
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of the halogenido ligands on the Kumada-coupling catalytic activity of [Ni{tBuN(PPh2)2-κ2P}X2], X = Cl, Br, I, complexes†
Polydoros-Chrysovalantis Ioannoua,
Radek Coufalbc,
Kalliopi Kakridia,
Catherine P. Raptopoulou d,
Olga Trhlíkováe,
Vassilis Psycharis*d,
Jiří Zedník*c,
Panayotis Kyritsis*a and
Jiří Vohlídalc
d,
Olga Trhlíkováe,
Vassilis Psycharis*d,
Jiří Zedník*c,
Panayotis Kyritsis*a and
Jiří Vohlídalc
aInorganic Chemistry Laboratory, Department of Chemistry, National and Kapodistrian, University of Athens, Panepistimiopolis, Zografou, 15771 Athens, Greece. E-mail: kyritsis@chem.uoa.gr
bTechnical University of Liberec, Faculty of Health Studies, Department of Science and Research, Studentska 1402/2, CZ-461 17 Liberec 1, Czech Republic
cCharles University, Faculty of Science, Department of Physical and Macromolecular Chemistry, Albertov 2030, CZ-128 43, Prague 2, Czech Republic. E-mail: zednik@natur.cuni.cz
dInstitute of Nanoscience and Nanotechnolgy, N.C.S.R. “Demokritos”, Aghia Paraskevi 15310, Attiki, Greece. E-mail: v.psycharis@inn.demokritos.gr
eInstitute of Macromolecular Chemistry, v.v.i., Academy of Sciences of the Czech Republic, Heyrovský Sq. 2, 16206 Prague 6, Czech Republic
First published on 14th January 2022
Abstract
Novel nickel(II) complexes bearing (tbutyl)bis(diphenylphosphanyl)amine and different halogenido ligands, [Ni(P,P)X2] = [Ni{tBuN(PPh2)2-κ2P}X2], (X = Cl, Br, I) are prepared, characterized by IR and NMR spectroscopy, mass spectrometry and X-ray crystallography, and tested as catalysts in the Kumada cross-coupling reaction of model substituted iodobenzenes and p-tolylmagnesium bromide. The data obtained together with DFT calculations indicate that these new catalysts operate in the Ni(I)–Ni(III) mode. The highest catalytic activity and selectivity are exhibited by [Ni(P,P)Cl2], which is most easily reduced by the used Grignard reagent to the Ni(I) state. This process is much more energy demanding in the case of the bromido and iodido complexes, causing the appearance of the induction period. [Ni(P,P)Cl2] is also very active in the cross-couplings of substrates with iodine atoms sterically shielded by ortho substituents. The data obtained are in good accordance with the described positive effect of the increased electron-releasing power of N-substituents R′ on the overall catalytic performance of [Ni{R′N(PPh2)2-κ2P}X2] complexes.
In recent years, the chemical and catalytic properties of transition metal complexes bearing N-functionalized bis(diphenylphosphanyl)amine ligands, R′N(PPh2)2, have been under consideration.1,2 For instance, chromium complexes with this type of ligand are known to oligomerize various olefins.3–8 In addition, a large number of [M{R′N(PPh2)2-κ2P}X2] complexes, M = Ni, Pd, Pt; X = Cl, Br, I (see Scheme 1), exhibiting small P–M–P bite angles, were recently reviewed.2 Selected palladium(II) complexes bearing X = Cl,9–14 Br,14,15 I,14,16 catalyze the Suzuki–Miyaura and Heck coupling reactions. Some structurally characterized Ni(II) analogous complexes bearing X = Cl,17–28 Br,18,29–37 I,18,36,38 catalyze polymerization of norbornene20,21 or oligomerization (X = Br,32,34 I,38) and polymerization (X = Br29) of ethene. It should be stressed that nickel(II) complexes of this family are only moderately active catalysts in the Suzuki–Miyaura reaction.35 On the other hand, they exhibit a considerable catalytic activity and acceptable selectivity in the Kumada coupling reaction.23,35
 | ||
| Scheme 1 General structure of the studied complexes [M(P,P)X2], M = Ni, Pd, Pt; X = Cl, Br, I; R' = ((S)-CHMePh), (CH2)3Si(OCH3)3, tBu. | ||
Kumada coupling is one of the most important C–C coupling reactions39 for a wide range of purposes, including pharmaceutical applications.40 Although palladium-based complexes are mostly the first choice catalysts for this coupling,41–43 complexes of other transition metals such as iron,44,45 and nickel46,47 are also used. We have already investigated the catalytic activity of [Ni{R′N(PPh2)2-κ2P}X2], R′ = (S)-CHMePh; X = Cl, Br,35 and R′ = (CH2)3Si(OMe)3; X = Cl,23 in homogeneous systems to extend the scope of nickel(II) catalysts in this reaction. The latter catalyst has also been anchored onto mesoporous molecular sieves, thus providing an active heterogenized catalyst.23 In homogeneous catalytic reactions, both catalysts bearing R′ = (S)-CHMePh) showed a substrate conversion (68% for X = Cl and 63% for X = Br),35 significantly lower compared to that of the catalyst with R′ = (CH2)3Si(OMe)3 and X = Cl (79%).23 These results suggested that the increased electronegativity of coordinated halogenido ligands and the increased electron-donating power of the R′ moiety have positive effects on the catalytic efficiency of this type of nickel(II) complexes. In the work presented herein, the effects exerted by the identity of halogenido ligands X− and the R′ moiety on catalytic activity and selectivity were further assessed by exploring three novel complexes, [Ni{tBuN(PPh2)2-κ2P}X2], X = Cl, Br, I, henceforth referred to as [Ni(P,P)X2], bearing the strongly electron-releasing tbutyl (tBu) group as R′.
Results and discussion
Synthesis, spectroscopic and mass spectrometric characterization of nickel(II) complexes
Complexes [Ni(P,P)X2], X = Cl, Br were prepared by mixing equimolar amounts of tBuN(PPh2)2 ligand48 with a dichloromethane solution of [Ni(PPh3)2Cl2] or [Ni(DME)Br2] (DME = 1,2-dimethoxyethane), respectively (see Scheme 2). Complex [Ni(P,P)I2] was prepared by adding a four-fold stoichiometric excess of NaI to complex [Ni(P,P)Cl2], in acetone, to replace the chlorido ligand by iodide (Scheme 2). The three complexes were characterized by IR, UV/vis and 1H, 13C and 31P NMR spectroscopy, mass spectrometry, as well as by UV/vis spectroscopy (see spectra in ESI†). | ||
| Scheme 2 Synthesis of complexes [Ni(P,P)X2], DME = 1,2-dimethoxyethane. | ||
The 31P NMR spectra of complexes [Ni(P,P)X2], X = Cl, Br, I, each exhibits a singlet at δ 46.6, 53.0 and 63.7 ppm, respectively, due to their coordinated (P,P) = tBuN(PPh2)2 ligand. The signal of the free ligand at δ 57.8 ppm48 is absent in all spectra. These observations demonstrate that, in all complexes, the ligand is coordinated to nickel(II) via both Phosphorus atoms that are equivalent to each other, in agreement with recently reported data on the analogous [Pd(P,P)X2] complexes.14 In addition, the spectra monitored during the six-month storage of samples did not show any significant change, which confirmed the long-term stability of all three complexes when exposed to air at room temperature. 13C NMR spectra are affected by the presence of paramagnetic impurities formed in the presence of the air and moisture. It should be stressed that the amount of impurities affected only the 13C NMR line shape.
The broadening of 13C NMR lines remained constant for at least six months, confirming the sufficient stability of all complexes in the environment.
Based on the comparison with similar [Ni{R′N(PPh2)2-κ2P}X2] complexes reported in the literature,17–38 as well as with the corresponding [Pd(P,P)X2] complexes,14 the IR bands of [Ni(P,P)X2], occurring at 1437, 1433 and 1437 cm−1 for X = Cl, Br and I, respectively, should be assigned to the ν(P–Ph) stretching modes, whereas the band occurring uniformly at 868 cm−1 is assigned to the ν(P–N–P) stretching mode. These data indicate nearly identical conformation of the P–N–P backbone of the ligand in all three complexes, as confirmed by X-ray crystallography analysis (vide infra).
The LDI-MS spectra of the [Ni(P,P)X2] samples (see ESI†) contain molecular peaks at 592.09, 679.98, and 775.95 Da (for X = Cl, Br, I, respectively) that correspond to the structures ionized with Na+ ion.
The respective masses observed at 534.13, 578.07, and 626.05 Da correspond to the charged molecular fragments formed by a loss of one halogenido ligand during the ionization process.
X-ray crystallography
Single crystals suitable for X-ray crystallographic studies of [Ni(P,P)X2] were obtained by the layering method employing a CH2Cl2/n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 by vol.) solvent system. Selected bond lengths and angles are listed in Table 1. As can be seen in Fig. 1, the chlorido complex crystallized in the P212121 space group as disolvate [Ni(P,P)Cl2]·2CH2Cl2 (Fig. 1A), while the bromido and iodido complexes crystallized as pure substances in the P21cn and P21/c space group, respectively. The structure of the [Ni(P,P)X2], X = Cl, Br, complexes is shown in Fig. S13 (ESI†). The overlapped crystal-structure representations of molecules of the three complexes indicate a high structural similarity (Fig. 1B).
:3 by vol.) solvent system. Selected bond lengths and angles are listed in Table 1. As can be seen in Fig. 1, the chlorido complex crystallized in the P212121 space group as disolvate [Ni(P,P)Cl2]·2CH2Cl2 (Fig. 1A), while the bromido and iodido complexes crystallized as pure substances in the P21cn and P21/c space group, respectively. The structure of the [Ni(P,P)X2], X = Cl, Br, complexes is shown in Fig. S13 (ESI†). The overlapped crystal-structure representations of molecules of the three complexes indicate a high structural similarity (Fig. 1B).
| Bonds | [Ni(P,P)Cl2]·2CH2Cl2 | [Ni(P,P)Br2] | [Ni(P,P)I2] |
|---|---|---|---|
| Ni1–X1 | 2.2020 (6) | 2.3404 (5) | 2.5173 (2) |
| Ni1–X2 | 2.2098 (6) | 2.3398 (5) | 2.5132 (2) |
| Ni1–P2 | 2.1232 (6) | 2.1221 (9) | 2.1368 (4) |
| Ni1–P1 | 2.1246 (6) | 2.1347 (10) | 2.1333 (4) |
| P1–N1 | 1.7127 (19) | 1.711 (3) | 1.7181 (13) |
| P1–C1 | 1.808 (2) | 1.815 (3) | 1.8138 (16) |
| P1–C7 | 1.818 (2) | 1.809 (3) | 1.8078 (15) |
| P2–N1 | 1.7045 (19) | 1.709 (3) | 1.7096 (13) |
| P2–C13 | 1.811 (2) | 1.811 (3) | 1.8142 (16) |
| P2–C19 | 1.816 (2) | 1.806 (3) | 1.8139 (16) |
| Cl1–Ni1–X2 | 98.64 (2) | 98.30 (2) | 98.621 (7) |
| P1–Ni1–X1 | 92.42 (2) | 95.77 (3) | 93.389 (13) |
| P2–Ni1–X1 | 165.62 (2) | 167.35 (3) | 166.932 (14) |
| P2–Ni1–X2 | 95.73 (2) | 93.35 (3) | 94.313 (13) |
| P1–Ni1–X2 | 167.57 (3) | 164.52 (3) | 167.918 (14) |
| P2–Ni1–P1 | 73.32 (2) | 73.29 (4) | 73.731 (16) |
| N1–P1–Ni1 | 95.22 (7) | 95.11 (10) | 94.58 (5) |
| P2–N1–P1 | 95.84 (10) | 95.95 (15) | 96.73 (7) |
| N1–P2–Ni1 | 95.52 (7) | 95.65 (10) | 94.70 (5) |
| N1–P1–C1 | 111.39 (10) | 111.36 (16) | 111.46 (7) |
| N1–P1–C7 | 112.07 (10) | 110.15 (16) | 110.53 (7) |
| C1–P1–C7 | 107.38 (12) | 105.69 (16) | 107.06 (7) |
| C1–P1–Ni1 | 114.47 (8) | 112.76 (13) | 117.10 (5) |
| C7–P1–Ni1 | 116.02 (8) | 121.43 (12) | 115.62 (5) |
| N1–P2–C13 | 110.78 (10) | 111.45 (15) | 109.67 (7) |
| N1–P2–C19 | 110.79 (10) | 111.20 (16) | 110.20 (7) |
| C13–P2–C19 | 106.96 (11) | 106.08 (16) | 107.09 (7) |
| C13–P2–Ni1 | 116.70 (8) | 118.88 (12) | 116.72 (5) |
| C19–P2–Ni1 | 115.68 (8) | 113.38 (12) | 117.68 (5) |
 | ||
| Fig. 1 (A) Molecular structure of [Ni(P,P)Cl2]·2CH2Cl2 (hydrogen atoms are omitted for clarity). (B) Overlapped mode presentations of the chlorido (blue), bromido (red) and iodido (green) complexes. | ||
Individual molecules of all three complexes exhibit a nearly square planar geometry of their NiP2X2 core units (with the Ni atom in the center) (Fig. 1). The NiP2X2 core shows a mean deviation from the best plane defined by the Ni, P and X atoms equal to 0.0598 Å for X = Cl, 0.1086 Å for X = Br, and 0.0311 Å for X = I, with the P1 atom exhibiting the largest deviation (−0.0809 Å for X = Cl and −0.1408 Å for X = Br), and the P2 atom (−0.0417 Å) for X = I. The NiP2X2 core is slightly tetrahedrally distorted, with the X1–Ni–X2 and P1–Ni–P2 planes exhibiting a dihedral angle of 6.06°, 9.59°, and 2.67° for X = Cl, Br, and I, respectively.49 The nickel(II) center is coordinated to the (P,P) chelating ligand, forming a four-membered Ni–P–N–P ring. The mean deviation from the best plane defined by all four atoms is 0.0187 Å, 0.0065 Å and 0.0314 Å for X = Cl, Br and I, respectively.
The Ni–P and Ni–X bond lengths, as well as the P–Ni–P endocyclic angles of [Ni(P,P)X2], X = Cl, Br, I complexes (73.32°, 73.29° and 73.73°, respectively, Table 1) are typical of [Ni{R′N(PPh2)2-κ2P}X2] complexes reported in the literature.17–38 The narrow range of the observed endocyclic bite P–Ni–P angles is attributed to the formation of the four-membered Ni–P–N–P ring. The P1–N–P2 angles of the three complexes are also similar (Table 1), confirming the proposed similar conformation of their P–N–P backbone based on the conserved ν(P–N–P) IR transitions of the three complexes (868 cm−1, vide supra). As expected, the Ni–X bond lengths of [Ni(P,P)X2] increase with the increasing size of the halogen atom X, whereas the respective Ni–P bond lengths remain rather conserved (Table 1).
Catalytic activity of [Ni(P,P)X2] in Kumada cross-coupling
The catalytic activity of new complexes in Kumada coupling was tested using 4-(tbutyl)-1-iodobenzene (4-tBu) as the substrate and p-tolylmagnesium bromide (T) as the reagent (Scheme 3). Grignard reagent T was used in a stoichiometric excess of 20% to compensate for the decay of T due to its homo-coupling caused by traces of oxygen. (Up to 12% homocoupling of the product: 4,4′-dimethylbiphenyl, was detected even in the supplied reagents). | ||
| Scheme 3 Substrates, reagent, catalyst and key products of the performed catalytic tests (1 mmol of iododerivative, 1.2 mmol of the Grignard reagent T, 1 mol% of [Ni(P,P)X2], 3 mL THF), 25 °C. | ||
Experimental data indicating the effect of halogenido ligand on the catalytic activity of [Ni(P,P)X2] complexes are presented in Fig. 4. As can be seen, [Ni(P,P)Cl2] showed not only clearly the highest initial and overall (final) yield of the cross-coupling product, YCROSS, but also high product selectivity. Further, it is seen that the bromido and iodido complexes showed a very low initial activity (YCROSS of 1%), but a reasonably high final yield (YCROSS of 60–65%), albeit significantly lower compared to [Ni(P,P)Cl2] (90%). Such data indicate a difficult start of propagation (the presence of an induction period) but a reasonably long lifetime of the active species (vide infra).
A nearly constant yield of the substrate homo-coupling product, YHOMO, up to 2% occurred in all experiments. This side product most likely resulted from the Ullmann type reaction that took place during degassing of the mixed solutions of substrate and catalyst before adding the Grignard reagent T. Nickel compounds are well known to catalyze Ullmann-type reactions.50–52
The observed difference in the catalytic activity [Ni(P,P)Cl2] and its bromido and iodido counterparts underscores the role of the halogenido ligands in the catalytic activity of nickel(II) complexes. Therefore, the catalytically most active complex [Ni(P,P)Cl2] was in addition tested in couplings of T with substrates in which the reacting iodine atom is sterically hindered by one or two methyl groups in ortho positions (see Scheme 3). These experiments were aimed to assess how this shielding affects the catalytic activity and product selectivity of the catalyst. As can be seen in Fig. 3, the increase in iodine atom shielding significantly but not substantially decreased the cross-coupling conversion (YCROSS) and increased the substrate homo-coupling conversion (YHOMO). Interestingly, the final total substrate conversion was nearly the same (YCROSS + YHOMO = 90 ± 2%) for all three substrates, regardless of shielding (see also Tables S1 and S2 in the ESI†).
Useful information for understanding the mechanism of the catalytic action of Ni(PNP) complexes provides the comparison of the data shown in Fig. 2 with literature data (see Table 2). The literature data show that [Ni(P,P)X2] complexes bearing R' = tBu exhibit a significantly higher catalytic efficiency and selectivity in the Kumada cross-coupling as compared to the earlier reported [Ni{R′N(PPh2)2-κ2P}X2] complexes with different N-substituents R′. The chlorido complex with R′ = (CH2)3Si(OMe)3 showed an overall activity (YCROSS ≈ 64%) comparable to that of [Ni(P,P)X2], X = Br, I, but significantly lower cross-coupling selectivity (YHOMO ≈ 13% vs. 2%, respectively). The X = Cl, Br complexes with R′ = (S)-CHMePh showed lower activity (YCROSS of 48 and 42%, respectively) as well as selectivity (YHOMO ≈ 20%). These data pointed to the important role of the N-substituent (R′ group) of the (P,P) ligand. The electron-donating power of the N-substituents decreases in the sequence: tBu > (CH2)3Si(OMe)3 > (S)-CHMePh. The data presented in Fig. 2 and 3 and Table 2 show that the catalytic activity and selectivity decrease in the same order. Hence it is clear that the increased electron-releasing power of the N-substituent in [Ni(P,P)Cl2] increases its catalytic activity and selectivity in the Kumada cross-coupling reactions of aromatic compounds. The reason for this observation might be that electron density released by the tBu group towards the nickel(II) center facilitates the oxidative addition step of the overall reaction mechanism.
 | ||
| Fig. 2 Results of catalytic test of [Ni(P,P)X2] complexes in Kumada coupling of 4-tBu substrate with Grignard reagent T. YCROSS and YHOMO stands for the yield of cross-coupling and homo-coupling product, respectively. | ||
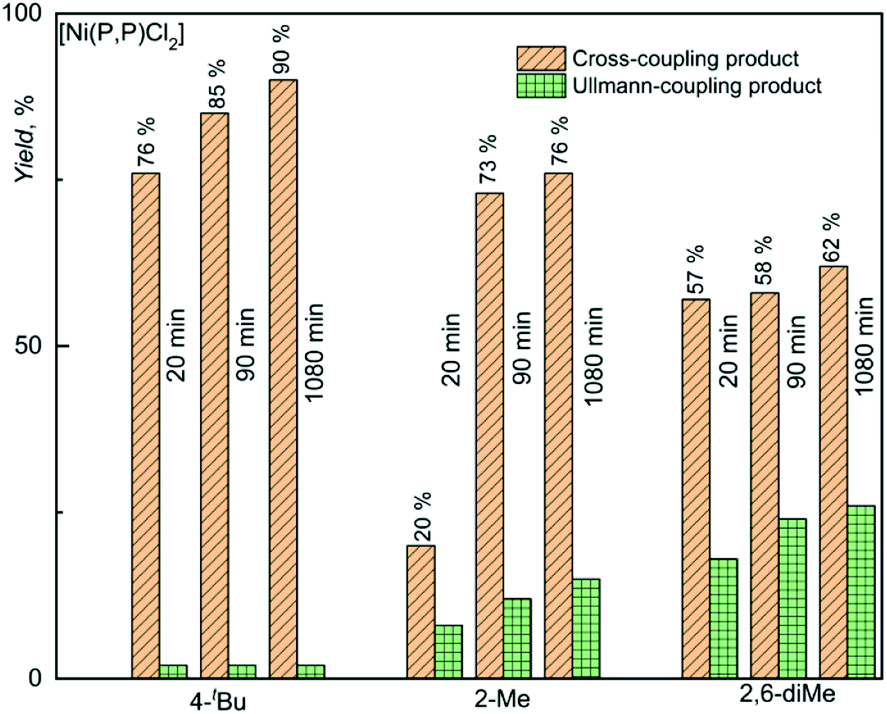 | ||
| Fig. 3 Results of Kumada coupling tests carried out with the most active catalyst [Ni(P,P)Cl2] and substrates differing in the extent of shielding of the iodine atom of the substrate. Numerical data are presented in Table S2 (ESI†). | ||
 | ||
| Fig. 4 UV/vis spectral changes due to the addition of 4-tBu and T (A) and T and 4-tBu (B) to a THF solution of [Ni(P,P)Cl2]. In A and B, the curves [Ni(P,P)Cl2] + 4-tBu, and [Ni(P,P)Cl2] + T + 4-tBu, respectively, were graphically shifted along Y-axis to prevent overlapping. | ||
Mechanistic considerations
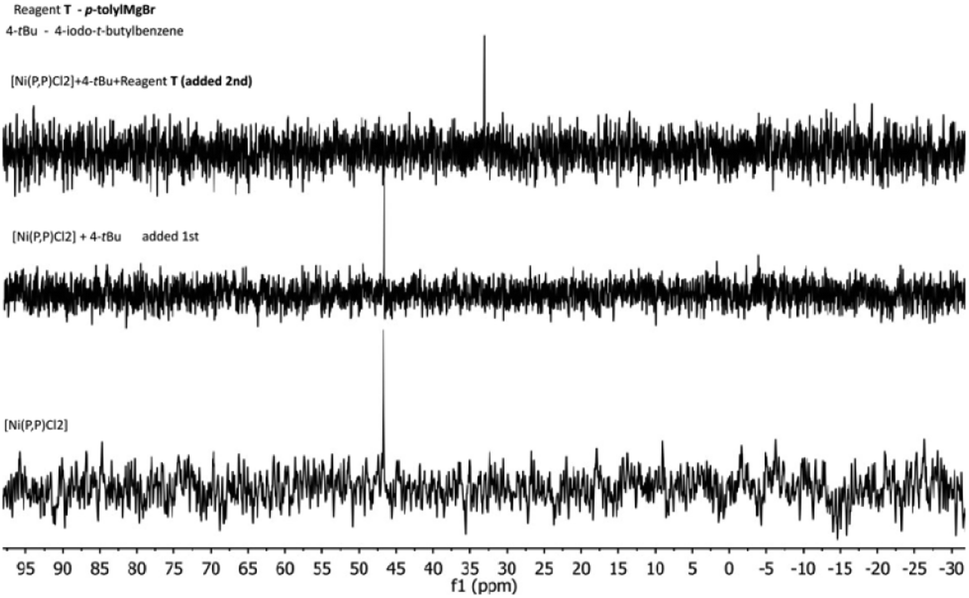
In an effort to probe the operative mechanism, the following experiments were carried out. Specifically, to a THF solution of [Ni(P,P)Cl2], the reagents 4-(tbutyl)-1-iodobenzene (4-tBu) and p-tolylmagnesium bromide (T) were added in this order, with concomitant recording of UV-vis spectra (Fig. 4A). In a second experiment, the above order of adding the two reagents was reversed (Fig. 4B).The spectra showed in Fig. 4 provide evidence that the addition of the Grignard reagent (T) may lead to the reduction of the nickel(II) pristine catalyst to catalytically active nickel(I) species, as previously described in the literature.53,54 The reaction mixture shown in Fig. 4A was also studied by 1H and 31P NMR spectroscopy (Fig. 5 and 6, respectively).
 | ||
| Fig. 5 1H NMR spectral changes in the reaction mixture of Fig. 4A. | ||
 | ||
| Fig. 6 31P NMR spectral changes in the reaction mixture of Fig. 4A. | ||
Two multiplets around 8.4 and 7.6 ppm are assigned to the [Ni(P,P)Cl2] complex, whereas those at 7.59 and 7.18 ppm are ascribed to 4-tBu. Upon the addition of 4-tBu, the catalyst's multiplet at 8.4 ppm persists unchanged. After the addition of T, a reaction is taking place, observable in the region 7.6–6.9 ppm. Conversely, when T was added first to the solution of [Ni(P,P)Cl2], the latter changes were not observed.
The NMR spectra for the reaction mixture of Fig. 4B (i.e. first addition of T and then of 4-tBu), are presented in the ESI (Fig. S17 and S18,† respectively). The 1H NMR signals ascribed to the catalyst disappeared in both cases, irrespective of the order of the reagents' addition. The 31P NMR signal was shifted from 46.6 ppm to 33 ppm upon addition of 4-tBu first, and disappeared for the reversed order of reagents' addition. The signal disappearance is most probably caused by the reduction of the diamagnetic nickel(II) precatalyst to the paramagnetic nickel(I) species, and hence broadening of the corresponding peaks. The observed 31P NMR signal shift (when 4-tBu was added first) is probably related to a species resulting from the pristine complex (vide infra), but the identity of this species cannot be conclusively determined. Moreover, we do not have a plausible explanation for the difference in behavior upon the reverse order of the reagents' addition (mixture of Fig. 4B). Based on the above experimental evidence, the catalytic cycle may be operative via the Ni(I)–Ni(III) mode (vide infra), however, the Ni(0)–Ni(II) mode cannot be entirely ruled out. Additional investigations would be needed for such an endeavor, but this is beyond the scope of the current work.55–58
DFT calculations
X-ray crystallographic and DFT-calculated structures of complexes [Ni(P,P)X2] are depicted in Fig. 7. The significant structural similarity indicates an adequate accuracy of the performed DFT calculations. | ||
| Fig. 7 Comparison of the DFT-calculated (gas phase, green) and X-ray determined (red) structures of [Ni(P,P)X2]. | ||
The DFT-calculated HOMO and LUMO orbitals of [Ni(P,P)X2], X = Cl, Br, I, are depicted in Fig. S16 (ESI†). As can be seen, the LUMOs are similar for all three complexes and exhibit antibonding character. However, the HOMOs differ significantly. The HOMOs of the bromido and iodido complexes dominantly comprise non-bonding orbitals located on the halogenido ligands (lone pairs), whereas the HOMO of [Ni(P,P)Cl2] is significantly contributed with the electrons of d orbitals of nickel(II). In turn, the opposite trend is observed in HOMO-2 orbitals of [Ni(P,P)Cl2], with almost no contribution of d orbitals of nickel(II).
It is known that in the palladium-catalyzed C–C bond-forming couplings (including Kumada), palladium alternates exclusively between oxidation states Pd(0) (when entering the oxidative addition) and Pd(II) (at reductive elimination). In contrast, the nickel-based catalysts are known to operate either in the Ni(0)–Ni(II) or the Ni(I)–Ni(III) mode.53–58 Consequently, the nickel(II) precatalyst (pristine nickel complex) undergoes reduction to the nickel(I) state (initiation), before entering to the propagation cycle operating in the Ni(I)–Ni(III) mode. The reduction occurs by reaction with the Grignard reagent. Based on literature screening and our experimental observation, we presume that initiation provides nickel(I)-based catalytic active species, which, subsequently, operate in the Ni(I)–Ni(III) mode, as depicted in Fig. 8.
 | ||
| Fig. 8 Proposed mechanism of the Kumada coupling catalyzed by [Ni(P,P)X2] complexes, X = Cl, Br, I. | ||
In order to shed more light on the halogenido effect, we investigate the transmetalation step of the catalytic cycle in detail, Fig. 9. First, the interaction of [Ni(P,P)X] intermediate with Grignard reagent leads to transient species (TS), with significantly lower ΔG values. These transient species can be regarded as true transition states with inherent imaginary frequency corresponding to the reaction coordinate. Then, the respective MgBrX molecule is released and an aryl derivative [Ni(P,P)Ar] enters the oxidative addition step.
 | ||
| Fig. 9 Relative Gibbs free energies (in bulk THF), concerning the sum of the isolated reactants, on the potential energy surface of the reaction intermediates of the transmetalation step of the catalytic cycle. The corresponding values in the gas phase are shown in parentheses. | ||
It can be presumed that the initiation step, being a radical process, is rate-determining, however, it was computationally very time-consuming to locate the corresponding transition state. Nevertheless, as shown in Fig. 9, the corresponding ΔG values are systematically lower for the chlorido intermediates with respect to bromido and iodido intermediates. Building on the fact that the initiation step comprises interaction with the Grignard reagent, it can be presumed that this step will follow a similar trend as shown for the transmetalation step.
Consequently, the DFT calculations on the nickel(II) precatalysts [Ni(P,P)X2] revealed differences in the electronic structure of the chlorido complex compared to the bromido and iodido complexes. These differences, together with the aforementioned mechanistic considerations (vide supra), probably explain the differences in the catalytic activity as well as in the observed induction periods for the bromido and iodido complexes. The full set of the DFT-optimized structures investigated in this study is gathered in the ESI2 as a text file.† Noteworthy, magnesium halides (reaction byproducts) optimized in the bulk THF showed bent structures (linear in the gas-phase), an effect that was observed for water microsolvation of magnesium chloride.59
Conclusions
The work presented herein demonstrates that the catalytic activity and product selectivity of [Ni{R′N(PPh2)2-κ2P}X2] complexes in Kumada cross-coupling reactions depend on two tunable factors: (i) the identity of the halogenido ligand, and (ii) the nature of the N-substituent R′ moiety of the (P,P) ligand. All three [Ni(P,P)X2] complexes showed excellent product selectivity in the Kumada cross-coupling of Grignard reagent T with 4-tBu substrate bearing the spatially unshielded reacting iodine atom. Iodine atom shielding by ortho-substituents significantly decreases the cross-coupling selectivity of [Ni(P,P)Cl2]. The yield of the Kumada cross-coupling catalyzed by [Ni(P,P)X2] complexes decreases in line Cl > Br > I. The comparison of the activity of [Ni(P,P)X2] with that of [Ni{R′N(PPh2)2-κ2P}X2] complexes bearing different R′ moieties indicates that increasing the electron-releasing power of R′ increases both the catalytic activity and the cross-coupling selectivity of this family of nickel(II) catalysts. We outline the transformation of the precatalyst complex to a nickel(I) active species via reduction with Grignard reagent accompanied by a one halogenido ligand loss. DFT calculations reveal that [Ni(P,P)X] active species formation is thermodynamically preferred for chlorido complex, as well as, the transmetalation step of the catalytic cycle is smoother for this species than for bromido and iodidio counterparts. Consequently, calculated results explain the difference in the catalytic activity of the chlorido complex compared to the bromido and iodido complexes.Experimental section
Syntheses were performed under argon atmosphere using standard Schlenk techniques. Solvents were dried and distilled according to procedures reported in the literature.60 The tBuN(Ph2P)2 ligand was synthesized according to the literature procedure.48 Determination of C, H, and N contents: FlashSmart™ Elemental Analyzer (Thermo Fisher Scientific) was used for the concurrent determination of C, H and N content. Approximately 1.5 mg of the sample was weighed to a tin capsule using the precision weighing balance Sartorius SE 2-OCE. The capsule was sealed and inserted into the autosampler of the Analyzer and the standard operational procedures were employed. 1H and 31P NMR spectra were monitored on a Varian NMR Systems 300 MHz spectrometer at 300 MHz for 1H NMR spectra referenced to the solvent signal δ = 7.25 ppm and Varian UNITY Inova 400 MHz spectrometer for 31P NMR spectra at 162 MHz using 1H decoupling, referenced to the external standard 85% H3PO4 δ = 0.00 ppm, in CHCl3-d at 25 °C. First-order analysis was used for spectra deciphering. All NMR spectra are shown in the ESI.† 13C NMR spectra were recorded on BRUKER Avance 600 MHz (151 MHz, CDCl3) and Varian Unity Inova 400 MHz (101 MHz, CDCl3). FTIR spectra were obtained on a Shimadzu IR Affinity-1 instrument using KBr pellets in transmission mode. All FTIR spectra are shown in the ESI.† The samples for Laser Desorption Ionization – Time Of Flight Mass spectrometry (LDI-TOF MS) were prepared using a modified dried droplet method: the sample with sodium trifluoroacetate as ionization agent was dissolved in CHCl3. The solution, without any matrix, was deposited on the target and left to dry before measurement. Mass spectra were recorded on an ultrafleXtreme ToF–ToF mass spectrometer (Bruker Daltonics) equipped with a 2000 Hz smartbeam-II laser (355 nm) using the positive-ion reflectron mode and panoramic pulsed ion extraction, after external calibration with poly(ethyleneglycol).61 The MS spectra are shown in the ESI in Fig. S14.† UV-vis spectra in THF solution are presented in ESI in Fig. S15.†Synthesis of complexes
X-ray crystallography
A crystal of [Ni(P,P)Cl2]·2CH2Cl2 (0.25 × 0.34 × 0.42 mm), [Ni(P,P)Br2] (0.09 × 0.19 × 0.25 mm) and [Ni(P,P)I2] (0.12 × 0.26 × 0.37 mm) were taken from the mother liquor and immediately cooled to −113 °C. Diffraction measurements were taken on a Rigaku R-AXIS SPIDER Image Plate diffractometer using graphite monochromated Mo Kα radiation. Data collection (ω-scans) and processing (cell refinement, data reduction and Empirical absorption correction) were performed using the Crystal Clear program package.62 Important crystallographic data are listed in Table 3. The structures were solved using direct methods and SHELXS v.2013/1 and then refined using full-matrix least-squares techniques on F2 with SHELXL ver.2014/6.63 Further experimental crystallographic details for [Ni(P,P)Cl2]·2CH2Cl2: 2θmax = 54°; reflections collected/unique/used, 32034/7324 [Rint = 0.0268]/7324; 493 parameters refined; (Δ/σ)max = 0.002; (Δρ)max/(Δρ)min = 0.383/−0.254 e Å−3; R1/wR2 (for all data), 0.0232/0.0531, Flack parameter x = 0.009(4) using 3031 quotients.62 Further experimental crystallographic details for [Ni(P,P)Br2]: 2θmax = 54°; reflections collected/unique/used, 25149/5972 [Rint = 0.0455]/5972; 423 parameters refined; (Δ/σ)max = 0.002; (Δρ)max/(Δρ)min = 0.318/−0.269 e Å3−; R1/wR2 (for all data), 0.0272/0.0465, Flack parameter x = 0.004(5) using 2428 quotients.64 Further experimental crystallographic details for [Ni(P,P)I2]: 2θmax = 54°; reflections collected/unique/used, 49930/6206 [Rint = 0.0200]/6206; 423 parameters refined; (Δ/σ)max = 0.001; (Δρ)max/(Δρ)min = 0.467/−0.330 e Å−3; R1/wR2 (for all data), 0.0165/0.0394. All hydrogen atoms were located in difference density maps and refined isotropically. All non-hydrogen atoms were refined anisotropically. Structural plots were drawn using the Diamond 3 program package.65
| [Ni(P,P)Cl2]·2CH2Cl2 | [Ni(P,P)Br2] | [Ni(P,P)I2] | |
|---|---|---|---|
| a R1 = Σ(|Fo| − |Fc|)/Σ(|Fo|) and wR2 = [Σ{w(Fo2 − Fc2)2]/Σ[w(Fo2)2}]1/2. w = 1/[σ2(Fo2) + (αP)2 + bP] and P = [max (Fo2,0)+2Fc2]/3, α = 0.0287, b = 0.5803 for [Ni(P,P)Cl2]·2CH2Cl2; α = 0.0112, b = 0.00 for [Ni(P,P)Br2]; α = 0.0181, b = 1.67 for [Ni(P,P)I2]. | |||
| Formula | C30H33Cl6NNiP2 | C28H29Br2NNiP2 | C28H29I2NNiP2 |
| Fw | 740.92 | 659.99 | 753.97 |
| Space group | P212121 | P21cn | P21/c |
| a (Å) | 10.6796(4) | 9.8001(2) | 15.4142(3) |
| b (Å) | 14.9649(1) | 13.6022(3) | 10.0508(2) |
| c (Å) | 20.9874(4) | 20.5470(4) | 18.3727(3) |
| α (°) | 90.0 | 90.0 | 90.0 |
| β (°) | 90.0 | 90.0 | 92.727(1) |
| γ (°) | 90.0 | 90.0 | 90.0 |
| V (Å3) | 3354.19(14) | 2738.98(10) | 2843.17(9) |
| Z | 4 | 4 | 4 |
| T (°C) | −113 | −113 | −113 |
| Radiation | Mo Kα | Mo Kα | Mo Kα |
| ρcalcd (g cm−3) | 1.467 | 1.601 | 1.761 |
| μ (mm−1) | 1.174 | 3.760 | 2.984 |
| Reflections with I > 2σ(I) | 7074 | 5591 | 5996 |
| R1a | 0.0220 | 0.0239 | 0.0156 |
| wR2a | 0.0525 | 0.0455 | 0.0390 |
General experimental procedure for the Kumada coupling
Catalytic Kumada coupling reactions were performed using a modified literature procedure.35 In a Schlenk flask, equipped with rubber septum, 1-iodoaryl derivative ((a) 4-iodo-tbutylbenzene, (b) 2-iodotoluene, (c) 2-iodo-1,3-dimethylbenzene (1.0 mmol)), 10 μmol of complex [Ni(P,P)X2] and 0.5 mmol of mesitylene were dissolved in 3 mL of freshly distilled THF. The mixture was cooled down with liquid nitrogen (10 min). Schlenk flask was connected to the vacuum-argon manifold and evacuated (0.5 mbar). The sidearm stopcock of the Schlenk flask was closed, removing the cooling, and mixture was left standing to reach room temperature. The Schlenk flask was flushed with argon in the final degassing cycle stage. The degassing cycle was repeated three times. Finally, a hexane solution p-tolylMgBr (1.2 mmol) was carefully added under argon. The mixture was left to react at room temperature. Pick outs (50 μL) were performed under argon at 20, 90, 1080 min. Withdrawn samples were diluted in 2 mL of pure methanol (quenching of the reaction). Each sample was analyzed on a Shimadzu GC-2010 chromatograph, equipped with an FID detector and a 30 m, 0.25 mm (0.25 μm film of anchored phase) Zebron DB-5 capillary column.Author contributions
Polydoros-Chrysovalantis Ioannou – investigation, writing – original draft; Kalliopi Kakridi – investigation; Radek Coufal – investigation, Catherine P. Raptopoulou – investigation, methodology, software, visualization; Olga Trhlíková – investigation, methodology, software, visualization; Vassilis Psycharis – investigation, methodology, software, visualization, writing – original draft; Jiří Zedník – investigation, methodology, software, visualization, writing – review & editing; Panayotis Kyritsis – conceptualization, project administration, resources, supervision, writing – review & editing, Jiří Vohlídal – supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported, in part, by the Special Account for Research Grants of the National and Kapodistrian University of Athens. We thank Drs A. Grigoropoulos and I. Stamatopoulos for helpful discussions.References
- T. Appleby and J. D. Woollins, Coord. Chem. Rev., 2002, 235, 121–140 CrossRef CAS.
- C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116, 9237–9304 CrossRef CAS PubMed.
- L. E. Bowen, M. Charernsuk, T. W. Hey, C. L. McMullin, A. G. Orpen and D. F. Wass, Dalton Trans., 2010, 39, 560–567 RSC.
- G. J. P. Britovsek, D. S. McGuinness, T. S. Wierenga and C. T. Young, ACS Catal., 2015, 5, 4152–4166 CrossRef CAS.
- L. H. Do, J. A. Labinger and J. E. Bercaw, ACS Catal., 2013, 3, 2582–2585 CrossRef CAS PubMed.
- E. Killian, K. Blann, A. Bollmann, J. T. Dixon, S. Kuhlmann, M. C. Maumela, H. Maumela, D. H. Morgan, P. Nongodlwana, M. J. Overett, M. Pretorius, K. Höfener and P. Wasserscheid, J. Mol. Catal. A: Chem., 2007, 270, 214–218 CrossRef CAS.
- S. Sa, S. M. Lee and S. Y. Kim, J. Mol. Catal. A: Chem., 2013, 378, 17–21 CrossRef CAS.
- G. J. P. Britovsek, D. S. McGuinness and A. K. Tomov, Catal. Sci. Technol., 2016, 6, 8234–8241 RSC.
- I. M. Aladzheva, O. V. Bykhovskaya, A. A. Vasil'ev, Y. V. Nelyubina and Z. S. Klemenkova, Russ. Chem. Bull., 2015, 64, 909–913 CrossRef CAS.
- C. Kayan, N. Biricik, M. Aydemir and R. Scopelliti, Inorg. Chim. Acta, 2012, 385, 164–169 CrossRef CAS.
- C. Kayan, N. Biricik and M. Aydemir, Transition Met. Chem., 2011, 36, 513–520 CrossRef CAS.
- N. Biricik, C. Kayan, B. Gumgum, Z. F. Fei, R. Scopelliti, P. J. Dyson, N. Gurbuz and I. Ozdemir, Inorg. Chim. Acta, 2010, 363, 1039–1047 CrossRef CAS.
- N. Biricik, F. Durap, C. Kayan, B. Gümgüm, N. Gürbüz, İ. Özdemir, W. H. Ang, Z. Fei and R. Scopelliti, J. Organomet. Chem., 2008, 693, 2693–2699 CrossRef CAS.
- P. C. Ioannou, C. Arbez-Gindre, M. Zoumpanioti, C. P. Raptopoulou, V. Psycharis, I. D. Kostas and P. Kyritsis, J. Organomet. Chem., 2019, 879, 40–46 CrossRef CAS.
- I. Stamatopoulos, M. Roulia, K. A. Vallianatou, C. P. Raptopoulou, V. Psycharis, M. Carravetta, C. Papachristodoulou, E. Hey-Hawkins, I. D. Kostas and P. Kyritsis, ChemistrySelect, 2017, 2, 12051–12059 CrossRef CAS.
- I. K. Stamatopoulos, M. Kapsi, M. Roulia, G. C. Vougioukalakis, C. P. Raptopoulou, V. Psycharis, I. D. Kostas, L. Kollár and P. Kyritsis, Polyhedron, 2018, 151, 292–298 CrossRef CAS.
- H. T. Al-Masri and Z. Moussa, Z. Anorg. Allg. Chem., 2016, 642, 914–920 CrossRef CAS.
- D. A. Dickie, B. E. Chacon, A. Issabekov, K. Lam and R. A. Kemp, Inorg. Chim. Acta, 2016, 453, 42–50 CrossRef CAS.
- L.-C. Song, J.-P. Li, Z.-J. Xie and H.-B. Song, Inorg. Chem., 2013, 52, 11618–11626 CrossRef CAS PubMed.
- G. C. Vougioukalakis, I. Stamatopoulos, N. Petzetakis, C. P. Raptopoulou, V. Psycharis, A. Terzis, P. Kyritsis, M. Pitsikalis and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5241–5250 CrossRef CAS.
- Z. Sun, F. Zhu, Q. Wu and S.-a. Lin, Appl. Organomet. Chem., 2006, 20, 175–180 CrossRef CAS.
- A. Ghisolfi, C. Fliedel, V. Rosa, R. Pattacini, A. Thibon, K. Y. Monakhov and P. Braunstein, Chem.–Asian J., 2013, 8, 1795–1805 CrossRef CAS PubMed.
- I. Stamatopoulos, D. Giannitsios, V. Psycharis, C. P. Raptopoulou, H. Balcar, A. Zukal, J. Svoboda, P. Kyritsis and J. Vohlídal, Eur. J. Inorg. Chem., 2015, 3038–3044 CrossRef CAS.
- B. R. Aluri, N. Peulecke, S. Peitz, A. Spannenberg, B. H. Muller, S. Schulz, H. J. Drexler, D. Heller, M. H. Al-Hazmi, F. M. Mosa, A. Wohl, W. Muller and U. Rosenthal, Dalton Trans., 2010, 39, 7911–7920 RSC.
- B.-S. Yin, T.-B. Li and M.-S. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1572 CrossRef CAS PubMed.
- B.-S. Yin, T.-B. Li and M.-S. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1571 CrossRef CAS PubMed.
- X.-F. Liu, Inorg. Chim. Acta, 2014, 421, 10–17 CrossRef CAS.
- A. Ghisolfi, C. Fliedel, V. Rosa, K. Y. Monakhov and P. Braunstein, Organometallics, 2014, 33, 2523–2534 CrossRef CAS.
- N. A. Cooley, S. M. Green, D. F. Wass, K. Heslop, A. G. Orpen and P. G. Pringle, Organometallics, 2001, 20, 4769–4771 CrossRef CAS.
- V. V. Sushev, A. N. Kornev, Y. A. Kurskii, O. V. Kuznetsova, G. K. Fukin, Y. H. Budnikova and G. A. Abakumov, J. Organomet. Chem., 2005, 690, 1814–1821 CrossRef CAS.
- M. Hapke, A. Wohl, S. Peitz, B. H. Muller, A. Spannenberg and U. Rosenthal, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m252 CrossRef CAS PubMed.
- K. M. Song, H. Y. Gao, F. S. Liu, J. Pan, L. H. Guo, S. B. Zai and Q. Wu, Eur. J. Inorg. Chem., 2009, 3016–3024 CrossRef CAS.
- C. Ganesamoorthy, J. T. Mague and M. S. Balakrishna, J. Organomet. Chem., 2007, 692, 3400–3408 CrossRef CAS.
- P. Boulens, M. Lutz, E. Jeanneau, H. Olivier-Bourbigou, J. N. H. Reek and P. A. R. Breuil, Eur. J. Inorg. Chem., 2014, 3754–3762 CrossRef CAS.
- I. Stamatopoulos, M. Placek, V. Psycharis, A. Terzis, J. Svoboda, P. Kyritsis and J. Vohlidal, Inorg. Chim. Acta, 2012, 387, 390–395 CrossRef CAS.
- L.-C. Song, X.-F. Han, W. Chen, J.-P. Li and X.-Y. Wang, Dalton Trans., 2017, 46, 10003–11013 RSC.
- L. Lavanant, A. S. Rodrigues, E. Kirillov, J. F. Carpentier and R. F. Jordan, Organometallics, 2008, 27, 2107–2117 CrossRef CAS.
- I. Stamatopoulos, C. P. Raptopoulou, V. Psycharis and P. Kyritsis, Open Chem., 2016, 14, 351–356 CAS.
- K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS.
- J. G. de Vries, Palladium-Catalysed Coupling Reactions, in Organometallics as Catalysts in the Fine Chemical Industry, ed. M. Beller and H.-U. Blaser, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 1–34 Search PubMed.
- J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC.
- A. A. Dahadha and M. M. Aldhoun, Arkivoc, 2018, 234–253 Search PubMed.
- C. E. I. Knappke and A. Jacobi von Wangelin, Chem. Soc. Rev., 2011, 40, 4948–4962 RSC.
- M. L. Neidig, S. H. Carpenter, D. J. Curran, J. C. DeMuth, V. E. Fleischauer, T. E. Iannuzzi, P. G. N. Neate, J. D. Sears and N. J. Wolford, Acc. Chem. Res., 2019, 52, 140–150 CrossRef CAS PubMed.
- T. Parchomyk and K. Koszinowski, Synthesis, 2017, 49, 3269–3280 CrossRef CAS.
- J. Magano and S. Monfette, ACS Catal., 2015, 5, 3120–3123 CrossRef CAS.
- Z. X. Wang and N. Liu, Eur. J. Inorg. Chem., 2012, 901–911 CrossRef CAS.
- M. C. Maumela, K. Blann, H. de Bod, J. T. Dixon, W. F. Gabrielli and D. B. G. Williams, Synthesis, 2007, 3863–3867 CrossRef CAS.
- G. Sciortino, G. Lubinu, J. D. Marechal and E. Garribba, Magnetochemistry, 2018, 4, 55 CrossRef CAS.
- T. D. Nelson and R. D. Crouch, Org. React., 2004, 63, 265–555 CrossRef CAS.
- W. W. Chen, Q. Zhao, M. H. Xu and C. Q. Lin, Org. Lett., 2010, 12, 1072–1075 CrossRef CAS PubMed.
- H. R. Hoen, J. Zhang and G. Lin, Synlett, 2001, 1527–1530 Search PubMed.
- L. Iffland, A. Petuker, M. van Gastel and U.-P. Apfel, Inorganics, 2017, 5, 78 CrossRef.
- D. D. Dawson, V. F. Oswald, A. S. Borovik and E. R. Jarvo, Chem.–Eur. J., 2020, 26, 3044–3048 CrossRef CAS PubMed.
- J. Berding, T. F. van Dijkman, M. Lutz, A. L. Spek and E. Bouwman, Dalton Trans., 2009, 6948–6955 RSC.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- M. E. Greaves, T. O. Ronson, F. Maseras and D. J. Nelson, Organometallics, 2021, 402, 1997–2007 CrossRef PubMed.
- J. Breitenfeld, J. Ruiz, M. D. Wodrich and X. Hu, J. Am. Chem. Soc., 2013, 135, 12004–12012 CrossRef CAS PubMed.
- G. Feng, C.-W. Liu, Z. Zeng, G.-L. Hou, H.-G. Xu and W.-J. Zheng, Phys. Chem. Chem. Phys., 2017, 19, 15562–15569 RSC.
- Purification of Laboratory Chemicals, ed. W. L. F. Armarego and C. L. L. Chai, Butterworth–Heinmann, Oxford, UK, 2003 Search PubMed.
- A. A. Rakic, M. Vukomanovic, S. Trifunovic, J. Travas-Sejdic, O. J. Chaudhary, J. Horský and G. Ciric-Marjanovic, Synth. Met., 2015, 209, 79–296 CrossRef.
- Rigaku/MSC, CrystalClear, Rigaku/MSC Inc., The Woodlands, Texas, USA, 2005 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249–259 CrossRef CAS PubMed.
- DIAMOND – Crystal and Molecular Structure Visualization, Ver. 3.1, Crystal Impact, Rathausgasse 30, 53111, Bonn, Germany Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin;K. Morokuma, V. G. Zakrzewski, A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and J. D. Fox, Gaussian 09, RevisionD.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR, NMR, X-ray crystallography, MS, and computational data of the three complexes. CCDC 1969669, 1969670, 1969671, for [Ni(P,P)Cl2]·2CH2Cl2, [Ni(P,P)Br2] and [Ni(P,P)I2], respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra04572e |
| This journal is © The Royal Society of Chemistry 2022 |