
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sharing the salt bowl: counterion identity drives N-alkyl resorcinarene affinity for pyrophosphate in water†
Kwaku
Twum
 a,
Seyed
Iraj Sadraei
b,
Jordan
Feder
a,
S. Maryamdokht
Taimoory
*bc,
Kari
Rissanen
*d,
John F.
Trant
*b and
Ngong Kodiah
Beyeh
*a
a,
Seyed
Iraj Sadraei
b,
Jordan
Feder
a,
S. Maryamdokht
Taimoory
*bc,
Kari
Rissanen
*d,
John F.
Trant
*b and
Ngong Kodiah
Beyeh
*a
aDepartment of Chemistry, Oakland University, 146 Library Drive, Rochester, MI 48309-4479, USA. E-mail: beyeh@oakland.edu
bDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Avenue, Windsor, ON N9B 3P4, Canada. E-mail: Nazanin.Taimoory@uwindsor.ca; j.trant@uwindsor.ca
cUniversity of Michigan, Department of Chemistry, Ann Arbor, MI, USA
dUniversity of Jyvaskyla, Department of Chemistry, P. O. Box 35, 40014 Jyvaskyla, Finland. E-mail: kari.t.rissanen@jyu.fi
First published on 14th January 2022
Abstract
N-Alkyl ammonium resorcinarene chloride receptors, NARX4, have been shown to act as high-sensitivity detectors of pyrophosphate (PPi), a biomarker of disease, in aqueous media through the chloride-to-PPi exchange [NAR(Cl)4 to NARPPi]. The nature of the anion of the macrocyclic NARX4 (X = Cl−, Br−, triflate OTf−) receptor greatly influences the PPi-affinity in aqueous media. The binding affinity for [NAR (Cl)4] is 3.61 × 105 M−1, while the NAR (Br)4 and NAR (OTf)4 show stronger binding of 5.30 × 105 M−1, and 6.10 × 105 M−1, respectively. The effects of upper rim ammonium cation, –N+H2R substituents (R = 3-hydroxypropyl, cyclohexyl, benzyl, or napththalen-1-ylmethyl), of the macrocyclic resorcinarene hosts have also been evaluated. The highest affinity was obtained using 3-hydroxypropyl groups due to the additional hydrogen bonds and the naphthyl upper-rim group that provides a larger hydrophobic surface area and favorable stacking interaction (i.e., π–π and CH–π). We note that two PPi molecules can bind to the more selective receptors through an additional interaction with the lower rim hydroxyls, making the resorcinarene a divalent binder. Comparing PPi with other phosphate anions (PO43−, AMP, ADP, and ATP) shows that the receptors are more selective for PPi due to the size and charge complementarity. Experimental (1H, 31P NMR, and isothermal titration calorimetry), and computational analyses support the reported trends for PPi selectivity even in highly competing aqueous media.
Introduction
A significant challenge in supramolecular chemistry is developing high-affinity receptors for anions in biologically relevant solvents,1–5 and the design of such receptors has elicited considerable effort from the research community.6–15 Primarily, this challenge has been addressed by leveraging the cooperative effect of non-covalent attractions.16–18 One such anion is pyrophosphate (PPi), produced as a side product during ATP metabolism.14 Physiological levels of PPi are used as a clinical indicator for disease diagnosis and prognosis.19–22 For example, low PPi levels are common in hemodialysis patients23 as it correlates inversely with the levels of vascular calcification in patients with chronic kidney diseases.24 As a result, considerable effort has gone into developing chemosensors for analytical detection of anions. Most of these sensors are based on metal complexes of europium,25 palladium,26 iron,27 copper,28 cadmium,29 and iron.30 Other classical receptors based on polyammoniums are also used for the anion recognition in water. A naphthalimide-based receptor has been reported for pyrophosphate anion sensing in buffered solutions with around 103–105 M−1 affinities.31Our preliminary contributions reported on N-alkyl ammonium resorcinarene salts (NARX4), consisting of a macrocyclic resorcinarene tetra-ammonium cation and four anions. Chloride, viz. NAR (Cl)4, is preferred for resorcinarene conformational stability in organic media over the bromide, nitrate, triflate, or picrate.32–34 This binding preference is due to the chloride's suitable size, the snug fit between two adjacent ammonium moieties, and the strong hydrogen bond circular seam [NH2+⋯–Cl−⋯NH2+⋯–Cl−⋯]2 along the upper rim of the macrocycle that holds it in place. The tetra-cationic NARX4s are, on even a cursory inspection, excellent potential hosts for tetra-anionic pyrophosphate due to size-charge complementarity and chelate cooperativity. In 2018, we reported that an N-alkyl ammonium resorcinarene chloride receptor is capable of selective high-affinity (107 M−1) binding of PPi in pure water.35 Binding is driven by the entropically favorable displacement of the four chloride counter ions upon complexation of PPi. Based on the mechanism, we recognized the potential to improve the receptor's affinity by employing more weakly coordinating counter ions. The upper rim of the ammonium salt receptor is available for structural modification. In our previous study, a seemingly minor change, incorporating an additional methylene group converting the chain from ethanol to propanol, greatly improved PPi affinity.35 In these cooperative systems, even minor differences can drastically affect the thermodynamics of complexation.
We now report the application of less strongly coordinating counter anions, intending to significantly increase NARX4's affinity towards PPi. We synthesized ten new R-NARX4's (R = upper rim substituent, X = counter anion) receptors with three different counter anions: chloride, bromide, and triflate (OTf); and four different upper rim substituents: one with a flexible terminal hydroxyl propyl group (C3OHNARX4, X = Cl, Br, and OTf), the second with a rigid cyclohexyl group at the upper rim (CyNARX4, X = Cl and Br), the third with a flexible benzyl group (BnNARX, X = Cl and Br), and the fourth with a flexible and fluorescent napththalen-1-ylmethyl group (NpNARX4, X = Cl, Br and OTf, Fig. 1). In addition to PPi, the binding properties of the receptors towards a tribasic monophosphate (K3PO4), and a dibasic monophosphate (AMP), diphosphate (ADP), and triphosphate (ATP) are also investigated (Fig. 1). Binding was confirmed through 1H and 31P NMR experiments. Quantifying the thermodynamics of binding was accomplished using a series of isothermal titration calorimetry (ITC) and computational studies. The binding modes were justified using density functional theory (DFT) at the (wB96XD/6-311-G(d,p)) level of theory which was supported by molecular dynamic (MD) simulations.
 | ||
| Fig. 1 The resorcinarene salt receptors C3OHNARX4, CyNARX4, BnNARX4, NpNARX4 (X = Cl−, Br− and CF3SO3−), and the phosphate guests K3PO4, Na4PPi, Na2AMP, Na2ADP, and Na2ATP. | ||
Results and discussion
The Mannich condensation between aliphatic amines and C-hydroxyl-resorcinarene in the presence of excess formaldehyde generates tetrabenzoxazines.36 Ring-opening of the six-membered tetrabenzoxazine ring is effected by refluxing in the presence of an acid (HCl, HBr, and triflic acid) to provide crude R-NARX4s (Fig. 1); these are purified through recrystallization to give the final products in 50–85% yields (see ESI† for details).Isothermal titration calorimetry (ITC)
Thermodynamic parameters (K, ΔH, ΔS, and ΔG) between the R-NARX4 receptors and the different phosphate anions were measured by a series of ITC experiments in 90% H2O/10% DMSO. A mixed solvent system was necessary to ensure the complete dissolution of the variable hydrophobic groups on the different R-NARX4 receptors at the concentrations used for the study (0.25 mM).Probing different functionalization of the upper rim alkyl (R) substituent was necessary to underscore its importance in defining phosphate anion affinity (Table 1). The switch to a combination of solvent systems is a likely explanation for lower Ka values than we earlier reported.35 The NARX4 with naphthalene groups show higher binding when compared to benzyl or cyclohexyl analogs, potentially due to the presence of a series of stacking interactions (Table 1). However, our previous C3OH chains provided the best affinity constant due to the extra hydrogen bond potential of the hydroxyl groups. On all scaffolds, counter anion identity showed the same trend; the K1 value for the complexation of PPi to Np-NARX4 increased by 52.5% and 68.2% (Table 1) upon switching the chloride counterion to bromide and triflate, respectively. The same trend remained when the water was replaced with pH 7.4 Tris buffer, confirming that the observed isotherms do not arise from (de)protonation but host–guest binding processes. Negative ΔG values in all cases confirm that association is spontaneous at 298 K. The ΔH and TΔS results also indicate the first binding event is both enthalpically and entropically favorable. This is slightly unexpected; entropically-driven complexation is the norm as solvent and counterions are liberated upon binding, but the energy of dehydrating pyrophosphate was expected to be substantial. The favorable ΔH term hints at the significant strength of the salt bridges formed in the binding pocket.37,38 Affinity is maintained in these new receptors: the K1 values for PPi are generally more than for AMP, ADP, and ATP (Fig. S23–S33 and Tables S1–S11†).
| Complex | K 1 (×105) M−1 | ΔH1 kcal mol−1 | TΔS1 kcal mol−1 | ΔG1 kcal mol−1 | Complex | K 1 (×105) M−1 | ΔH1 kcal mol−1 | TΔS1 kcal mol−1 | ΔG1 kcal mol−1 |
|---|---|---|---|---|---|---|---|---|---|
| PPi@C3OHNAR(Br)4 | 5.3 ± 1.1 | −6.7 ± 2.2 | 1.09 | −7.81 | PPi@NpNAR(Cl)4 | 2.0 ± 0.5 | −3.3 ± 2.0 | 3.97 | −7.23 |
| PPi@CyNAR(Br)4 | 2.6 ± 0.5 | −1.1 ± 0.5 | 6.29 | −7.38 | PPi@NpNAR(Br)4 | 3.0 ± 0.2 | −4.9 ± 0.6 | 2.55 | −7.47 |
| PPi@BnNAR(Br)4 | 3.1 ± 0.2 | −0.4 ± 0.1 | 7.06 | −7.48 | PPi@NpNAR(OTf)4 | 3.3 ± 0.9 | 7.0 ± 0.7 | 14.5 | −7.51 |
| PPi@NpNAR(Br)4 | 3.0 ± 0.2 | −4.9 ± 0.6 | 2.55 | −7.47 |
| Complex | K 2 (×105) M−1 | ΔH2 kcal mol−1 | TΔS2 kcal mol−1 | ΔG2 kcal mol−1 | Complex | K 2 (×105) M−1 | ΔH2 kcal mol−1 | TΔS2 kcal mol−1 | ΔG2 kcal mol−1 |
|---|---|---|---|---|---|---|---|---|---|
| a ITC was done in H2O (90%) /DMSO (10%) at 298 K. K1 and K2 represent the first and second binding constants. | |||||||||
| PPi@C3OHNAR(Br)4 | 1.40 ± 0.03 | 8.4 ± 1.7 | 15.3 | −6.94 | PPi@NpNAR(Cl)4 | 0.59 ± 0.08 | 11.1 ± 1.8 | 17.6 | −6.50 |
| PPi@CyNAR(Br)4 | 0.18 ± 0.01 | 26.7 ± 0.8 | 32.2 | −5.60 | PPi@NpNAR(Br)4 | 1.13 ± 0.01 | 32.6 ± 2.1 | 39.6 | −7.00 |
| PPi@BnNAR(Br)4 | 0.29 ± 0.03 | 16.0 ± 1.5 | 22.1 | −6.10 | PPi@NpNAR(OTf)4 | 4.6 ± 2.1 | −1.4 ± 0.2 | 7.72 | −9.09 |
| PPi@NpNAR(Br)4 | 1.13 ± 0.01 | 32.6 ± 2.1 | 39.6 | −7.00 | |||||
All the data, with few exceptions, were fitted to a two-set-of-sites binding model. As the cavity is committed to binding the first PPi molecule, the second interaction in C3OH-NARX may be allosteric exo binding with the hydroxyls of the top rim, as we previously speculated.35 However, as the other hydrophobic R-NARXs, lacking these hydroxyl hydrogen bond participants, present a similar two binding site event in this solvent mixture, this seems unlikely. There is, however, another allosteric binding site formed from the four dangling hydroxyl chains of the lower rim. To investigate this possibility, we turned to NMR spectroscopy.
NMR spectroscopy
To probe these receptors’ binding capability, we performed a series of 1H NMR measurements on the phosphate guests alone. The methylene (Ar–CH2N) signals of the host alone confirm that a host–guest interaction between the NARX4s and the PPi occurs when the two compounds are mixed (Fig. 2 and Fig. S35–43†). We used the same solvent mixture of D2O/[D6]DMSO (90%/10%) at 298 K. The binding processes are fast on the NMR timescale; however, clear changes in the hosts’ signals demonstrate that binding occurs. Hydrogen/Deuterium exchange in this solvent mixture prevents the –OH and –NH2 signals from being monitored. However, we observed changes in the chemical environment of the non-exchangeable methylene protons (Ar–CH2N) closest to the –OH and –NH2 groups. We also observed, by NMR, that the resorcinarene core distorts its bowl conformation as observed from the Ar–H signals to accommodate PPi better. We first performed a representative NMR titration with C3OH-NAR(OTf)4 and C3OH-NAR(Br)4 in D2O to confirm the binding event. A Job's plot of the Aryl-H and R-CH2-NH2 proton signals that participate in the cavitand binding pocket reveals a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry (Fig. S34†).39 Moreover, a careful examination of the proton R-C(H2)OH signals of the lower rim reveal a gradual shielding due to this secondary binding event (Fig. S35†). From the NMRs, the –OH and –NH2 protons of the cationic core of the receptors form hydrogen bonds with the counter anions. When the R-NARX4 receptors bind to PPi, the counter anions are displaced to accommodate PPi. This reorganization produces observable differences in the 1H resonances of the –OH, –NH2, and neighboring –CH2– protons.
:2 stoichiometry (Fig. S34†).39 Moreover, a careful examination of the proton R-C(H2)OH signals of the lower rim reveal a gradual shielding due to this secondary binding event (Fig. S35†). From the NMRs, the –OH and –NH2 protons of the cationic core of the receptors form hydrogen bonds with the counter anions. When the R-NARX4 receptors bind to PPi, the counter anions are displaced to accommodate PPi. This reorganization produces observable differences in the 1H resonances of the –OH, –NH2, and neighboring –CH2– protons.
 | ||
| Fig. 2 Sections of the 1H NMR spectra in D2O/[D6]DMSO (90/10 v/v) at 298 K of (a) C3OHNARCl4 and (b) the equimolar mixture C3OHNARCl4 and PPi, (c) C3OHNARBr4, and (d) the equimolar mixture C3OHNARBr4 and PPi, (e) C3OHNAROTf4 and (f) the equimolar mixture C3OHNAROTf and PPi. The dashed lines indicate the signal changes in ppm. Star represents the residual D2O solvent. | ||
Taking the binding of PPi by C3OHNARX4s as an example, up to 0.09 ppm upfield shifts are realized by the methylene protons, and up to 0.22 ppm upfield shifts are observed for the aromatic protons of the resorcinarene core. The changes in the host's signals support the host re-organizing the cavity during the binding process. No significant differences in the 1H NMR of the receptor in PPi@R-NAR(X)4 complex was observed between the chloride, bromide, or triflate counter ions once the counter anions are displaced to accommodate pyrophosphate. The final assembly is expected to be the same PPi@C3OHNAR(X)4, where X represents the initial counterion that has now been fully displaced (Fig. 2 and Fig. S36–S43†). An even better way to access the binding process is to monitor the 31P NMR signals of the phosphate guests upon complexation with the NARX4 receptors. A series of 31P NMR experiments of the phosphate anions and equimolar concentrations of pure phosphates and receptors were used to qualitatively probe the binding processes (Fig. 3 and Fig. S44–S56†). The magnitude of the upfield shift of the phosphorus isotopic signals upon binding the R-NARX4 receptors qualitatively infers how deep the guests’ sit in the host's binding pocket. Of the different R-NARBr4 receptors, PPi experienced the most significant upfield movement with R = naphthalene (Fig. 3). This was followed closely by the benzyl (Bn)–NARBr4 receptor and 1-propanol (C3OH)–NARBr4. Qualitatively, the absence of an aromatic environment or a hydrogen bond potential of hydroxyl groups in the cyclohexyl upper rim modification may explain why it has the smallest effect on the PPi resonance (Fig. 3, Table 1 and Fig. S44–S56†).
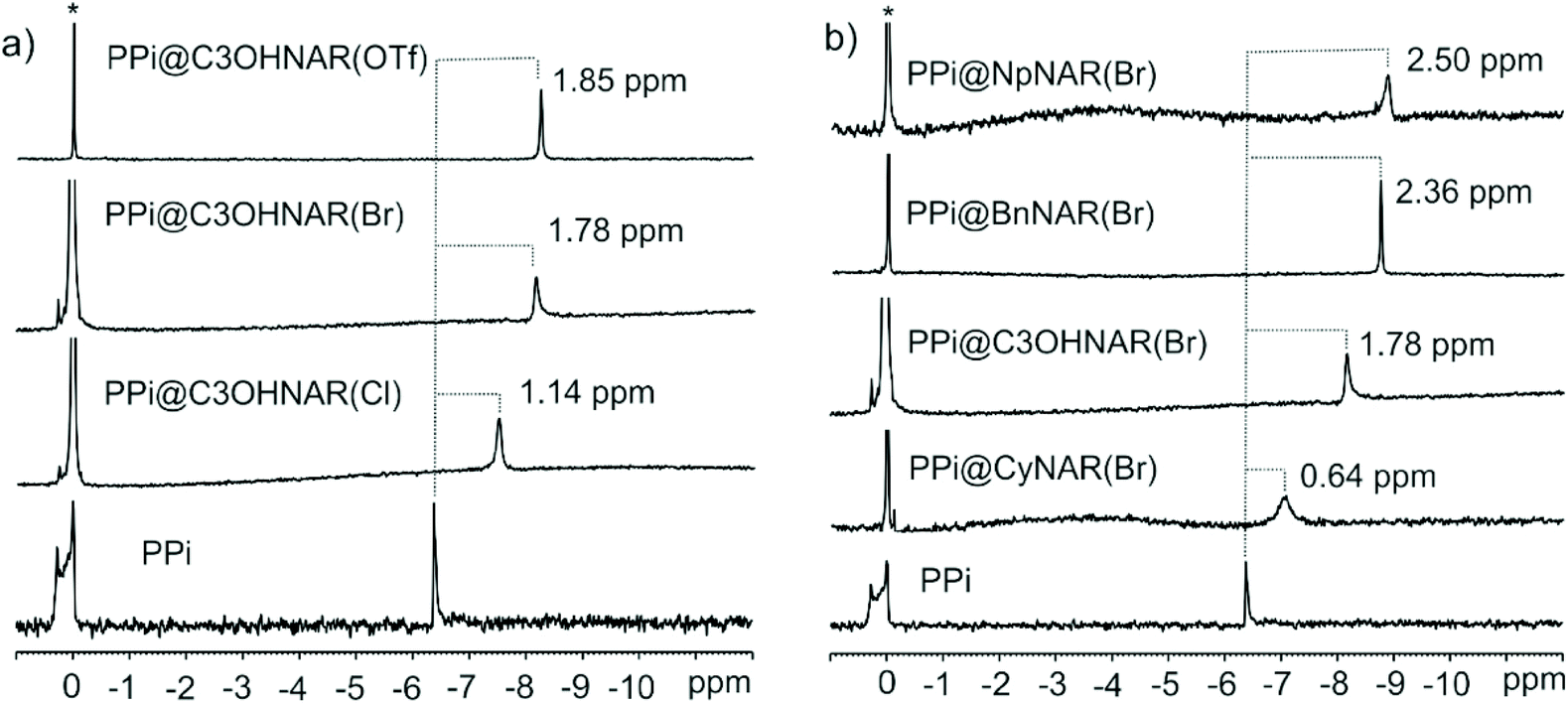 | ||
| Fig. 3 Sections of the 31P NMR spectra in D2O/[D6]DMSO (90/10 v/v) at 298 K. (a) Equimolar mixtures of PPi and NpNARX4s (X = Cl, Br, and OTf) as compared to pure PPi. (b) Equimolar mixtures of PPi and CyNARBr4, PPi and C3OHNARBr4, PPi and BnNARBr4, PPi and NpNARBr4 compared to pure PPi. The dashed lines indicate the signal changes in ppm. Star represents the residual H3PO4 as an external standard. Note on nomenclature: for clarity, the original counter anion is indicated in parenthesis. | ||
Additionally, it is evident how the different counter ions contribute to the shielding of the pyrophosphate anions. The PPi signal was shielded by 1.14 ppm with C3OHNAR(Cl)4, 1.76 ppm with C3OHNAR(Br)4, and 1.85 ppm with C3OHNAR(OTf)4. Larger upfield shifts of 2.42, 2.50, and 2.72 ppm were observed upon switching from chloride to bromide or triflate with the NpNARX4 receptors. The magnitude of these shifts is a qualitative representation of the different affinities. As the product is identical in each case, the changes are better interpreted as a measurement of the population ratios between bound (upfield shifted) and free (unchanged) PPi. This is further complicated by the second binding interaction of the guest (NaPPi) with the lower rim. Computational results suggest that PPi phosphorous atoms in the cavity have a higher point-charge electron density (Natural charge = 2.53e) than a PPi bound to the lower rim (Natural charge = 2.48e, Fig. S57†). The weaker the coordinating anion, the greater the binding affinity for the upper rim. Thus, one would expect the instantaneous relative population of PPi in the upper vs. lower binding sites to grow as the anion coordinating ability drops. The increased shielding observed for phosphorus in the triflate pro-receptor compared to the chloride or bromide strongly supports this inference. The most significant shifts are observed for largely dissociative triflate as this counterion is easiest to displace. This same differential effect is not immediately apparent in the 1H NMR spectra, where we note that the absolute shifts are similar. But they start from different starting points: the chemical shift of the indicated resonances in the parent resorcinarenes are not identical (although the peak shapes are), but upon binding, we do see differences in peak broadness, suggesting that although the PPi-bound complexes are expected to be all identical, we might be observing indicators of the dynamism of the binding process.
Similar 31P experiments were used to probe the binding of the receptors towards other phosphates: PO43−, AMP, ADP, and ATP. PO43− showed similar but much weaker upfield movement than PPi, while AMP showed greater field changes than PPi. Moderate but measurable deshielding was observed for ADP and ATP. The interaction of ADP and ATP with the host might be through an exo-binding mode, explaining the weaker effects; their larger size could be the main factor in this behavior. Table 1 summarizes the 31P signal changes upon complexation with the different R–NARX4s receptors.
Computational studies
The structural features of selected host–guest complexes, NpNARX4, C3OHNARX4, PPi@NpNAR, PPi@C3OHNAR, were then computationally evaluated in a 90% H2O/10% DMSO (SCRF = SMD solvation model) using ωB97X-D/6-311-G(d,p) method.40 For details of the methods, please see the ESI.† When X = Cl, the four chlorides position between the four-upper rim NH2+ groups, forming a plane parallel to the resorcinarene base (Fig. 4) as expected. Both C3OHNARCl4 and NpNARCl4 adopt superficially similar conformations but with different energetic behavior. The calculated binding energy of the host-counterion complex in this solvent system shows that the Cl in NpNARCl4 (ΔEb (1DMSO:9H2O) = −62.4 kcal mol−1) is far less tightly bound than those in C3OHNARCl4 ((ΔEb (1DMSO:9H2O) = −88.1 kcal mol−1). This would imply that NpNARX should more readily lose its counterions, allowing for easier binding with the incoming phosphate, all else being equal. In the OTf-coordinated complexes, the bulky counterions distort the host geometry, forcing it further open, a decidedly unfavorable conformation. The C3OHNAROTf4 complex has a positive host-anion binding energy (ΔEb (1DMSO:9H2O) = 7.6 kcal mol−1), suggesting that it should spontaneously dissociate. This unfavorable form was highlighted for the NpNAROTf4 complex, where all attempts to optimize the structure failed: the ions simply do not form a defined, stable salt complex. This can be due to the high hydration energy in the 1DMSO/9H2O solvent system and strong competition between the host and the solvent for the counterions’ attention. To better understand this relationship, a molecular dynamics (MD) simulation was performed on NpNAROTf4 using OPLS-200528,29 for the counterions and the receptor to reflect the experimental conditions.41 The optimized geometry of the counterion-coordinated host system was initially relaxed, then the relaxed structure was immersed within the solvent box using the disordered system builder implemented in Schrödinger's Materials module.30 This solvent coordinated system was then subjected to MD simulations, with an initial production run of 5 ns (see ESI† for more details). The dynamic behavior of the counterion was then traced through four replicate MD simulations of 5 to 10 ns, each with different starting vectors. Due to this observed dynamic behavior of the counterion in the solvent system, the MD sampling was further extended with four more replicates of 20 ns each. The starting point and end result of representative MD trajectories are shown in Fig. 5. In all the simulated trajectories, two of the four OTf anions displace from their original positions between the pendant upper arms of host, leaving all the ions to be stabilized by solvent instead of each other. This explains why we were unable to optimize the structure using DFT method. This also prophesizes an incredibly favorable dissociation of triflate and helps explain the high affinity for PPi.
 | ||
| Fig. 4 The optimized geometries and the calculated binding energies of three representative complexes of the chloride and triflate salts. NpNARTf4 could not be converged. | ||
 | ||
| Fig. 5 MD simulation cell of the system containing disordered DMSO, H2O, and two Np-8OTf systems, showing the position and displacement of the OTf counteranions in the beginning (left) and at the end of the equilibration state (right). The 2Np-8OTf complexes are shown in space-filling mode, and the solvent systems are shown in lines. | ||
This arrangement is maintained throughout the full 20 ns of our MD sampling. This bulky and more weakly coordinating counterion, by readily dissociating, should greatly increase the binding affinity of incoming PPi for the host. With this initial view of host-counteranions, we next investigated the complex formation with PPi.
The relative value of the predicted solvated binding energy of complex formation for PPi@C3OHNAR was found to be 1.8 kcal mol−1 (ΔΔG) more favorable than for the PPi@NpNAR complex. The very strong hydrogen bonds in the former are met by the formation of new intra-host upper rim π–π interactions between the naphthyl groups. It can be seen from the calculated host–guest and intra-host bond lengths (Fig. 7) that in PPi@NpNAR, four of PPi's oxygen atoms sit deep in the NpNAR cavity, forming strong interactions with the trimethyl ammonium and phenols, leaving only two PPi oxygen atoms to face the solvent. In PPi@C3OHNAR, the oxygen atoms of PPi are also immersed in the C3OHNAR host cavity, but less deeply and potentially more easily interrupted by the solvent. The deeper positioning of the guest in the PPi@NpNAR complex facilitates the intra-host interactions between upper rim OH⋯OH and the π–π interactions between the Np groups, which are obviously not available to the PPi@C3OHNAR complex. The interactions block solvent from almost half of the circumference of the complex, protecting the host–guest interactions from interference.
However, all these calculations simply look at the 1:1 system. With it becoming increasingly clear that a 2:1 PPi:host stoichiometry exists, we reoptimized the trimeric complexes (Fig. 8). As we had considered, the optimal structure does have the second PPi unit, in the form of Na4PPi, localized to the bottom rim of the cavitand. It does not adopt the same conformation for both systems; with C3OHNAR, it sits perpendicular to the axis of the cavitand, roughly parallel with the PPi in the upper cavity, while for NpNAR it sits aligned with the cavitand's axis. This difference is likely because these are both low-lying interactions, and in neither case can all four rim hydroxyls engage with the PPi. However, it would be sensible that the degree of entropic contribution (provided by PPi and cavitand desolvation) would drive this forward. The overall ΔG of binding to form this ternary complex, −60.6 kcal mol−1 for C3OHNAR and −61.6 kcal mol−1 for NpNAR, is roughly double that of the dimer above; binding to the lower rim is less favorable than the initial interaction with the cavity but is still highly exogenic. The reaction actually becomes endothermic on the ΔH term (sum of the binding energy and the zero-point correction energy), with the dimer (Fig. 6) being more favorable than the trimer; trimer formation is driven entirely by entropy. The entropy terms are overestimated in these calculations. This binding could likely be further improved through cooperative anion–cation interactions.
 | ||
| Fig. 6 The optimized geometry of PPi@NpNAR, and PPi@C3OHNAR complexes. BE is binding energy, Ezpe is the zero-point energy; these terms together equate to ΔH. | ||
 | ||
| Fig. 7 The predicted structural parameters of the optimized PPi@NpNAR, and PPi@C3OHNAR complexes. | ||
 | ||
| Fig. 8 The optimized geometry of 2PPi@NpNAR and 2PPi@C3OHNAR complexes. Side view, top-down view, bottom-up view. BE is binding energy, Ezpe is the zero-point energy; these terms together equate to ΔH. | ||
In all, the computational data parallels the relative results of the experimental ITC data: binding is favorable for a two-site model, and the NpNAR forms the stronger interactions with PPi.
Conclusions
In conclusion, an extensive study in solution of ten N-alkyl ammonium resorcinarene salts with varying counter anions using 1H and 31P NMR, and ITC show the resorcinarene salts to be high-affinity receptors for PPi in aqueous media. The receptor with the terminal propyl hydroxyl groups C3OHNAR(X)4 and the napththalen-1-ylmethyl group at the upper rim, both with weakly coordinating triflate counter anions, gave the best results with PPi. The results show that both the nature of the upper rim substituent and the coordinating strength of the counter anions play crucial roles in determining binding affinity. The C3OHNAR(X)4 receptors provide an extra hydrogen bond for enhanced binding, while the NpNAR(X)4 provides a larger surface area for binding. Weakly coordinating counter anions such as triflate enhanced the binding affinity over more coordinating anions such as chloride. Our results also show good binding for AMP while PO43− due to its smaller size was only weakly bound. The larger sizes of ADP and ATP suggest the binding to the receptor to be exo-cavity.A detailed series of molecular dynamic simulations (MD) and the density functional theory (DFT) study highlighted the significance of the upper rim substituents and the counter anion as crucial factors for establishing the high binding affinity. These results show that modifying the upper rim and counter anions of the R-NAR(X)4 enhances their affinity and sensor ability towards PPi. Similarly, they support the contention that the second binding interaction with PPi is competitive and occurs with a specific conformation with the lower rim hydroxyls rather than as an exo-interaction with the cavity, as we had previously speculated. These results pave the way to using a supramolecular approach using cavity containing resorcinarene salts receptors as qualitative sensors for PPi in biological media.
Author contributions
Conceptualization, NKB, JFT, SMT, KR; methodology, KT, SIS, JF, SMT; software, SMT, validation, KT, SIS, JF, SMT; writing—original draft preparation, KT, NKB, JFT, SIS, JF, SMT; writing—review and editing, KT, SMT, JFT, NKB, supervision, NKB, JFT, KR, SMT, project administration, NKB, JFT, KR; funding acquisition, NKB, KT, JFT, KR All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge financial support from the Provost Graduate Research Scholarship (KT, JF), Oakland University, MI, USA, and the University of Windsor, ON, Canada. This work was made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: http://www.sharcnet.ca) and Compute/Calcul Canada. This work was funded in part by the Natural Sciences and Engineering Research Council of Canada (Grant # 2018-06338 to JFT), the Windsor Cancer Centre Foundation Seeds4Hope Seed Grant Funding Program (Grant # 2017-03 to JFT), and an Ontario Early Researcher Award (Grant # ER18-14-114 to JFT).Notes and references
- M. A. Palacios, R. Nishiyabu, M. Marquez and P. Anzenbacher, Supramolecular chemistry approach to the design of a high-resolution sensor array for multianion detection in water, J. Am. Chem. Soc., 2007, 129(24), 7538–7544 CrossRef CAS PubMed.
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Artificial Receptors for the Recognition of Phosphorylated Molecules, Chem. Rev., 2011, 111(11), 6603–6782 CrossRef CAS PubMed.
- A. Rostami, C. J. Wei, G. Guérin and M. S. Taylor, Anion Detection by a Fluorescent Poly(squaramide): Self-Assembly of Anion-Binding Sites by Polymer Aggregation, Angew. Chem., Int. Ed., 2011, 50, 2059–2062 CrossRef CAS PubMed.
- T. Egawa, K. Hirabayashi, Y. Koide, C. Kobayashi, N. Takahashi, T. Mineno, T. Terai, T. Ueno, T. Komatsu, Y. Ikegaya, N. Matsuki, T. Nagano and K. Hanaoka, Red fluorescent probe for monitoring the dynamics of cytoplasmic calcium ions, Angew. Chem., Int. Ed., 2013, 52, 3874–3877 CrossRef CAS PubMed.
- F. Zapata, A. Caballero, N. G. White, T. D. W. Claridge, P. J. Costa, V. Félix and P. D. Beer, Fluorescent charge-assisted halogen-bonding macrocyclic halo-imidazolium receptors for anion recognition and sensing in aqueous media, J. Am. Chem. Soc., 2012, 134(28), 11533–11541 CrossRef CAS PubMed.
- X. Ji, R. T. Wu, L. Long, C. Guo, N. M. Khashab, F. Huang and J. L. Sessler, Physical Removal of Anions from Aqueous Media by Means of a Macrocycle-Containing Polymeric Network, J. Am. Chem. Soc., 2018, 140(8), 2777–2780 CrossRef CAS PubMed.
- F. Pan, N. K. Beyeh and K. Rissanen, Dimeric resorcinarene salt capsules with very tight encapsulation of anions and guest molecules, RSC Adv., 2015, 5, 57912–57916 RSC.
- A. Borissov, I. Marques, J. Y. C. Lim, V. Félix, M. D. Smith and P. D. Beer, Anion Recognition in Water by Charge-Neutral Halogen and Chalcogen Bonding Foldamer Receptors, J. Am. Chem. Soc., 2019, 141(9), 4119–4129 CrossRef CAS PubMed.
- D. H. Lee, S. Y. Kim and J. I. Hong, A fluorescent pyrophosphate sensor with high selectivity over ATP in water, Angew. Chem., Int. Ed., 2004, 43(36), 4777–4780 CrossRef CAS PubMed.
- N. G. White, S. Carvalho, V. Félix and P. D. Beer, Anion binding in aqueous media by a tetra-triazolium macrocycle, Org. Biomol. Chem., 2012, 10, 6951–6959 RSC.
- P. A. Gale, J. T. Davis and R. Quesada, Anion transport and supramolecular medicinal chemistry, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Applications of Supramolecular Anion Recognition, Chem. Rev., 2015, 115(15), 8038–8155 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Halogen bonding in water results in enhanced anion recognition in acyclic and rotaxane hosts, Nat. Chem., 2014, 6, 1039–1043 CrossRef CAS PubMed.
- S. K. Kim, D. H. Lee, J.-I. Hong and J. Yoon, Chemosensors for pyrophosphate, Acc. Chem. Res., 2009, 42(1), 23–31 CrossRef CAS PubMed.
- V. Amendola, L. Fabbrizzi and L. Mosca, Anion recognition by hydrogen bonding: urea-based receptors, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, A biomimetic receptor for glucose, Nat. Chem., 2019, 11, 52–56 CrossRef CAS PubMed.
- Z. Zhang and P. R. Schreiner, (Thio)urea organocatalysis—what can be learnt from anion recognition?, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC.
- S. Kubik, Anion recognition in water, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC.
- L. Li Tong, Z. Z. Chen, Z. Y. Jiang, M. M. Sun, L. Li, J. Liu and B. Tang, Fluorescent sensing of pyrophosphate anion in synovial fluid based on DNA-attached magnetic nanoparticles, Biosens. Bioelectron., 2015, 72, 51–55 CrossRef CAS PubMed.
- A. Micheli, J. Po and G. H. Fallet, Measurement of soluble pyrophosphate in plasma and synovial fluid of patients with various rheumatic diseases, Scand. J. Rheumatol., 1981, 10, 237–240 CrossRef CAS PubMed.
- R. G. Russell, S. Bisaz, A. Donath, D. B. Morgan and H. Fleisch, Inorganic pyrophosphate in plasma in normal persons and in patients with hypophosphatasia, osteogenesis imperfecta, and other disorders of bone, J. Clin. Invest., 1971, 50, 961–969 CrossRef CAS PubMed.
- M. Doherty, C. Belcher, M. Regan, A. Jones and J. Ledingham, Association between synovial fluid levels of inorganic pyrophosphate and short term radiographic outcome of knee osteoarthritis, Ann. Rheum. Dis., 1996, 55, 432–436 CrossRef CAS PubMed.
- K. A. Lomashvili, W. Khawandi and W. C. O'Neill, Reduced Plasma Pyrophosphate Levels in Hemodialysis Patients, J. Am. Soc. Nephrol., 2005, 16(8), 2495–2500 CrossRef CAS PubMed.
- W. C. O'Neill, M. K. Sigrist and C. W. McIntyre, Plasma pyrophosphate and vascular calcification in chronic kidney disease, Nephrol., Dial., Transplant., 2010, 25(1), 187–191 CrossRef PubMed.
- S.-H. Li, C.-W. Yu, W.-T. Yuan and J.-G. Xu, A Lanthanide Hybrid Cluster as a Selective Optical Chemosensor for Phosphate-containing Anions in Aqueous Solution, Anal. Sci., 2004, 20(10), 1375–1377 CrossRef CAS PubMed.
- J. Gao, T. Riis-Johannessen, R. Scopelliti, X. Qian and K. Severin, A fluorescent sensor for pyrophosphate based on a Pd(ii) complex, Dalton Trans., 2010, 39, 7114–7118 RSC.
- W. Wang, H. Zhao, B. Zhao, H. Liu, Q. Liu and Y. Gao, Highly Selective Recognition of Pyrophosphate by a Novel Coumarin-Iron(III) Complex and the Application in Living Cells, Chemosensors, 2021, 9(3), 48 CrossRef CAS.
- J. Qiang, C. Chang, Z. Zhu, T. Wei, W. Yu, F. Wang, J. Yin, Y. Wang, W. Zhang, J. Xie and X. Chen, A dinuclear-copper(II) complex-based sensor for pyrophosphate and its applications to detecting pyrophosphatase activity and monitoring polymerase chain reaction, Sens. Actuators, B, 2016, 233, 591–598 CrossRef CAS.
- J. X. Liu and S. N. Ding, Monitoring pyrophosphate anions via cobalt(II)-modulated fluorescence of cadmium sulfide quantum dots, Anal. Methods, 2016, 8, 2170–2175 RSC.
- N. Kumari, H. Huang, H. Chao, G. Gasser and F. Zelder, A Disassembly Strategy for Imaging Endogenous Pyrophosphate in Mitochondria by Using an FeIII–salen Complex, ChemBioChem, 2016, 17(13), 1211–1215 CrossRef CAS PubMed.
- A. S. Oshchepkov, R. R. Mittapalli, O. A. Fedorova and E. A. Kataev, Naphthalimide-Based Polyammonium Chemosensors for Anions: Study of Binding Properties and Sensing Mechanisms, Chem. – Eur. J., 2017, 23(40), 9657–9665 CrossRef CAS PubMed.
- N. K. Beyeh, F. Pan, S. Bhowmik, T. Mäkelä, R. H. A. Ras and K. Rissanen, N-Alkyl Ammonium Resorcinarene Salts as High-Affinity Tetravalent Chloride Receptors, Chem. – Eur. J., 2016, 22(4), 1355–1361 CrossRef CAS PubMed.
- N. K. Beyeh, A. Valkonen, S. Bhowmik, F. Pan and K. Rissanen, N-Alkyl ammonium resorcinarene salts: Multivalent halogen-bonded deep-cavity cavitands, Org. Chem. Front., 2015, 2, 340–345 RSC.
- N. K. Beyeh, M. Cetina and K. Rissanen, Halogen bonded analogues of deep cavity cavitands, Chem. Commun., 2014, 50, 1959–1961 RSC.
- N. K. Beyeh, I. Díez, S. M. Taimoory, D. Meister, A. I. Feig, J. F. Trant, R. H. A. Ras and K. Rissanen, High-affinity and selective detection of pyrophosphate in water by a resorcinarene salt receptor, Chem. Sci., 2018, 9, 1358–1367 RSC.
- N. K. Beyeh and K. Rissanen, N-Alkyl Ammonium Resorcinarene Salts: A Versatile Family of Calixarene-Related Host Molecules, in Calixarenes and Beyond, P. Neri, J. Sessler and M. X. Wang, 2016, pp. 255–284 Search PubMed.
- D. H. Williams, E. Stephens, D. P. O'Brien and M. Zhou, Understanding Noncovalent Interactions: Ligand Binding Energy and Catalytic Efficiency from Ligand-Induced Reductions in Motion within Receptors and Enzymes, Angew. Chem., Int. Ed., 2004, 43(48), 6596–6616 CrossRef CAS PubMed.
- H. Saint-Martin, I. Ortega-Blake, A. Leś and L. Adamowicz, The role of hydration in the hydrolysis of pyrophosphate. A Monte Carlo simulation with polarizable-type interaction potentials, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1994, 1207(1), 12–23 CrossRef CAS.
- K. A. Connors, Binding constants: the measurement of molecular complex stability, Wiley, 1987 Search PubMed.
- J. da Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- D. E. Shaw, Desmond Molecular Dynamics System, Schrödinger, New York, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, analytical methods, experimental details, NMR, ITC and UV-Vis/fluorescence data and detailed DFT. See DOI: 10.1039/d1qo01877a |
| This journal is © the Partner Organisations 2022 |