
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into the photoelectrical properties of metal adsorption on a two-dimensional organic–inorganic hybrid perovskite surface: theoretical and experimental research†
Liping Peng *,
Yulin Xie and
Changquan Yang
*,
Yulin Xie and
Changquan Yang
School of Physics and Telecommunications, Huanggang Normal University, Huangzhou, Hubei 43800, P. R. China. E-mail: pengliping@hgnu.edu.cn
First published on 16th February 2022
Abstract
In order to study the photoelectric properties of the adsorption of different metal atoms on a two-dimensional (2D) perovskite surface, in this article, we built many models of Ag, Au, and Bi atoms adsorbed on 2D perovskite. We studied the rules influencing 2D perovskite adsorbing metal atoms with different n values (the n value is the number of inorganic layers of 2D perovskite; here n = 1, 2, and 3). Based on n = 2 2D perovskite, we successively used Ag, Au, and Bi metal atoms to adsorb on the 2D perovskite surface. Firstly, we calculated their adsorption energies. Based on the lowest energy principle, we found that Bi atom adsorption on the 2D perovskite surface gave the most stable structure among the three metal adsorptions because the energy of the Bi adsorption system was the smallest. Secondly, the electron transport process takes place from the s to the p orbital when Au and Ag atoms adsorb on the 2D perovskite surface, but in the Bi atom adsorption, the electron transport process takes place from the p to the p orbital, because the p–p orbital transport energy is lower than that of the s–p orbital. Therefore, Bi atom adsorption on the 2D perovskite surface can improve charge carrier transfer. Thirdly, we calculated the bond angles and bond energies of different metal adsorptions on 2D perovskite. Bi adsorption has greater interaction with the surface atoms of 2D perovskite than Ag or Au atom adsorption, which effectively enhances the surface polarization effects, and enhances the photoelectric properties of 2D perovskite. The light absorption spectrum further confirms that Bi atom adsorption has a greater impact on the 2D perovskite than the action of Ag or Au adsorption. Finally, in an experiment, we fabricated a 2D perovskite solar cell with an ITO/PEDOT:PSS/2D perovskite/PEI/Ag (Au, Bi) structure. The Bi electrode solar cell achieves the highest photoelectric conversion efficiency (PCE) of 15.16% among the three cells with forward scanning, which is consistent with the theoretical analysis. We believe that the adsorption of metals like Bi on a 2D perovskite surface as an electrode is conducive to improving the charge transport performance.
1. Introduction
Recently, the photoelectric conversion efficiency (PCE) of an organic–inorganic hybrid perovskite solar cell with three-dimensional (3D) structure has been as high as 25.5%,1 but poor stability is a bottleneck restricting its commercial application.2 In order to improve its stability, most research groups have introduced a large organic cation into the perovskite material body, enhancing its stability by reducing the dimension from 3D perovskite to two-dimensional perovskite (hereinafter referred to as 2D perovskite).3–5 But this is at the expense of light absorption performance and efficiency.6 Therefore, there is a need for a balance between structural dimensionality and efficiency.7 In 2D perovskite, the n value represents the number of inorganic layers. With an increase in n value, the structure of the perovskite will get close to the 3D structure.8 Simultaneously, the quantum effect gradually disappears in the perovskite material. A low-dimensional 2D perovskite, such as n = 1, has an extremely large exciton binding energy, where the value is usually between 100 and 700 meV. The large exciton binding energy originates from the quantum confinement effect, and it decreases dielectric shielding.9 When the n value further increases, the exciton binding energy begins to decrease. Therefore, the n value is one of the main factors to determine the PCE of a 2D perovskite solar cell, and it is important to continuously search for a suitable n value for a 2D perovskite material. On the other hand, for improving the PCE of solar cells and maintaining the stability of a solar cell, the stability of the device is another important problem.10 The decline in performance of a perovskite solar cell comes from three main aspects:11 (1) organic cation volatilization; (2) external H2O or O2 molecules entering into the perovskite layer across the hole transport layer, leading to a degradation in the quality of the perovskite material; (3) the reactions taking place between the metal electrode and the halogen ion of the perovskite. Among these reasons, there has been little research on the PCE degradation of perovskite solar cells caused by metal electrodes.12Kato et al. found that a perovskite solar cell with an Ag electrode became yellow after a period of time in a device with the structure glass/FTO/TiO2/MAPbI3/spiro-MeOTAD/Ag, accompanied by a decrease in device efficiency. X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) results found that the hole transport layer of spiro-MeOTAD had many pinholes. H2O can enter into the perovskite layer through these pinholes, making the perovskite performance recede, then producing volatile I ions in the Ag electrode, eventually forming AgI, leading to a decline in the perovskite device performance.13 Domanski et al. also found that the metal electrodes could enter into the perovskite layer across the hole transport layer of spiro-MeOTAD at 70 °C, so their open circuit voltage (VOC), fill factor (FF) and short circuit current (JSC) were all decreased. However, when a chromium (Cr) layer was introduced between the hole transport layer and the Au electrode, with a rise in temperature, the solar cell could maintain its performance and not degrade.14 When Wu et al. inserted a Bi metal layer between the hole transport layer and the Ag electrode layer, they found the unpackaged devices retained their initial efficiency of 88% for 6000 h in the dark under atmospheric conditions. Even at 85 °C, 95% of the efficiency was retained. When subjected to light for 500 h under N2 conditions, 97% efficiency was retained.11 Xiong et al. reported that Bi doping greatly improves the optical stability and thermal stability of perovskite materials.15
All the above research indicates that choosing an appropriate n value and metal electrode are conducive to improving the PCE and stability of the 2D perovskite. But it only analyzed the protection of perovskite materials. Essentially, metal electrode deposition onto the perovskite material surface can seep into the perovskite materials to a certain depth,16 and form an interface layer between the electrode and perovskite layer, which will improve the charge transfer between the metal electrode and perovskite material, enhancing the efficiency of this kind of device. This is another important factor but, at present, there are few studies on the subject.
In this article, we take fluorinated 2D perovskite as the research object (R–P phase structure), whose general formula is: (4-FBA)2(MA)n−1PbnI3n+1 (n = 1, 2, or 3). We study the changes in energy parameters and polarization parameters of different metal atoms of Ag/Au/Bi adsorbed on the surface of the organic terminal and inorganic terminal of the fluorinated 2D perovskite surface, to analyze their photoelectric properties. In an experiment, we fabricated three kinds of devices of ITO/PEDOT:PSS/2D perovskite/PEI/Ag (Au, Bi), and we achieved the highest PCE (15.6%) for the Bi electrode device. We believe that the adsorption of metals like Bi on the organic–inorganic two-dimensional perovskite surface as electrodes is conducive to improving the charge transport performance of 2D perovskite photoelectric devices.
2. Calculation methods
2.1 Theoretical parameter setting
We used first-principles density functional theory (DFT) to calculate the adsorption properties of three different metal atoms adsorbed on the surface of 2D perovskite materials. The software package used was CASTEP,17,18 and the Generalized Gradient Approximation from the Perdew–Wang 1991 (GGA-PW91) and vdW-DF methods19,20 was adopted in the calculation method, and the plane wave cut-off was 380 eV. The GGA has so far been successfully used in the calculation of density of states and optical properties. The vdW-DF method was selected as a widely applicable method that has been proved to describe interatomic interactions, at short and medium distances (5 Å), more accurately and reliably than traditional GGA methods. GGA+U with PW91 includes an empirical correction term proposed by Grimme. The geometry of 2D perovskite was optimized by DFT using the global hybrid Generalized Gradient Approximation (GGA) functional PW91. The structure was optimized at 298 K without any geometric restrictions. The nature of the stationary points was determined by performing frequency analysis: the equilibrium geometries have no imaginary vibrations. The two-dimensional crystal structures of perovskite were obtained from the Protein Data Bank (PDB ID: 1B09). Materials Studio 8.0 was used to prepare perovskite for docking by removing the number of 3D perovskites. The vdW correction of Grimme's DFT + D2 was included in all calculations, because the vdW correction plays an important role in describing the weak interactions within the perovskite material.In addition, we know that the spin–orbit coupling effect has an effect on the band gap, especially in materials systems containing heavy metal elements.21 Therefore, the energy band, density of states, and optical properties of the perovskite were calculated using the GGA-PBE and vdW-DF methods with consideration of the spin–orbit coupling effect. The NBO program was used to obtain the natural atomic orbital (NAO) charges for atoms and overlap-weighted NAO bond order.22 Ag, Au, and Bi atoms were adsorbed on the 2D perovskite surface with n = 1, 2, and 3, and the (001) face was selected for the 2D perovskite with n = 1 or 2, and the (100) face was selected for the n = 3 perovskite. Here, we considered the organic terminal and inorganic terminal of the 2D perovskite surface adsorption conditions, where the surface vacuum layer thickness of the perovskite was set at 15 Å. The limit of convergence was 2 × 10−5 eV per atom. The maximum force, maximum pressure and maximum displacement were 0.05 eV Å−1, 0.1 GPA and 0.002 Å, respectively.
After optimization of the perovskite phase structure for n = 1, 2, and 3, the band gap values obtained were 2.008 eV, 2.156 eV, and 1.793 eV, respectively, which are similar to those reported in the literature.23 The K point in the Brillouin region was set as 3 × 3 × 1. For the n = 1, 2, and 3 perovskites, the valence electrons of the atoms involved are: H 1s1, C 2s2 2p2, N 2s2 2p3, I 5s2 5p5, F 2s2 2p5, Pb 5d10 6s2 6p2, Bi 5d10 6s2 6p3, Ag 4d10 5s1, Au 5d10 6s1.
2.2 Materials and device fabrication
Unless otherwise stated, all materials were purchased from Sigma-Aldrich or Acros Organics Company and used as received. (4-FBA)2MAPb2I7 was synthesized according to the reported procedure.24 (4-FBA)2, Pb2I4 and MAI3 (1 M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 M:1 M) were dissolved in a mixed solvent of N,N-dimethylformamide (DMF, Aldrich Co.) and dimethyl sulfoxide (DMSO, Aldrich Co.) as the precursor solution, and stirred at 80 °C for 2 h.
:1 M:1 M) were dissolved in a mixed solvent of N,N-dimethylformamide (DMF, Aldrich Co.) and dimethyl sulfoxide (DMSO, Aldrich Co.) as the precursor solution, and stirred at 80 °C for 2 h.
For device fabrication: firstly, laser-patterned and ITO-coated glass substrates were cleaned by ultrasonic oscillation in an alkaline aqueous washing solution for 15 min, followed by rinsing with deionized water, ethanol, and acetone, respectively, and then the ITO glass was placed under O3 conditions under ultraviolet treatment for 30 min. PEDOT:PSS (Sigma-Aldrich) was deposited by spin coating at 5000 rpm for 30 s to give a thickness of 40 nm. Afterwards, the thin film preparation of perovskite ((4-FBA)2MAPb2I7) was spin coated on the PEDOT:PSS thin film at 1000 rpm for 10 s, followed by 4000 rpm for 35 s. Then the thin film was immediately annealed at 70 °C for 80 min. After cooling down, poly(ethylene imine) (PEI, 0.1 wt%, in 2-propanol anhydride) was applied on top of the perovskite film at 5000 rpm for 60 s. Finally, an 80 nm thick silver layer (or Bi or Au) was thermally deposited as the top electrode under a vacuum of 4 × 10−4 Pa to fabricate perovskite solar cells with an area of 9 mm2. All devices were encapsulated by epoxy resin in a glove box for the experimental measurements.
2.3 Device measurements
Photovoltaic measurements were employed using an AM 1.5 solar simulator equipped with a 450 W xenon lamp (Newport), and the power output was adjusted to match AM 1.5 global sunlight (100 mW cm−2 by using a reference Si solar cell). Current density J–V curves were obtained by applying an external bias to the cell and measuring the generated photocurrent with a Keithley model 2400 digital source meter. The voltage step and delay time of the photocurrent were set to 10 mV and 40 ms, respectively. A similar data acquisition system was used to determine the monochromatic incident photon to electric current conversion efficiency. Under full computer control, the light emitted by a 300 W xenon lamp passes through a Gemini-180 double monochromator and focuses on the photovoltaic cell to be tested. The monochrome photoelectric conversion efficiency IPCE(λ) is generated in the visible spectrum with the increase of the monochromator, which is defined as IPCE(λ) = 12400 (JSC/λφ), where λ is the wavelength, JSC is the short-circuit photocurrent density (mA cm−2), and φ is the incident radiative flux (mW cm−2). The photovoltaic performance was measured by using a metal mask with an aperture area of 9 mm2. All the measurements were performed under the ambient light of bias light. The cross sections of the solar cells were measured with a Zeiss Gemini FEG-SEM instrument, using 5 kV at a magnification of 250 K.
2.4 Model building of Ag, Au, and Bi adsorption on the surface of 2D perovskite
In our calculations, we selected the (001), (001) and (100) faces of the 2D perovskite with n = 1, 2, and 3 as the adsorption surfaces, respectively. Usually, two terminal faces of 2D perovskite are exposed to the air, and they can connect with the metal atom: one is the inorganic terminal (defined as the I-T end), and the other is the organic terminal (defined as the O-T end).23 The top atoms of the two surface are selected as the adsorption sites. Fig. 1 shows that the different terminal faces of n = 1, 2, and 3 adsorbed three kinds of models of metal atoms; they were the most stable models of Ag, Au, and Bi adsorbed on the organic and inorganic terminals on the perovskite surfaces with n = 1, 2, and 3. | ||
| Fig. 1 Surface adsorption models of (a) C1: Ag/Au/Bi atoms adsorbed on the (001) surface of the n = 1 O-T end face, respectively; (b) C2: Ag/Au/Bi atoms absorbed on the (001) surface of the n = 1 I-T end face, respectively; (c) C3: Ag/Au/Bi atom adsorbed on the (001) surface of the n = 2 O-T end face, respectively; (d) C4: Ag/Au/Bi atom adsorbed on the (001) surface of the n = 2 I-T end face, respectively; (e) C5: Ag/Au/Bi atom adsorbed on the (100) surface of the n = 3 O-T end face, respectively; (f) C6: Ag/Au/Bi atoms adsorbed on the (100) surface of the n = 3 I-T end face, respectively. | ||
As shown in Fig. 1, for the n = 1 2D perovskite, the adsorption sites of the O-T and I-T ends on the (001) surface were the I1, I2, F1 and F2 positions, respectively. For the n = 2 2D perovskite, the adsorption sites of the O-T and I-T ends on the (001) surface were the F1, F2, I1 and I7 positions, respectively. Similarly, for the n = 3 2D perovskite, the adsorption sites of the O-T and I-T ends on the surface of (100) were the F1 and I3 positions, respectively. Three kinds of Ag, Au, and Bi atoms were adsorbed at different adsorption sites with different n values. According to the energy of the system, the adsorption energy could be obtained, and the most stable system of each atom adsorbed on a certain surface was selected (see Tables 1–3). The lattice and energy parameters were calculated before and after the metal atoms were adsorbed on the surface of the two terminals of 2D perovskites with n = 1, 2, and 3, respectively. The structure with the smallest adsorption energy was selected as the object of subsequent analysis.
| Clean surface/adsorption model | Lattice parameters of plate model (Å) | Total energy, Etot (eV) | Adsorption energy, Eads (eV) | |||
|---|---|---|---|---|---|---|
| a | b | c | ||||
| n = 1, (001) surfaces I-T end (I1) | Clean surface | 8.76910 | 9.34340 | 30.5428 | −14475.9614 |
— |
| Ag@I1 | 8.76910 | 9.34340 | 30.5428 | −15502.9039 |
−2.22883 | |
| Au@I1 | 8.76910 | 9.34340 | 30.5428 | −15388.3838 |
−2.48374 | |
| Bi@I1 | 8.76910 | 9.34340 | 30.5428 | −14625.9070 |
−2.54415 | |
| n = 1, (001) surfaces O-T end (F1) | Clean surface | 8.79610 | 9.34340 | 31.9668 | −14482.9707 |
— |
| Ag@F1 | 8.76910 | 9.34340 | 31.9668 | −15507.9725 |
−0.28813 | |
| Au@F1 | 8.76910 | 9.34340 | 31.9668 | −15393.6079 |
−0.69854 | |
| Bi@F1 | 8.76910 | 9.34340 | 31.9668 | −14630.3636 |
−0.00855 | |
| Clean surface/adsorption model | Lattice parameters of plate model (Å) | Total energy, Etot (eV) | Adsorption energy, Eads (eV) | |||
|---|---|---|---|---|---|---|
| a | b | c | ||||
| n = 2, (001) surface I-T end (I1) | Clean surface | 9.29002 | 9.59282 | 33.8652 | −16403.4151 |
— |
| Ag@I1 | 9.29002 | 9.59282 | 33.8652 | −17431.1187 |
−2.98993 | |
| Au@I1 | 9.29002 | 9.59282 | 33.8652 | −17317.9158 |
−4.56204 | |
| Bi@I1 | 9.29002 | 9.59282 | 33.8652 | −16553.9146 |
−3.09805 | |
| n = 2, (001) surface O-T end (F1) | Clean surface | 9.29002 | 9.59282 | 33.8652 | −16403.4255 |
— |
| Ag@F1 | 9.29002 | 9.59282 | 33.8652 | −17428.7892 |
−4.65003 | |
| Au@F1 | 9.29002 | 9.59282 | 33.8652 | −17314.2299 |
−0.865742 | |
| Bi@F1 | 9.29002 | 9.59282 | 33.8652 | −16551.1870 |
−0.36005 | |
| Clean surface/adsorption model | Lattice parameters of plate model (Å) | Total energy, Etot (eV) | Adsorption energy, Eads (eV) | |||
|---|---|---|---|---|---|---|
| a | b | c | ||||
| n = 3, (100) surface I-T end (I3) | Clean surface | 9.28802 | 9.07040 | 35.3060 | −18324.7244 |
— |
| Ag@I3 | 9.28802 | 9.07040 | 35.3060 | −19352.4547 |
−3.01663 | |
| Au@I3 | 9.28802 | 9.07040 | 35.3060 | −19237.9904 |
−3.32734 | |
| Bi@I3 | 9.28802 | 9.07040 | 35.3060 | −18475.5829 |
−3.45705 | |
| n = 3, (100) surface O-T end (F1) | Clean surface | 9.28802 | 9.07040 | 40.9471 | −21603.7820 |
— |
| Ag@F1 | 9.28802 | 9.07040 | 40.9471 | −22629.2909 |
−0.79523 | |
| Au@F1 | 9.28802 | 9.07040 | 40.9471 | −22514.7482 |
−1.02754 | |
| Bi@F1 | 9.28802 | 9.07040 | 40.9471 | −21751.6849 |
−0.50145 | |
According to Tables 1–3, it could be concluded that for the n = 1 2D perovskite system, on the (001) surface the most stable structure included: Ag adsorbed at the F1 position, defined as Ag@F1; Au adsorbed at the F1 position, defined as Au@F1; Bi adsorbed at the F1 position, defined as Bi@F1; Ag adsorbed at the I2 position, defined as Ag@I2; Au adsorbed at the I2 position, defined as Au@I2; Bi adsorbed at the I2 position, defined as Bi@I2. Similarly, for n = 2 perovskite, on the (001) surface the most stable structure included: Ag@F1, Au@F1, Bi@F1, Ag@I1, Au@I1, Bi@I1. As for the n = 3 perovskite, on the (100) surface, because there is only one adsorption site on the O-T and I-T end faces, all of them can be discussed as being the most stable structures: Ag@F1, Au@F1, Bi@F1, and Ag@I3, Au@I3, Bi@I3.
3. Results and discussion
3.1 Adsorption energy calculation
The adsorption model is shown in Fig. 1, where the energies of single atoms of Ag, Au, and Bi are −1024.713(67) eV, −909.938(658) eV and −147.401(450) eV, respectively. The adsorption energy formula is:25
 | (1) |
Tables 1–3 shows the adsorption energies of different atomic adsorptions on the 2D perovskite (n = 1, 2, and 3) surface at the stable position. The calculated results were: (1) when n = 1, on the (001) surface of the I-T end, the Ag, Au, and Bi adsorbing at the I1 position have lower energies, which are −2.228(83) eV, −2.483(74) eV, −2.544(15) eV, indicating that the structure of the system is most stable when the adsorption locates at the I1 position. In addition, among the adsorptions of the three metals, the lowest adsorption energy is for the Ag atom adsorption conditions, and the system structure is most stable after Ag surface adsorption. For the (001) surface of the O-T end, when Ag, Au, and Bi adsorb at the F1 position, the system has low energy; meanwhile, the energy of the system after Au atom adsorption is the highest, at −0.731(14) eV, and the Bi adsorption energy is the lowest, at −0.00855 eV. Therefore, according to the calculation results, this condition of O-T end exposure in the air and contact with adsorbed metal atoms is the stable structure of Bi adsorption on the O-T end. (2) When n = 2, for the (001) surface of the I-T end in contact with the metal atoms, they still adsorb at the I1 location, when the system has the minimum adsorption energy and the most stable structure, among the adsorptions of the three metals atoms. After Ag adsorption, the energy is the minimum, which shows that the structure of the system is most stable after absorption of Ag at the (001) face of the I-T end. For the (001) surface of the O-T end, Ag, Au, and Bi atoms adsorb at the F2 position, when the system has a lower energy, which shows that Bi adsorption has the minimum adsorption energy. (3) For n = 3 2D perovskite, for the condition of metal atoms adsorbing at the (100) surface of the I-T end, when the adsorption locates at the I3 position, the system possesses a lower adsorption energy, and a relatively stable structure. Ag adsorption possesses the minimum absorption energy among adsorption of the three atoms, which shows that the structure of the Ag adsorption system is most stable on the (100) face of the O-T end. For the O-T end surface adsorption, the Ag, Au, and Bi adsorptions locate at the F1 position, and the system has lower energy compared with the F2 adsorption position, so the adsorption energy of Bi adsorbed on the (100) face system is still the lowest among adsorptions of the three metal atoms.
Therefore, to summarize these results, we found that for 2D perovskite with n = 1, 2, or 3, when the metal atom adsorbs at the I1 position of the I-T end, the adsorption energy of Ag atoms (Ag@I1) is the lowest, at −2.228(83) eV for n = 1 (see Tables 1–3). When the metal atom adsorbs at the F1 position of the O-T end, the adsorption energy of Bi atoms (Bi@F1) is the lowest, at −0.008(55) eV for n = 1. These adsorption energies are negative, which means it is an exothermic reaction, and the adsorption structure is relatively stable.
In addition, the change in adsorption energy of adsorption of Au and Bi is the smallest. When the Au atom adsorbs at the F1 position on the I-T end face of 2D perovskite with n = 1, 2, and 3, the change in adsorption energy like the Bi atom adsorption is small, and it shows relative stability. However, for Ag adsorption on the I-T end of 2D-perovskite with n = 1, 2, and 3, the change in adsorption energy is very obvious, as shown in Fig. 2. When n = 2, in the Ag@F1 system, the adsorption energy is a minimum, at −4.650(03) eV; when n = 1 or 3, the adsorption energy of Ag@F1 is located between the adsorption energies of Au@F1 and Bi@F1 for adsorption at the O-T end, which indicates that Ag has a greater influence on the energy of the O-T end than Au or Bi atom adsorption, because Ag can react with I ions to form the AgI compound. Why is there is a sudden drop in the energy at n = 2, which then goes up again at n = 3? We think there are several reasons: (1) we think that it may be associated with the corresponding surface adsorption. We know that the n value represents the number of inorganic layers in the 2D perovskite: n = 1 where there is one inorganic layer for an organic layer, n = 2 for two inorganic layers, n = 3 for three inorganic layers; therefore, the two-dimensional perovskites are arranged in organic–inorganic intervals. When n = 1 or 3, the corresponding adsorption surface is the inorganic layer; when n = 2, the metal atom adsorption on the two-dimensional surface is by covalent bonding interaction, where the dangling bonds form covalent bonds on the surface atoms of 2D perovskite, so there is a strong chemical reaction between Ag and the surface atoms on the perovskite, because the activity of Ag metal is stronger than the activity of Au or Bi metal. On the other hand, the Au and Bi atoms possess a passivation effect, so their reactions with the perovskite surface ions are much more stable than those of the Ag atoms. When the n = 1 or 3, the adsorption action is ionic bonding, and the atomic force in the adsorption is different. Therefore, there is a sudden drop in the energy at n = 2, which then goes up again at n = 3.26 (2) The adsorption energy is used to connect with the changes in charge densities as well as the changes in bonding. Since the metal atom adsorption on the perovskite surface forms the charge density transfer from the electron acceptor (halide ion) to the electron donor (metal ion), the amount of charge transfer to the donor and the other bonds is a crucial point for the elongation and contraction of the X (X = Au, Ag, Bi)–I(F) bond, and different metal atom adsorptions on the perovskite surface are different, so the charge transport is also different, and so the adsorption energy is different.27 In addition, the result shows that the relative energy value of Ag adsorption is much larger than that of Au or Bi metal adsorption on perovskite. When an Au or Bi atom adsorbs on the surface of perovskite, the adsorption energy decreases, which can be ascribed to the increasing repulsive force between the Au or Bi atom and the I or F atom on the surface of the perovskite. Meng and co-workers28 studied the adsorption site of one metal atom adsorbed on the (001) surface of a 1 × 1 cell of tetragonal MAPbI3, and found that the adsorption energy is lowest when Au or Bi is adsorbed at the hollow site (between two MA+) on the MAI-termination surface. (3) Ag forms AgI with iodide ions which come from the diffusion in the perovskite layer at the interface, and reacts with perovskite more easily than Au or Bi.29 As a result, the stability of the perovskite is reduced, as the energy increases greatly, so the relative energy value of Ag is much larger than that of Au or Bi metal adsorption on perovskite.
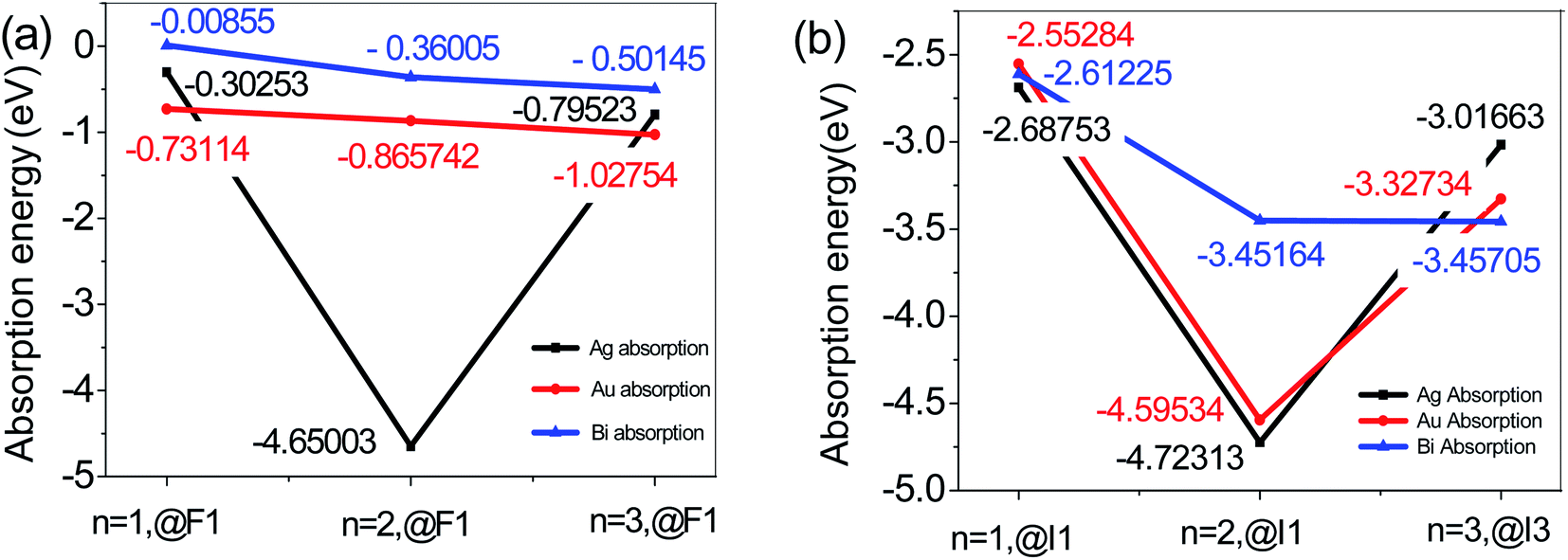 | ||
| Fig. 2 Adsorption energies of (a) metal atoms adsorbed on the O-T end face of n = 1, 2, and 3. (b) Metal atoms adsorbed on the I-T end face of n = 1, 2, and 3. | ||
To sum up, for two-dimensional perovskites, on the one hand, the probability of exposure of the O-T end to air is greater than that of the I-T end to air. On the other hand, the average absorption energy of metal atom adsorption on the O-T end is smaller than that on the I-T end. Therefore, we mainly pay attention to the adsorption situation on the O-T end face, and the adsorption situation of the I-T end face is used mainly for comparative analysis. We believe that the (001) face of the O-T terminal exposed in the air is much better at making contact with metal atoms than the (001) face of the I-T terminal for 2D perovskite with n = 1, 2, or 3. With this condition of O-T end adsorption, the structural stability of a Bi atom adsorbed on its surface is better than that of Ag or Au atoms. On the I-T end adsorption, when Ag adsorbs on the 2D perovskite surface, the system has a lower energy. For total absorption, the adsorption energy of Ag and Bi atoms adsorbed at the I1 and F1 positions are biggest for the n = 2 2D perovskite. When n = 2, the Ag@I1 and Bi@I1 system adsorption energies can reach −4.562(04) and −3.098(05) eV, respectively. This indicates that the interaction action between Ag and Bi atoms and the surface of n = 2 perovskite is increased.
In order to further explore the interaction between metal atoms and the surface of 2D perovskite, we next discuss the metal atom adsorption properties and other physical properties, such as energy band structure, density of states, bonding situation, and light absorption spectra. In view of the above characteristics of adsorption energy, here we selected n = 2 2D perovskite as the object of study.
3.2 Energy parameters
 | ||
| Fig. 3 Energy band structures of n = 2 2D perovskite: (a) (001) surface of the clean O-T end; (b) (001) surface of the clean I-T end; (c), (d) and (e) are Ag/Au/Bi atoms adsorbed at the F1 atom of the O-T end, corresponding to Ag@ F1, Au@F1, and Bi@F1, respectively; (f) (g) (h) are Ag/Au/Bi atoms adsorbed at the I1 atom of the I-T end, corresponding to Ag@I1, Au@I1, and Bi@I1, respectively. | ||
 | ||
| Fig. 4 Density of states (DOS) and partial density of states (PDOS), (a) n = 2, the clean surface in the O-T end face; (b) n = 2, the clean surface in the I-T end face; (c) n = 2, Bi atom adsorbed at the F1 atom of the O-T end, corresponding to Bi@F1; (d) n = 2, Bi atom adsorbed at the I1 atom of the O-T end, corresponding to Bi@I1. | ||
From Fig. 4c, when n = 2, in the Bi@F1 system, we found that the impurity level in the energy band mainly comes from the electrons on the F1 2p and Bi 6p orbitals, and at the F1 position atoms come from the organic ion of 4-FBA, which indicates that there is an interaction between the electrons on the F1 2p and Bi 6p orbitals. When n = 2 in the Bi@I1 system, from Fig. 4e, it can be seen that the impurity level in the energy band mainly comes from the electrons in the I 5p and Bi 6p orbitals, indicating that the electrons interact with each other on the I 5p and the Bi 6p orbitals. Similarly, from analysis it also can be shown that the impurity level of n = 3 in the Bi@F1 system is occupied by the F1 2p and Bi 6p orbital electrons. The impurity level in the energy band of Ag@I3 mainly comes from Ag 5s and I 5p orbital electrons, and contains a few Pb 6s and 6p orbital electrons. The new energy level in Au@I3 mainly comes from Au 6s and I 5p orbital electrons. The impurity level in the BI@I3 system mainly comes from the I 5p and Bi 6p orbital electrons.
From the above analysis, it can be seen that the impurity levels in the energy band of the adsorbed metal system are occupied by the orbital electrons in the adsorptive atoms and metal atoms. For the n = 1 2D case (see Fig. S1 in the ESI†), Ag/Au/Bi atoms adsorb at the top of the F atom position on the O-T end, and the impurity level is generated from the outermost electron of the metal atom, such as Ag 5s, Au 6s and Bi 6p. In the Bi@F1 system with n = 2 or 3, both levels are occupied by the electrons on the F1 2p and Bi 6p orbitals near the top of the valence band, forming a deep principal energy level. In the n = 2 Bi@I1 system, and n = 3 Ag@I3 and Bi@I3 systems, the I 5p and Bi 6p orbitals occupy the bottom of the conduction band. In the n = 3 Au@I3 system, the impurity level is located in the middle of the band gap. This shows that the interaction between adsorptive atoms and metal atoms is strengthened as the n value increases, the quantization characteristics of the 2D perovskite structure are weakened, and the electron transport is enhanced. Comparing the energy bands of Ag/Au/Bi adsorption on the O-T and I-T ends of the n = 3 2D perovskite, it can also be found that Bi adsorption generates many new energy levels in the two end face adsorptions. The impurity level is the donor level in the Bi@F1 system, binding the electrons on the F1 2p orbital, but in the Bi@I3 system, the impurity level is the acceptor level, binding a few Pb 6p orbital electrons. Therefore, from the energy bands of all adsorption systems of n = 1, 2, and 3, Bi atom adsorption can introduce deep energy levels into the O-T and I-T ends of 2D perovskite with n = 1, 2, and 3, which indicates that Bi adsorption is beneficial to the carrier transport process.
In order to further explore the interaction between metal atoms and adsorbed atoms, we next discuss two problems from the aspects of bond length and bond angle.
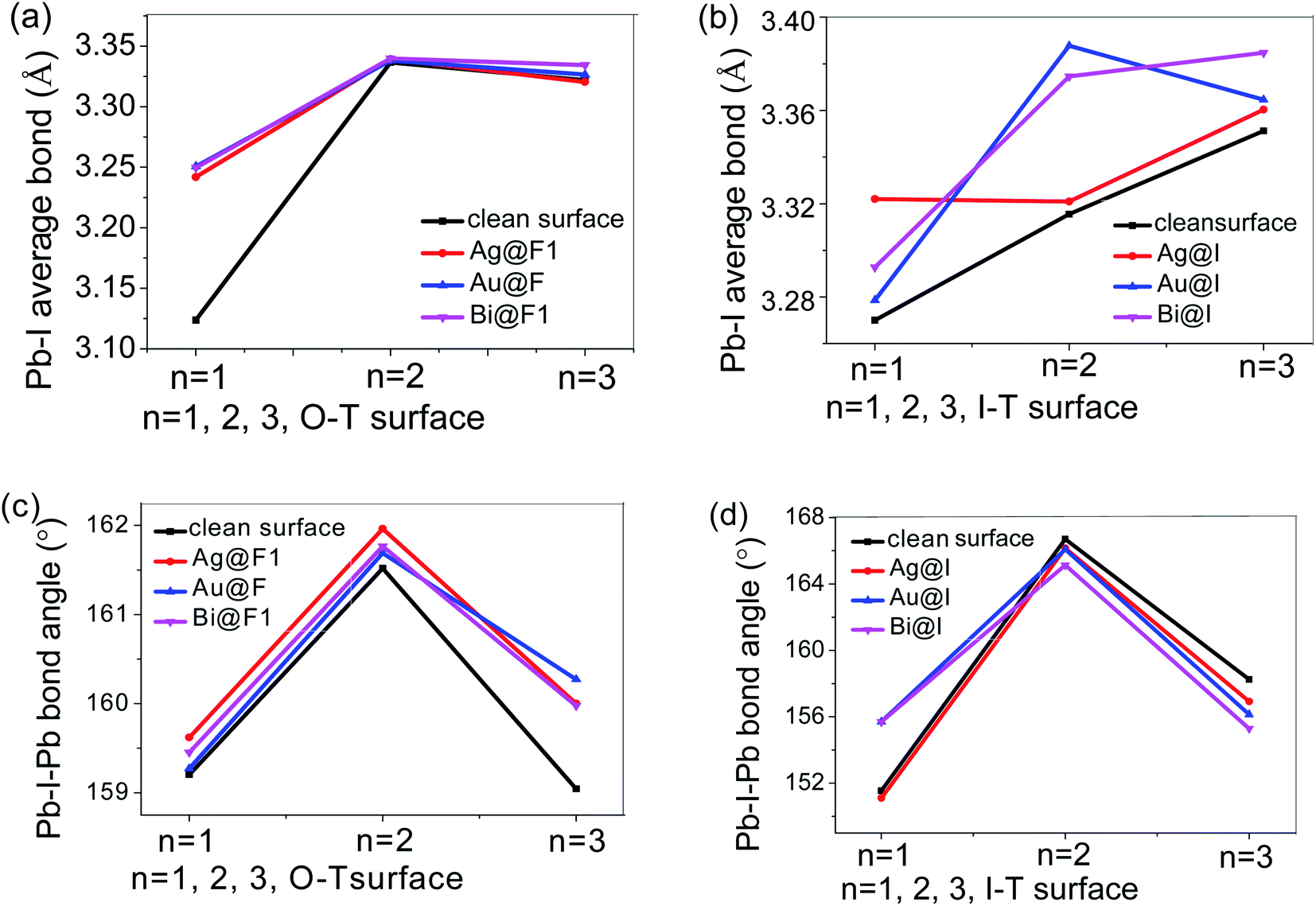 | ||
| Fig. 5 (a and b) Represent the average lengths of the Pb–I bond when Ag/Au/Bi atoms are adsorbed at the O-T and I-T ends, respectively. (c and d) Represent the average bond angles of Pb–I–Pb after metal atom adsorption on O-T and I-T ends, respectively. | ||
The NBO program was used to obtain bond length, population, charges and stabilization energy for atoms and the overlap-weighted bond order. From Table 4, we can see that the Bi (Ag or Au)–F bond and Bi (Ag or Au)–I bond played an important role on the 2D perovskite (001) surface, producing strong interaction between the I or F atom and the metal atom. For n = 1 Bi adsorption on a 2D perovskite (001) surface on the O-T end, the bond length of Bi–F decreased and the F atom lost a charge of 0.02e, while the I atom lost 0.04e of charge on the (001) surface of the I-T end.
| cases | No. | Bond | Bond length | Population | Atom | Charge/e |
|---|---|---|---|---|---|---|
| N = 1 of O-T end surface | 1 | Bi–F bond | 2.429 | −0.24 | F1 | −0.20 |
| 2 | Ag–F bond | 3.134 | −0.28 | F1 | −0.20 | |
| 3 | Au–F bond | 3.419 | −0.34 | F1 | −0.20 | |
| N = 1 of I-T end surface | 4 | Bi–I bond | 2.755 | −0.86 | I1 | −0.22 |
| 5 | Ag–I bond | 2.625 | −0.95 | I1 | −0.22 | |
| 6 | Au–I bond | 2.544 | −1.01 | I1 | −0.22 | |
| N = 2 of O-T end surface | 7 | Bi–F bond | 2.386 | −0.11 | F2 | −0.15 |
| 8 | Ag–F bond | 2.853 | −0.14 | F2 | −0.16 | |
| 9 | Au–F bond | 3.369 | −0.16 | F2 | −0.16 | |
| N = 2 of I-T end surface | 10 | Bi–I bond | 2.747 | −0.66 | I1 | −0.23 |
| 11 | Ag–I bond | 2.639 | −0.74 | I1 | −0.23 | |
| 12 | Au–I bond | 2.581 | −0.78 | I1 | −0.23 | |
| N = 3 of O-T end surface | 13 | Bi–F bond | 3.362 | −0.42 | F1 | −0.33 |
| 14 | Ag–F bond | 2.743 | −0.44 | F1 | −0.33 | |
| 15 | Au–F bond | 3.319 | −0.48 | F1 | −0.33 | |
| N = 3 of I-T end surface | 16 | Bi–I bond | 2.731 | −1.15 | I3 | −0.35 |
| 17 | Ag–I bond | 2.581 | −1.25 | I3 | −0.35 | |
| 18 | Au–I bond | 2.564 | −1.25 | I3 | −0.35 |
For the n = 2 (001) surface of the O-T end, the bond length of Bi–F became shorter and the F atom gained 0.05e charge, while I lost 0.03e charge in the (001) surface of the I-T end, implying that the Bi–I bond strengthened under n = 2 Bi adsorption conditions. This indicated that Bi adsorption on the F position was more stable than Bi adsorption on the I position at n = 2. By contrast, for n = 3, no matter what the Bi adsorption on the F position or Ag or Au atom adsorption on the F position was, the bond length of Bi (Ag or Au)–I is longer in I-T surface adsorption than the Bi (Ag or Au)–F bond in O-T end surface adsorption. In the meantime, the charge of the I atom lost 0.15e and the F atom lost 0.12e, meaning that the charge lost by the F atom was less than that lost by the I atom under the same conditions, because the metal–F bond was elevated, but the metal–I bond was not enhanced. Overall, for n = 1, 2, and 3, Bi adsorption on the F position was more stable than other adsorption conditions.
We also calculated the stabilization energy. The stabilization energy is connected with the bond length and bond energy. The association of stabilization energy E with i–j delocalization is given by the following equation:
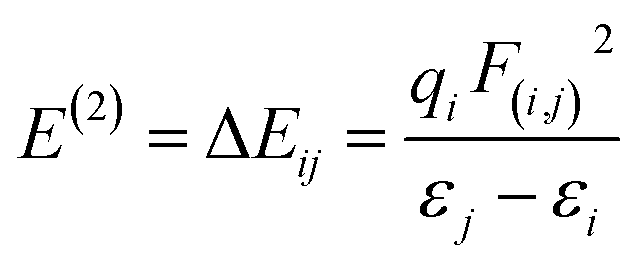 | (2) |
| Case | No. | Bond | Donor | Acceptor | E(2)(n(Y) → σ*(X–H)) | |
|---|---|---|---|---|---|---|
| E(σ*(metal–F)) | E(σ*(metal–I)) | E(n(Y)) | ||||
| N = 1 of O-T end surface | 1 | Bi–F bond | 0.0039 | — | −0.8772 | 2.07 |
| 2 | Ag–F bond | 0.0045 | — | −0.7632 | 3.72 | |
| 3 | Au–F bond | 0.0048 | — | −0.7830 | 4.22 | |
| N = 1 of I-T end surface | 4 | Bi–I bond | — | 0.0036 | −0.5042 | 2.11 |
| 5 | Ag–I bond | — | 0.0038 | −0.5236 | 2.15 | |
| 6 | Au–I bond | — | 0.0041 | −0.5255 | 2.17 | |
| N = 2 of O-T end surface | 7 | Bi–F bond | 0.0035 | — | −0.8470 | 5.14 |
| 8 | Ag–F bond | 0.0055 | — | −0.8652 | 6.09 | |
| 9 | Au–F bond | 0.0063 | — | −0.9253 | 8.23 | |
| N = 2 of I-T end surface | 10 | Bi–I bond | — | 0.0039 | −0.7543 | 3.19 |
| 11 | Ag–I bond | — | 0.0041 | −0.7768 | 3.22 | |
| 12 | Au–I bond | — | 0.0046 | −0.7953 | 3.28 | |
| N = 3 of O-T end surface | 13 | Bi–F bond | 0.0042 | — | −0.7691 | 8.11 |
| 14 | Ag–F bond | 0.00798 | — | −0.9385 | 8.83 | |
| 15 | Au–F bond | 0.0065 | — | −0.8952 | 8.56 | |
| N = 3 of I-T end surface | 16 | Bi–I bond | — | 0.00746 | −0.7401 | 3.18 |
| 17 | Ag–I bond | — | 0.0042 | −0.7544 | 3.20 | |
| 18 | Au–I bond | — | 0.0078 | −0.8254 | 3.83 | |
The stabilization energies for metal adsorption on the I atom are found to be less than the metal adsorption on the F atom. In the six different adsorption cases, contraction of the metal–I bond occurs on the 2D perovskite surface. From Table 5, it can be observed that for metal adsorption on the F atom the charge transfer to the antibonding orbital of the Bi–F bond of the n = 1 O-T end surface is found to be less in all cases and, hence, it causes a blue shift in the vibrational frequency. In cases 3, 9, and 13 the charge is transferred from ‘Y’ to the metal–F bond and, hence, there is an increase in the charge transfer to the antibonding orbital, which results in elongation of the metal–F bond, but contradictorily, contraction in the metal–I bond is observed. Hobza et al. reported that for improper hydrogen bonding, the charge is transferred from the electron acceptor to the remote part of the electron-donor molecule. In the present study, it has been noted that this is not the case for all the systems. As previously mentioned, for some systems (such as 1, 7, and 14), the metal–F bond has contracted even after more charge is transferred to the antibonding orbital of the C–H bond. This is due to a secondary effect. In others cases, such as 4, 5, 6, 10, 11, 12, 16, 17, and 18, the other atom which is attached to the hydrogen atom is a halogen atom which is electronegative. Furthermore, it has been observed that there is only a marginal charge transfer to the antibonding orbitals of the metal–I(F) bond. For these different adsorption conditions, the major charge transfer takes place within the donor molecule itself, i.e. from the lone-pair electrons of the halogen atom to the halogen atom, making the halogen atom electronegative, which in turn attracts the hydrogen atom towards it, thereby resulting in the contraction of the H–F and H–I bonds. This has been supported by the stabilization energy, i.e. n(N) → Ry*(C) in cases 1, 7, and 13 are 2.07, 5.14 and 8.11 kcal mol−1, respectively. For cases 1 and 7, it is found that for all the other bonds that are attached to the hydrogen atom, the charge transfer to the antibonding orbital is less. Hence the contraction in the Bi–F bond in these two cases is very small and they exhibit a very weak hydrogen bond. Even so, there is a significant charge transfer during intermolecular interaction, and the intramolecular charge transfer interactions restrict the elongation of the C–H bond, leading to a decrease in the length of the Bi–F bond during adsorption and hence result in a blue shift in the stretching vibrational frequency.
From Fig. 5a and b, we found that the Ag/Au/Bi metal atom adsorption can stretch the average length of the Pb–I bond in the O-T end of n = 1 perovskite, but in n = 2 2D perovskite, adsorption of the three kinds of metal atom does not affect the Pb–I bond length. For n = 3, Bi adsorption is on the F atom position, which shows a small increase in the Pb–I bond. After Au adsorption, the Pb–I bond length changes less than for Bi atom absorption on the O-T end. As for the I-T end absorption conditions, the situation is the opposite: when n = 1, the Pb–I bond tensile rate is increased under Ag adsorption conditions, and with Au absorption it is the weakest. When n = 2, Ag has the least influence on Pb–I bond length, while Au atom adsorption shows the greatest tensile strength. When n = 3, Bi has the maximum Pb–I bond stretching, and Ag has the minimum influence on Pb–I bond length. Therefore, by analyzing the bond length, when n = 1, the intensity of influence of metal absorption on the Pb–I bond can be sorted as: Ag > Au > Bi. Simultaneously, the main adsorption condition is adsorption of the three metal atoms on the I-T end; the adsorption on the O-T end is similar, but it is weak compared with the I-T end face adsorption. When n = 2, the influence of Ag/Au/Bi adsorption on the 2D perovskite surface is: Au > Bi > Ag, for which the main influence is also on the I-T end, and the influence of the three metal absorptions is the same as the O-T end absorption. When n = 3, the order of metal influence on the Pb–I bond is: Bi > Au > Ag, and this conclusion is true for both the I-T and O-T ends. It can be found that when the n value increases, Bi has a more obvious stretching effect on the Pb–I bond; specifically, Bi adsorption made the Pb–I bond longer on the surface of the perovskite.
According to Fig. 5c and d, for the O-T end face, it can be found that when Ag, Au, and Bi adsorb on the surfaces with n = 1 or 2, the influence of metal absorption on the bond angle of Pb–I–Pb is as follows: Ag > Bi > Au, and adsorption of all three metals shows an increase in this bond angle, and Au adsorption shows the maximum value of the Pb–I–Pb bond angle. When n = 3, the influence of adsorption of the three metals on the bond angle is as follows: Au > Ag = Bi. When Ag, Au, and Bi adsorb on the n = 3 O-T end, the Pb–I–Pb bond angle increases. As for the I-T end, when n = 1, Au and Bi adsorption can expand the Pb–I–Pb bond angle, while Ag atom adsorption slightly reduces the bond angle. When n = 2, adsorption of all three metals atoms can make the bond angle smaller, and the bond angle after Bi atom adsorption changes to the smallest value among the three atom adsorptions. When n = 3, the Bi atom also reduces the bond angle the most, followed by Au, and Ag has the least influence on the bond angle of Pb–I–Pb.
To sum up, from the analysis of bond length and bond angle, Bi atom adsorption has the greatest influence on the bond length and bond angle, with the increase in n value: the Bi atom makes the Pb–I bond length increase, while the Pb–I–Pb bond angle becomes smaller. From the structural stability of 2D perovskite, Bi atom adsorption has a greater influence on the octahedral structure [PbI6]4− than Ag or Au atom adsorption: the bond length elongated, indicating that the Pb–I bond energy was weakened; the bond angle reduced, indicating that the octahedron was more distorted, and the distorted octahedron structure could affect the polarization characteristic of 2D perovskite, affecting the charge transport.
The above discussion on the bond length and bond angle shows the change in the 2D perovskite structure itself after metal adsorption. This brings us to the second problem: the interaction effect between a 2D perovskite surface atom and the metal atom of Ag, Au, or Bi. It is necessary to discuss the bonding between metal atoms and adsorbed atoms on the surface of 2D perovskite. From Fig. 6, we can obtain bond length information between metal atoms and perovskite surface atoms.
 | ||
| Fig. 6 (a) The lengths of the F and metal bonds when Ag/Au/Bi are adsorbed at the F atom of the O-T end face. (b) The lengths of the I and metal bonds when Ag/Au/Bi are adsorbed at the I atom of the I-T end face. | ||
As shown in Fig. 6a, for the I-T end, for metal atom adsorption in the 2D perovskite surface with different n = 1, 2, and 3, the Ag and Au atoms are adsorbed on the F atom position. When the n value increases, the Ag–F and Au–I bonds are both shortened, which shows that the interaction effect is increased between the metal and halogen atoms. For the adsorption of Bi atoms, when the n value increases, the Bi–F bond first shortens and then lengthens. In particular, when n = 3, the Bi–F bond length increases sharply, suggesting the interaction effect is sharply decreased between Bi atoms and F atoms in the n = 3 2D perovskite. As for the I-T end face, as shown in Fig. 6b, the bond length of the Ag–I and Au–I bonds first increases and then decreases with an increase in n value. After Bi adsorption, the length of the BI–I bond is slowly reduced, which indicates that the interaction effect between the Bi atom and the I atom on the I-T end of perovskite increases with an increase in the n value.
In addition, from Fig. 6, we further find that Ag and Au not only adsorb on the O-T end but also adsorb on the I-T end. With an increase in n value, the Ag–I and Ag–F bond lengths and Au–I and Au–F bond lengths both decrease, which shows that the absorption effect is increased between Au and Ag and the I and F atoms on the 2D perovskite surface adsorption sites. However, the change in bond length for Bi adsorption on the I-T and O-T end faces are different compared with the Ag and Au adsorption: for adsorption on O-T end, the Bi–F bond length increases, and the Bi–I bond is shortened with an increase in the value of n. This indicates that on the O-T end, as the n value increases, the Bi–F bond is weakened, while the Bi–I bond is strengthened. On the whole, when Bi adsorbs on the O-T end, the interaction is weak between the Bi atom and the F atom on the O-T end. From the perspective of bond length and bond angle, with an increase in n value, Bi adsorption can elongate the Pb–I bond in the perovskite octahedron [PbI6]4−, and reduce the Pb–I–Pb bond angle. This indicates that Bi atom adsorption has a stronger influence on the structure of perovskite than Ag or Au adsorption, and its interaction with F atoms on the surface of perovskite is relatively weak. In summary, it can be concluded that Bi atom adsorption has a great influence on the surface of perovskite.
3.3 Optical parameters
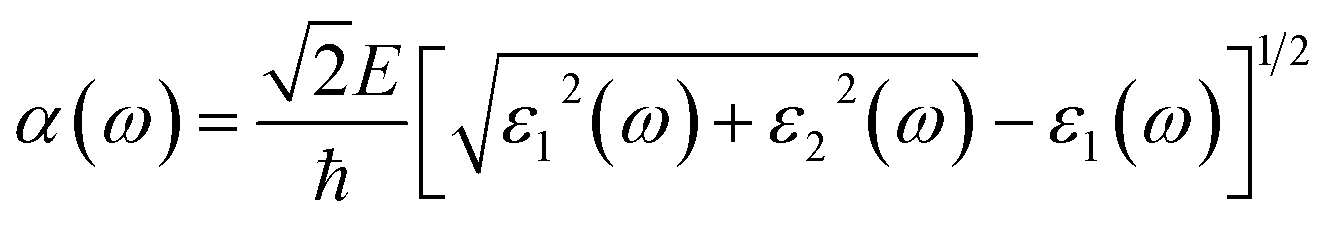 | (3) |
In order to gain a deeper understanding of the optical properties of Ag, Au, and Bi adsorbing on 2D perovskite surfaces, the light absorption spectrum was analyzed, as shown in Fig. 7. For metal adsorption on the O-T end when n = 1, as shown in Fig. 7c, the order of the effect of Ag/Au/Bi metal atom adsorption on 2D perovskite surface peak strength was: Au@F1 = Ag@F1 > O-T > Bi@F1, but the peak did not drift significantly before or after metal atom adsorption. When n = 2, as shown in Fig. 7d, the order of the effect of Ag/Au/Bi metal atom adsorption on 2D perovskite surface peak strength was: Au@F1 > O-T > Bi@F1 > Au@F1. The absorption peaks after adsorption move toward longer wavelength relative to those before adsorption, and the order of peak drift amplitude is: Bi@F1 > Ag@F1 > Au@F1. When n = 3, the O-T end adsorbs Ag/Au/Bi, as shown in Fig. 7e. According to the rising edge, the order of the absorption peak strength is: Bi@F1 > Au@F1 > Ag@F1 > O-T. After the metal atom adsorption, the peak position moves towards the direction of longer wavelength, and the order of drift amplitude is: Bi@F1 > Ag@F1 > Au@F1. Therefore, from the optical spectra of O-T end adsorption for 2-D perovskite, when n = 1, the absorption peak positions of the three metals are unchanged, while for Bi atom adsorption on 2D perovskite (n = 2 or 3), the positions of most of the absorption peaks move towards the direction of longer wavelength. This result is consistent with the reduction in band gap values in the Bi@F1 and Bi@I1 systems of n = 2 2D perovskite, and the n = 3 Bi@F1 system.
 | ||
| Fig. 7 Absorption spectra: (a and b) of O-T and I-T, respectively; (c), (d) and (e) of metal (Ag/Au/Bi) adsorbing on the O-T end; (f), (g) and (h) of metal (Ag/Au/Bi) adsorbing at the I-T end (n = 1, 2, and 3). | ||
For metal adsorption on the I-T end, as shown in Fig. 7f, when n = 1, after adsorption of the metal atoms, the order of the peak values of absorption is: Ag@I1 > I-T > Bi@I2 > Au@I2. It is quite obvious that the absorption peak of the BI@I1 system moves towards the direction of longer wavelength, while the other three peaks do not differ much. When n = 2, as shown in Fig. 7g, the peak value of absorption in the four cases follows the order: Ag@I1 > Au@I1 > I-T > Bi@I1, and in all three cases, all the absorption peaks move toward the direction of longer wavelength, and the drift amplitude follows the order: Bi@I1 > Au@I1 > Ag@I1. When n = 3, as shown in Fig. 7h, the peak values of absorption in the four cases follow the order: Au@I3 > O-T > Ag@I3. The absorption peaks of the I-T end move towards the direction of longer wavelength compared to the clean surface, and the order of drift amplitude is: Bi@I3 > Ag@I3 > Au@I3. From the perspective of I-T end adsorption on the inorganic surface, for n = 1 2D perovskite, Ag adsorption enhances the light absorption intensity, while for 2D perovskite with n = 2 or 3, when the Bi atom is adsorbed on the I atom, the shift in amplitude of the absorption peak value is larger than that of Ag or Au adsorption, moving in the direction of longer wavelength.
In addition, by observing the light absorption spectrum of metal atom adsorption for n = 1, 2, and 3, we find that the half peak width widens as the value of n increases. At n = 1, the light absorption spectrum is narrow, and the characteristic of quantization is obvious, mainly because the potential well between the organic layer and inorganic layer atoms is deep, leading to limited electron transition. Therefore, the 2D perovskite energy bands of n = 1, 2, and 3 show that the outermost electrons of Ag/Au/Bi were in the local area of the Fermi level, and the interaction between the adsorbed atom and the F atom is weak.
In summary, under Bi adsorption, its light absorption spectrum has a greater impact on the 2D perovskite than Ag or Au atom adsorption, and the absorption peaks of Bi-adsorbed on perovskite move towards the direction of longer wavelength.
 | (4) |
 | ||
| Fig. 8 The I–V curves characteristic of ITO/PEDOT:PSS/perovskite/PEI/Ag (Au and Bi) solar cells based on (4-FBA)2MAPb2I7 2D perovskite materials with forward and reverse scanning. | ||
| Device structure | Scanning direction | VOC (V) | JSC (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| Ag electrode | Forward | 0.85 | 19.44 | 0.70 | 11.57 |
| Reverse | 0.84 | 18.42 | 0.68 | 10.52 | |
| Au electrode | Forward | 0.90 | 20.79 | 0.75 | 14.03 |
| Reverse | 0.89 | 20.19 | 0.73 | 13.12 | |
| Bi electrode | Forward | 0.95 | 21.57 | 0.74 | 15.16 |
| Reverse | 0.93 | 21.17 | 0.72 | 14.18 |
4. Summary
In this paper, the Ag, Au, and Bi metal atoms adsorbed on the organic and inorganic terminals of 2D perovskite were discussed from the points of view of adsorption energy, energy band structure, density of states, bond length, bond angle and the light absorption spectrum, respectively, and the conclusions are as follows.(1) By adsorption energy calculation, we found with n = 1, the adsorption energy of the Bi@F1 system was the lowest, at −0.008(55) eV. When n = 2 or 3, the metal atom adsorption energy is also negative, which shows that between the metal atoms and perovskite there exists an attraction effect. Comparing the different metal adsorption energies with different n values, the adsorption of the Bi system has minimal impact on the system energy. Therefore, from the perspective of energy, when Bi is adsorbed on the 2D perovskite surface, the whole system has an adsorption energy minimum, and this structure is the most stable.
(2) According to the energy band structure and density of states, when n = 1, Ag, Au, and Bi adsorb on the organic end face (O-T), the generated impurity level locates at the Fermi level, and the impurity level binds the outermost valence electrons of the metal atoms, such as the electrons on the Ag 5s, Au 6s and Bi 6p orbitals. When n = 2, only the Bi@F1 and Bi@I1 systems produce impurity levels. When n = 3, impurity levels are generated in organic and inorganic end surface adsorption, including Bi–F1, Ag@I3, Au@I3 and Bi–I3. We found that the impurity levels are mainly occupied by the outer valence electrons of the adsorptive atoms and metal atoms. According to the results of energy bands and density of states, Bi atom adsorption can introduce deep energy levels into the energy bands of the O-T and I-T ends of 2D perovskite with n = 1, 2, or 3, which indicates that Bi adsorption is beneficial to carrier transmission.
(3) Through bond length and bond angle analysis, we found that Bi adsorption can enhance the Pb–I bond length in the perovskite octahedron [PbI6]4−, and reduce the Pb–I–Pb bond angle, and Bi has a stronger influence on the intrinsic structure of perovskite than Ag or Au. The intrinsic structural change of Bi absorption on the 2D perovskite surface atom can affect the polarization characteristic of 2D perovskite material, affecting its charge transport.
(4) Under Bi adsorption, its light absorption spectrum has a greater impact on the 2D perovskite than Ag or Au absorption, and the absorption peaks of Bi adsorbed on perovskite moves towards the direction of longer wavelength.
(5) We used the Ag, Au, and Bi as electrodes in a 2D perovskite solar cell, and achieved the highest PCE of 15.16% (forward scanning) in the Bi electrode of the 2D perovskite solar cell, which is consistent with the theoretical analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Central Government Guide Local Funds for Scientific and Technological Development (Grant No. ZYYD2020000045), the National Natural Science Foundation of China (Grant No. 62074068), the Intergovernmental Cooperation Project, National Key Research and Development Program, Ministry of Science and Technology, PRC (Grant No. 2019YFE0108400), and the Scientific Research Project of Hubei Education Department (Grant No. D20182901).References
- NREL, Best Research-Cell Efficiency Chart, 2020, p. 11, https://www.nrel.gov/pv/cell-396-efficiency.html Search PubMed.
- M. F. M. Noh, N. A. Arzaee, I. N. N. Mumthas, N. A. Mohamed, S. N. F. M. Nasir, J. Safaei, A. R. b. M. Yusoff, M. K. Nazeeruddin and M. A. M. Teridi, High-humidity processed perovskite solar cells, J. Mater. Chem. A, 2020, 8, 10481–10518 RSC.
- X. Xiao, J. Dai, Y. J. Fang, J. J. Zhao, X. P. Zheng, S. Tang, P. N. Rudd, X. C. Zeng and J. S. Huang, Suppressed ion migration along the in-plane direction in layered perovskites, ACS Energy Lett., 2018, 3, 684–688 CrossRef CAS.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, A layered hybrid perovskite solar-cell absorber with enhanced moisture stability, Angew. Chem., Int. Ed. Engl., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- S. Ahmad, P. Fu, S. Yu, Q. Yang, X. Liu, X. Wang, X. Wang, X. Guo and C. Li, Dion-Jacobson phase 2D layered perovskites for solar cells with ultrahigh stability, Joule, 2019, 3, 794 CrossRef CAS.
- D. T. Gangadharan and D. L. Ma, Searching for stability at lower dimensions: current trends and future prospects of layered perovskite solar cells, Energy Environ. Sci., 2019, 12, 2860 RSC.
- P. Gao, A. R. B. M. Yusoff and M. K. Nazeeruddin, Dimensionality engineering of hybrid halide perovskite light absorbers, Nat. Commun., 2018, 9, 5028 CrossRef PubMed.
- J. Hu, L. Yan and W. You, Two-Dimensional Organic-Inorganic Hybrid Perovskites: A New Platform for Optoelectronic Applications, Adv. Mater., 2018, 28, 1802041 CrossRef PubMed.
- M. D. Smith, L. Pedesseau, M. Kepenekian, I. C. Smith, C. Katan, J. Even and H. I. Karunadasa, Two-dimensional organic-inorganic hybrid Perovskites: a new platform for optoelectronic applications, Chem. Sci., 2017, 8, 1960–1968 RSC.
- N. H. Tiep, Z. L. Ku and H. J. Fan, Recent advances in improving the stability of perovskite solar cells, Adv. Energy Mater., 2016, 4, 1501420 CrossRef.
- S. Wu, R. Chen, S. Zhang, L. Y. Han and W. Chen, et al., A chemically inert bismuth interlayer enhances long-term stability of inverted perovskite solar cells, Nat. Commun., 2019, 10, 1–10 Search PubMed.
- W. Ming, D. Yang and T. Li, et al., Formation and diffusion of metal impurities in perovskite solar cell material CH3NH3PbI3: implications on solar cell degradation and choice of electrode, Adv. Sci., 2018, 5, 1700662 CrossRef PubMed.
- Y. Kato, L. K. Ono and M. V. Lee, et al., Silver iodide formation in methyl ammonium lead iodide perovskite solar cells with silver top electrodes, Adv. Mater. Interfaces, 2015, 2, 1500195 CrossRef.
- K. Domanski, J. P. Correa and N. Mine, et al., Not all that glitters is gold:metal-migration-induced degradation in perovskite solar cells, ACS Nano, 2016, 10, 6306–6314 CrossRef CAS PubMed.
- Y. Xiong, L. Xu, P. Wu, L. Sun, G. Xie and B. Hu, Bismuth Doping-Induced Stable Seebeck Effect Based on MAPbI3 Polycrystalline Thin Films, Adv. Funct. Mater., 2019, 29, 1900615 CrossRef.
- K. Refson, P. R. Tulip and S. J. Clark, Variational density-functional perturbation theory for dielectrics and lattice dynamics, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 155114 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- M. D. Patey and C. E. H. Dessent, APW91 density functional study of conformational choice in 2-phenylethanol, n-butylbenzene, and their cations: problems for density functional theory?, Russ. J. Phys. Chem. A, 2002, 106, 4623–4631 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- Y. Hao, Z. Qiu and X. Zhang, et al., Series of 2D multilayered perovskites constructed by slicing the 3D [(CH3NH3)PbI3] with 4-fluorobenzylamine, Inorg. Chem. Commun., 2018, 97, 134–138 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 98, Revision A.9, Gaussian Inc., Pittsburgh, PA, 1998 Search PubMed.
- C. Bernal and K. Yang, First-principles hybrid functional study of the organic–inorganic perovskites CH3NH3SnBr3 and CH3NH3SnI3, J. Phys. Chem. C, 2014, 118, 24383 CrossRef CAS.
- I. Zimmermann, S. Aghazada and M. K. Nazeeruddin, Lead and HTM free stable two-dimensional tin perovskites with suitable band gap for solar cell applications, Angew. Chem., Int. Ed., 2019, 131, 1084–1088 CrossRef.
- G. Wang, S. Mei, J. Liao, W. Wang, Y. Tang, Q. Zhang, Z. Tang, B. Wu and G. Xing, Advances of nonlinear photonics in low-dimensional halide perovskites, Small, 2021, 2100809 CrossRef CAS PubMed.
- A. Pivrikas, G. Juška, A. J. Mozer, M. Scharber, K. Arlauskas, N. S. Sariciftci, H. Stubb and R. Österbacka, Bimolecular recombination coefficient as a sensitive testing parameter for low-mobility solar-cell materials, Phys. Rev. Lett., 2005, 94, 176806 CrossRef CAS PubMed.
- J. Hu, L. Yan and W. You, Two-dimensional organic-inorganic hybrid perovskites: A new platform for Optoelectronic Applications, Adv. Mater., 2018, 30, 1802041 CrossRef PubMed.
- P. Kolandaivel and V. Nirmala, Study of proper and improper hydrogen bonding using Bader's atoms in molecules (AIM) theory and NBO analysis, J. Mol. Struct., 2004, 694, 33–38 CrossRef CAS.
- Y. Jia, Y. Zeng, X. Li and L. Meng, Effect of Sr substitution on the property and stability of CH3NH3SnI3 perovskite: A first-principles investigation, Int. J. Energy Res., 2020, 44, 5765–5778 CrossRef CAS.
- C. T. Lin, et al., Enhancing the operational stability of unencapsulated perovskite solar cells through Cu-Ag bilayer electrode incorporation, J. Mater. Chem. A, 2020, 8, 8684–8691 RSC.
- L. J. A. Koster, V. D. Mihailetchi and P. W. M. Blom, Bimolecular recombination in polymer/fullerene bulk heterojunction solar cells, Appl. Phys. Lett., 2006, 88, 052104 CrossRef.
- W.-J. Yin, T. Shi and Y. Yan, Unique properties of halide perovskites as possible origins of the superior solar cell performance, Adv. Mater., 2014, 26, 4653 CrossRef CAS PubMed.
- R. J. Bondi, S. Lee and G. S. Hwang, Role of structural disorder in optical absorption in silicon, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 115214 CrossRef.
- Z. Li, C. X. Xiao, Y. Yang, S. P. Harvey, D. H. Kim, J. A. Christians, M. J. Yang, P. Schulz, S. U. Nanayakkara, C. S. Jiang, J. M. Luther, J. J. Berry, M. C. Beard, M. M. Al-Jassima and K. Zhu, Extrinsic ion migration in perovskite solar cells, Energy Environ. Sci., 2017, 10, 1234–1242 RSC.
- H. Yu, Y. Xie, J. Zhang, J. Duan, X. Chen, Y. Liang, K. Wang and L. Xu, Thermal and Humidity Stability of Mixed Spacer Cations 2D Perovskite Solar Cells, Adv. Sci., 2021, 8, 2004510 CrossRef CAS PubMed.
- Z. Y. Cheng and J. Lin, Layered organic-inorganic hybrid perovskites: structure, optical properties, film preparation, patterning and templating engineering, CrystEngComm, 2010, 12, 2646–2662 RSC.
- G. Farley, Energy absorption of composite materials, J. Compos. Mater., 1983, 17, 267–279 CrossRef CAS.
- M.-S. Kim, B.-G. Kim and J. Kim, Effective variables to control the fill factor of organic photovoltaic cells, ACS Appl. Mater. Interfaces, 2009, 1(6), 1264–1269 CrossRef CAS PubMed.
- J. L. Miao and F. J. Zhang, Recent progress on highly sensitive perovskite photodetectors, J. Mater. Chem. C, 2019, 7, 1741 RSC.
- K. Hong, Q. V. Le, S. Y. Kim and H. W. Jang, Low-dimensional halide perovskites: review and issues, J. Mater. Chem. C, 2018, 6, 2189–2209 RSC.
- Y. Takahashi, H. Hasegawa, Y. Takahashi and T. Inabe, Hall mobility in tin iodide perovskite CH3NH3SnI3: Evidence for a doped semiconductor, J. Solid State Chem., 2013, 205, 39 CrossRef CAS.
- A. Pivrikas, N. S. Sariciftci, G. Juška and R. Österbacka, A review of charge transport and recombination in polymer/fullerene organic solar cells, Prog. Photovoltaics, 2007, 15, 677 CAS.
- C. G. Shuttle, B. O'Regan, A. M. Ballantyne, J. Nelson, D. D. C. Bradley and J. R. Durrant, Bimolecular recombination losses in polythiophene: Fullerene solar cells, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 113201 CrossRef.
- F. Gao, Y. Liu, Y. Xiong, P. Wu, B. Hu and L. Xu, Fabricate organic thermoelectric modules use modified PCBM and PEDOT:PSS materials, Front. Optoelectron., 2017, 10, 117 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04557a |
| This journal is © The Royal Society of Chemistry 2022 |