
Molecular rotor based on an oxidized resorcinarene†
Daniel T.
Payne
 *ab,
Jan
Labuta
*a,
Zdeněk
Futera
c,
Václav
Březina
d,
Lenka
Hanyková
d,
Mandeep K.
Chahal
a and
Jonathan P.
Hill
*a
*ab,
Jan
Labuta
*a,
Zdeněk
Futera
c,
Václav
Březina
d,
Lenka
Hanyková
d,
Mandeep K.
Chahal
a and
Jonathan P.
Hill
*a
aInternational Center for Materials Nanoarchitectonics, National Institute for Materials Science, Namiki 1-1, Tsukuba, Ibaraki 305-0044, Japan. E-mail: PAYNE.Daniel@nims.go.jp; LABUTA.Jan@nims.go.jp; Jonathan.HILL@nims.go.jp
bInternational Center for Young Scientists, National Institute for Materials Science, Namiki 1-1, Tsukuba, Ibaraki 305-0044, Japan
cInstitute of Physics, Faculty of Science, University of South Bohemia, Branišovská 1760, České Budějovice 370 05, Czech Republic
dFaculty of Mathematics and Physics, Charles University, V Holešovičkách 2, 180 00 Prague 8, Czech Republic
First published on 2nd November 2021
Abstract
Molecular rotors are an important class of dynamic molecules which have been studied not only for their possible uses as components of molecular machines but also because of potential applications as probes of local viscosity in biological media, especially self-assembled membranes. For the former, factors affecting rotational motility are critical while for the latter the rotor activity must be complexed with an output signal (often fluorescence) for reporting of local conditions. Molecular single stator-double rotor activity of an oxidized resorcinarene (fuchsonarene) macrocycle containing unsaturated hemiquinonoid groups at its meso positions was investigated. Fuchsonarenes contain two hemiquinonoid substituents at diagonally-opposed meso-positions with two electron rich phenol groups at the remaining meso-positions between the hemiquinonoid groups. All meso-substituents are in proximity at one side of the resorcinarene macrocycle (so-called rccc-type isomer) with rotational activity of the phenol meso-substituents. Rotation rates of the phenol moieties can be controlled by varying temperature, solvent polarity and acidity of the medium of study with rotation being thermally activated in neutral and acidic media and tunable in the range from 2 s−1 to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1. Experimental and computational data indicate that rotation of the mobile phenol meso-substituents is remotely affected by interactions with acidic solvents at the carbonyl C
000 s−1. Experimental and computational data indicate that rotation of the mobile phenol meso-substituents is remotely affected by interactions with acidic solvents at the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of macrocyclic acetyloxy groups, which occurs with the emergence of a lower energy electronic absorption band whose intensity is correlated with both the acidity of the medium and the rotation rate of the phenol substituents. Time-dependent DFT calculations suggest that the low energy band is due to a molecular conformational adjustment affecting electronic conjugation caused by strong interaction of macrocyclic acetyloxy carbonyl groups with the acid medium. The work presents a molecular mechanical model for estimating solution acidity and also gives insight into a possible method for modulating rotor activity in molecular machines.
O groups of macrocyclic acetyloxy groups, which occurs with the emergence of a lower energy electronic absorption band whose intensity is correlated with both the acidity of the medium and the rotation rate of the phenol substituents. Time-dependent DFT calculations suggest that the low energy band is due to a molecular conformational adjustment affecting electronic conjugation caused by strong interaction of macrocyclic acetyloxy carbonyl groups with the acid medium. The work presents a molecular mechanical model for estimating solution acidity and also gives insight into a possible method for modulating rotor activity in molecular machines.
Introduction
Molecular rotors1–7 are a class of molecular machine8–13 containing two key components, the stator and the rotor, which undergo facile relative rotation.14 The operation of rotors might be an important factor in the development of synthetic molecular machines where they would be integrated with other molecular components having different operational activities.15,16 Also, there exist several examples of biological molecular machine where a rotor molecular element operates based on chemical and physical stimuli.17–19 These can be treated as archetypes for the development of molecular- and nanomolecular-level machine architectures. Although substantial progress has been made in the area of molecular machine synthesis,20,21 and there have been demonstrations of important features of rotors such as unidirectional motion22–24 or supramolecular activity,25 there remains the requirement to investigate different molecular geometries and molecule-level activity in order to establish design rules for each active element of any possible molecular machine assemblies. A useful medium in this respect has been the metal–organic-frameworks (MOFs), where the rotation especially of the organic linker components presents the possibility to study substituent rotation under low density conditions.26–28 Apart from the molecular machine aspects of rotors, there already exist several applications of molecular rotors as probes of local conditions in biological systems, or as sensors.29–31 Rotor activity signified by variation in fluorescence output wavelength of certain molecule types can be harnessed to reveal local viscosity in biological membranes or models thereof,32–35 while rotation rate modulation also signalled by fluorescence emission characteristics can also be used to indicate the presence of analytes.36–38 Molecules having several proximal component moieties are suitable archetypes of rotors because of intramolecular interactions that govern the mutual motion of substituents. However, although macrocycles such as porphyrins39–41 and calixarenes42 or highly-substituted hydrocarbons43,44 have been extensively used, resorcinarenes45–49 have hardly been studied for this purpose, and in cases where they have been used, they are usually operating only as a supramolecular host where an encapsulated guest rotates.50,51Resorcinarenes (Fig. 1) are an intensively studied class of macrocycles, which can be readily synthesized in high yields by the simple condensation of resorcinol with an aldehyde.52 They are sterically crowded molecules and can exist in many different isomeric forms due to the relative orientations of their hindered substituents.53 Of the common isomers denoted rctt, rccc, rcct, rtct, the rccc forms have been studied due to their propensity to form oligo-molecular capsules with host–guest45–49,54,55 and catalytic properties.56–58 However, the effect of meso-substituent identity on the chemical properties of resorcinarene macrocycles has not been investigated in any depth despite the ease of synthesis of the relevant compounds and the possibility of assessing inter- and intramolecular processes based on the isomeric structures of the compounds. This has been due to the widespread interest in the chemistry of the carcerand structures and the essential chemistry of the resorcinarene macrocycle itself has been largely neglected.59,60 We have recently reported a class of resorcinarenes containing unsaturated meso-positions for which we have applied the term ‘Fuchsonarenes’ (Fig. 1) since these macrocycles contain components similar to fuchsone dyes.61 The compounds have so far been studied for their activity as multimodal molecular switches62 or as photosensitizers for singlet oxygen generation.63 Synthesis of the fuchsonarenes is enabled by the introduction of several oxidizable groups at their meso-positions, i.e., anti-oxidant 3,5-di-tert-butyl-4-hydroxyphenyl groups (DtBHP), which allows hybridization at the meso-positions to be transformed to sp2 following oxidation, introducing electronic conjugation between macrocycle and the resulting hemiquinonoid meso-substituents, and substantially rigidifying their structures. We had previously applied this principle to the meso-tetraphenylporphine system.64–72 The nanometric dimensions, rigid conformations and chemical stability of the resulting fuchsonarene molecules suggest their potential as supramolecular components of functional nanostructures. This potential is enhanced by the already demonstrated possibility of synthesising different isomer forms (e.g., rctt and rccc) or by functionalisation at the macrocyclic resorcinol moieties.
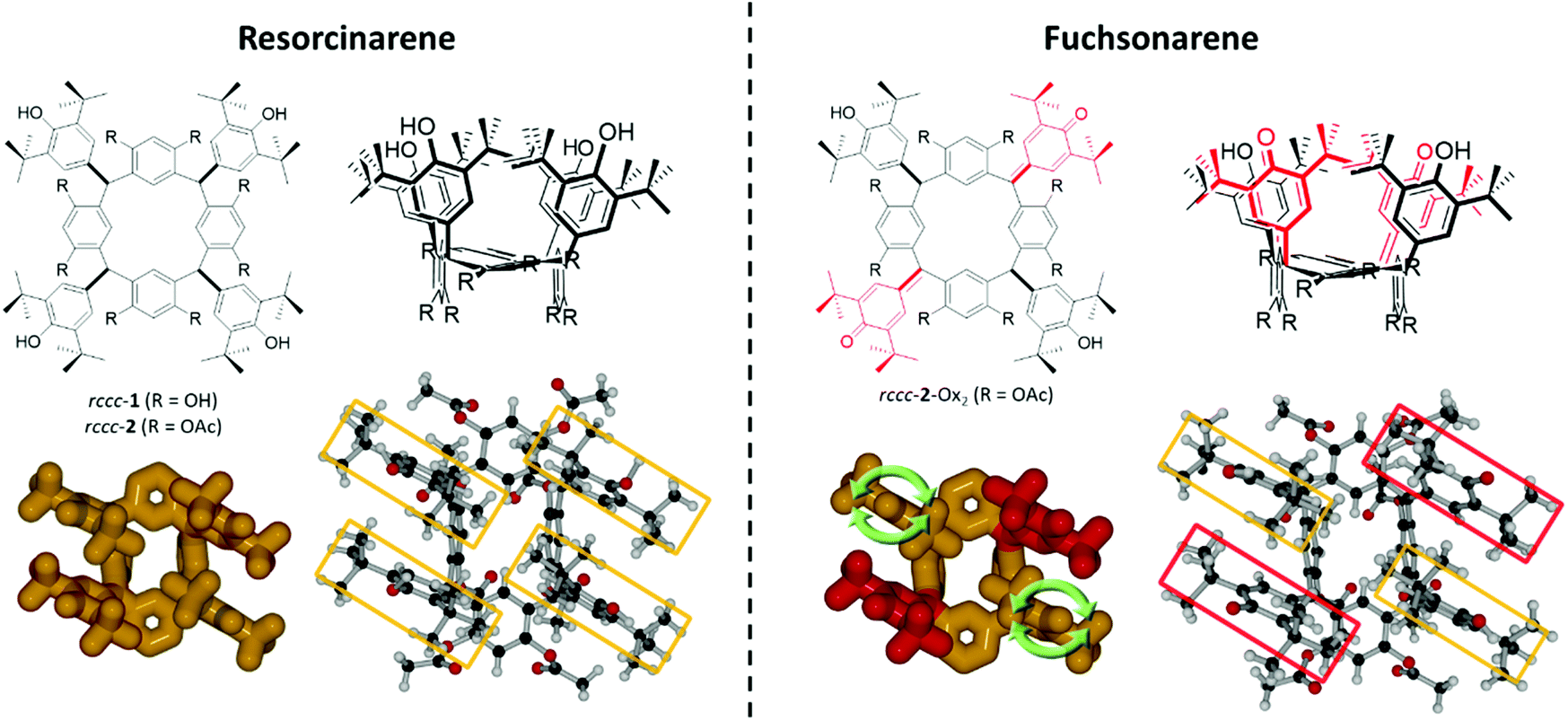 | ||
| Fig. 1 Chemical structures of resorcinarenes (left) and the corresponding fuchsonarenes (right) used in this work. Top: 2D and 3D line structures of the compounds. Bottom: 3D depictions of rccc-2 and rccc-2-Ox2 derived from X-ray crystallographic data. Green arrows denote rotational activity of the phenolic meso-substituents in rccc-2-Ox2. Antioxidant DtBHP substituents are indicated in yellow frames, while hemiquinonoid groups are denoted in red frames. | ||
Here we describe a system that exploits not only the rigidification of the resorcinarene macrocycle but also the rccc conformer in the formation of a fuchsonarene-based double-rotor/stator where the specific configuration of its substituents allows observation and study of the rotational motility of those substituents. Rotation rates of the rotors relative to the stator can be controlled between 2 s−1 and >20000 s−1 by varying the solvent, temperature or acidity. These effects were studied by 1H NMR spectroscopy and electronic absorption (UV-vis) spectrophotometry. The latter was used to demonstrate that rotation rate can be probed optically where acidity of the solvent was applied as the controlling factor. Rotation rates and mechanisms were also investigated computationally using density functional theory (DFT), revealing good agreement between experimental and computed scenarios.
Results
The resorcinarene synthesis involving condensation between resorcinol and a benzaldehyde, the rccc isomer of 1 is the thermodynamically favoured product and is available in increasing proportion over the kinetically favoured product (rctt isomer) at depressed reaction temperatures.52 For 1, a 2.6:1 mixture of rctt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :rccc was obtained when carrying out the synthesis at room temperature. Isomers were separated by per-O-acetylation (selectively at resorcinol-type hydroxyl groups) followed by fractional crystallization; rctt-2 precipitates rapidly from the acetylation reaction mixture while rccc-2 crystallizes more slowly over a period of several days with further subsequent purification performed by using column chromatography. A full description of the synthetic methods can be found in the ESI.† Oxidation of rccc-2 follows the same trend as previous studies,62,63 where double oxidation of rccc isomers is preferred, yielding the fuchsonarene derivative rccc-2-Ox2, whose properties as a double-rotor/stator are described here.
:rccc was obtained when carrying out the synthesis at room temperature. Isomers were separated by per-O-acetylation (selectively at resorcinol-type hydroxyl groups) followed by fractional crystallization; rctt-2 precipitates rapidly from the acetylation reaction mixture while rccc-2 crystallizes more slowly over a period of several days with further subsequent purification performed by using column chromatography. A full description of the synthetic methods can be found in the ESI.† Oxidation of rccc-2 follows the same trend as previous studies,62,63 where double oxidation of rccc isomers is preferred, yielding the fuchsonarene derivative rccc-2-Ox2, whose properties as a double-rotor/stator are described here.
Significant line broadening of the resonances due to certain meso substituent t-butyl groups in 1H NMR spectra obtained at room temperature in chloroform-d (CDCl3) was observed (Fig. 2a and b). This is assigned as being due to restricted rotation of the remaining non-oxidized phenolic meso-substituents of rccc-2-Ox2. This had not previously been observed in 1H NMR spectra of other fuchsonarene molecules and is so far a unique observation for this isomer structure and oxidation state, prompting our further investigation. An assignment of the NMR spectra of rccc-2-Ox2 in CDCl3 (with TMS reference at room temperature) was achieved by a combination of 2D NMR spectra (at 50 °C) with 1H NMR spectra measured at various temperatures (1H VT-NMR) (see ESI†).
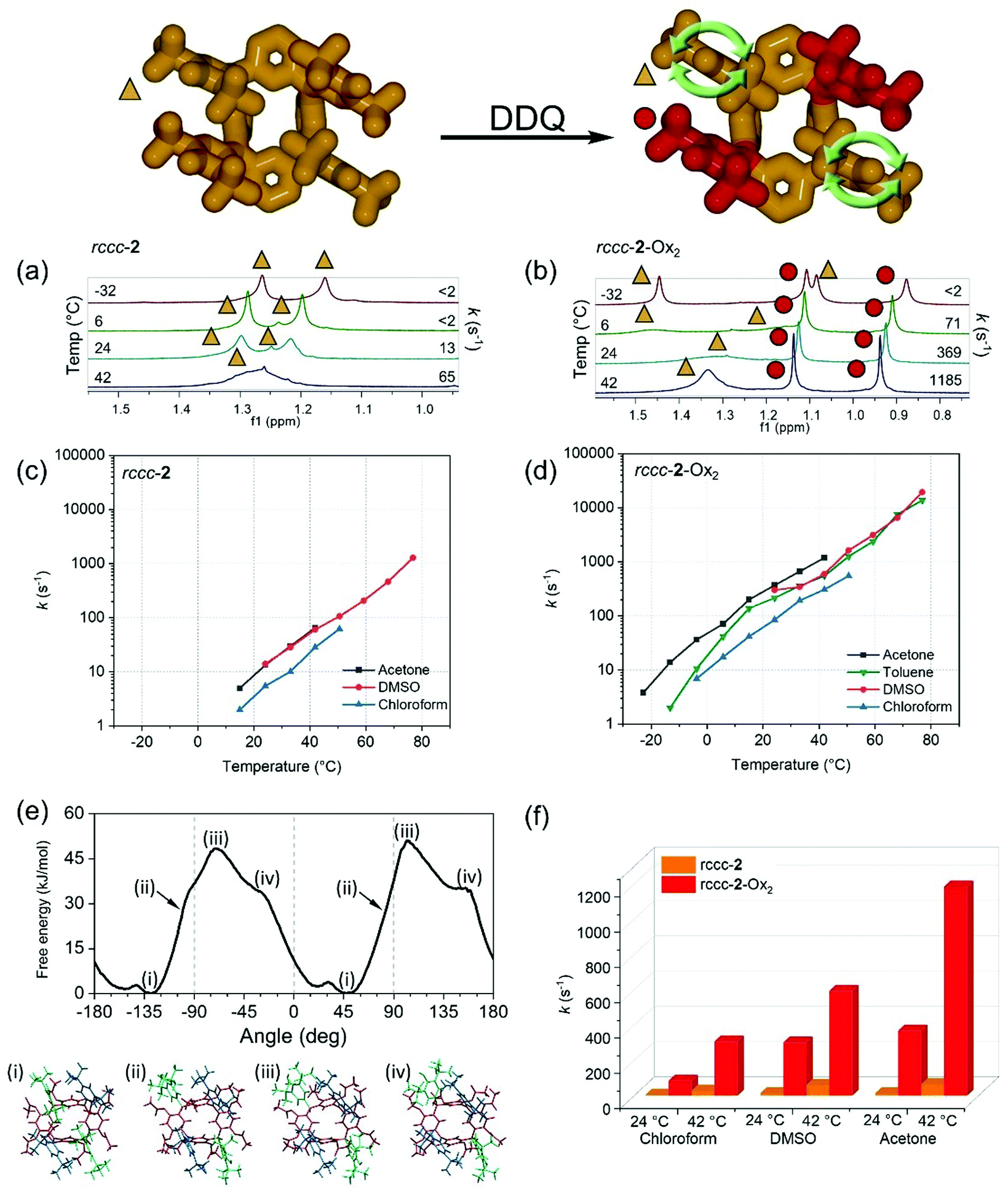 | ||
| Fig. 2 Relative rotation of the meso-substituents of rccc-2-Ox2. The model at the top depicts the oxidation of meso-substituents where hemiquinonoid groups are shown in red in the model at right. Regions of the molecule unchanged by the oxidation process are shown in yellow in the scheme at the top (DDQ: 2,3-dichloro-5,6-dicyano-1,4-benzoquinone). (a and b) High field (t-butyl) region of 1H NMR spectra in acetone-d6 at different temperatures of (a) rccc-2 and (b) rccc-2-Ox2. (c and d) Log-linear plot of rotation rates in different solvents at accessible temperatures of phenolic meso substituents in (c) rccc-2 and (d) rccc-2-Ox2. (e) Free energy profile of rotation of one of the phenolic meso substituent (the top-left one in shown structures) of rccc-2-Ox2 as obtained from MD simulations at 25 °C. Computational snapshots of several of the structures in acetone are included. (f) Comparison of rotation rates of phenolic meso substituents in rccc-2 and rccc-2-Ox2 in different solvents at 24 °C and 42 °C. | ||
1H VT-NMR spectroscopy, in conjunction with spectral line-shape analysis, allows for a detailed characterization of the rotation rate (k) of phenolic meso substituents in the compounds studied here. The broadening and coalescence of spectral lines (e.g.Fig. 2a and b) can be described by invoking a two-state symmetrical exchange between A and B states of phenolic t-butyl groups close to and remote from the centre of the macrocycle, respectively (Fig. 2, top). The spectrum can then be derived from the exchange between two non-interacting spin singlets and fitted using the spectral line shape formula S(ω) expressed in eqn (1).70,73,74
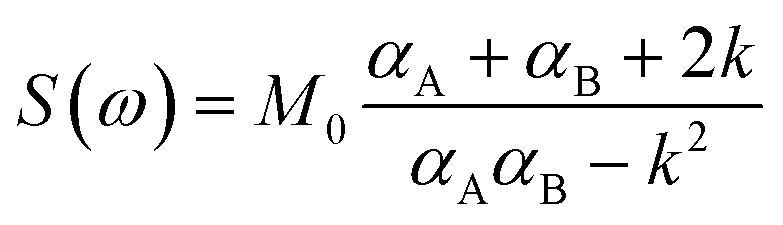 | (1) |
| αA = R2A + k − i(ω − ωA) |
| αB = R2B + k − i(ω − ωB) |
There is a connection between the rate constant k and the Gibbs energy of activation ΔG‡ (i.e., the barrier height) in the form of the Eyring equation (eqn (2)).75
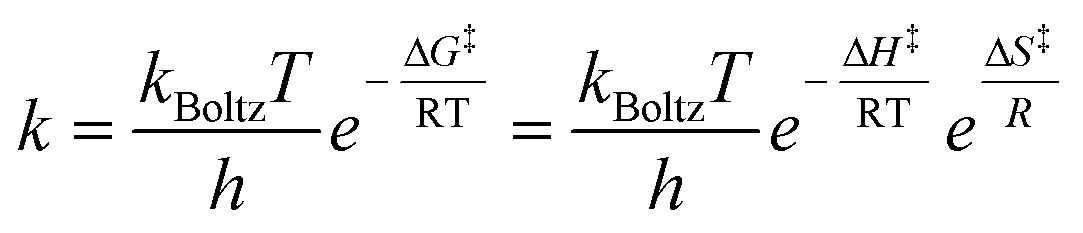 | (2) |
1H VT-NMR studies on rccc-2 (Fig. 2a, Table S1 and S2, Fig. S3–S6†) revealed that 180° rotation of the meso substituents is slow (13 s−1) at room temperature in acetone-d6 and that the barrier to rotation is high (ΔG‡ = 66.5 kJ mol−1) at 24.1 °C. The rate increases with temperature exponentially (i.e. straight lines in log-linear plot), and changing the solvent leads to variation in the rotation rates within a half order of magnitude, as shown in Fig. 2c. For rccc-2-Ox2 under identical conditions (Fig. 2b, Table S3 and S4, Fig. S7–S11†), a similar pattern of rotation rate dependencies was found (Fig. 2d). However, the rotation rate of the remaining unoxidized phenol substituents in rccc-2-Ox2 is significantly (more than one order of magnitude) greater than that of phenols in rccc-2 with, for example, a 28-fold rate increase observed at 24 °C in acetone-d6 (compare Fig. 2c and d or see Fig. 2f). This behaviour is counterintuitive considering the highly sterically hindered environments of the substituents since the unoxidized phenol groups are apparently blocked by the hemiquinonoid groups whose rotation is prevented by the double-bound connection of these substituents to the macrocyclic meso-positions. The barrier ΔG‡ to the rotation of the phenolic meso substituents of rccc-2-Ox2 was determined from NMR data to be 58.3 kJ mol−1 (in acetone-d6, at 24 °C, Table S4†), which is in good agreement with the value obtained from its free energy profile (ca. 50 kJ mol−1 at 25 °C, Fig. 2e) obtained using molecular dynamics (MD) simulations (see ESI section 1.3, Computational methods and Fig. S14†).76–85 These values are similar to those found for 2-substituted biphenyls and meso-tetraarylporphyrins systems.86,87 These results demonstrate that oxidation of two meso-substituents of resorcinarene rccc-2 to fuchsonarene rccc-2-Ox2 has the effect of promoting the rotation of the unoxidized phenolic meso substituents over those in rccc-2 and that the rate of rotation can be controlled by varying both the solvent used and the temperature, with rotation rates ranging from <2 s−1 to >20000 s−1 depending on conditions (see Fig. 2c, d and f, Fig. S15†). The promoted rotation rate caused by oxidation at the meso-substituents is an interesting feature of this system which can be assigned to variation in the conformation of the macrocycle caused by sp2 hybridization at its meso positions. It is also an interesting possible design parameter when considering the preparation of rotor systems in similarly substituted molecules.
While variations in temperature and solvent can be used to control the rotation rate of meso-substituents in rccc-2-Ox2, it was also found that exposure to trifluoroacetic acid-d (TFA-d) leads to a decrease in the rate of rotation of the unoxidized phenol meso substituents (370 s−1 in acetone-d6vs. 25 s−1 in TFA-d, Fig. 3a) denoted by the appearance of two individual t-butyl resonances in the 1H NMR spectrum. Interestingly, solutions of rccc-2-Ox2 are orange in TFA, and the colour change is accompanied by a new absorption band centred at 517 nm in its UV-vis spectrum (Fig. 3b). 1H VT-NMR studies revealed that the rate of rotation in the presence of TFA can again be controlled by varying temperature, where rotation is essentially arrested below 6 °C but increases to 460 s−1 at 59 °C (Fig. 3c, Table S3, Fig. S10 and S15†). This suggests that a third input for controlling the phenolic meso substituents rotation rate can be used, where exposure of the compounds to an acidic environment leads to a decrease in the rate of rotation.
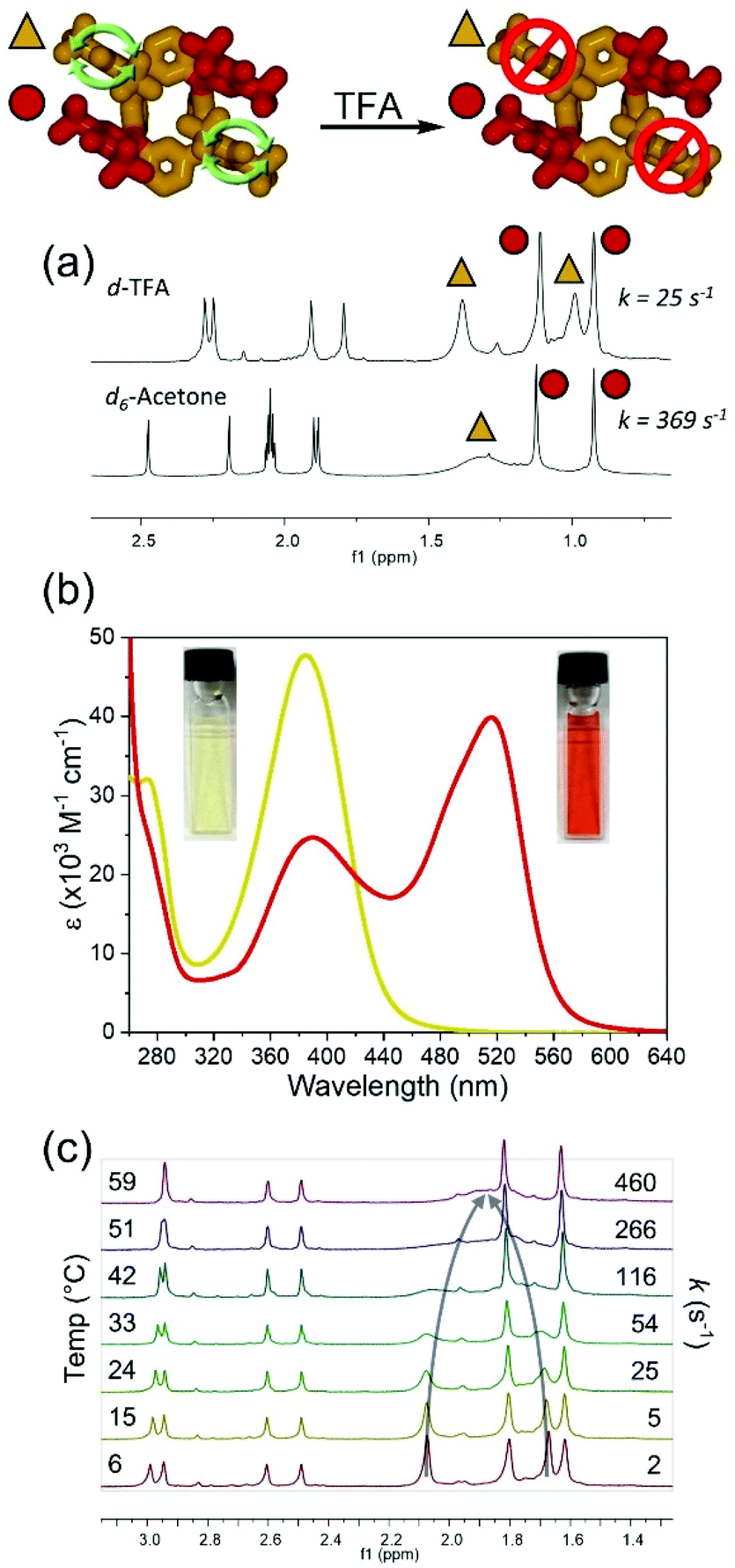 | ||
| Fig. 3 Effect of trifluoroacetic acid on the rate of rotation of phenolic meso substituents in rccc-2-Ox2. (a) 1H NMR spectra of rccc-2-Ox2 in acetone-d6 and TFA-d at 24 °C with symbols (triangle and circle) denoting different types of meso substituents. (b) UV-vis spectra of rccc-2-Ox2 in chloroform (yellow) and TFA (red) at 3.05 × 10−4 M in a 1 cm pathlength cell. (c) 1H VT-NMR spectra of rccc-2-Ox2 in TFA-d with spectra referenced to chemical shift of the first acetate resonance (2.48 ppm). Arrows indicate the coalescing resonances due to phenolic meso substituent. | ||
The rotation rate of the unoxidized meso-substituents can be tuned under isothermal conditions by varying the composition of a binary mixture of TFA-d/acetone-d6 (Fig. 4, Table S5, Fig. S12 and S13†). Increasing the ratio of TFA-d in the binary mixture slows the rotation rate from ca. 470 s−1 to 10 s−1 (Fig. 4a, Table S5†) with a concomitant gradual change in the solution colour (Fig. 4b) and variation in the UV-vis spectrum (Fig. 4c). It should be noted that the barrier to the rotation ΔG‡ of the meso-substituent increases in pure TFA to 65.6 kJ mol−1 (determined from NMR at 24 °C, Table S4†), which is again in reasonable agreement with the value of ca. 61 kJ mol−1 (at 25 °C, Fig. S14†) obtained from the free energy profile using MD simulations.
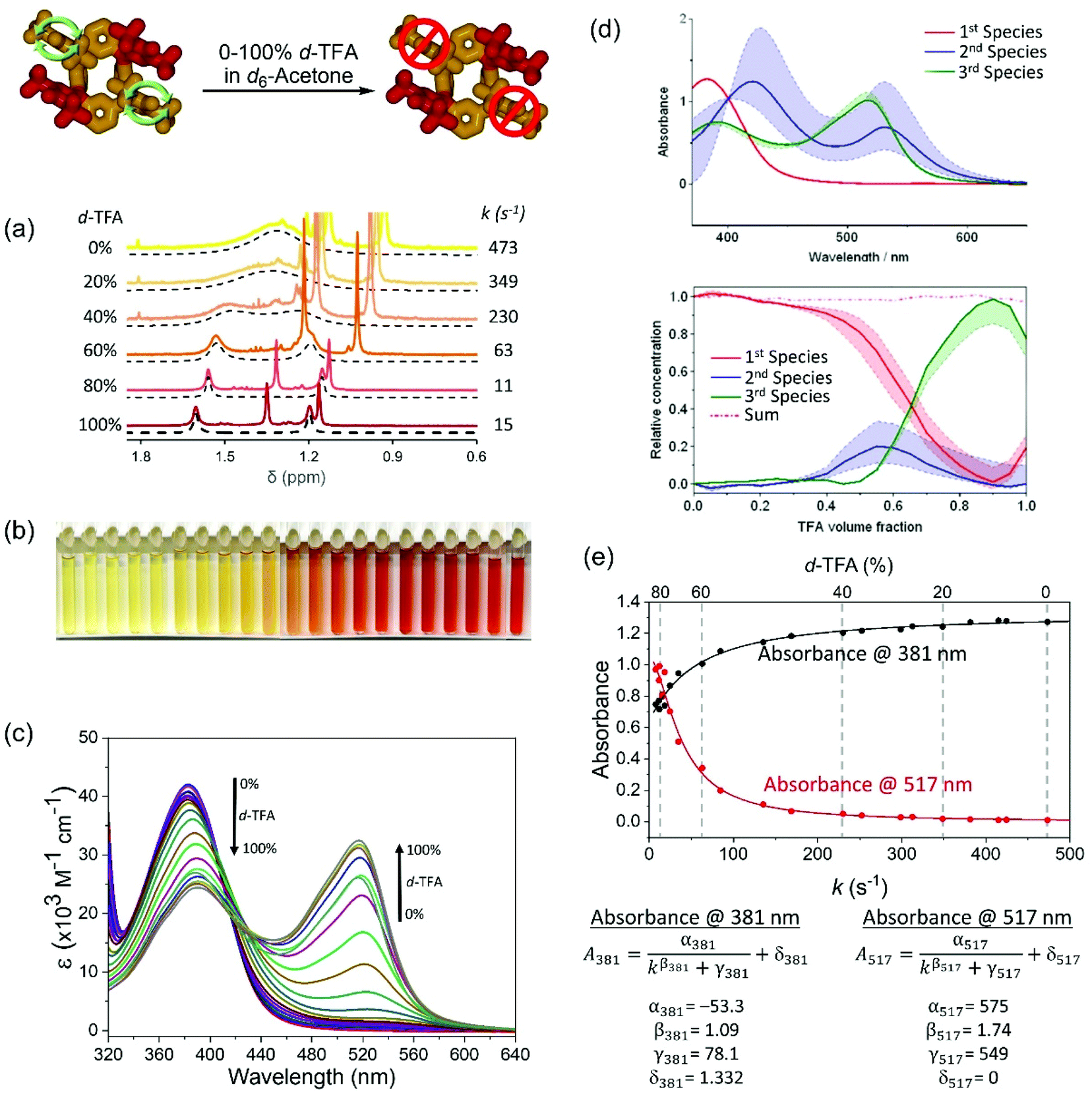 | ||
| Fig. 4 Effect of TFA-d volume fraction in acetone-d6 on the rate of rotation of phenolic meso substituents in rccc-2-Ox2. (a) 1H NMR spectra of rccc-2-Ox2 at 3.05 × 10−4 M in TFA-d/acetone-d6 mixtures at 24 °C. (b) Photographs of the samples prepared for this study indicating a gradual colour change. (c) UV-vis spectra of rccc-2-Ox2 in TFA-d/acetone-d6 mixtures at 3.05 × 10−4 M in a 0.1 cm pathlength cell. (d) Summary of the SVD analysis indicating the presence of 3 different species (for details, see ESI, section 5†). (e) Calibration curves and formulae constructed by correlating UV-vis and 1H NMR data (for details, see ESI, section 6†). | ||
The presence of two isosbestic points at 401 and 427 nm (Fig. 4c) in UV-vis spectra of solutions of varying TFA-d/acetone-d6 compositions and the results of singular value decomposition analysis (SVD)88–94 (Fig. 4d, for details see section 5 in ESI†) indicate the presence of a third intermediate species. The spectra of second and third species qualitatively resemble those obtained by time-dependent density functional theory (TD-DFT) calculations (Fig. 5b), which indicates some degree of interaction of the acid medium with rccc-2-Ox2 (for more details, see Discussion). The predictable variation in UV-vis profile dependent on solvent composition (in this case, acid content) presents a molecular system whose rotation rate can be indirectly estimated using UV-vis spectrophotometry, allowing for the construction of calibration curves if 1H NMR data can be correlated with the UV-vis data (Fig. 4e). Based on the correlation, a set of calibration formulae can be generated and used to determine indirectly the rate of rotation of the phenolic meso substituents based on the absorbance data (Fig. 4e; for details see section 6 in ESI†).
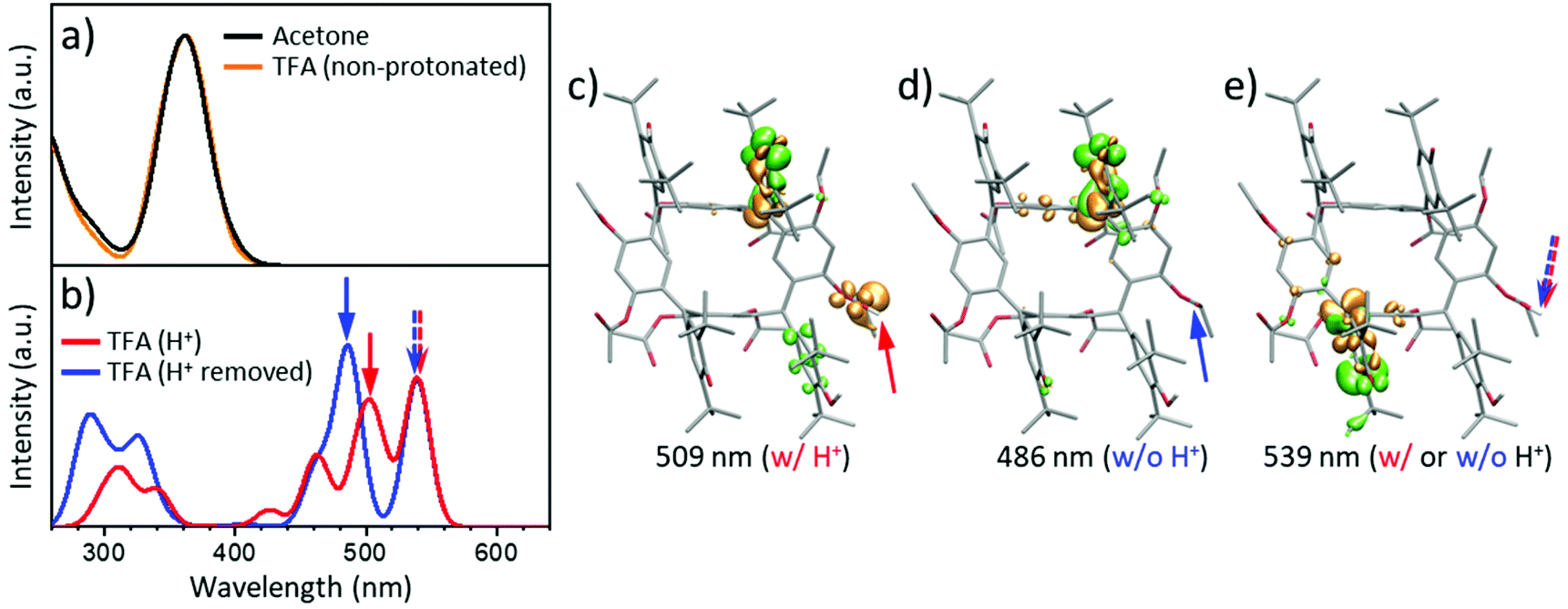 | ||
| Fig. 5 Calculated UV-vis spectra and related molecular conformations. TD-DFT (ωB97XD 6-311G(d)) UV-vis spectra of rccc-2-Ox2 calculated from 20 averaged statistically-independent snapshots from MD simulations performed under various conditions: (a) Non-protonated rccc-2-Ox2 in neat acetone and TFA solvents. (b) Spectrum of rccc-2-Ox2 in TFA with protonation constrained at one acetyloxy group and the spectrum after removal of the proton (without structure relaxation). MD snapshots of differential excitation density (DED) of various states of rccc-2-Ox2 in TFA. Green isodensities show the negative regions from which electrons are excited while the positive orange isodensities indicate the regions to which the electrons are excited. Transitions shown are responsible for UV-vis absorptions at wavelengths indicated below each structure. (c) DED of acetyloxy protonated (location denoted by red arrow) rccc-2-Ox2 leading to absorption at 509 nm. (d) DED of rccc-2-Ox2 after removal of proton from acetyloxy group in (c) (location denoted by blue arrow) shifts the absorption from 509 nm to 486 nm. (e) DED of rccc-2-Ox2 absorbance band at 539 nm indicates no change with or without protonation of the acetyloxy group (dashed red/blue arrows). The actual structure represents a protonated form. | ||
Discussion
There are several interesting aspects of the rccc-2-Ox2 system presented here that have connotations for the properties of other similar molecular constructs. For example, many compounds containing multiple, especially conjugated, hemiquinonoid groups can also exist with an open shell electronic structure95,96 with unpaired electron density present at unsaturated oxygen atoms. In this case, we have excluded this possibility based on the only weak ESR response of rccc-2-Ox2 (due to small amounts of phenoxyl radical always present in samples of these compounds, see Fig. S16†) and the thermally induced coalescence of the relevant 1H NMR resonances, which must be a symptom of moiety rotation. Other work62,63 has shown that carbonyl CO bond lengths are consistent with closed-shell forms. Also, the changes in the hue of solutions where the acetone/TFA ratio is varied is reminiscent of solvatochromism. However, the variation is merely due to increases in absorbance of the new band at 520 nm, and no shifts in absorption wavelength are observed, implying the emergence of another chemical entity with a different electronic structure.
Of the solvents employed in the present study, trifluoroacetic acid causes the most notable effects based on the emergent UV-vis band and rotation rates determined by NMR spectroscopy. Therefore, we must consider the effects of acid and possible protonation scenarios most closely. Protonation of fuchsone dyes such as 2,6-di-tert-butyl-4-(diphenylmethylene)cyclohexadien-1-one97 initially occurs at the carbonyl oxygen atom with subsequent rearrangement to a resonance stabilized methanide carbocation that has a characteristic 13C NMR resonance at ∼200 ppm. However, for rccc-2-Ox2 in TFA-d, no such low field carbocation resonance appears and the chemical shift of its hemiquinonoid carbonyl carbon atom is similar to that in CDCl3 solution (see ESI, Fig. S17 & S30† peak at low field: 186.5 ppm in both cases) indicating that protonation does not occur at hemiquinonoid groups of rccc-2-Ox2 (for assignments of the NMR spectra of rccc-2-Ox2 in different media see ESI†). Resistance to protonation by rccc-2-Ox2 is most likely due to the low electron donating power of the resorcinarene acetyloxy ester groups so that rearrangement from protonated hemiquinone to meso-carbocation (which certainly occurs in the case of simple fuchsone97) is not supported, and the corresponding 13C NMR resonance at low field is not found. 13C NMR spectra of rccc-2-Ox2 in TFA-d do, however, reveal significant shifts in carbonyl resonances due to acetyloxy carbonyl groups (see Fig. S17 & S30;† group of 4 peaks at 167–169 ppm (CDCl3) shifts to 173–175 ppm (TFA-d); see Fig. S29† for the corresponding 1H NMR spectra). There are accompanying 13C NMR shifts for the sp2-type meso-positions and the neighbouring cyclohexadienylidene substituent proton and carbon atoms (see section 8, ESI†) indicating that some structural change occurs close to the oxidized meso-positions, which is associated with acetyloxy/TFA interactions. These are the most important variations in the NMR spectra occurring in passing from non-polar (CDCl3) to acidic (TFA-d) solvent. This approximately 6 ppm downfield shift of the acetyloxy CO 13C resonances (and associated NMR variations of oxidized meso-substituents) suggests some interaction with the acidic medium. Therefore, we have also investigated this using time-dependent density functional theory (TD-DFT) methods.
In TD-DFT, notably, while simple H-bonding interactions were observed not to generate low energy electronic absorption in rccc-2-Ox2 (Fig. 5a), the constrained protonation of one of its acetyloxy CO group(s) was found to lead to the appearance of new lower energy bands in its TD-DFT calculated UV-vis spectrum similar to those observed experimentally for solutions of rccc-2-Ox2 in TFA (compare Fig. 5b and 3b).98–101 UV-vis spectra of rccc-2-Ox2 in acetone or TFA without constrained protonation calculated using TD-DFT77,102,103 are shown in Fig. 5a (for details, see section 1.3: Computational methods in ESI†). The UV-vis spectra calculated for rccc-2-Ox2 in TFA with initial constrained protonation of an acetyloxy CO group (Fig. 5b, red line) followed by removal of the proton without structure relaxation (Fig. 5b, blue line) show very good agreement with the experimentally obtained data (compare to Fig. 3b). The small difference between the spectra in Fig. 5b indicates that any charge located at the acetyloxy group present due to protonation makes only a minor contribution to the resulting low-energy absorbance band in UV-vis. This effect is also supported by the plots of differential excitation densities (DED) prior to and after removal of the proton from the protonated form of rccc-2-Ox2, (Fig. 5c–e). The low energy absorption bands in the 450 to 580 nm region originate from the hemiquinone moieties of rccc-2-Ox2. The effects of solvent environment were also tested using TD-DFT resulting in consistent significant absorptive transitions in the 410 to 580 nm region (Fig. S18†). A further assessment of the effects of acetyloxy protonation on the structure was carried out by MD simulations. The results indicate that protonation at an acetyloxy CO group causes structural variation of the rccc-2-Ox2 macrocycle so that planes of adjacent phenol-hemiquinone substituent pairs become almost parallel (Fig. S19a†) allowing their closer approach causing reduction in rotation rate with the resorcinarene macrocycle being concurrently increasingly skewed (Fig. S19b†). These structural changes are responsible for the emergence of the new low-energy absorption bands due to the resulting increase in π-electronic conjugation between hemiquinone and macrocycle. It should be noted that protonation on an acetyloxy group would be the first step in their acid promoted hydrolytic cleavage, although we found no evidence for decomposition of rccc-2-Ox2 by this route under the conditions used (treatment with strong mineral acids does result in decomposition to intractable products). Therefore, while protonation of acetyloxy groups in this system can influence the molecular conformation so that new low energy bands appear in UV-vis spectra, these protonated states are not sufficiently persistent to lead to deacetylation of the macrocycle.
The calculations suggest that in TFA there is a degree of protonation or at least strong H-bonding of acetyloxy CO groups of rccc-2-Ox2, which influences its electronic structure, although the corresponding experimental observations suggest only incomplete or equilibrium processes. Also, the addition of stronger (than TFA) organic acids causes a further increase in the intensity of the absorption centred at 520 nm in experimentally observed spectra. Other pertinent experimental observations include infrared spectra (see ESI, Fig. S20†), where absorptions due to acetyloxy carbonyl stretching are shifted to lower frequency when exposed to TFA vapour, which can be associated with hydrogen bonding. There is also a weak ESR signal in rccc-2-Ox2 (see ESI, Fig. S16†) although compounds containing multiple DtBHP groups usually contain small amounts of phenoxyl radical due to handling under aerobic conditions. The spectroscopic changes, including shifts in the 13C resonances of the acetyloxy carbonyl groups and the appearance of the low energy UV-vis absorption, suggest that there is a significant protic process occurring between TFA and the acetyloxy CO groups. The strength of this interaction is sufficient to change the conformation of rccc-2-Ox2, which cannot be detected directly. It also occurs in the proximity of a fuchsone moiety which is usually susceptible to protonation and rearrangement. In rccc-2-Ox2, it seems then that two effects operate: (1) macrocyclic acetyloxy substituents are sufficiently electron-withdrawing to destabilize prospective meso-carbocations thus disfavouring protonation at the hemiquinonoid CO, and (2) multiple acetyloxy groups of rccc-2-Ox2 are available for interaction with the acid medium, which leads to modification of its chromophore structure and emergence of electronic absorption bands in the visible region. It is currently unclear whether or not (2) influences (1) so that protonation at hemiquinonoid is even more strongly disfavoured at higher acidities.
We note that the simple variation of macrocyclic substituent from benzyloxy (ether) to acetyloxy (ester) leads to a dramatic change in the response of the compounds to their environments. For the former (ether), meso-substituent protonation affects the properties of the compounds, while for the latter (ester) described here, interaction with the medium is limited to the macrocyclic acetyloxy (i.e., not meso) substituents. This is an important point as we learn about the factors affecting the chemistry of resorcinarenes and, in particular, the fuchsonarenes. As mentioned previously, the resorcinarenes have largely been considered for their propensity to form large nanocapsular assemblies. In this and previous work, we have found that complex chemical relationships exist, and these can occur not only intramolecularly between different types of substituents (e.g., macrocylic benzyloxy groups support meso-carbocation formation in contrast to acetyloxy) but also intermolecularly involving the solvent medium. It is interesting to note that in this case, the resistance to protonation of hemiquinonoid groups probably caused by macrocyclic acetyloxy groups is countered by a corresponding perhaps counterintuitive interaction of the acetyloxy groups with the acidic medium. Also, coincidentally, interactions of acid with fuchsonarenes at hemiquinonoid or acetyloxy groups lead to the emergence of UV-vis bands in the visible region despite the origins of these bands being very different.
Conclusions
In conclusion, we have demonstrated the activity of a fuchsonarene as a molecular single stator-double rotor. In terms of its structure, this is a rare example where rotary action can be controlled by varying solvent, temperature, or chemical inputs yielding a controllable system whose rotation rate can be tuned in the range of 2 s−1 to 20000 s−1 depending upon the combination of the aforementioned inputs. Rotation speed governed by the TFA concentration can be monitored using UV-vis absorption data due to its correlation with the rotation rate determined by 1H NMR spectroscopy, and suitable formulae for this purpose have been presented. TD-DFT was used to investigate features of this system and indicates that strong interactions of the acetyloxy groups with the solvent (most likely H-bonding) occur in acidic solution and are responsible for the emergence of new low energy bands in the UV-vis spectrum of rccc-2-Ox2. Resorcinarenes are more often considered from the point-of-view of supramolecular capsule formation and related catalytic activities. Here, we have further expanded the chemistry of the resorcinarenes through the study of the rotor activity of their substituents – a previously unknown aspect of these well-studied compounds. The resorcinarene macrocycle presents a new scaffold for molecular machines where the orientation of rotor-active substituents can be controlled within a compact 3-dimensional space, and rotation rate is modulated according to temperature, solvent polarity and acidity. Overall, this work suggests a molecular mechanical method for indirect estimation of solution acidity by simple monitoring of UV-vis spectra, and also gives insight into a possible method for modulating rotary activity in molecular machines.
Data availability
General experimental details, description of synthetic procedures, rotation data and 1H-NMR spectra from which they were derived, supplementary figures, description of singular value decomposition methods, calibration curves, additional characterization data, assignment of the NMR spectra of rccc-2-Ox2 inclduing extensive 2D-NMR spectra. These items are available in the ESI.†Author contributions
D. T. P., J. L. and J. P. H. designed the experiments, led the project and composed and wrote the manuscript with scientific discussions and contributions from all co-authors. D. T. P. performed the synthesis and chemical analyses of the compounds and collected analytical data. J. L. analysed the VT 1H NMR data, extracted the rate constants and constructed the calibration curves. Z. F. undertook the MD and DFT computational work. V. B. and L. H. performed SVD analysis. M. K. C. undertook UV-vis measurements and data processing. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by World Premier International Research Center Initiative (WPI Initiative), MEXT, Japan. D. T. P is grateful to the Japan Society for the Promotion of Science (JSPS) for a JSPS Fellowship and to the International Center for Young Scientists (ICYS-Namiki), NIMS for continued financial support. This work was also partially supported by JSPS KAKENHI Grant No. 19K05229. Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures.Notes and references
- V. Garcia-Lopez, D. Liu and J. M. Tour, Light-activated organic molecular motors and their applications, Chem. Rev., 2020, 120, 79–124 CrossRef CAS.
- J. Michl and E. C. H. Sykes, Molecular rotors and motors: Recent advances and future challenges, ACS Nano, 2009, 3, 1042–1048 CrossRef CAS.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Artificial molecular rotors, Chem. Rev., 2005, 105, 1285–1376 CrossRef.
- G. Rapenne and C. Joachim, Single rotating molecule-machines: nanovehicles and molecular motors, Top. Curr. Chem., 2014, 354, 253–277 CrossRef CAS PubMed.
- D. Lensen and J. A. A. W. Elemans, Artificial molecular rotors and motors on surfaces: STM reveals and triggers, Soft Matter, 2012, 8, 9053–9063 RSC.
- C. Ni and J.-Z. Wang, STM studies on molecular rotors and motors, Surf. Rev. Lett., 2018, 25(Supp01), 1841004 CrossRef CAS.
- F. Cherioux, O. Galangau, F. Palmino and G. Rapenne, Controlled directional motions of molecular vehicles, rotors, and motors: From metallic to silicon surfaces, a strategy to operate at higher temperatures, ChemPhysChem, 2016, 17, 1742–1751 CrossRef CAS.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial molecular machines, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Artificial molecular motors, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- F. Lancia, A. Ryabchun and N. Katsonis, Life-like motion driven by artificial molecular machines, Nat. Rev. Chem., 2019, 3, 536–551 CrossRef CAS.
- R. D. Astumian, Trajectory and cycle-based thermodynamics and kinetics of molecular machinesL The importance of microscopic reversibility, Acc. Chem. Res., 2018, 51, 2653–2661 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, Wiley VCH Weinheim, Germany, 2008 Search PubMed.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Artificial molecular machines, Angew. Chem., Int. Ed. Engl., 2000, 39, 3348–3391 CrossRef CAS.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Synthetic molecular motors and mechanical machines, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS.
- M. Schmittel, Dynamic functional molecular systems: From supramolecular structures to multi-component machinery and to molecular cybernetics, Isr. J. Chem., 2019, 59, 197–208 CrossRef CAS.
- R. Herges, Molecular assemblers: molecular machines performing chemical synthesis, Chem. Sci., 2020, 11, 9048–9055 RSC.
- A. B. Kolomeisky, Motor proteins and, molecular motors: how to operate machines at the nanoscale, J. Phys.: Condens. Matter, 2013, 25, 463101 CrossRef.
- M. Baba, K. Iwamoto, R. Iino, H. Ueno, M. Hara, A. Nakanishi, J.-I. Kishikawa, H. Noji and K. Yokoyama, Rotation of artificial rotor axles in rotary molecular motors, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11214–11219 CrossRef CAS.
- M. Baroncini, S. Silvi and A. Credi, Photo- and redox-driven artificial molecular motors, Chem. Rev., 2020, 120, 200–268 CrossRef CAS.
- C. Cheng and J. F. Stoddart, Wholly synthetic molecular machines, ChemPhysChem, 2016, 17, 1780–1793 CrossRef CAS.
- M. Baroncini, L. Casimiro, C. de Vet, J. Groppi, S. Silvi and A. Credi, Making and operating molecular machines: A multidisciplinary challenge, ChemistryOpen, 2018, 7, 169–179 CrossRef CAS PubMed.
- H. A. Kistemaker, P. Stacko, J. Visser and B. L. Feringa, Unidirectional rotary motion in achiral molecular motors, Nat. Chem., 2015, 7, 890–896 CrossRef PubMed.
- A. Gerwien, P. Mayer and H. Dube, Green light powered molecular state motor enabling eight-shaped unidirectional rotation, Nat. Commun., 2019, 10, 4449 CrossRef PubMed.
- J. Echeverria, S. Monturet and C. Joachim, One-way rotation of a molecule-rotor driven by a shot noise, Nanoscale, 2014, 6, 2793–2799 RSC.
- K. Sun, J.-L. Luo, X. Zhang, Z.-J. Wu, Y. Wang, H.-K. Yuan, Z.-H. Xiong, S.-C. Li, Q.-K. Xue and J.-Z. Wang, Supramolecular motors on graphite surface stabilized by charge states and hydrogen bonds, ACS Nano, 2017, 11, 10236–10242 CrossRef CAS PubMed.
- M. E. Howe and M. A. Garcia-Garibay, The roles of intrinsic barriers and crystal fluidity in determining the dynamics of crystalline molecular rotors and molecular machines, J. Org. Chem., 2019, 84, 9835–9849 CrossRef CAS.
- A. Comotti, S. Bracco and P. Sozzani, Molecular rotors built in porous materials, Acc. Chem. Res., 2016, 49, 1701–1710 CrossRef PubMed.
- P. Martinez-Bulit, A. J. Stirk and S. J. Loeb, Rotors, Motors, and Machines Inside Metal–Organic Frameworks, Trends Chem., 2019, 1, 588–600 CrossRef CAS.
- C. H. Wolstenholme, H. Hu, S. Ye, B. E. Funk, D. Jain, C.-H. Hsiung, G. Ning, Y. Liu, X. Li and X. Zhang, AggFluor: Fluorogenic toolbox enables direct visualization of the multi-step protein aggregation process in live cells, J. Am. Chem. Soc., 2020, 142, 17515–17523 CrossRef CAS.
- W. L. Goh, M. Y. Lee, T. L. Joseph, S. T. Quah, C. J. Brown, C. Verma, S. Brenner, F. J. Ghadessy and Y. N. Teo, Molecular rotors as conditionally fluorescent labels for rapid detection of biomolecular interactions, J. Am. Chem. Soc., 2014, 136, 6159–6162 CrossRef CAS PubMed.
- M. A. Haidekker and E. A. Theodorakis, Environment-sensitive behavior of fluorescent molecular rotors, J. Biol. Eng., 2010, 4, 11 CrossRef CAS PubMed.
- A. S. Kashirina, I. Lopez-Duarte, M. Kubankova, A. A. Gulin, V. V. Dudenkova, S. A. Rodimova, H. G. Torgomyan, E. V. Zagaynova, A. V. Meleshina and M. K. Kuimova, Monitoring Membrane Viscosity in Differentiating Stem Cells Using BODIPY-Based Molecular Rotors and FLIM, Sci. Rep., 2020, 10, 14063 CrossRef CAS.
- X. Liu, W. Chi, Q. Qiao, S. V. Kokate, E. P. Cabrera, Z. Xu, X. Liu and Y.-T. Chang, Molecular mechanism of viscosity sensitivity in BODIPY rotors and application to motion-based fluorescent sensors, ACS Sens., 2020, 5, 731–739 CrossRef CAS PubMed.
- T. Mori, H. Chin, K. Kawashima, H. T. Ngo, N.-J. Cho, W. Nakanishi, J. P. Hill and K. Ariga, Dynamic control of intramolecular rotation by tuning the surrounding two-dimensional matrix field, ACS Nano, 2019, 13, 2410–2419 CAS.
- T. Mori, H. Komatsu, N. Sakamoto, K. Suzuki, J. P. Hill, M. Matsumoto, H. Sakai, K. Ariga and W. Nakanishi, Molecular rotors confined at an ordered 2D interface, Phys. Chem. Chem. Phys., 2018, 20, 3073–3078 RSC.
- L. Feng, Y. Xie, S. K. Au-Yeung, H. B. Hailu, Z. Liu, Q. Chen, J. Zhang, Q. Pang, X. Yao, M. Yang, L. Zhang and H. Sun, A fluorescent molecular rotor probe for tracking plasma membranes and exosomes in living cells, Chem. Commun., 2020, 56, 8480–8483 RSC.
- A. M. Desai and P. K. Singh, An ultrafast molecular-rotor-based fluorescent turn-on sensor for the perrhenate anion in aqueous solution, Chem. – Eur. J., 2019, 25, 2035–2042 CrossRef CAS PubMed.
- Y. Wu, G. Wang, Q. Li, J. Xiang, H. Jiang and Y. Wang, A multistage rotational speed changing molecular rotor regulated by pH and metal cations, Nat. Commun., 2018, 9, 1953 CrossRef PubMed.
- P. K. Biswas, S. Saha, S. Gaikwad and M. Schmittel, Multitasking with chemical fuel: dissipative formation of a pseudorotaxane rotor from five distinct components, J. Am. Chem. Soc., 2020, 142, 7889–7897 CrossRef CAS PubMed.
- P. K. Biswas, S. Saha, Y. Nanaji, A. Rana and M. Schmittel, Influence of rotor design on the speed of self-assembled four-component nanorotors: coordinative versus dispersive interactions, Inorg. Chem., 2017, 56, 6662–6670 CrossRef CAS PubMed.
- T. Ikeda, T. Tsukahara, R. Iino, M. Takeuchi and H. Noji, Motion capture and manipulation of a single synthetic rotor by optical microscopy, Angew. Chem., Int. Ed., 2014, 53, 10082–10085 CrossRef CAS PubMed.
- Z. Asfari, C. Naumann, G. Kaufmann and J. Vicens, Synthesis of a molecular mill designed from a calix[4]-bis-crown, Tetrahedron Lett., 1998, 39, 9007–9010 CrossRef CAS.
- J. Sturala, M. K. Etherington, A. N. Bismillah, H. E. Higginbotham, W. Trewby, J. A. Aguilar, E. H. C. Bromley, A.-J. Avestro, A. P. Monkman and P. R. McGonigal, Excited-state aromatic interactions in the aggregation-induced emission of molecular toros, J. Am. Chem. Soc., 2017, 139, 17882–17889 CrossRef CAS.
- K. K. Kartha, A. Takai, Z. Futera, J. Labuta and M. Takeuchi, Dynamics of meso—chiral interconversion in a butterfly-shape overcrowded alkene rotor tunable by solvent properties, Angew. Chem., Int. Ed., 2021, 60, 16466–16471 CrossRef CAS PubMed.
- C. M. A. Gangemi, A. Pappalardo and G. T. Sfrazzetto, Assembling of supramolecular capsules with resorin[4]arene and calix[4]arene building blocks, Curr. Org. Chem., 2015, 19, 2281–2308 CrossRef CAS.
- P. Jin, S. J. Dalgarno and J. L. Atwood, Mixed metal-organic nanocapsules, Coord. Chem. Rev., 2010, 254, 1760–1768 CrossRef CAS.
- A. M. A. van Wageningen, W. Verboom and D. N. Reinhoudt, Synthesis of supramolecular structures via combination of calix[4]arenes with other medium-sized building blocks, Pure Appl. Chem., 1996, 68, 1273–1277 CAS.
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Resorcinarenes, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS.
- H. Erdtman, S. Högberg, S. Abrahamsson and B. Nilsson, Cyclooligomeric phenol-aldehyde condensation products I, Tetrahedron Lett., 1968, 9, 1679–1682 CrossRef.
- H. Kitagawa, Y. Kobori, M. Yamanaka, K. Yoza and K. Kobayashi, Encapsulated-guest rotation in a self-assembled heterocapsule directed toward a supramolecular gyroscope, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10444–10448 CrossRef CAS PubMed.
- S. Harthong, B. Dubessy, J. Vachon, C. Aronica, J.-C. Mulatier and J.-P. Dutasta, Restricted guest tumbling in phosphorylated self-assembled capsules, J. Am. Chem. Soc., 2010, 132, 15637–15643 CrossRef CAS.
- A. G. S. Högberg, Cyclooligomeric phenol-aldehyde condensation products. 2. Stereoselective synthesis and DNMR study of two 1,8,15,22-tetrphenyl[14]metacyclophan-3,5,10,12,17,19,24,26-octols, J. Am. Chem. Soc., 1980, 102, 6046–6050 CrossRef.
- R. H. Patil, C. Zhang and J. L. Atwood, Process development for separation of conformers from derivatives of resorcin[4]arenes and pyrogallol[4]arenes, Chem. – Eur. J., 2016, 22, 15202–15207 CrossRef CAS PubMed.
- L. R. MacGillivray and J. L. Atwood, A chiral spherical molecular assembly held together by 60 hydrogen bonds, Nature, 1997, 389, 469–472 CrossRef CAS.
- J. L. Atwood, L. J. Barbour and A. Jerga, Organization of the interior of molecular capsules by hydrogen bonding, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4837–4841 CrossRef CAS.
- Q. Zhang, L. Catti and K. Tiefenbacher, Catalysis inside the hexameric resorcinarene capsule, Acc. Chem. Res., 2018, 51, 2107–2114 CrossRef CAS.
- A. Pappalardo, R. Puglisi and G. T. Sfrazzetto, Catalysis inside supramolecular capsules, Catalysts, 2019, 9, 630 CrossRef CAS.
- B. W. Purse and J. Rebek Jr., Functional cavitands: chemical reactivity in structured environments, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10777–10782 CrossRef CAS.
- S. Pascal, L. Lavaud, C. Azarias, A. Varlot, G. Canard, M. Giorgi, D. Jacquemin and O. Siri, Azacalixquinarenes: from canonical to (poly-)zwitterionic macrocycles, J. Org. Chem., 2019, 84, 1387–1397 CrossRef CAS PubMed.
- W. Maes and W. Dehaen, Oxacalix[n](het)arenes, Chem. Soc. Rev., 2008, 37, 2393–2402 RSC.
- H.-D. Becker, Preparation of fuchsones, J. Org. Chem., 1967, 32, 2943–2947 CrossRef CAS.
- D. T. Payne, W. A. Webre, Y. Matsushita, N. Zhu, Z. Futera, J. Labuta, W. Jevasuwan, N. Fukata, J. S. Fossey, F. D'Souza, K. Ariga, W. Schmitt and J. P. Hill, Multimodal switching of a redox-active macrocycle, Nat. Commun., 2019, 10, 1007 CrossRef.
- D. T. Payne, W. A. Webre, H. Gobese, Y. Matsushita, P. A. Karr, M. K. Chahal, J. Labuta, J. S. Fossey, K. Ariga, F. D'Souza and J. P. Hill, Nanomolecular singlet oxygen photosensitizers based on hemiquinonoid-resorcinarenes, the fuchsonarenes, Chem. Sci., 2020, 11, 2614–2620 RSC.
- L. R. Milgrom, The facile aerial oxidation of a porphyrin, Tetrahedron, 1983, 39, 3895–3898 CrossRef CAS.
- A. J. Golder, L. R. Milgrom, K. B. Nolan and D. C. Povey, 5,10,15,20-Meso-tetrakis(3,5-di-t-butyl-4-quinomethide) porphyrinogen: A highly puckered tetrapyrrolic macrocycle from the facile aerial oxidation of a phenolic porphyrin, J. Chem. Soc., Chem. Commun., 1989, 1751–1753 RSC.
- L. R. Milgrom and J. P. Hill, Aerial oxidation kinetics of a phenolic porphyrin, J. Heterocycl. Chem., 1993, 30, 1629–1633 CrossRef CAS.
- J. P. Hill, I. J. Hewitt, C. E. Anson, A. K. Powell, A. L. McCarthy, P. Karr, M. Zandler and F. D'Souza, Highly planar, electron deficient, N-substituted tetra-oxocyclohexadienylidene porphyrinogens: structural, computational, and electrochemical investigations, J. Org. Chem., 2004, 69, 5861–5869 CrossRef CAS.
- F. D'Souza, N. K. Subbaiyan, Y. Xie, J. P. Hill, K. Ariga, K. Ohkubo and S. Fukuzumi, Anion-complexation-induced stabilization of charge separation, J. Am. Chem. Soc., 2009, 131, 16138–16146 CrossRef.
- S. Ishihara, J. P. Hill, A. Shundo, K. Ohkubo, S. Fukuzumi, M. R. J. Elsegood, S. J. Teat and K. Ariga, Reversible photoredox switching of porphyrin-bridged bis-2,6-di-tert-butylphenols, J. Am. Chem. Soc., 2011, 133, 16119–16126 CrossRef CAS.
- J. Labuta, Z. Futera, S. Ishihara, H. Kourilova, Y. Tateyama, K. Ariga and J. P. Hill, Chiral guest binding as a probe of macrocycle dynamics and tautomerism in a conjugated terapyrrole, J. Am. Chem. Soc., 2014, 136, 2112–2118 CrossRef CAS PubMed.
- M. K. Chahal, D. T. Payne, J. Labuta, P. A. Karr, F. D'Souza, K. Ariga and J. P. Hill, Selective phase transfer reagents (OxP-crowns) for chromogenic detection of nitrates especially ammonium nitrate, Chem. – Eur. J., 2020, 26, 13177–13183 CrossRef CAS.
- M. K. Chahal, A. Liyanage, A. Z. Alsaleh, P. A. Karr, J. P. Hill and F. D'Souza, Anion-enhanced excited state charge separation in a spiro-locked N-heterocycle-fused push-pull zinc porphyrin, Chem. Sci., 2021, 12, 4925–4930 RSC.
- H. S. Gutowsky and A. Saika, Dissociation, chemical exchange, and the proton magnetic resonance in some aqueous electrolytes, J. Chem. Phys., 1953, 21, 1688–1694 CrossRef CAS.
- V. Římal, H. Štěpánková and J. Štěpánek, Analysis of NMR spectra in case of temperature-dependent chemical exchange between two unequally populated states, Concepts Magn. Reson., Part A, 2011, 38, 117–127 CrossRef.
- S. Glasstone, K. J. Laidler and H. Eyring, The Theory of Rate Processes, McGraw-Hill Book Company, New York and London, 1941, p. 199 Search PubMed.
- J. Wang, R. M. Wolf, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, GROMACS: A message-passing molecular dynamics implementation, Comp. Phys. Comm., 1995, 91, 43–56 CrossRef CAS.
- E. Lindahl, M. J. Abraham, B. Hess and D. van der Spoel, GROMACS 2020 Manual, 2020, DOI:10.5281/zenodo.3562512.
- S. Nose, A unified formulation of the constant yemperature molecular dynamics methods, J. Chem. Phys., 1984, 81, 511–519 CrossRef CAS.
- W. G. Hoover, Canonical dynamics: Equilibrium phase-space distributions, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef.
- T. Darden, D. York and L. Pedersen, Particle mesh: An N·log(N) method for Ewald sums large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera and M. L. Berkowitz, A smooth particle mesh Ewald method, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, Nonphysical sampling distributions in Monte Carlo free-energy estimation Umbrella sampling, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, D. Bouzida, R. H. Swendsen, P. A. Kollman and J. M. Rosenberg, THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- A. Mazzanti, L. Lunazzi, M. Minzoni and J. E. Anderson, Rotation in biphenyls with a single ortho-substituent, J. Org. Chem., 2006, 71, 5474–5481 CrossRef CAS PubMed.
- C. J. Medforth, R. E. Haddad, C. M. Muzzi, N. R. Dooley, L. Jaquinod, D. C. Shyr, D. J. Nurco, M. M. Olmstead, K. M. Smith, J.-G. Ma and J. A. Shelnutt, UnusuaL aryl-porphyrin rotational barriers in peripherally crowded porphyrins, Inorg. Chem., 2003, 42, 2227–2241 CrossRef CAS PubMed.
- E. R. Henry and J. Hofrichter, Numerical Computer Methods, in Methods in Enzymology, ed. L. Brand and M. L. Johnson, Academic Press, San Diego, CA, 1992, vol. 210, pp. 129–192 Search PubMed.
- E. R. Malinowski, Factor Analysis in Chemistry, Wiley, 2002 Search PubMed.
- P. Mojzes, P. Praus, V. Baumruk, P.-Y. Turpin, P. Matousek and M. Towrie, Structural features of two distinct molecular complexes of copper(II) cationic porphyrin and deoxyribonucleotides, Biopolymers, 2002, 67, 278–281 CrossRef PubMed.
- J. Palacký, P. Mojzeš and J. Bok, SVD-based method for intensity normalization, background correction and solvent subtraction in Raman spectroscopy exploiting the properties of water stretching vibrations, J. Raman Spectrosc., 2011, 42, 1528–1539 CrossRef.
- V. Březina, S. Ishihara, J. Lang, L. Hanyková, K. Ariga, J. P. Hill and J. Labuta, Structural modulation of chromic response: effects of binding-site blocking in a conjugated calix[4]pyrrole chromophore, ChemistryOpen, 2018, 7, 323–335 CrossRef.
- D. T. Payne, M. K. Chahal, V. Březina, W. A. Webre, K. Ariga, F. D'Souza, J. Labuta and J. P. Hill, Diporphyrin tweezer for multichannel spectroscopic analysis of enantiomeric excess, Front. Chem. Sci. Eng., 2020, 14, 28–40 CrossRef CAS.
- M. K. Chahal, J. Labuta, V. Březina, P. A. Karr, Y. Matsushita, W. A. Webre, D. T. Payne, K. Ariga, F. D'Souza and J. P. Hill, Knock-on synthesis of tritopic calix[4]pyrrole host for enhanced anion interactions, Dalton Trans., 2019, 48, 15583–15596 RSC.
- K. Naoda, D. Shimizu, J. O. Kim, K. Furukawa, D. Kim and A. Osuka, Thienylquinonoidal porphyrins and hexaphyrins with singlet diradical ground states, Chem. – Eur. J., 2017, 23, 8969–8979 CrossRef CAS.
- D. Shimizu and A. Osuka, Porphyrinoids as a platform of stable radicals, Chem. Sci., 2018, 9, 1408–1423 RSC.
- C. Fraser and R. D. Young, Stable carbocation generated via 2,5-cyclohexadien-1-one protonation, J. Org. Chem., 2018, 83, 505–509 CrossRef CAS PubMed.
- B. Kim, G. Storch, G. Banerjee, B. Q. Mercado, J. Castillo-Lora, G. W. Brudvig, J. M. Mayer and S. J. Miller, Stereodynamic quinone-hydroquinone molecules that enatiomerize at sp3-carbon via redox-interconversion, J. Am. Chem. Soc., 2017, 139, 15239–15244 CrossRef CAS PubMed.
- O. B. Youchret-Zallez, S. Besbes-Hentati, M. Bouvet and H. Said, Electrochemical and spectral studies of auto-assembled arrays of calix[4]arenequinhydrone charge-transfer complex on indium-tin ocide (ITO) glass, J. Inclusion Phenom. Macrocyclic Chem., 2014, 79, 383–390 CrossRef.
- S. Meddeb-Limem, S. Besbes-Hentati, H. Said and M. Bouvet, From the electrochemical generation of cali[4]arenequinhydrone to the electrodeposition of calix[4]arenediquinone-calix[4]arenedihydroquinone assembly, Electrochim. Acta, 2011, 58, 372–382 CrossRef CAS.
- P. R. Bangal, Hydrogen-bonding and protonation effects on the formation of charge transfer complex between para-benzoquinone and 2,6-dimethoxyphenol, Chem. Phys. Lett., 2005, 401, 200–204 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: characterization and correction of the time-dependent local density approximation ionization threshold, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo01479j |
| This journal is © the Partner Organisations 2022 |