
“The red cage”: implementation of pH-responsiveness within a macrobicyclic pyridinium-based molecular host†
Pablo
Cortón‡
 a,
Hongye
Wang‡
a,
Iago
Neira
a,
Arturo
Blanco-Gómez
a,
Elena
Pazos
a,
Carlos
Peinador
a,
Hao
Li
*b and
Marcos D.
García
*a
a,
Hongye
Wang‡
a,
Iago
Neira
a,
Arturo
Blanco-Gómez
a,
Elena
Pazos
a,
Carlos
Peinador
a,
Hao
Li
*b and
Marcos D.
García
*a
aDepartamento de Química and Centro de Investigacións Científicas Avanzadas (CICA), Facultade de Ciencias, Universidade da Coruña, 15071, A Coruña, Spain. E-mail: marcos.garcia1@udc.es
bDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: lihao2015@zju.edu.cn
First published on 9th November 2021
Abstract
We present herein the implementation of pH-responsiveness into a new polycationic macrobicyclic structure, namely what we have termed the “red cage”. The hydrolytically-stable cryptand-like compound has been prepared in a relatively high yield in aqueous media by a kinetically-controlled hydrazone-exchange reaction, promoted by the unusual high stability of the new hydrazone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds formed. In organic media the macrobicycle was found not able to complex model aromatic substrates. In buffered aqueous solutions, as a comparison, the “red cage” was found able to recognize them, but the binding was observed to be more efficient in acidic form of the cyclophane compared with its conjugate base.
N bonds formed. In organic media the macrobicycle was found not able to complex model aromatic substrates. In buffered aqueous solutions, as a comparison, the “red cage” was found able to recognize them, but the binding was observed to be more efficient in acidic form of the cyclophane compared with its conjugate base.
1. Introduction
Molecular switches1 have become the weapon of choice for chemists aiming to control the, otherwise erratic, intrinsic dynamism of chemical systems.2 These entities regulate chemical behavior in a reversible fashion, by swapping their structure between different stable states in response to external stimuli such as light, electric current, pH, or chemical effectors.3 In recent years, these responsive molecules have extensively shown their applicability on a myriad of man-controlled chemical functions such as catalysis,4 information processing,5 macroscopic motion,6 biomolecular modification,7 or phototherapy.8 Furthermore, molecular switches have demonstrated as well an incredible potential when introduced into more host–guest entities,9 in essence as control units within supramolecular10 or mechanically-interlocked switches.11 Nevertheless, the introduction of responsive capabilities into molecular receptors is not trivial.12 That is especially true for macrocyclic hosts,13 which suffer in many cases from challenging syntheses that hamper their availability14 and the fine-tuning of their function.8–11Recently, we and others have reported on the use of N-substituted imine-based chemistry for the aqueous synthesis of pyridinium-based macrocycles, cages, catenanes and molecular knots.15 Not only does this strategy allow to prepare macrocycle-containing species of adjustable kinetic stability in a predictable and modular fashion, but it opens the door as well to the implementation of dynamic characteristics inherent to hydrazone or acyl hydrazone bonds.16 In particular, some of us have recently reported on the development of the “red box” (R4+, Scheme 1),17 an hydrazone-containing analogue of the well-known “blue box” redox-responsive macrocycle (cyclobis(paraquat-p-phenylene), first developed by Stoddart et al.18R4+ was not only found to act as a pH-based molecular switch in both aqueous and organic media, but to translate this behaviour into supramolecular responsiveness on the corresponding host–guest aggregates. Crucially, the accessible pH-responsiveness of R4+ (pKa ≈ 8.7), could be correlated with the anomalous high stability of the imine bonds within the structure, provoked in turn by the high degree electronic delocalization of the π-system on each of the large sides of the molecular rectangle.
 | ||
| Scheme 1 Synthetic conditions for the preparation of the “red box” R4+, and those intended for its bicyclic analogue “red cage” C6+. | ||
Encouraged by these previous results, and the excellent 83% isolated yield obtained for the “red box” R·4PF6 despite the observed kinetic control, we decided to explore the synthesis, pH-responsiveness and host–guest chemistry of the cryptand-like analogue “red cage” C6+ (Scheme 1).§ Hence, important issues on this type of host–guest chemistry could be analyzed by comparing the obtained results for C6+ with those of the model compound R4+ (e.g. binding site flexibility, increased number of charges, etc.). Furthermore, the higher structural complexity of C6+ it is not only appealing as a synthetic challenge, but would open the door for the creation of more intricate imine-based cages (e.g. [2 + 3] or [4 + 6] cages), by using appropriate counterparts with unmatched number of aldehyde and amine reacting groups.19
2. Results and discussion
2.1. Synthesis and characterization of the “red cage”
Regarding the synthesis of the cryptand-like compound C6+, the two precursors (H1–23+, Fig. 1), were synthesized in good yields from commercially available materials.20 Whereas H13+ owns three formyl groups masked as hydrates, H23+ contains the three complementary reactive acetone-protected hydrazide units. Following the standard methodology used for the synthesis of this type of stabilized imine bonds,17 an equimolar mixture of H13+ and H23+, both at 2.5 mM in D2O, was heated at 70 °C overnight in the presence of a catalytic amount of either CF3COOH or DCl.20 These acids were used to assure the hydrolysis of hydrazone protecting groups of the hydrazine units within H23+, and to catalyze the forming of the new kinetically inert hydrazone bonds between the building blocks.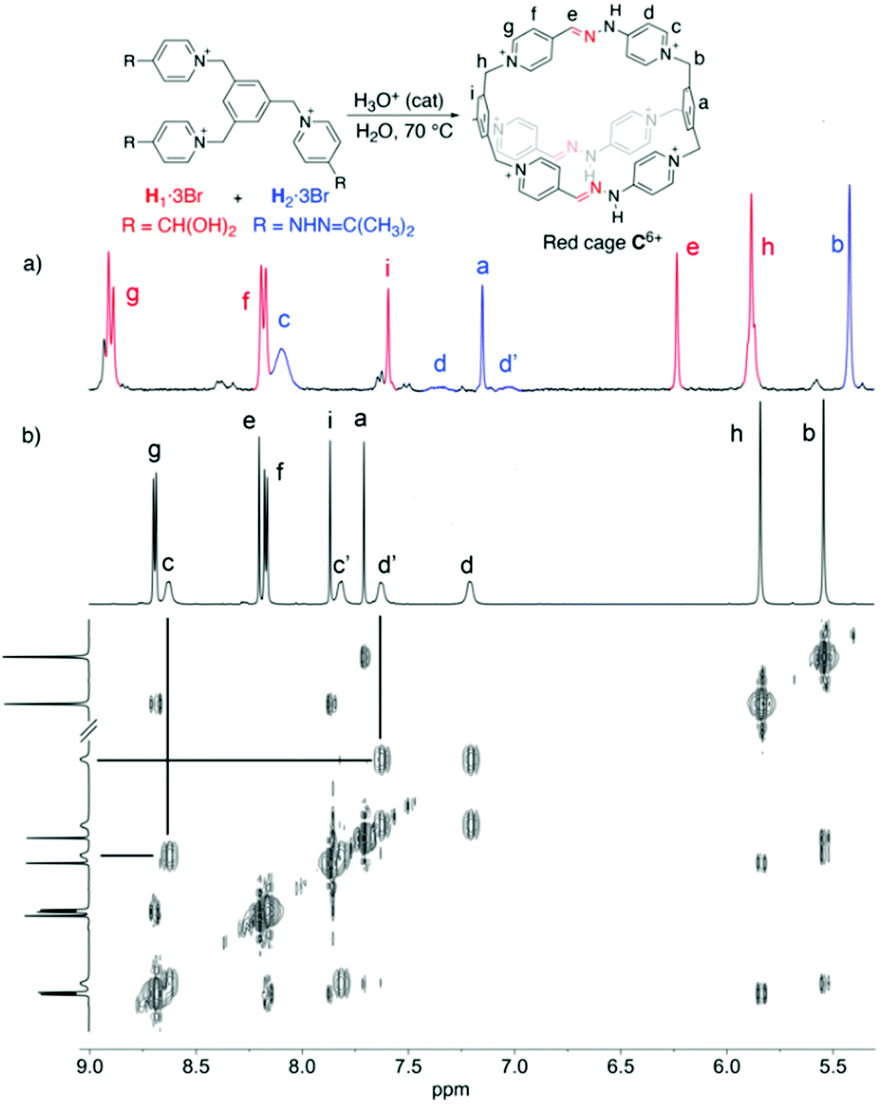 | ||
| Fig. 1 Partial 1H NMR spectra (D2O, 500 MHz) of a mixture of equimolar 2.5 mM H1/H2·3Br and a catalytic amount of CF3COOH (a) at t = 0, (b) after being heated at 70 °C overnight. 2D ROESY NMR showing exchange EXSY peaks for Hc–d/c′–d′. | ||
As shown in Fig. 1 for the CF3COOH-catalysed process, the 1H NMR spectrum recorded for the reaction mixture clearly shows the complete disappearance of the starting materials, as well as their apparent complete conversion into a new highly symmetric structure in accordance with the D3h cage C6+. In essence, the formation of the macrobicycle can be inferred by the appearance of a new imino resonance at 8.1 ppm, along with two different sets of signals for the two chemically non-equivalent pyridinium rings on the structure: one set of two well-resolved doublets for Hf–g, and four resonances in a situation of moderately slow exchange on the NMR timescale that translates in four near coalescence signals (Hc–d/c′–d′), accounting for the well-known restricted rotation around the C(Pyr)–N bond in this type of pyridinium derivatives,21 and corroborated by the corresponding EXSY exchange peaks on a 2D ROESY experiment (Fig. 1b).
The reaction was then carried out on a preparative scale,20† using the very same reaction conditions described above. After completion, isolation of crude C·6PF6 could be easily achieved, simply by adding an excess of KPF6 to the corresponding aqueous reaction mixture until no further precipitation was observed. The crude compound could be then conveniently purified by flash column chromatography, obtaining pure C·6PF6 in a 52% yield. Alternatively, semipreparative HPLC allowed as well for the obtention of pure C·6CF3COO on a similar 48% yield, with the trifluoroacetate salt being conveniently soluble both in organic and aqueous media. In turn, C·6Cl, a water-soluble counterpart of C·6PF6, could be obtained quantitatively by treatment of the hexafluorophosphate salt of the cage with excess of TBACl in CH3CN.
Extensive 1D/2D NMR experiments were recorded for both salts C·6PF6/6CF3COO in CD3CN,20 allowed us to fully assign the nuclei on the cationic cage (Fig. 2). Both compounds show similar spectroscopic features as those commented above for the crude reaction product in D2O. Nevertheless, a strong hydrogen bond between the –HeCN-NHam moieties and the CF3CO2− counterions results evident on the 1H-NMR spectrum of C·6CF3COO. In this case, both He (δ = 8.58 ppm) and Ham (δ = 14.94 ppm) are considerably deshielded because of the interaction when compared with C·6PF6 or, crucially, when compared with the 1H NMR of C·6CF3COO in more polar protic solvents, such as D2O or MeOD.
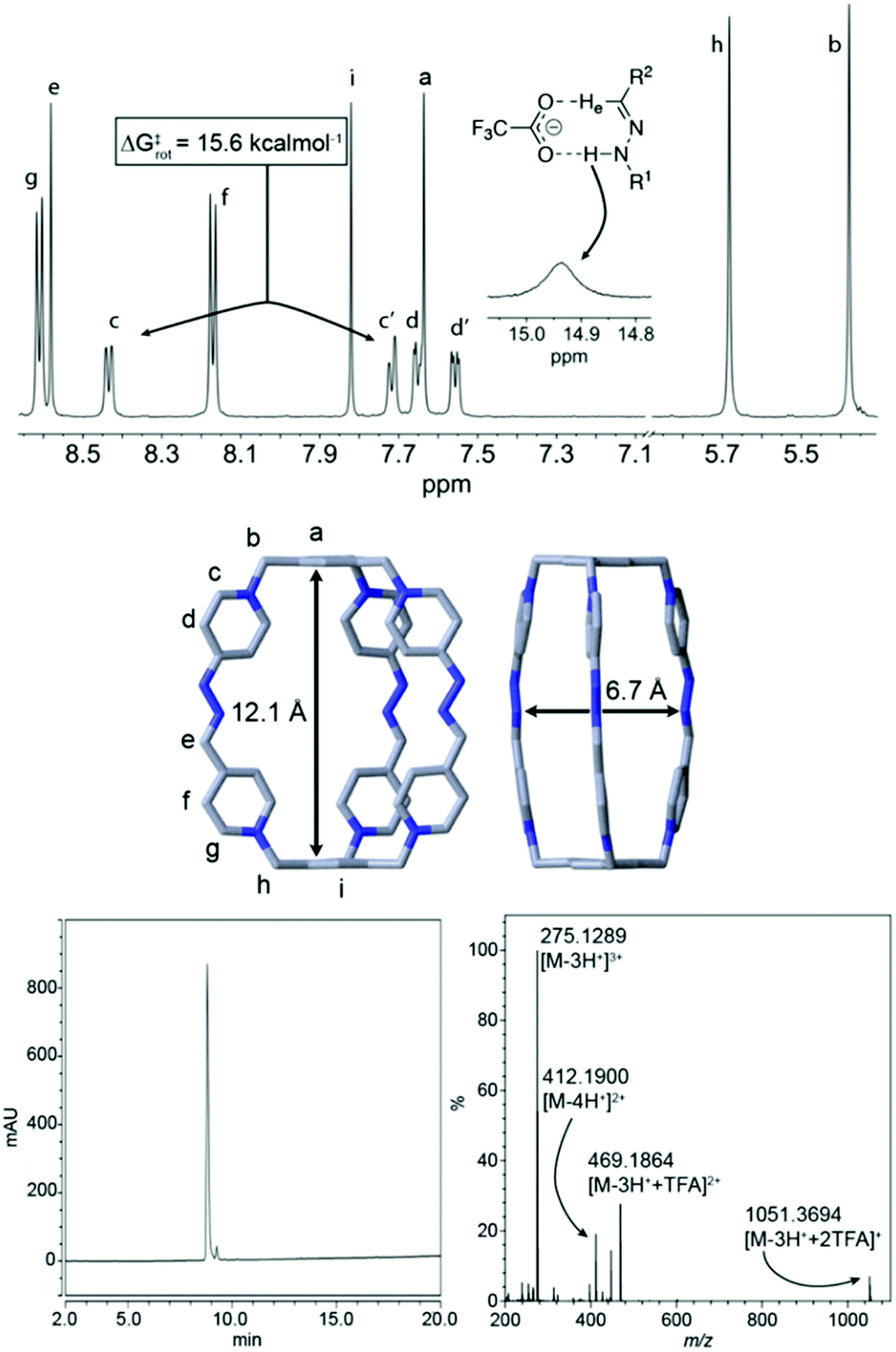 | ||
| Fig. 2 (Top) Partial 1H NMR spectra (CD3CN, 500 MHz) for C·6CF3COO; insets: ΔG‡rot calculated from the VT 1H NMR experiments by the coalescence method, for the exchange between Hc and Hc′. Proposed mode of hydrogen bonding interaction observed between the hydrazone moiety and a TFA anion. (Middle) Solid-state structure of C·6Cl obtained from single-crystal X-ray diffraction analysis. Carbon, grey; nitrogen, blue. Hydrogen atoms and chloride anions in cage frameworks are omitted for clarity. The CCDC numbers corresponding to this structure is 1988842.† (Bottom) HPLC chromatogram at 220 nm and HR ESI-MS spectrum for the isolated peak of C·6CF3COO, showing the loss of CF3CO2− counterions and protons. | ||
Another interesting feature of the 1H-NMR spectrum of C·6CF3COO in CD3CN at rt, is the appearance of 4 well-resolved resonances for the non-equivalent protons Hc–d/c′–d′, which allowed us to estimate a ΔG‡rot ≈ 15.6 kcal mol−1 for the restricted rotation of the hydrazino pyridinium moieties, from the VT 1H NMR experiments and by using the coalescence method,20 a value in good agreement with that observed in similar systems.17 The NMR data obtained in D2O for the water soluble salts C·6CF3COO/6Cl was also consistent with the proposed macrobicyclic structure. ESI-MS data recorded for C·6CF3COO similarly corroborated this end, showing the typical loss of CF3CO2− counterions and protons typical for this type of structures, and being correlated with the abnormal acidity of the NH protons on the cage (vide infra). Finally, diffraction grade single crystals (Fig. 2) of C·6Cl were obtained by slow diffusion of acetone into an aqueous solution of the salt, which provided unambiguously the architecture of the cage C6+, and shows a cavity within the cation of 12.1 × 6.2 Å2, dimensions appropriate for the inclusion of aromatic electron donors (vide infra).
2.2. Acid–base responsiveness of the “red cage”
A series of experiments were conducted in order to test the pH-responsiveness of C6+. Firstly, this was assessed in aqueous media, by performing an UV-Vis acid–base titration. On increasing the pH, the appearance of the conjugated base C3+ clearly results on a substantial decrease of the originally observed main absorption band for the bipyridinium chromophores within C6+ (λmax = 358 nm, ε = (132.4 ± 0.3) × 103 L mol−1 cm−1), associated to π–π* transitions, and the concomitant manifestation of a new band at λmax = 448 nm (ε = (148.6 ± 0.8) × 103 L mol−1 cm−1), which clearly indicates an increased charge delocalization over the pyridinium rings upon deprotonation (Fig. 3a). Although a unique clear isosbestic point is not observed on the titration experiment, hampering an accurate determination of the pKa for C·6CF3COO by UV-vis, an approximate value of 8.7 could be estimated, which is in decent agreement with the experimental data previously reported.17 This estimation allowed us to establish safe pD windows to study the NMR spectroscopic features of both C6+ (pD = 2) and its conjugate base C3+ (pD = 12). Whilst the main features of C6+ are comparable to those described for the compound in CD3CN, those obtained for the conjugate base are consistent with a drastic enhancement of the electron density on the cage, which results in the shielding of all the resonances of the pseudoviologen chromophores (Fig. 3b).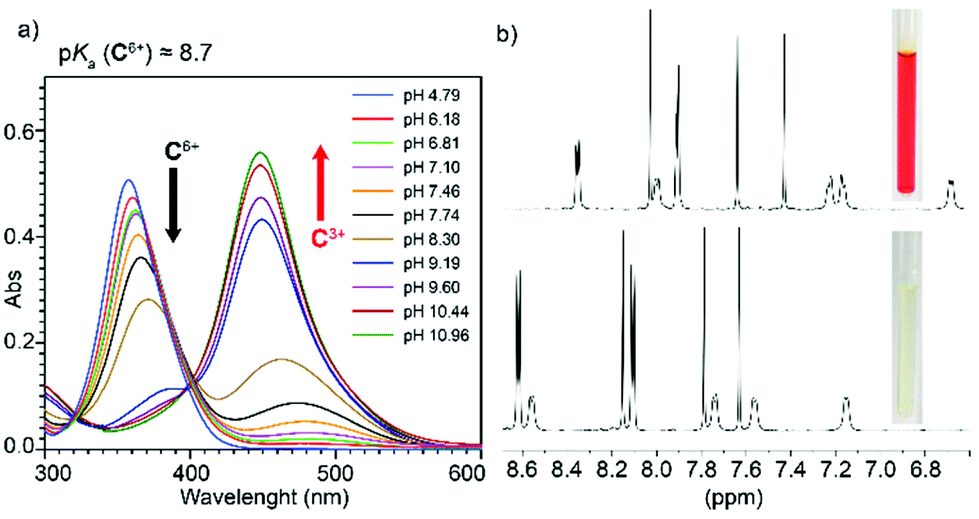 | ||
| Fig. 3 (a) UV-vis spectra for the titration of a 4.5 μM C·6CF3COO solution in H2O with aliquots of appropriate solutions of NaH2PO4/Na2HPO4 and NaHCO3/Na2CO3 buffers of increasing pH. (b) 1H NMR (D2O, r.t., 400 MHz) of C·6CF3COO at pD = 12 (top), pD = 2 (bottom); insets: photographs of the corresponding NMR solutions. | ||
The pH-responsiveness of C6+ was also qualitatively assessed in organic media.20 Addition of 1 eq. of Et3N to a 1.5 mM solution of C·6PF6 in CD3CN produced substantial changes on the 1H NMR of the macrocycle, in good agreement with its deprotonation (i.e. disappearance of the amine signal Ham, and substantial shielding of the remaining resonances due to the increased electronic density on the chromophores). The observed changes could be efficiently restored by addition of 1 eq. of CF3COOD to the organic solution. Finally, the pH-responsive behavior of the compound in CH3CN was also monitored by UV-Vis, rendering similar results as those observed in water. Accordingly, the main absorption band for C·6PF6 at λmax = 356 nm (ε = (130.5 ± 5.2) × 103 L mol−1 cm−1), significantly disappears upon deprotonation, resulting in the appearance of a new main band centered at λmax = 478 nm (ε = (147.2 ± 0.1) × 103 L mol−1 cm−1).
2.2. Host![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :guest chemistry
:guest chemistry
The host–guest chemistry of the red cage was firstly studied by means of NMR techniques in D2O. We envisioned that a series of dihydroxynaphthalenes (DHNs) would represent appropriate electron rich aromatic substrates, given the cationic and π-acceptor nature of the cage. Essentially, the complexation induced chemical shifts (CISs) observed in equimolar 0.5 mM solutions of C·6Cl and the selected guests within the aggregates in this media (Table 1), were fully consistent with the macrobicycle being able to sequester those from the aqueous media at a measured pD = 6.1. As expected for the formation of the corresponding 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complexes, the substrates showed the archetypical shielding provoked by π–π and C–H⋯π interactions.22 Furthermore, the host part of the assembly exhibited fast exchange regimes on the NMR timescale. DOSY NMR recorded for the host–guest mixtures confirmed the association in each case, with all the 1H nuclei on the complex diffusing as a whole.20 Finally, association constants for the complexes prepared from C·6Cl and the DHN derivatives could be determined by NMR titrations, yielding Ka values on the 103–104 M−1 range in good agreement with previously reported complexes (Table 1).15
:1 inclusion complexes, the substrates showed the archetypical shielding provoked by π–π and C–H⋯π interactions.22 Furthermore, the host part of the assembly exhibited fast exchange regimes on the NMR timescale. DOSY NMR recorded for the host–guest mixtures confirmed the association in each case, with all the 1H nuclei on the complex diffusing as a whole.20 Finally, association constants for the complexes prepared from C·6Cl and the DHN derivatives could be determined by NMR titrations, yielding Ka values on the 103–104 M−1 range in good agreement with previously reported complexes (Table 1).15
|
|
|||||
|---|---|---|---|---|---|
| Guest | Δδ (D2O, ppm, 0.5 mM C·6Cl + DHN) | ||||
| H1 | H2 | H3 | H4 | K a (103 M−1) | |
| a Signals of the substrate are broadened due to a near coalescence situation on the NMR timescale. | |||||
| 1,5-DHN | −1.42 | −1.60 | −1.71 | 4.9 (±0.1) | |
| 2,6-DHN | −1.60 | −1.29 | −1.22 | 1.1 (±0.1) | |
| 2,7-DHN | −1.66 | −1.26 | −1.00 | 3.9 (±0.2) | |
| Δδ (D2O, pD = 2, ppm, 0.5 mM C·6Cl + DHN) | |||||
| 1,5-DHNca | — | — | — | 20 (±2) | |
| Δδ (D2O, pD = 12, ppm, 0.5 mM C·6Cl + DHN) | |||||
| 1,5-DHNc | −1.20 | −1.87 | −1.41 | 5.3 (±0.6) | |
Diffraction grade single crystals (Fig. 4) of the complex 2,7-DHN⊂C·6Cl could be obtained, by slow vapor diffusion of acetone into an aqueous solution of an equimolar mixture of C·6Cl and 2,7-DHN. Surprisingly, the obtained structure was not that expected for the inclusion complex 2,7-DHN⊂C·6Cl, but instead the pair 2,7-DHN2−⊂C·4Cl, which would imply the deprotonation of the substrate upon complexation. Considering the weak acidic nature of both the red cage (pKa ≈ 8.7) and 2,7-DHN (pKa ≈ 9.1), the dissociation of both compounds to their conjugate bases in D2O, although not very significant, can lead to the obtention of the observed salt 2,7-DHN2−⊂C·4Cl as a kinetically-trapped species in the solid state. Regarding the structure obtained for 2,7-DHN2−⊂C·4Cl (Fig. 4), that shows the deprotonated diol not completely inserted within the cavity of the red cage, and establishing π–π interactions with two of the walls of the macrobicycle. Interestingly, four chloride anions are clustered within the cage, with geometrical parameters for two of them in perfect agreement with idealized anion–π interactions23 (Cl2/Cl4: dcentroid = dplane ≈ 3.9 Å; α ≈ 90.4°).
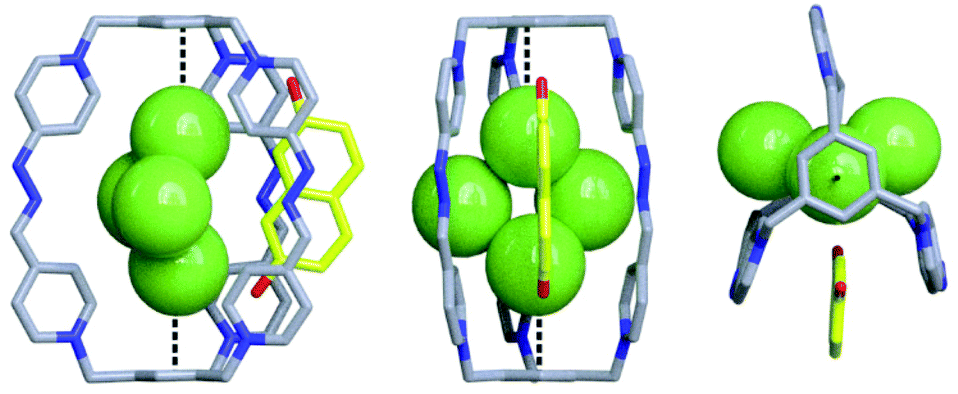 | ||
| Fig. 4 Solid-state structure of 2,7-DHN2−⊂C·4Cl obtained from single-crystal X-ray diffraction analysis. Dotted black lines used to depict the observed anion–π interactions. Carbon, grey (host) and yellow (guest); nitrogen, blue; oxygen, red; chlorine, green. Hydrogen atoms in cage frameworks are omitted for clarity. The CCDC numbers corresponding to this structure is 1988362.† | ||
In order to clarify the influence of the pH on the ability of the red cage as a molecular receptor, we decided to study the host–guest chemistry of the compound by 1H NMR spectroscopy in buffered solutions using 1,5-DHNc (see structure in Table 1), as an appropriate water-soluble and pH-insensitive electron donor. To assure sufficiently acidic or basic conditions to study either the protonated C6+ form or its conjugated base, these studies were carried out in buffered aqueous media at pD = 2 (C6+) < pKa ≈ 8.7 < pD = 12 (C3+). In both cases, the results point out to the formation of the corresponding inclusion complexes, with notable differences being observed on the corresponding NMR experiments. Nevertheless, only at pD = 12 well-resolved resonances are observed both for host and guest, with CISs in good agreement with the insertion of the substrate within the cavity of the macrobyciclic host (Table 1). On the contrary, at pD = 2, a less clear situation is observed, with only few diagnostic signals on the host (i.e. the Hg signal, Fig. 5), but not the guest, escaping from a near coalescence state on the NMR timescale. Titration experiments allowed us to estimate the association constants, in good agreement with 1:1 complexes. The Ka of 1,5-DHNc⊂C6+ and 1,5-DHNc⊂C3+ are 2.0 (±0.2) × 104 and 5.3 (±0.6) × 103 M−1, respectively (Fig. 5b). These values nicely agree with the increased electron acceptor character of C6+ compared with its conjugate base, namely C3+, and with the hydrophobic effect being the main driven force of the complexation. This end was further corroborated by studying the interaction between 1,5-DHNc and C·6CF3COO in CD3CN, which resulted in no appreciable complexation of the guest in this media.
 | ||
| Fig. 5 (a) Schematic representation of the equilibria involved on the complexation in aqueous media of 1,5-DHNc by C6+ and its conjugate base C3+. (b) Fitting of the 1H titration data of Hg for 1,5-DHNc⊂C·6CF3COO (top) and 1,5-DHNc⊂C·3CF3COO (bottom). | ||
3. Conclusions
We have described here the extension of our previously reported hydrazone condensation methodology for the synthesis of new pyridinium hosts, by developing a new hexacationic cryptand-like macrocycle termed the “red cage”. This host molecule was obtained in an excellent yield considering the challenging macrocyclization kinetically-controlled process that implies. As its model monocyclic congener, the “red cage” has shown a remarkable pH-responsiveness and the ability to complex model aromatic substrates based on π–π/C–H⋯π interactions and, mainly, due to the hydrophobic effect in aqueous media. This importance is corroborated by the observation of the cage being not able to complex the pH-insensitive substrate 1,5-DHNc in organic medium. Furthermore, in buffered aqueous solutions, a difference of just an order of magnitude was found between the association constants of 1,5-DHNc and, respectively, the hexacationic cage and its tricationic conjugated base. In summary, we believe that results discussed herein further establish the reliability of imine bonding for the synthesis of new pyridinium-based macrocycles, compounds able to display remarkable differences in their abilities as hosts in response to modifications on the reaction media (polarity, pH, etc.).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the funding received from the Agencia Estatal de Investigación and FEDER (PID2019-105272GB-I00 and CTQ2017-89166-R), the Xunta de Galicia (ED431C 2018/39 and 508/2020), and the European Research Council (grant agreement no. 851179). I. N. and P. C. thank, respectively, the MECD (FPU program, FPU2016/02347) and Xunta de Galicia (Axudas de Apoio á Etapa de Formación Predoutoral, ED481A-2020/019) for financial support. A. B. G. thanks Xunta de Galicia for a Postdoctoral Fellowship (ED481B-2021-099). E. P. thanks the Agencia Estatal de Investigación for her Ramón y Cajal contract (RYC2019-027199-I). H. Li wants to thank the financial supports from the National Natural Science Foundation of China (no. 91856116, 21772173, and 21922108), and the Natural Science Foundation of Zhejiang Province (no. LR18B020001). The authors are deeply grateful to professor Jonathan L. Sessler, for promoting and encouraging the collaboration reported herein: “we should be competing with Mother Nature to extract her secrets, not with one another!”.Notes and references
- Molecular switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed.
- J.-M. Lehn, Perspectives in Chemistry—Aspects of Adaptive Chemistry and Materials, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- For selected recent reviews on molecular switches see, for instance: (a) M.-M. Russew and S. Hecht, Photoswitches: From Molecules to Materials, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS; (b) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed; (c) K. Rafal, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43, 148–184 RSC; (d) D. Bleger and S. Hecht, Visible-Light-Activated Molecular Switches, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef CAS; (e) A. Fihey, A. Perrier, W. R. Browned and D. Jacquemin, Multiphotochromic molecular systems, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC; (f) I. Aprahamian, Hydrazone switches and things in between, Chem. Commun., 2017, 53, 6674–6684 RSC; (g) J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, Viologen-inspired functional materials: synthetic strategies and applications, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- (a) V. Blanco, D. A. Leigh and V. Marcos, Artificial switchable catalysts, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC; (b) R. Dorel and B. L. Feringa, Photoswitchable catalysis based on the isomerisation of double bonds, Chem. Commun., 2019, 55, 6477–6486 RSC.
- J. Andreasson and U. Pischel, Light-stimulated molecular and supramolecular systems for information processing and beyond, Coord. Chem. Rev., 2021, 429, 213695 CrossRef CAS.
- (a) F. Lancia, A. Ryabchun and N. Katsonis, Life-like motion driven by artificial molecular machines, Nat. Rev. Chem., 2019, 3, 536–551 CrossRef CAS; (b) Q. Zhang, D.-H. Qu, H. Tian and B. L. Feringa, Bottom-Up: Can Supramolecular Tools Deliver Responsiveness from Molecular Motors to Macroscopic Materials?, Matter, 2020, 3, 355–370 CrossRef.
- K. Hull, J. Morstein and D. Trauner, In Vivo Photopharmacology, Chem. Rev., 2018, 118, 10710–10747 CrossRef CAS.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymanski, Photoresponsive molecular tools for emerging applications of light in medicine, Chem. Sci., 2020, 11, 11672–11691 RSC.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Nanoparticles functionalised with reversible molecular and supramolecular switches, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC.
- For some selected reviews on supramolecular switches and their applications, see: (a) S. Dong, B. Zheng, F. Wang and F. Huang, Supramolecular Polymers Constructed from Macrocycle-Based Host–Guest Molecular Recognition Motifs, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS; (b) N. Songa and Y.-W. Yang, Molecular and supramolecular switches on mesoporous silica nanoparticles, Chem. Soc. Rev., 2015, 44, 3474–3504 RSC; (c) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Photoresponsive Host–Guest Functional Systems, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS; (d) X. Ma and Y. Zhao, Biomedical Applications of Supramolecular Systems Based on Host–Guest Interactions, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS; (e) E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. García, Cucurbit[8]uril (CB[8])-Based Supramolecular Switches, Angew. Chem., Int. Ed., 2019, 58, 403–416 CrossRef CAS.
- For some selected reviews on mechanically-interlocked molecular switches and their applications, see: (a) W. Yang, Y. Li, H. Liu, L. Chi and Y. Li, Design and Assembly of Rotaxane-Based Molecular Switches and Machines, Small, 2012, 8, 504–516 CrossRef CAS; (b) M. J. Langton and P. D. Beer, Rotaxane and Catenane Host Structures for Sensing Charged Guest Species, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed; (c) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Stimuli-Responsive Metal–Ligand Assemblies, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed; (d) J. F. Stoddart, Mechanically Interlocked Molecules (MIMs)—Molecular Shuttles, Switches, and Machines (Nobel Lecture), Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS; (e) N. Pairault and J. Niemeyer, Chiral Mechanically Interlocked Molecules – Applications of Rotaxanes, Catenanes and Molecular Knots in Stereoselective Chemosensing and Catalysis, Synlett, 2018, 29, 689–698 CrossRef CAS.
- (a) M. Natalia and S. Giordani, Molecular switches as photocontrollable “smart” receptors, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC; (b) A. Blanco-Gómez, P. Cortón, L. Barravecchia, I. Neira, E. Pazos, C. Peinador and M. D. García, Controlled binding of organic guests by stimuli-responsive macrocycles, Chem. Soc. Rev., 2020, 49, 3834–3862 RSC.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Surveying macrocyclic chemistry: from flexible crown ethers to rigid cyclophanes, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed.
- For two recent reviews on the topic, see: (a) T. Jiao, G. Wu, Y. Zhang, L. Shen, Y. Lei, C.-Y. Wang, A. C. Fahrenbach and H. Li, Self–Assembly in Water with N-Substituted Imines, Angew. Chem., Int. Ed., 2020, 59, 18350–18367 CrossRef CAS PubMed; I. Neira, A. Blanco-Gómez, J. M. Quintela, Marcos D. García and C. Peinador, Dissecting the “Blue Box”: Self-Assembly Strategies for the Construction of Multipurpose Polycationic Cyclophanes, Acc. Chem. Res., 2020, 53, 2336–2346 Search PubMed.
- (a) X. Su and I. Aprahamian, Hydrazone-based switches, metallo-assemblies and sensors, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC; (b) B. Shao and I. Aprahamian, Hydrazones as new molecular tools, Chem, 2020, 6, 2162–2173 CrossRef CAS.
- (a) A. Blanco-Gómez, Á. Fernández-Blanco, V. Blanco, J. Rodríguez, C. Peinador and M. D. García, Thinking outside the “Blue Box”: induced fit within a unique self-assembled polycationic cyclophane, J. Am. Chem. Soc., 2019, 141, 3959–3964 CrossRef PubMed; (b) A. Blanco-Gómez, I. Neira, J. L. Barriada, M. Melle-Franco, C. Peinador and M. D. García, Thinking outside the “Blue Box”: from molecular to supramolecular pH-responsiveness, Chem. Sci., 2019, 10, 10680–10686 RSC.
- For a recent account on the development of the “blue box” and analogues, see: E. J. Dale, N. A. Vermeulen, M. Juríček, J. C. Barnes, R. M. Young, M. R. Wasielewski and J. F. Stoddart, Supramolecular Explorations: Exhibiting the Extent of Extended Cationic Cyclophanes, Acc. Chem. Res., 2016, 49, 262–273 CrossRef CAS PubMed.
- K. Acharyya and P. S. Mukherjee, Organic Imine Cages: Molecular Marriage and Applications, Angew. Chem., Int. Ed., 2019, 58, 8640–8653 CrossRef CAS PubMed.
- See the ESI† for further details.
- See, for instance: (a) M. Prinz, S. Parlar, G. Bayraktar, V. Alptuzun, E. Erciyas, A. Fallarero, D. Karlsson, P. Vuorela, M. Burek, C. Forster, E. Turunc, G. Armagan, A. Yalcin, C. Schiller, K. Leuner, M. Krug, C. A. Sotriffer and U. Holzgrabe, 1, 4-Substituted 4-(1H)-pyridylene-hydrazone-type inhibitors of AChE, BuChE, and amyloid-β aggregation crossing the blood–brain barrier, Eur. J. Pharm. Sci., 2013, 49, 603–613 CrossRef CAS PubMed; (b) S. Parlar, Y. Erzurumlu, R. Ilhan, P. B. Kırmızıbayrak, V. Alptüzün and E. Erciyas, Synthesis and evaluation of pyridinium–hydrazone derivatives as potential antitumoral agents, Chem. Biol. Drug Des., 2018, 92, 1198–1205 CrossRef CAS PubMed.
- See, for instance: (a) C. Alvariño, E. Pía, M. D. García, V. Blanco, A. Fernández, C. Peinador and J. M. Quintela, Chem. – Eur. J., 2013, 19, 15329–15335 CrossRef; (b) T. Rama, E. M. López-Vidal, M. D. García, C. Peinador and J. M. Quintela, Dimensional Matching of Polycyclic Aromatics with Rectangular Metallacycles: Insertion Modes Determined by [C–H⋯π] Interactions, Chem. – Eur. J., 2015, 21, 9482–9487 CrossRef CAS.
- According to Hay et al.,24 the interaction mode between anions and π-deficient arenes can be semiquantitatively evaluated by the following parameters: (1) dcentroid: the distance between the anion atom(s) and the center of the ring, (2) dplane the distance from the halide to the ring plane, (3) doffset: the distance (dcentroid2 − dplane2)1/2, and (4) the angle α. The doffset parameter would display a value of 0 Å for an idealized centered anion–π complex.
- (a) O. B. Berryman, V. S. Bryantsev, D. P. Stay, D. W. Johnson and B. P. Hay, Structural criteria for the design of anion receptors: the interaction of halides with electron-deficient arenes, J. Am. Chem. Soc., 2007, 129, 48–58 CrossRef CAS PubMed; (b) B. P. Hay and R. Custelcean, Anion− π interactions in crystal structures: commonplace or extraordinary?, Cryst. Growth Des., 2009, 9, 2539–2545 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1988362 and 1988842. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo01331a |
| ‡ These two authors contributed equally to the work presented herein. |
| § By analogy with the term “blue box”, used by Stoddart et al. to describe the characteristic blue color displayed by the cation radical produced upon reduction of the viologen-based moieties,18 we have termed our hydrazone-containing analogue as the “red cage”, due to the strong red coloration shown by the conjugate bases of the corresponding bipyridinium moieties.17 |
| This journal is © the Partner Organisations 2022 |