
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of H-bonding on network junction and macroscopic elastomer properties in photocured polyacrylate films†
Bing
Wu
 *abc,
Walter
Chassé
d,
Andreas
Heise
b,
Arno P. M.
Kentgens
e,
Dermot F.
Brougham
*b and
Victor M.
Litvinov
*f
*abc,
Walter
Chassé
d,
Andreas
Heise
b,
Arno P. M.
Kentgens
e,
Dermot F.
Brougham
*b and
Victor M.
Litvinov
*f
aDutch-Belgian Beamline (DUBBLE), ESRF- The European Synchrotron Radiation Facility, CS 40220, 38043 Grenoble Cedex 9, France. E-mail: friedrichbing.wu@utoronto.ca
bSchool of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland
cDepartment of Chemistry, RCSI University of Health Sciences, 123 St. Stephen's Green, Dublin 2, Ireland
dInstitut für Physikalische Chemie, University of Münster, Corrensstr. 28/30, 48149 Münster, Germany
eInstitute for Molecules and Materials, Radboud University Nijmegen, Heyendaalsweg 135, 6525 AJ Nijmegen, The Netherlands
fV. Lit Consult, Gozewijnstraat 4, 6191WV, Beek, The Netherlands
First published on 24th January 2022
Abstract
Two series of polyacrylate films with different H-bonding capable monoacrylate copolymers were synthesized by UV-initiated photo-polymerization. Detailed IR analyses on cured samples show that networks made from structurally distinguishable mono-acrylate have significantly different extent of H-bonded monoacrylate present. We found that this is induced by differences in the relative reaction rate between homo-polymerization and copolymerization of the relevant monoacrylates and crosslinker (PEGDA). Although ‘pre-organization’ of H-bond capable monoacrylates was observed for those tested formulations prior to crosslinking, which should significantly increase the homo-polymerization rate, it was found that the co-polymerization rate was also significantly increased. Furthermore, DQ NMR analyses on these networks pinpoints the presence of H-bonding clusters, which decreases mobility of chain segments near the H-bonding functional groups in the monoacrylate sidechains (dangling chain segments of monoacrylate). However, the overall network topology does not alter significantly upon forming H-bonding clusters in the monoacrylate sidechains. Finally it was also found that larger H-bonding clusters increase the apparent network junction functionality. These findings are discussed in the context of utilizing the H-bonding capable monoacrylates to tailor the microscopic topological properties as well as macroscopic physical properties of photocured polyacrylate polymer films.
1. Introduction
Polyacrylate-based films have been widely used for biomedical applications ranging from tissue engineering to drug delivery due to their excellent biocompatibility and substantial durability. Among the available methods for producing polyacrylates, photo-initiated curing is the most popular approach implemented in industrial manufacture due to its ease of application and fast production.1–4 Modification of photo-cured crosslinked networks can be easily achieved by copolymerizing functionalized (e.g. hydrogen bonding capable) mono-vinyl monomers with divinyl crosslinkers, enabling tailoring of their macroscopic properties. However, as a consequence of the uncontrollable nature of free radical polymerization, the resulting materials often have complicated microscopic structures and network topologies which in turn strongly affects their characteristics in bulk.5,6 Hence, insights into the effects of including functionalized monoacrylates on the local structure and chain dynamics within photo-cured polyacrylate networks are of fundamental interest for designing new functional materials.H-bond capable monomers have been frequently introduced into polymer networks to alter their apparent network density in order to change their macroscopic physical properties. Burattini et al. constructed a polyimide based tough elastomeric material with self-healable properties arising from H-bonding and π–π stacking interactions between di-imide and pyrenyl groups.7 Bhattarai et al. created a chitosan-based tunable polymer network for localized drug delivery where the release mechanism was controlled by the size of domains arising due to H-bonding groups.8 In general, the glass transition temperature (Tg),9,10 network morphology,11,12 viscoelastic property13 as well as the mechanical strength14 of modified networks have been shown to be strongly influenced by the presence of H-bonding, both directly by altering chain interactions and indirectly through cluster formation. UV-curable (meth)acrylate monomers with H-bonding capability typically involve hydroxyl (–OH) or amine (–NH2) groups. This seemingly simple modification has been found to induce significant changes in the bulk properties. Peppas et al. analyzed a series of poly(ethylene glycol) based polyacrylate networks and found H-bonding strongly affects solute diffusion through the networks.15 Lewis et al. described a strong correlation between monomer H-bonding strength and the viscoelastic properties of loosely crosslinked H-bond capable polyacrylates.13 We have previously described how H-bonding can induce changes in mesh size, as assessed by NMR diffusometry and DSC thermoporomety.16 Hence, while H-bonding capable monomers can in principle be used to regulate the final properties, the potential to exploit this approach to provide materials with tunable properties for biomedical application is limited by the lack of understanding into how changes at a molecular level can induce local and intermediate-range structural modifications that determine the emergent physical properties of the cured polymer networks.
Multiple techniques are used in combination to probe the complex chemical structure and molecular dynamics in crosslinked polymeric network. Nuclear magnetic resonance (NMR) owing to its non-destructive and informative nature has been widely applied in analyzing these systems.17 While the conventional NMR spectral analysis provides highly selective structure determination,18–20 NMR relaxometry provides further insights into polymer chain dynamics.21,22T2 relaxometry is more commonly used for polymeric materials due to greater sensitivity to small differences23,24 in crosslink density and chain mobility. The ongoing developments in this important field have been reviewed on several occasions.25,26 By implementing Hahn-echo-based T2 analysis, Litvinov et al. successfully evaluated crosslink density of UV-cured PEG-DA networks.6 Recently, application of high resolution multiple quantum, multi-quantum (MQ) NMR, techniques to polymeric materials has been implemented, with chemical shift-specified residual dipolar coupling constants, Dres, now obtainable through accepted experimental and regularization procedure.27,28 We have previously shown that T2 and MQ techniques can be particularly useful when combined for extracting information about network crosslink density and its relation to chain dynamics, as modulated by functional group type and relative composition.5
In this study two series of UV-polymerized poly(ethylene glycol)-based polyacrylate networks were synthesized by copolymerizing the diacrylate (PEGDA-700) with monoacrylates, bearing either terminal hydroxy (t-OH) or non-terminal amide (nt-NH) H-bond capable functional groups on their side chains (see Table 1) forming networks with multifunctional acrylic network junctions interconnected by PEG chains with a fraction of dangling monoacrylate sidechains. The weight percentage (w%) of the PEGDA-700 in the curing formulation was systematically varied to evaluate the effect of gradually increasing the H-bonding capability on the networks. After confirming high double bond conversion by FTIR and 13C NMR spectroscopy, the networks were studied by a series of analytical techniques. The H-bonded fractions were estimated from the O–H and N–H IR stretching bands in detailed FTIR analyses, while the polymer chain dynamics were evaluated by dynamic mechanical analyses (DMA), low field 1H NMR T2 relaxometry and high field DQ NMR. We demonstrate how strikingly different properties can emerge due to the formation of clusters of H-bonded mono-arylate units, illustrated schematically in Fig. 1, associated with the sidechains, whose sizes are regulated by the relative ratio between homo- and co-polymerization of the H-bonding capable monomers. The effects of H-bonding clusters on network chain mobility were further examined using chemical-shift specified residual dipolar coupling constants, Dres, extracted from high resolution DQ NMR. Finally, the impact of H-bonding on the curing kinetics, resulting in differences in cluster formation, and on the network chain dynamics of H-bonding capable side chains and polyacrylate network junctions are discussed. That comparative discussion is facilitated by the inclusion of relevant data from our earlier NMR and DMA study5 on similar networks that were formed using the hexyl acrylate (HA) monomer which contain no H-bond capable groups.
| Compounds | Structure | M w (g mol−1) |
|---|---|---|
| a HA is included here as data5 for networks formed using it are used in the comparisons analyses below. | ||
| Poly(ethylene glycol) diacrylate (PEGDA) |

|
700 |
| 4-Hydroxyethyl acrylate (HEA) |
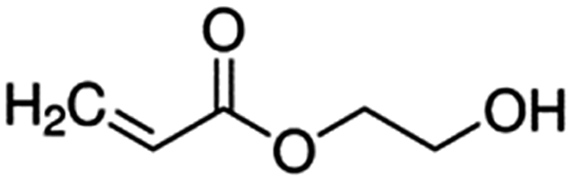
|
116.1 |
| 2-[[(Butylamino)carbonyl]oxy]ethyl acrylate (BAC) |
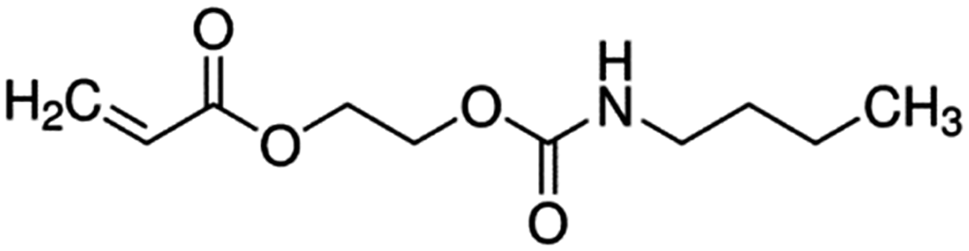
|
215.5 |
| Hexyl Acrylate (HA)a |
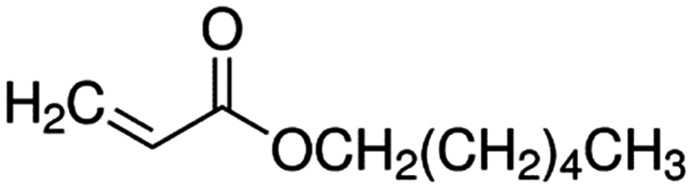
|
156.2 |
| 2,2-Dimethoxy-2-phenylacetophenone (DMPA) |

|
256.3 |
 | ||
| Fig. 1 Illustration of the formation of microscopic H-bonding clusters during bulk UV curing of a polyacrylate networks. | ||
2. Experimental
2.1. Sample preparation
Polyethylene glycol-700-diacrylate (PEGDA-700, Mn 700 g mol−1, nav 16, PDI: 1.05), Hexyl acrylate (HA), hydroxyethyl acrylate (HEA), 2-[[(butylamino)carbonyl]oxy]ethyl acrylate (BAC); photo-initiator: 2,2-dimethoxy-2-phenylacetophenone (DMPA) were used for the syntheses of networks. All the chemicals were obtained from Sigma Aldrich and used without any further purification.Networks were prepared by bulk UV-curing of the chemicals (typically 5 g of mixture) with the proportion described in Table 2 (Photo initiator: 10–20 mg 2,2-dimethoxy-2-phenylacetophenone (DMPA)). The formulation was poured into a Petri dish, and then the Petri dish was passed through the UV-rig (Heraeus Noblelight Fusion UV Inc.'s F300S model, with I300MB irradiator which has a lamp power of 300 W in−2 (120 W cm−2), 1800 W total) 30 times. Afterwards, the cured film was inverted, and was then passed through the UV-rig a further 30 times. The rim of the resulting film was cut off, and the remaining part with a thickness around 1 mm was used for all the tests.
| No. | MA Type | w%MAa (%) | Conversionb (%) | DMAc | GPCd | T 2 relaxometrye | DQ NMRf | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T g (K) | E′ (MPa) | M n (PAA) (kDa) | PDI (PAA) | n(PAA) | T dry2,av (μs) | T sw2,av(μs) | D avg/2π (kHz) | ||||
a The remaining w% in each case is PEGDA, summing to 100%.
b Determined by FTIR analyses of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching band (810 cm−1).
c
E′ is determined at 50 °C by DMA.
d Polyacrylic acid (PAA) derived from the backbone component following network hydrolysis, was characterized by gel permeation chromatography; Mn, number average molecular weight; PD, polydispersity; n(PAA), degree of polymerization. The error in Mn is estimated at c. 10%.
e
T
dry2,av, Tsw2,av, average T2 relaxation for dry and swollen samples at 70 °C, respectively; the standard deviation is c. 10%.
f
D
avg, average residual dipolar coupling constant recorded at 70 °C, the standard deviation is again estimated at c. 10%. C–H stretching band (810 cm−1).
c
E′ is determined at 50 °C by DMA.
d Polyacrylic acid (PAA) derived from the backbone component following network hydrolysis, was characterized by gel permeation chromatography; Mn, number average molecular weight; PD, polydispersity; n(PAA), degree of polymerization. The error in Mn is estimated at c. 10%.
e
T
dry2,av, Tsw2,av, average T2 relaxation for dry and swollen samples at 70 °C, respectively; the standard deviation is c. 10%.
f
D
avg, average residual dipolar coupling constant recorded at 70 °C, the standard deviation is again estimated at c. 10%.
|
|||||||||||
| 1 | HEA | 10 | 91 ± 5 | 246 | 28.3 | 9.5 | 2.2 | 131 | 154 | 160 | 2.26 |
| 2 | HEA | 30 | 95 ± 5 | 254 | 20.8 | 10.5 | 2.1 | 146 | 174 | 188 | 1.83 |
| 3 | HEA | 50 | 92 ± 5 | 263 | 13.5 | 11.8 | 2.5 | 164 | 212 | 237 | 1.51 |
| 4 | HEA | 70 | 91 ± 5 | 274 | 7.5 | 12.7 | 2.7 | 177 | 261 | 319 | 0.99 |
| 5 | HEA | 90 | 90 ± 5 | 289 | 2.8 | 11.3 | 2.6 | 157 | 324 | 565 | 0.43 |
| 6 | BAC | 10 | 92 ± 5 | 247 | 27.1 | 11.2 | 2.2 | 155 | 150 | 164 | 2.20 |
| 7 | BAC | 30 | 93 ± 5 | 252 | 19.5 | 10.2 | 2.7 | 142 | 172 | 188 | 1.71 |
| 8 | BAC | 50 | 91 ± 5 | 260 | 12.5 | 12.8 | 2.0 | 178 | 226 | 247 | 1.43 |
| 9 | BAC | 70 | 91 ± 5 | 268 | 7.0 | 11.5 | 2.6 | 160 | 332 | 418 | 0.89 |
| 10 | BAC | 90 | 89 ± 5 | 274 | 2.5 | 9.9 | 2.1 | 138 | 574 | 697 | 0.42 |
The double bond conversions of the films (which describes the completeness of crosslinking reaction) was measured by FTIR, and molecular weight of the hydrolyzed acrylate backbone chains was determined by chromatographic analyses.29 Results of network characterization are summarized in Table 2.
2.2. ATR-FTIR analyses
All the attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) analyses were performed on a Bruker Alpha-P FTIR spectrometer for the original uncured acrylate mixture and the cured polyacrylate films. The spectrometer consisted of a diamond crystal ATR with a KBr beamsplitter. The scanning range of the FT-IR used for this analysis was ranged from 360–4000 cm−1 with a resolution of 2 cm−1. 64 scans were collected for each sample in total. Examples of spectral deconvolution can be found in Fig. S10 (ESI†).2.3. Mechanical analyses
The glass transition temperature and storage modulus of all samples were measured (under elongation without pre-stretching) with a TA DMA Q800 Analyzer at a frequency of 1 Hz. Dog-bone test sample bars of 5 mm width along the working region, and 15 mm length were cut from cured films. The thickness of the test bars were measured with a calibrated micrometer. The DMA test were initiated at −90 °C and the temperature was gradually increased up to 150 °C with a ramp speed of 3 °C min−1. The oscillatory force was 13.28 N. The typical displacement (extension) was around 4–8 μm (depending on sample).2.4. Solid state NMR analyses of network structure
Solid-state magic angle spinning (MAS) 13C NMR spectra were recorded using a Bruker Avance III 600 MHz instrument equipped with a solid standard-bore Magic Angle Spinning (SBMAS) probe. A 4 mm MAS rotor (of 80 μL sample volume) was used with a spinning rate of 12 kHz. Inverse-gated decoupling and cross-polarization (CP) pulse sequences were applied. The 90° pulse length was calibrated to be 2 μs. The spectra were recorded with two different cross-polarization times (τcp) of 0.5 and 10 ms, which were selected to distinguish between rigid and soft parts of the material. The data recorded using 10 ms is shown in ESI.†2.5. 1H NMR T2 relaxation test
The 1H magnetization spin–spin relaxation (T2) times of the networks were measured using a Bruker Minispec MQ20 spectrometer operating at 20 MHz. The T2 relaxation decay was measured with the Hahn-echo (HE) pulse sequence, 90x°—τHE—180y°—τHE—(acquisition), where τHE ≥ 35 μs. In the HE experiment, an echo signal is formed after the second pulse in the HE with a maximum at time t = 2τHE after the first 90° pulse. By varying τHE, the amplitude of the transverse magnetization, A(t), is measured as a function of time t. The 1H NMR T2 relaxation experiments in this study were performed at 70 °C, which is >60 °C above the Tg value of the networks. For experiments on swollen samples, samples were swollen in the deuterated solvent C2D2Cl4 overnight before the measurement. Detailed description about the T2 fitting can be found in ESI† as well as our previous work.52.6. Homonuclear 1H–1H double quantum-single quantum (DQ-SQ) correlation NMR
The 2D 1H double-quantum (DQ) correlation experiments were carried out on a Varian VNMRS 850 MHz operating at a 1H Larmor frequency of 849.71 MHz equipped with a 1.6 mm triple resonance probe. The MAS frequency was set to 40 kHz and the sample temperature to 70 °C. The 90° pulse length was calibrated to 2 μs. The data for all network samples were acquired using a recently published improved version of the Back-to-Back (BaBa) sequence, BaBa-xy16, for broadband homonuclear DQ recoupling.28 The corresponding coupling constant distributions are obtained by the evaluation of the normalized build-up curves with ftikreg as describe in our previous work.30 The extracted residual dipolar coupling constants (Dres values) depend on the amplitude of the segmental dynamics, e.g. bond vector fluctuations in a large flexible molecule regarding to the angle between internuclear vector and the symmetry axis of motion.283. Results
3.1. Network chemical structure characterization
Two series of networks containing either t-OH (PEG-HEA) or nt-NH (PEG-BAC) groups with increasing weight fraction of monoacrylate (MA), w%MA (w%MA = 100 × wtMA/(wtMA + wtPEG), wtMA is the weight of monoacrylate, and wtPEG the weight of PEGDA), were prepared as described in Section 2. To confirm complete crosslinking polymerization of the formulations, 13C solid state NMR spectra and IR spectra were recorded for all the samples. 1H solution-state NMR was used for analysis of the sol fractions. The formulations and primary characterization for all samples in the two series are shown in Table 2, spectra and details of the analyses are included in the ESI† as well as in our previous publication.5 The double bond conversions for all the samples are found to be very high (>90%), and the amount of sol fraction determined by extraction was below 1 w%. No significant side reactions occurred during the curing procedure for either series, as confirmed by 13C solid state NMR analysis.According to the procedure described in our previous publication,5,29Mn and PDI values of the polyacrylic acid (PAA) present in the hydrolyzed networks were obtained by size exclusion chromatography (SEC) and listed in Table 2 (details of the analyses are in ESI†). PEG fraction was measured with mass spectrometry detection, assuming equal MS sensitivity, PEG has a Mn value of 488 Da. The molecular weight (Mn) of the acrylate backbone chains, a characteristic parameter for UV-cured PEGDA-based networks,29 was also measured. It was found there is no statistically significant change across the network series, given the estimated error of ∼10%, and the values are similar to those we reported when using a non H-bonding monomer.5 In general, these values are found to depend on the polymerization rate and on the bimolecular termination rate,31 which is apparently similar for the different samples.
3.2. Spectral analysis of hydrogen bonding in the networks
Several methods can demonstrate the presence of H-bonding in polymeric networks, with FT-IR most commonly used owing to its convenience.32,33 H-bonding usually significantly reduces the O–H (or N–H) bond strength, lowering the force constant and shifting the stretching modes to lower wavenumbers. For the PEG-HEA network series, it is observed that the O–H stretching band energy decreases progressively with increase in w%HEA (Fig. 2a). Typically, the stretching band of free hydroxyl groups connected to an aliphatic chain is found well above 3520 cm−1 within the blue area indicated.34 As illustrated in Fig. 2d, we assign the gradual shift to lower wavenumber and broadening of this band with increasing w%HEA to a greater fraction of OH groups residing within increasingly large H-bonding clusters, as opposed to those at their interfaces.35 We will return to the nature of these clusters in Section 4. Furthermore, across this series of samples the effect of curing on this region of the spectra is minimal as shown by the IR analyses of pre-curing formulation (Fig. S9, ESI†), demonstrating that most, if not all, of the HEA monomers participate in H-bonding before curing. This observation suggests that there is significant spatial re-arrangement of HEA monomers in the formulation, and curing does not alter this re-arrangement. | ||
| Fig. 2 Selected regions of the IR spectra of; (a) PEG-HEA networks; (b) PEG-BAC networks; (c) extent of H-bonding as a function of w%MA, the molar ratio of H-bonding capable monomer HEA or BAC in the formulations; (d) schematic representation of H-bonded monoacrylate clusters (t-OH case); (i) on the left a larger cluster with lower fraction of interfacial H-bonding groups; (ii) right, a smaller block with a higher of fraction of interfacial groups. Interfacial H-bonded OH groups are shown in light green, this inside cluster are in blue, and a ‘free’ OH group is shown in brown. Red line indicates PEG chain, orange line the polyacrylate main chain, and dark green lines the monoacrylate side chain. | ||
A different situation is observed for the PEG-BAC network series. The free N–H stretching band of secondary amide groups is normally observed above 3450 cm−1. The amide groups in the network series were found to gradually shift with increasing w%BAC from free to H-bonded forms.36,37 For the pre-curing samples the N–H stretching feature corresponds to the H-bonded form only (see Fig. S9, ESI†) and the spectra are identical for all PEG-BAC pre-curing formulations, while the cured PEG-BAC samples have an increasing contribution of stretching features in the characteristic range for H-bonded amides with increasing w%BAC. This suggests that curing induces different spatial arrangements for the different H-bonding-capable monoacrylates, perhaps due to the greater number of H-bonding possibilities for PEG-HEA, resulting in larger H-bonding cluster size, as compared to PEG-BAC (see below). Finally, for the two network series IR spectral deconvolution indicates that while >95 mol% of HEA sidechains are in their H-bonded form throughout this series, the fraction of H-bonded BAC side chains rises progressively with increasing w%BAC, and even at the highest fraction some BAC retain non H-bonded characteristics (Fig. 2c). These two rather different H-bonding clustering behaviors inferred following curing they may result in different morphological, topological and mechanical properties. These effects that have not been previously reported for H-bond capable acrylate networks. Control of this clustering behavior over the emergent properties through the H-bond capable mono-acrylate content and its impact are described in the later sections.
Conventional 1H NMR spectroscopy is also routinely used for evaluating the presence of H-bonding,16 in particular the temperature dependence of characteristic resonance shifts can provide information on H-bonding induced changes in local electron density. As shown in Fig. S5 and S6 (ESI†), changes in linewidth are apparent, but even more clearly a significant change in the chemical shift can be observed in both series for resonances assigned to the H-bonding groups (3.4 ppm for δ(OH) in HEA, 6.1 ppm for δ(NH) in BAC), confirming the presence of H-bonding in the fully cured samples.
3.3. Mechanical analysis of the networks
Dynamic mechanical analysis (DMA) provides viscoelastic parameters (e.g. storage and loss moduli) which can give insights into the macroscopic properties of polymer networks. The presence of a high temperature plateau and a single clean inflection for all the samples (Fig. 3), demonstrates that there is a single glass transition temperature, Tg, for all networks, indicating good mesoscopic mixing (on a length scale of smaller than several tens of nanometers) between PEGDA and the monoacrylate in all cases. The Tg values of the networks were determined from the temperature at the maximum of tan δ.4 The storage moduli (E′) of the two series show similar decreases with increasing monoacrylate content. According to classical rubber elasticity theory, the mean molar mass of network chains between chemical crosslinks and trapped chain entanglements, Mc+e, can be calculated using rubber elasticity theory (see ESI† for details). This relation is applicable to affine networks of the same chain length and without network defects. The Mc+e values are likely suffer from a large error for heterogeneous networks,38 so the extracted values are used for qualitative comparisons of network heterogeneity within the series in the discussion below.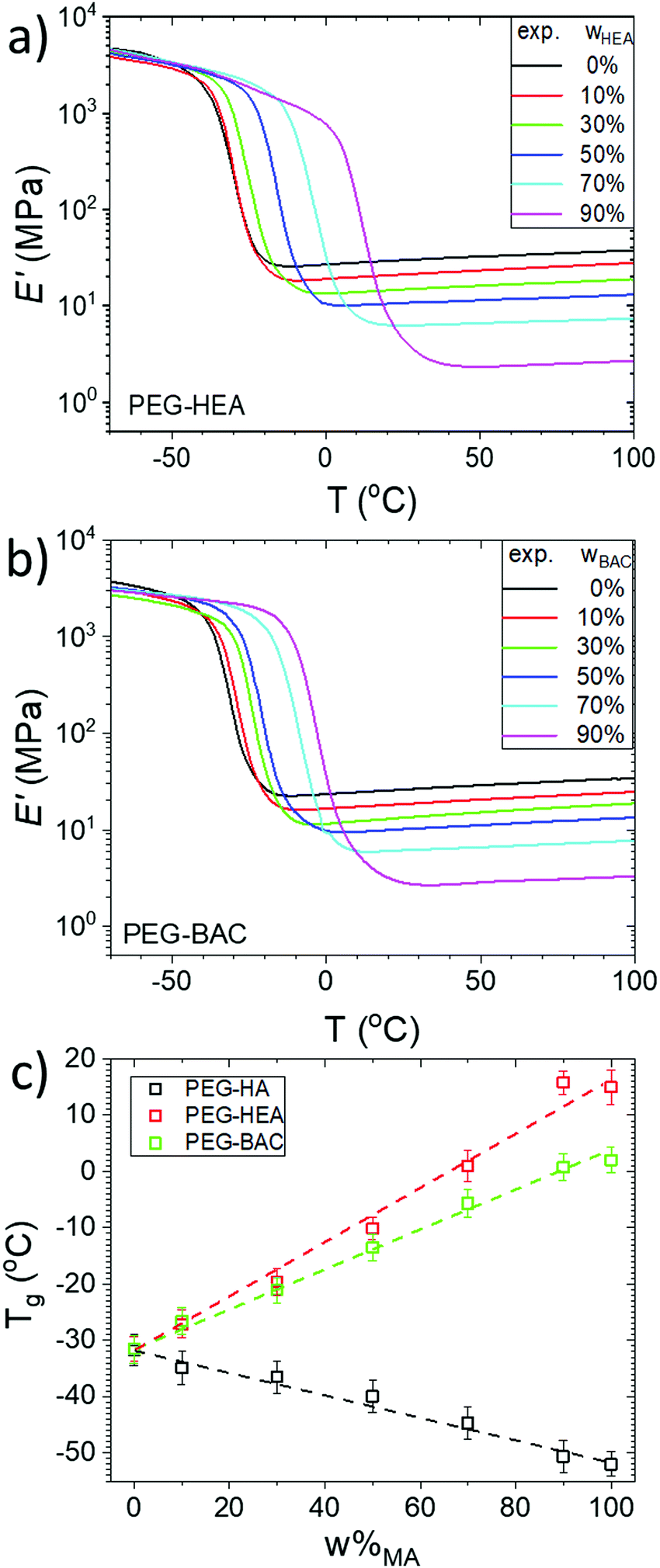 | ||
| Fig. 3 Storage modulus, E′, of cured; (a) PEG-HEA; (b) PEG-BAC networks measured by DMA with increasing wHEA or wBAC; (c) Tg of the cured all samples, determined from DMA analysis, as a function of the weight % monoacrylate (ω%MA). The PEG-HA data is extracted from our previous study.5 The data for 100% monoacrylate samples were obtained from analysis of samples polymerized from pure BAC, HEA and HA. | ||
The Fox-Loshaek relation (a modification of Flory-Fox approach) predicts a linear dependence of Tg on monoacrylate content, see ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39 As shown in Fig. 3c, a clear linear increase in Tg with w%MA is indeed observed for both series. Since a single sharp peak can be observed in the tan(δ) temperature dependencies of all the samples (Fig. S7, ESI†), it can again be taken that there is good mixing and no mesoscopic phase separation of mono- and diacrylate. The increase in Tg with increasing PEGDA content is the opposite to our previous observation for similar networks with the non H-bond capable monoacrylate HA.5 We suggest that inter-chain H-bonding interactions act as temporary cross-links that restrict segmental motion and decrease the free volume of the network.40,41 The slope of the w%MA dependence is greater for the HEA-than for the BAC-series, which, besides the significant larger molecular weight of BAC, is also a result of the larger ‘spacer’ (more atoms between the functional group and polyacrylate backbone) in the latter that is expected to increase molecular mobility and the available free volume, as previously reported for similar systems.13,42
39 As shown in Fig. 3c, a clear linear increase in Tg with w%MA is indeed observed for both series. Since a single sharp peak can be observed in the tan(δ) temperature dependencies of all the samples (Fig. S7, ESI†), it can again be taken that there is good mixing and no mesoscopic phase separation of mono- and diacrylate. The increase in Tg with increasing PEGDA content is the opposite to our previous observation for similar networks with the non H-bond capable monoacrylate HA.5 We suggest that inter-chain H-bonding interactions act as temporary cross-links that restrict segmental motion and decrease the free volume of the network.40,41 The slope of the w%MA dependence is greater for the HEA-than for the BAC-series, which, besides the significant larger molecular weight of BAC, is also a result of the larger ‘spacer’ (more atoms between the functional group and polyacrylate backbone) in the latter that is expected to increase molecular mobility and the available free volume, as previously reported for similar systems.13,42
3.4. Low field (LF) 1H NMR analysis of the networks
Polymer chain dynamics in crosslinked networks can be probed by 1H NMR spin–spin relaxation time analysis, which is very sensitive to even small differences in the crosslink density due to the strong influence of anisotropic segmental motions on relaxation at temperatures well above Tg (more than 60 °C above Tg).5 As shown in Fig. 4, increasing w%MA (HEA or BAC) in the formulation raises the overall transverse relaxation times of the networks which we ascribe to decreasing crosslink density, as several previous studies have found that crosslink density of the polymeric network is inversely proportional to the T2 value at high temperature.5,25,43 Swelling causes an increase in average T2, as it releases temporary physical chain entanglements and a decrease in the strength of inter-chain spin–spin interactions. The shorter Tsw2,av values across both series (Table 2), as compared to the non H-bonding capable (PEG-HA) networks we previously reported,5 demonstrates reduced network chain mobility due to the presence of H-bonding clusters. We have shown5,6 that it is also possible to estimate apparent Mc+e values from the T2 values (details in ESI†), the relative changes in the Mc+e values extracted will be discussed below. We note that the temperature of 70 °C, necessary for the LF NMR analysis, precludes direct comparison with the FT-IR data from Section 3.2. | ||
| Fig. 4 Normalized T2 relaxation decays, recorded at 70 °C and 20 MHz, of; (a and c) PEG-HEA networks; (b and d) PEG-BAC networks measured using Hahn echo pulse sequence (τCP: 10 ms) in dry state (a and b), and swollen state (c and d). Volume swelling ratio in C2D2Cl4, is ≈ 1.55). Vertical lines indicate the T2,av values. The points represent the measured data and the lines are the corresponding fits. Details on normalization and fitting are provided in ESI† Section 5. | ||
3.5. Network analyses by DQ NMR
1H DQ-NMR analysis offers more detailed insights into the effect of network structure on chain dynamics. The combination of fast sample spinning and high magnetic field strength provides spectral resolution permitting the acquisition of site-specific, chemical-shift resolved DQ build-up curves for the different chemical groups present, which is more informative than the averaged information provided by low field NMR (LF NMR) relaxometry, or indeed by the Tg analysis. The 1H spectra for both sample series are shown in Fig. S5 (ESI†) and representative normalized DQ build-up curves and fits are shown in Fig. S8 (ESI†).By evaluating the DQ build-up curves for the 1H resonance at 3.56 ppm, assigned to the CH2 groups in PEG chains (except for the CH2 next to the carbonyl group of the acrylate backbone), the conformational freedom of the PEGDA can be evaluated. As shown Fig. 5, the average residual dipolar coupling constant of the distributions (Davg), indicated by the vertical dotted lines (values in Table 2), also gradually decreases with w%MA for both series due to greater motional freedom arising from decreasing crosslink density. This coincides with the increase in Tsw2,av noted above which is also ascribed to increased motional freedom. These findings are similar to the observations for the PEG chain in the non H-bonding capable networks (PEG-HA),5 indicating that H-bonding has very limited impact on the PEG chain motion, consistent with observations in similar systems.16
 | ||
| Fig. 5 Residual dipolar coupling constant, Dres, distributions of PEG chains (δ 3.56 ppm) for; (a) PEG-HEA; (b) PEG-BAC networks at different w%MA. Data were recorded at 70 °C. Vertical doted lines indicate the (weighted) average coupling constant, Davg, of each distribution. | ||
As shown in Fig. 6a and c, the Davg values of all the chemical groups in the both series are found to decrease linearly with increasing w%MA, as observed for the non H-bonding networks.5 As expected, resonances related to the polyacrylate backbone in both series have greater Davg values than the PEG chains, with the highest values observed for PEG-HEA, indicating a stiffer polyacrylate backbone in this network series. The chain segments in the monoacrylate side chain (namely HEA or BAC) behaved differently depending on their position in relation to the H-bonding moiety. Since the polymer chain (PA and PEG) segments comprise from distinct local spin systems, direct comparison of the absolute Davg values is complicated. Therefore, the individual coupling constants have been normalized here with respect to those for w%MA (90%). The fact that the scaled values for the two resonances of the acrylic backbone are almost identical for both series demonstrates that this approach is at least internally consistent.
 | ||
| Fig. 6 Residual dipolar coupling, Dres, values of all 1H chemical shift specified network segments for; (a) PEG-HEA, and; (c) PEG-BAC networks, as a function of w%MA. In (b) and (d) the Davg values from (a) and (c) are normalized with respect to the appropriate Davg values determined for w%MA 90%. | ||
For the PEG-HEA networks, as shown in Fig. 6b, apart from the PEG units discussed before, all the resonances can be divided into two groups: group I, those belonging to the polyacrylate backbone, and group II, those located on PEG and mono-acrylate chains near the polyacrylate backbone or H-bonding functional group (–OH or –NH–). It is apparent from Fig. 6b that the change in normalized Davg with w%MA is larger for group I than for group II. Whereas stronger segmental motion restriction (higher Davg) in group II can be observed as compared to the equivalent sidechains in the non-H-bonding PEG-HA counterpart.5 This is also apparent from the absolute Davg values for H(f), which are two times larger than its counterpart (–(CH2)β–) in the PEG-HA system.5 For the PEG-BAC networks, the normalized Davg values for resonances in group I show a smaller variation in PA backbone stiffness with increasing w%MA than for PEG-HEA or PEG-HA, which may originate from the w%BAC dependence of the H-bonded fraction of BAC side chains (IR analysis, Fig. 2c). Moreover, a third group (group III) of resonances arising from methylene groups that are two bonds away from the H-bonding group and not adjacent to the polyacrylate backbone, can be further separated based on their less restrict segmental motions. The absolute and normalized Davg values for group III can be found to be similar to those from the side chains of PEG-HA,5 and have the weakest w%MA dependence.
4. Discussion
4.1. Effect of H-bonding on network structure
As proposed by Jansen et al. in their photo-induced polymerization kinetic studies, a so-called ‘pre-organization’ of monoacrylates containing H-bond capable groups in their side chains, typically occurs in the pre-curing formulations which can in turn increase the homo-polymerization rates.44,45 In this study, IR analysis of all the pre-curing formulations (see Fig S10, ESI†) shows that for both series the monomers are in H-bonded forms, suggesting that pre-organization indeed occurred in both systems. However, further IR analysis on the cured samples shows that a significant fraction of BAC in PEG-BAC networks is not H-bonded, which is not the case for PEG-HEA. For polymer networks with preferable homo-polymerization, a diagonal peak with stronger intensity than its corresponding cross peak would be expected in the DQ-SQ NMR analyses. Further DQ-SQ NMR analyses of PEG-BAC shows no preferable homo-polymerization of BAC, as the diagonal peak of BAC in 2D NMR spectra has similar intensity as the cross peak between BAC and PEGDA, again this is not the case for PEG-HEA (Fig. 7a and b). | ||
| Fig. 7 2D DQ-SQ correlation spectrum of; (a) PEG-HEA w%HEA (50); (b) PEG-BAC w%BAC (50). The red dotted lines connect cross-peak pairs between BAC side chain and PEG resonances The red box highlights the cross-peak between PEG and HEA/BAC. (c) Schematic representation of copolymerization of diacrylate and monoacrylate containing H-bond capable moieties, the real situation is more nuanced, see Section 4.3. Pre-organization occurs spontaneously on mixing. After initiation of the free radical polymerization, depending on the relative monoacrylate co-polymerization (kCP) and homo-polymerization (khp) propagation rates, the final network may have H-bonding clusters with variable size, i.e. H-bonding clusters are dynamic domains that are fixed by crosslinking. | ||
This observation is unexpected, as Jansen pointed out in his work,44 the homo-polymerization rates of H-bonding capable monomers are significantly (4- to 8-fold) enhanced, which should result in the formation of large clusters of H-bonded units. Hence, we propose a competing-reaction-rate model to explain this discrepancy for the two series. As illustrated in Fig. 7c using t-OH type mono-acrylate, upon mixing the diacrylate and H-bond capable monoacrylates, these monoacrylates spontaneously rearrange to form an ordered ‘pre-organized’ system. On initiation of photopolymerization, depending on the ratio of homo-(khp) and co-polymerization (kcp) propagation rates, the extent of retention of a H-bonded fraction can be significantly different. In scenario (i), khp > kcp and we suggest this faster monoacrylate homo-polymerization can generate a network with large H-bonded clusters, as in the case of PEG-HEA. On the other hand, in scenario (ii) higher kcp can result in a more homogeneous network with H-bonded clustering reduced, as in the case of PEG-BAC.
It is anticipated that the presence of H-bonding capable mono-acrylates should increase khp as well as kcp, as suggested by previous work.44 Hence, the above observation suggests that the induced increase in kcp is comparable to that in khp, which in turn indicates that there is no direct connection between the formation of larger H-bonded mono-acrylate clusters and the pre-organization phenomenon. Cluster formation can be considered as similar to other acrylate systems, such as the non-H-bonding capable PEG-HA networks,5 namely their formation is a kinetically driven process. To evaluate the differences in kinetic preference for homo-polymerization in HEA and BAC, a simple Alfrey-Price Q–e calculation of the relative reaction rate in these two networks was performed (details of these calculations are given in Section S7, ESI†). This analysis suggests a faster homo-polymerization rate for HEA than for its copolymerization with PEGDA, while the copolymerization reaction for PEG-BAC system is faster, which supports our interpretation.
In summary, as a result of faster homo-polymerization of HEA in PEG-HEA networks due to higher intrinsic homo-polymerization rate, larger H-bonded HEA clusters are formed. On the other hand, despite pre-organization also being present for PEG-BAC, faster copolymerization between BAC and PEG largely negates any pre-organization and the reorganization of dynamic H-bonded network structure produces a more homogenous network with minimal retention of structures comprised of H-bonded BAC units. Hence, the difference in H-bonding cluster size between the two series is mainly driven by the different copolymerization kinetics of the mono-acrylates.
4.2. Effect of H-bonding on apparent Mc+e of network chains
The crosslink/entanglement density, which is the inverse of Mc+e, is a key parameter linking microscopic network structures and macroscopic properties. As noted above, the apparent mean molecular mass, Mappc+e, can be calculated from the storge modulus E of the high temperature plateau in DMA, as well as from the Tsw2,av values from LF NMR relaxometry, as described previously.44 The data in Fig. 8 shows that the values from these two approaches agree well and are in the expected range, suggesting that they are at least sensitive to the same network density. Note that DQ NMR can in principle also be used to estimate Mappc+e, however we have not included it here as that value would only reflect the individual moieties. Furthermore, it is difficult to extrapolate them to obtain the average value for the overall network due to the lack of dref values for different moieties other than PEG (see description below). As expected, the Mappc+e value of the all the networks increases with increasing w%MA, an expected apparent decrease in crosslink density. Furthermore, as compared to the PEG-HA series, the values for PEG-BAC and PEG-HEA networks, are reduced across the w%MA range, due to the persistent effect of H-bonding induced reduction in the apparent network density. The data also suggests greater apparent crosslink/entanglement density in PEG-HEA, as compared to PEG-BAC, which is again consistent with stronger H-bonded effects for the former. However, whether this effect is equally distributed across all the network segments cannot be assessed by these volume average techniques. Hence, we will use the chemical shift specified DQ NMR analyses to evaluate H-bonding effects on polymer chain mobility in more detail below. | ||
| Fig. 8 Mean molar mass between crosslinks and trapped chain entanglements, Mappc+e, obtained from DMA (dry sample) and LF NMR analysis (swollen sample), as a function of monoacrylate weight fraction, wMA. The PEG-HA data is extracted from our previous study.5 | ||
4.3. Effect of H-bonding on network polymer chain mobility
In photocured acrylate networks, the sidechain of these acrylates are known to form in a zipper-like topology in the final polymer, and are often referred to as zipper-like junctions.46,47 This nano-scale network topology has far-reaching impacts on the macroscopic and in particular the mechanical properties.6 We have previously shown that for PEG-HA networks, the weighted-averaged Dres value (Davg) is sensitive to differences in segmental polymer chain dynamics induced by changes in network topology.5 As shown in Fig. 9a, the Davg of individual 1H resonances in PEG-BAC network does not monotonically decrease with their position along the BAC side chain from the polyacrylate (PA) backbone, which is unlike the situation for non H-bonding PEG-HA network.5 Here the units near the H-bonding moiety have a larger Davg value as a result of reduced chain segment mobility. By plotting Davg of the mono-acrylate side chain resonance groups assigned previously, see Fig. 9b, it can be seen that for PEG-BAC networks: (i) group II (1H in the chemical group adjacent to the PA backbone or N–H/O–H group) show very similar changes with increasing w%MA as group II in PEG-HEA series; (ii) for group III (1H in the chemical group two bonds away from N–H/O–H and not adjacent to the PA backbone) in PEG-BAC networks, the w%MA dependence is very similarly to that for group III in non-H-bonded PEG-HA series. Note that here the extent of this increase in Davg for 1H in chemical group adjacent to N–H/O–H groups is also dependent on the distance between the backbone and H-bonding group, as suggested by a similar test for a PEG-HBA (4-hydroxybutyl acrylate) network, see Fig. S11 (ESI†). The data suggests that, as shown schematically in Fig. 9c, the H-bonding capable groups impose a strongly localized restraint (within 1–2 bonds) on side chain mobility, while subsequent units along these chains follow the free chain model (progressively decreasing Davg along the sidechain) as found for non H-bonding networks. In a previous mechanical study13 it was suggested that side chains with multiple H-bonding groups significantly reduce chain mobility even in very loosely crosslinked networks. In this study we use monomers with single H-bonding sites. It is likely that H-bonding causes a strongly localized mobility restraint on nearby chain segments. | ||
| Fig. 9 (a) Average residual dipolar coupling constant Davg values of chemical groups along the BAC side chains in PEG-BAC networks, for orientation within the network see panel (c); (b) The Davg values for identified resonances of specified network segments in the monoacrylate side chains for PEG-HEA, PEG-BAC and PEG-HA as a function of w%MA; (c) Schematic representation of network dynamics for the PEG-BAC case, showing the polyacrylate backbone and sidechains and the PEG crosslinker with a H-bonding cluster highlighted in yellow. | ||
Furthermore, as suggested by Jansen's study on pre-organization effects,44 though polyacrylates produced by a free radical polymerization normally yield predominantly syndiotactic materials (i.e. zipper-like network junction), the pre-organization of H-bonding capable mono-acrylates results in an increasingly favorable isotactic structure (i.e. brush- or comb-like network junctions) in the clusters (see Fig. 9c), which will in turn decreases the PA backbone chain mobility.48 However, as shown in Fig. 6b, the normalized Davg values of PA backbone in PEG-HEA network have very similar values and w%HEA dependence as those in the PEG-HA series,5 suggesting the overall network junction topology (zipper-like network) did not change significantly in presence of H-bonding induced pre-organization. Note that as mentioned above, the lower normalized Davg values of the PA backbone for PEG-BAC arose from an increased overall reduction of network chain mobility due to increased ratio of H-bonded BAC monoacrylates, hence we do not comment here on the change in its topology as compared to the other two network types.
4.4. Effect of hydrogen bonding on network junction functionality
The network junction functionality, f, is defined as the number of elastically effective strands connected to a network junction, it is mainly determined by the chemistry, however, as free radical polymerization is uncontrollable, difference can also arise due to the change in the polymerization kinetics. For the networks in this study f is effectively the number of directly adjacent acrylate backbone monomer units bearing PEG chains, NPEG. For a pure PEGDA network (w%MA = 0), the functionality is theoretically equivalent to the polyacrylate backbone length which can be directly obtained from GPC analyses of the hydrolysates; n(PAA) and is c. 150, a similar value to that obtained for the PEG-HA networks in our previous study.5 The addition of mono-functional acrylates into the formulation generates topological defects in the zipper-like polyacrylate network junctions, which significantly reduces the network junction size (indicated in grey in Fig. 10c) and hence its functionality. Hence with increasing w%MA it is expected that feff (the effective volume average functionality) will gradually decrease and finally reach a value no bigger than 3, since the network junction functionality is f = NPEG+ 2 (or + 1, if the position of the crosslinking PEG is at the acrylic backbone terminal), and NPEG = 1 for extremely diluted PEGDA networks. The functionality of acrylic network junctions in PEGDA-MA networks (of any type) can be estimated by analyzing DQ-NMR results for PEG chains using a similar model (PEGDA-PETM) network with known functionality as a ref. 5. Based on these considerations, the Davg and Mc+e values of the PEG chains can be related by:49,50 | (1) |
 | ||
| Fig. 10 (a) Effective functionality of acrylic crosslinks in PEG-HEA and PEG-BAC networks as well as PEG-HA (extracted from our previous data5) derived from DQ-NMR analysis; (b) comparison of phantom and affine model pre-factors of all these networks as a function of crosslink functionality; (c) illustration of network junction with H-bonding clusters; (d) illustration of network junction without H-bonding clusters. | ||
5. Conclusions
In this study, two series of polyacrylate networks with different H-bonding capable monoacrylate copolymers were synthesized via UV-initiated photo-polymerization. It was found that different patterns of H-bonding clusters associated with the side chains were produced in the two network series. IR analysis and reactivity calculations demonstrate that preferable formation of clusters during UV-curing is mainly affected by differences in homo- and co-polymerization kinetics of the monoacrylates and PEGDA, hence we propose a competing-reaction-rate model to explain preferred cluster formation for PEG-HEA. Hence the competing roles of H-bonding preorganization and copolymerization kinetics are supported by ex situ FTIR analyses and indirectly by the reactivity calculations. Additional in situ FTIR- and NMR-based kinetic analyses, which can help us to track the evolution of individual compound over time, are currently undergoing that will allow us to develop this interpretation. Nevertheless, in this view thermodynamically favorable pre-organization is present for both PEG-HEA and PEG-BAC, significantly increasing the homo-polymerization rates, however the differences in H-bonding cluster size arise from differences in the co-polymerization rates. This is the first direct observation of H-bonding capable monoacrylate induced enhancement in co-polymerization between monoacrylate and crosslinker in UV-copolymerization.Additionally, the presence of H-bonding reduces the overall chain mobility in both networks, while different chain segments are strongly influenced (as indicated by the change in Dres along the side chain) by their position relative to the H-bonding group in the monoacrylate sidechain. Furthermore, though the preferable formation of H-bonding clusters does not change the overall polymer network junction (zipper-like) topology significantly, larger clusters indeed increase the size of the network junctions. This observation clearly demonstrates the important role of H-bonding type in the occurrence of monoacrylate pre-organization during UV initiated photo-polymerization processes. The microscopic picture that emerges is, H-bonding imposed dynamic restrictions during sample preparation and polymerization that coupled with the known monomer reactivities alter network organization, which also directly impact the emergent bulk properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research received financial support from EU FP7 Marie Curie Actions under the NEOGEL project (Grant No. 316973), the EU Horizon2020 Marie Curie Cofound project (Grant No. 713279). The Netherlands Organization of Scientific Research (NWO) is acknowledged for their support of the “solid-state NMR facility for advanced materials research” which is part of the μNMR-NL ROADMAP facility. We also acknowledge support from the Dutch Polymer Institute (DPI) and TI-COAST.References
- C. Decker, T. N. T. Viet, D. Decker and E. Weber-Koehl, UV-radiation curing of acrylate/epoxide systems, Polymer, 2001, 42, 5531–5541 CrossRef CAS.
- A. Endruweit, M. S. Johnson and A. C. Long, Curing of composite components by ultraviolet radiation: A review, Polym. Compos., 2006, 27, 119–128 CrossRef CAS.
- B. Wu, A. Sufi, R. Ghosh Biswas, A. Hisatsune, V. Moxley-Paquette, P. Ning, R. Soong, A. P. Dicks and A. J. Simpson, Direct Conversion of McDonald's Waste Cooking Oil into a Biodegradable High-Resolution 3D-Printing Resin, ACS Sustainable Chem. Eng., 2020, 8, 1171–1177 CrossRef CAS.
- F. Y. Gao, C. C. Jiao, B. Yu, H. L. Cong and Y. Q. Shen, Preparation and biomedical application of injectable hydrogels, Mater. Chem. Front., 2021, 5, 4912–4936 RSC.
- B. Wu, W. Chasse, R. Peters, T. Brooijmans, A. A. Dias, A. Heise, C. J. Duxbury, A. P. M. Kentgens, D. F. Brougham and V. M. Litvinov, Network Structure in Acrylate Systems: Effect of Junction Topology on Cross-Link Density and Macroscopic Gel Properties, Macromolecules, 2016, 49, 6531–6540 CrossRef CAS.
- V. M. Litvinov and A. A. Dias, Analysis of network structure of UV-cured acrylates by H-1 NMR relaxation, C-13 NMR spectroscopy, and dynamic mechanical experiments, Macromolecules, 2001, 34, 4051–4060 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. H. Merino, W. G. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, A Healable Supramolecular Polymer Blend Based on Aromatic pi-pi Stacking and Hydrogen-Bonding Interactions, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Q. Zhang, Chitosan-based hydrogels for controlled, localized drug delivery, Adv. Drug. Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- P. C. Painter, J. F. Graf and M. M. Coleman, Effect of Hydrogen-Bonding on the Enthalpy of Mixing and the Composition Dependence of the Glass-Transition Temperature in Polymer Blends, Macromolecules, 1991, 24, 5630–5638 CrossRef CAS.
- T. K. Kwei, The Effect of Hydrogen-Bonding on the Glass-Transition Temperatures of Polymer Mixtures, J. Polym. Sci., Polym. Lett. Ed., 1984, 22, 307–313 CrossRef CAS.
- J. R. Ilzhoefer and R. J. Spontak, Effect of Polymer Composition on the Morphology of Self-Assembled Dibenzylidene Sorbitol, Langmuir, 1995, 11, 3288–3291 CrossRef CAS.
- P. J. Woodward, D. H. Merino, B. W. Greenland, I. W. Hamley, Z. Light, A. T. Slark and W. Hayes, Hydrogen Bonded Supramolecular Elastomers: Correlating Hydrogen Bonding Strength with Morphology and Rheology, Macromolecules, 2010, 43, 2512–2517 CrossRef CAS.
- C. L. Lewis, K. Stewart and M. Anthamatten, The Influence of Hydrogen Bonding Side-Groups on Viscoelastic Behavior of Linear and Network Polymers, Macromolecules, 2014, 47, 729–740 CrossRef CAS.
- K. P. Nair, V. Breedveld and M. Weck, Modulating mechanical properties of self-assembled polymer networks by multi-functional complementary hydrogen bonding, Soft Matter, 2011, 7, 553–559 RSC.
- N. A. Peppas, K. B. Keys, M. Torres-Lugo and A. M. Lowman, Poly(ethylene glycol)-containing hydrogels in drug delivery, J. Controlled Release, 1999, 62, 81–87 CrossRef CAS PubMed.
- B. Wu, W. Chassé, K. Zick, M. D. Mantle, A. Heise, D. F. Brougham and V. M. Litvinov, The effect of hydrogen bonding on diffusion and permeability in UV-cured Polyacrylate-based networks for controlled release, J. Control Release, 2020, 327, 150–160 CrossRef CAS PubMed.
- K. Saalwachter, W. Chasse and J. U. Sommer, Structure and swelling of polymer networks: insights from NMR, Soft Matter, 2013, 9, 6587–6593 RSC.
- M. Gaborieau, S. P. S. Koo, P. Castignolles, T. Junkers and C. Barner-Kowollik, Reducing the Degree of Branching in Polyacrylates via Midchain Radical Patching: A Quantitative Melt-State NMR Study, Macromolecules, 2010, 43, 5492–5495 CrossRef CAS.
- B. Wu, S. von der Ecken, I. Swyer, C. Li, A. Jenne, F. Vincent, D. Schmidig, T. Kuehn, A. Beck, F. Busse, H. Stronks, R. Soong, A. R. Wheeler and A. Simpson, Rapid Chemical Reaction Monitoring by Digital Microfluidics-NMR: Proof of Principle Towards an Automated Synthetic Discovery Platform, Angew. Chem., Int. Ed., 2019, 58, 15372–15376 CrossRef CAS PubMed.
- B. Wu, E. I. Pascu, S. A. Brady, D. F. Brougham and O. M. Clarkin, Fine property-tuning through Ca content control in a facile synthesis of glasses-based self-setting injectable hydrogel, Mater. Des., 2021, 211 Search PubMed.
- I. Ando and T. Asakura, Solid state NMR of polymers, Elsevier, Amsterdam; New York, 1998 Search PubMed.
- B. Wu, M. E. Wiseman, M. E. Seitz, K. Tomić, A. Heise, D. F. Brougham and V. M. Litvinov, Impact of morphology on O2 permeability in silicone hydrogel membranes: new insights into domain percolation from experiments and simulations, J. Membr. Sci., 2021, 621, 118970 CrossRef CAS.
- C. Brosseau, A. Guillermo and J. P. Cohenaddad, Nuclear-Magnetic-Resonance Approach to Properties of Poly(Ethylene Oxide) Poly(Methyl Methacrylate) Blends. 1. Chain Dynamics and Free-Volume, Polymer, 1992, 33, 2076–2083 CrossRef CAS.
- M. Andreis and J. L. Koenig, Application of Nmr to Crosslinked Polymer Systems, Adv. Polym. Sci., 1989, 89, 69–160 CrossRef CAS.
- J. P. C. Addad, NMR and fractal properties of polymeric liquids and gels, Prog. Nucl. Magn. Reson. Spectrosc., 1993, 25, 1–316 CrossRef CAS.
- R. A. Kinsey, Solid-State NMR of Elastomers, Rubber Chem. Technol., 1990, 63, 407–425 CrossRef CAS.
- R. Graf, A. Heuer and H. W. Spiess, Chain-order effects in polymer melts probed by 1H double-quantum NMR spectroscopy, Phys. Rev. Lett., 1998, 80, 5738–5741 CrossRef CAS.
- K. Saalwachter, Robust NMR Approaches for the Determination of Homonuclear Dipole-Dipole Coupling Constants in Studies of Solid Materials and Biomolecules, Chemphyschem: a European journal of chemical physics and physical chemistry, 2013, 14, 3000–3014 CrossRef PubMed.
- R. Peters, V. M. Litvinov, P. Steeman, A. A. Dias, Y. Mengerink, R. van Benthem, C. G. de Koster, S. van der Waals and P. Schoenmakers, Characterisation of UV-cured acrylate networks by means of hydrolysis followed by aqueous size-exclusion combined with reversed-phase chromatography, J. Chromatogr. A, 2007, 1156, 111–123 CrossRef CAS PubMed.
- W. Chassé, J. L. Valentín, G. D. Genesky, C. Cohen and K. Saalwächter, Precise dipolar coupling constant distribution analysis in proton multiple-quantum NMR of elastomers, J. Chem. Phys., 2011, 134, 044907 CrossRef PubMed.
- J. A. Burdick, T. M. Lovestead and K. S. Anseth, Kinetic chain lengths in highly cross-linked networks formed by the photoinitiated polymerization of divinyl monomers: A gel permeation chromatography investigation, Biomacromolecules, 2003, 4, 149–156 CrossRef CAS PubMed.
- L. H. Guo, H. Sato, T. Hashimoto and Y. Ozaki, FTIR Study on Hydrogen-Bonding Interactions in Biodegradable Polymer Blends of Poly(3-hydroxybutyrate) and Poly(4-vinylphenol), Macromolecules, 2010, 43, 3897–3902 CrossRef CAS.
- T. Kondo and C. Sawatari, A Fourier transform infra-red spectroscopic analysis of the character of hydrogen bonds in amorphous cellulose, Polymer, 1996, 37, 393–399 CrossRef CAS.
- J. F. Kadla and S. Kubo, Lignin-based polymer blends: analysis of intermolecular interactions in lignin-synthetic polymer blends, Composites, Part A, 2004, 35, 395–400 CrossRef.
- F. Palombo, P. Sassi, M. Paolantoni, A. Morresi and R. S. Cataliotti, Comparison of Hydrogen Bonding in 1-Octanol and 2-Octanol as Probed by Spectroscopic Techniques, J. Phys. Chem. B, 2006, 110, 18017–18025 CrossRef CAS PubMed.
- D. J. Skrovanek, P. C. Painter and M. M. Coleman, Hydrogen bonding in polymers. 2. Infrared temperature studies of nylon 11, Macromolecules, 1986, 19, 699–705 CrossRef CAS.
- G. G. Suchkova and L. I. Maklakov, Amide bands in the IR spectra of urethanes, Vib. Spectrosc., 2009, 51, 333–339 CrossRef CAS.
- T. A. Vilgis and G. Heinrich, Statics and Dynamics of Heterogeneous Polymer Networks, Macromol. Theory Simul., 1994, 3, 271–293 CrossRef CAS.
- T. G. Fox and S. Loshaek, Influence of Molecular Weight and Degree of Crosslinking on the Specific Volume and Glass Temperature of Polymers, J. Polym. Sci., 1955, 15, 371–390 CrossRef CAS.
- S. A. Jenekhe and M. F. Roberts, Effects of Intermolecular Forces on the Glass-Transition of Polymers, Macromolecules, 1993, 26, 4981–4983 CrossRef CAS.
- A. T. Slark, The effect of intermolecular forces on the glass transition of solute-polymer blends, Polymer, 1997, 38, 2407–2414 CrossRef CAS.
- I. Ward and J. Sweeney, An Introduction to The Mechanical Properties of Solid Polymers, 2004 Search PubMed.
- C. G. Fry and A. C. Lind, Determination of Cross-Link Density in Thermoset Polymers by Use of Solid-State H-1-Nmr Techniques, Macromolecules, 1988, 21, 1292–1297 CrossRef CAS.
- J. F. G. A. Jansen, A. A. Dias, M. Dorschu and B. Coussens, Fast monomers: Factors affecting the inherent reactivity of acrylate monomers in photoinitiated acrylate polymerization, Macromolecules, 2003, 36, 3861–3873 CrossRef CAS.
- J. F. G. A. Jansen, A. A. Dias, M. Dorschu and B. Coussens, Effect of preorganization due to hydrogen bonding on the rate of photoinitiated acrylate polymerization, Photoinitiated Polymerization, 2003, 847, 127–139 CAS.
- G. de With, Polymer coatings: a guide to chemistry, characterization, and selected applications, Wiley-VCH Verlag, Weinheim, 2018 Search PubMed.
- V. M. Litvinov and P. P. De, Spectroscopy of Rubbers and Rubbery Materials, Rapra Technology, 2002 Search PubMed.
- C. S. Biswas, Y. H. Wu, Q. Wang, L. Du, K. Mitra, B. Ray, Z. C. Yan, B. Du and F. J. Stadler, Effect of tacticity and molecular weight on the rheological properties of poly(N-isopropylacrylamide) gels in benzyl alcohol, J. Rheol., 2017, 61, 1345–1357 CrossRef CAS.
- W. Chasse, M. Lang, J. U. Sommer and K. Saalwachter, Cross-Link Density Estimation of PDMS Networks with Precise Consideration of Networks Defects, Macromolecules, 2012, 45, 899–912 CrossRef CAS.
- S. Schlogl, M. L. Trutschel, W. Chasse, I. Letofsky-Papst, R. Schaller, A. Holzner, G. Riess, W. Kern and K. Saalwachter, Photo-vulcanization using thiol-ene chemistry: Film formation, morphology and network characteristics of UV crosslinked rubber latices, Polymer, 2014, 55, 5584–5595 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qm01535d |
| This journal is © the Partner Organisations 2022 |