chorkendorff and B. Seger, ACS Appl. Mater. Interfaces, 2019, 11, 41281 CrossRef PubMed.
- A. Patru, T. Binninger, B. Pribyl and T. J. Schmidt, J. Electrochem. Soc., 2019, 166(2), F34 CrossRef CAS.
- H. Pan and C. J. Barile, Energy Environ. Sci., 2020, 13, 3567 RSC.
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109 CrossRef CAS.
- X. Lu, D. Y. C. Leung, H. Wang, M. M. Maroto-Valer and J. A. Xuan, Renewable Energy, 2016, 95, 277 CrossRef CAS.
- F. P. G. D. Arquer, O. S. Bushuyev, P. D. Luna, C. T. Dinh, A. Seifitokaldani, M. I. Saidaminov, C. S. Tan, L. N. Quan, A. Proppe, M. G. Kibria, S. O. Kelley, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1802858 CrossRef PubMed.
- A. Del Castillo, M. Alvarez-Guerra, J. Solla-Gullón, A. Sáez, V. Montiel and A. Irabien, Appl. Energy, 2015, 157, 165 CrossRef CAS.
- B. De Mot, M. Ramdin, J. Hereijgers, T. J. H. Vlugt and T. Breugelmans, ChemElectroChem, 2020, 7, 3839 CrossRef CAS.
- J. Huang, X. Guo, J. Yang and L. Wang, J. CO2 Util., 2020, 38, 32 CrossRef CAS.
- Q. Lai, W. Yuan, W. Huang and G. Yuan, Appl. Surf. Sci., 2020, 508, 145221 CrossRef CAS.
- W. Ma, S. Xie, X. G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D. Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 1 CrossRef.
- R. Kortlever, I. Peters, S. Koper and M. T. M. Koper, ACS Catal., 2015, 5, 3916 CrossRef CAS.
- S. X. Guo, Y. Zhang, X. Zhang, C. D. Easton, D. R. MacFarlane and J. Zhang, ChemSusChem, 2019, 12, 1091 CrossRef CAS PubMed.
- S. R. Narayanan, B. Haines, J. Soler and T. I. Valdez, J. Electrochem. Soc., 2011, 158, 167 CrossRef.
- K. Bejtka, J. Zeng, A. Sacco, M. Castellino, S. Hernández, M. A. Farkhondehfal, U. Savino, S. Ansaloni, C. F. Pirri and A. Chiodoni, ACS Appl. Energy Mater., 2019, 2, 3081 CrossRef CAS.
- Q. Zhang, Y. Zhang, J. Mao, J. Liu, Y. Zhou, D. Guay and J. Qiao, ChemSusChem, 2019, 12, 1443 CrossRef CAS PubMed.
- N. Furuya, T. Yamazaki and M. Shibata, J. Electroanal. Chem., 1997, 431, 39 CrossRef CAS.
- J. Wang, J. Zou, X. Hu, S. Ning, X. Wang, X. Kang and S. Chen, J. Mater. Chem. A, 2019, 7, 27514 RSC.
- A. Del Castillo, M. Alvarez-Guerra, J. Solla-Gullón, A. Sáez, V. Montiel and A. Irabien, J. CO2 Util., 2017, 18, 222 CrossRef CAS.
- D. Kopljar, N. Wagner and E. Klemm, Chem. Eng. Technol., 2016, 39, 2042 CrossRef CAS.
- L. Ma, S. Fan, D. Zhen, X. Wu, S. Liu, J. Lin, S. Huang, W. Chen and G. He, Ind. Eng. Chem. Res., 2017, 56, 10242 CrossRef CAS.
- H. Xiang, H. A. Miller, M. Bellini, H. Christensen, K. Scott, S. Rasulac and E. H. Yu, Sustainable Energy Fuels, 2020, 4, 277 RSC.
- R. Sinha, A. Bisht, S. Rarotra and T. K. Mandal, Ind. Eng. Chem. Res., 2020, 59, 1737 CrossRef CAS.
- C. Liang, B. Kim, S. Yang, Y. Liu, C. F. Woellner, Z. Li, R. Vajtai, W. Yang, J. Wu, P. J. A. Kenis and P. M. Ajayan, J. Mater. Chem. A, 2018, 6, 10313 RSC.
- N. Gutierrez-Guerra, L. Moreno-Lopez, J. C. Serrano-Ruiz, J. L. Valverde and A. de Lucas- Consuegra, Appl. Catal., B, 2016, 188, 272 CrossRef CAS.
- A. Patru, T. Binninger, B. Pribyl and T. J. Schmidt, J. Electrochem. Soc., 2019, 166, 34 CrossRef.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li and T. E. Mallouk, ACS Energy Lett., 2018, 3, 149 CrossRef CAS.
- E. K. Unnikrishnan, S. D. Kumar and B. Maiti, J. Membr. Sci., 1997, 137, 133 CrossRef CAS.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1990, 137, 187 CrossRef CAS.
- H. Li and C. Oloman, J. Appl. Electrochem., 2007, 37, 1107 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49, 3361 CrossRef CAS.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977 RSC.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, Front. Chem., 2018, 6, 1 CrossRef PubMed.
- D. A. Vermaas and W. A. Smith, ACS Energy Lett., 2016, 1, 1143 CrossRef CAS.
- R. Simons, Electrochim. Acta, 1986, 31, 1175 CrossRef CAS.
- R. Simons, J. Membr. Sci., 1993, 78, 13 CrossRef CAS.
- Y. C. Li, Z. Yan, J. Hitt, R. Wycisk, P. N. Pintauro and T. E. Mallouk, Adv. Sustainable Syst., 2018, 2, 1700187 CrossRef.
- Y. C. Li, D. Zhou, Z. Yan, R. H. Goncalves, D. A. Salvatore and C. P. Berlinguette, ACS Energy Lett., 2016, 1, 1149 CrossRef CAS.
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage and S. A. Francis, ACS Energy Lett., 2016, 1, 764 CrossRef CAS.
- Y. Fu, Y. Li, X. Zhang, Y. Liu, J. Qiao and J. Zhang, Appl. Energy, 2016, 175, 536 CrossRef CAS.
- B. C. Marepally, C. Ampelli, C. Genovese, F. Tavella, E. A. Quadrelli and S. Perathoner, J. CO2 Util., 2020, 35, 194 CrossRef CAS.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, Front. Chem., 2018, 6, 1 CrossRef PubMed.
- X. Lu, D. Y. C. Leung, H. Wang and J. Xuan, Appl. Energy, 2017, 194, 549 CrossRef CAS.
- W. Lee, Y. E. Kim, M. H. Youn, S. K. Jeong and K. T. Park, Angew. Chem., Int. Ed., 2018, 57(23), 6883 CrossRef CAS PubMed.
- M. Moradzaman, C. S. Martınezb and G. Mul, Sustainable Energy Fuels, 2020, 4, 5195 RSC.
- J. J. Kaczur and D. W. Cawlfield, US Pat., 1992, 5,092,970 Search PubMed.
- L. Su, S. Pei, L. Li, H. Li, Y. Zhang, W. Yu and C. Zhou, Int. J. Hydrogen Energy, 2009, 34, 6892 CrossRef CAS.
- R. I. Masel, Q. Chen, Z. Liu and R. Kutz, US Pat., 2016, 9,370,773 Search PubMed.
- F. Wang, B. Xue, S. Zhou, J. Zheng, S. Li, S. Zhang and T. A. Sherazi, J. Membr. Sci., 2019, 591, 117334 CrossRef.
- S. Chabi, A. G. Wright, S. Holdcroft and M. S. Freund, ACS Appl. Mater. Interfaces, 2017, 9(32), 26749 CrossRef CAS PubMed.
- J. Pan, L. Hou, Q. Wang, Y. He, L. Wu, A. N. Mondal and T. Xu, Mater. Chem. Phys., 2017, 186, 484 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833 CrossRef CAS.
- N. Kumari, M. A. Haider and S. Baisu, Mechanism of Catalytic and Electrocatalytic CO2 Reduction to Fuels and Chemicals, in Electrochemical Reduction of Carbon Dioxide: Fundamentals and Technologies, ed. J. Qiao, Y. Liu and J. Zhang, CRC Press, 2016, ch. 2 Search PubMed.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2019, 1902338, 1 Search PubMed.
- H. Noda, S. Ikeda and Y. Oda, Bull. Chem. Soc. Jpn., 1990, 63, 2459 CrossRef CAS.
- F. Köleli, T. Atilan, N. Palamut, A. M. Gizir, R. Aydin and C. H. Hamann, J. Appl. Electrochem., 2003, 33, 447 CrossRef.
- Z. Zhang, C. Liu, J. T. Brosnahan, H. Zhou, W. Xu and S. Zhang, J. Mater. Chem. A, 2019, 7, 23775 RSC.
- Y. Xing, M. Cui, P. Fan, J. Ren, C. Zhang, N. Li, X. Wen and X. Ji, Mater. Chem. Phys., 2019, 237, 121826 CrossRef CAS.
- P. Widiatmoko, I. Nurdin, H. Devianto, B. Prakarsa and H. Hudoyo, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 823, 012053 CAS.
- S. Basu, D. Biswal and A. Shegonkar, J. CO2 Util., 2017, 18, 80 CrossRef CAS.
- T. Tsujiguchi, Y. Kawabe, S. Jeong, T. Ohto, S. Kukunuri, H. Kuramochi, Y. Takahashi, T. Nishiuchi, H. Masuda, M. Wakisaka, K. Hu, G. Elumalai, J. Fujita and Y. Ito, ACS Catal., 2021, 11, 3310 CrossRef CAS.
- G. Wen, D. U. Lee, B. Ren, F. M. Hassan, G. Jiang, Z. P. Cano, J. Gostick, E. Croiset, Z. Bai, L. Yang and Z. Chen, Adv. Energy Mater., 2018, 1802427 CrossRef.
- S. Ning, J. Wang, D. Xiang, S. Huang, W. Chen, S. Chen and X. Kang, J. Catal., 2021, 399, 67 CrossRef CAS.
- Y. Ito, S. Kukunuri, S. Jeong, G. Elumalai, A. A. H. Tajuddin, Z. Xi, K. Hu and T. Ohto, ACS Appl. Energy Mater., 2021, 4, 7122 CrossRef CAS.
- S. Y. Komatsu, T. Y. Hiraga, M. Tanaka and A. Kunugi, Denki Kagaku, 1995, 63, 217 CrossRef CAS.
- W. Ma, J. Bu, Z. Liu, C. Yan, Y. Yao, N. Chang, H. Zhang, T. Wang and J. Zhang, Adv. Funct. Mater., 2020, 2006704 Search PubMed.
- Q. Li, Y. Zhang, X. Zhang, H. Wang, Q. Li, J. Sheng, J. Yi, Y. Liu and J. Zhang, Ind. Eng. Chem. Res., 2020, 59, 6806 CrossRef CAS.
- Z. Wu, H. Wu, W. Cai, Z. Wen, B. Jia, L. Wang, W. Jin and T. Ma, Angew. Chem., Int. Ed., 2021, 60, 12554 CrossRef CAS PubMed.
- P. Hou, X. Wang and P. Kang, J. CO2 Util., 2021, 45, 101449 CrossRef CAS.
- Z. Bitar, A. Fecant, E. Trela-Baudot, S. Chardon-Noblat and D. Pasquier, Appl. Catal., B, 2016, 189, 172 CrossRef CAS.
- X. Teng, Y. Niu, S. Gong, M. Xu, X. Liu, L. Ji and Z. Chen, Mater. Chem. Front., 2021, 5, 6618 RSC.
- J. Li, J. Li, X. Liu, J. Chen, P. Tian, S. Dai, M. Zhu and Y. F. Han, Appl. Catal., B, 2021, 298, 120581 CrossRef CAS.
- R. Masel, R. Ni, Z. Liu, Q. Chen, R. Kutz, L. Nereng and K. Lewinski, Energy Procedia, 2014, 63, 7959 CrossRef CAS.
- Mantra Venture Group, ERC and MRFC Technology, Mantra Venture Group, Surrey, Canada, 2014, retrieved from https://1stdirectory.co.uk/_assets/files_comp/9a5be6a3-bb42-4244-9780-2f9a2a7dc3dc.pdf Search PubMed.
- Korea Carbon Capture and Storage Association, Analysis of domestic and overseas CO2 utilization technology for creating carbon dioxide utilization industry, Korea Carbon Capture and Storage Association, Republic of Korea, 2015, retrieved from http://www.prism.go.kr/homepage/researchCommon/downloadResearchAttachFile.do;jsessionid=D1ECC5637B87A8758E5D61977F33E73E.node02?work_key=001%26file_type=CPR%26seq_no=001%26pdf_conv_yn=N%26research_id=1450000-201500157 Search PubMed.
- N. Sridhar, D. Hill, A. Agarwal, Y. Zhai and E. Hektor, Carbon dioxide utilization, Electrochemical conversion of CO2-opportunities and challenges, Position paper 07, Det Norske Veritas, Høvik, Norway, 2011 Search PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 1 CrossRef PubMed.
- G. O. Larrazábal, A. J. Martín and J. P. Ramírez, J. Phys. Chem. Lett., 2017, 8, 3933 CrossRef PubMed.
- O. G. Sánchez, Y. Y. Birdja, M. Bulut, J. Vaes, T. Breugelmans and D. Pant, Curr. Opin. Green Sustain. Chem., 2019, 16, 47 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Mamata Mohapatra
,
Mamata Mohapatra