
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoconfinement in miniemulsion increases reaction rates of thiol–ene photopolymerization and yields high molecular weight polymers†
Lorena
Infante Teixeira
a,
Katharina
Landfester
 *a and
Héloïse
Thérien-Aubin
*ab
*a and
Héloïse
Thérien-Aubin
*ab
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: landfester@mpip-mainz.mpg.de
bDepartment of Chemistry, Memorial University of Newfoundland, St John's, Newfoundland and Labrador A1B 3X7, Canada. E-mail: htherienaubin@mun.ca
First published on 25th April 2022
Abstract
Thiol–ene polymerization is a powerful synthetic platform for the preparation of a variety of polymer materials but is often plagued by the formation of low molecular weight polymers. This is typical of step-growth polymerization, where high molecular weights are achieved only at nearly complete monomer conversions. However, experimental results suggest that it is possible to produce step-growth polymers with a high degree of polymerization by performing the reaction in a miniemulsion, where the dispersed droplets act as nanoreactors. Here, we investigate the effect of confinement arising from the reduction of the reaction loci from bulk to a nanoreactor and how it affects the thiol–ene reaction and the resulting polymers. The polymerization rates observed for the reaction in the miniemulsion were up to 35-fold higher than the rates observed in bulk. Different monomer pairs were evaluated using either a diallyl, divinyl, or diacrylate monomer as dienes. The reaction was followed by Raman spectroscopy to simultaneously quantify the conversion of thiols and enes in the system, which enabled the detection of side reactions, such as homopolymerization. Mixtures with a non-stoichiometric ratio of dithiol and diene monomer also benefited from the polymerization in nanoconfinement. In such cases, the polymerizations in bulk were limited to very low degrees of polymerization. However, when the polymerization was performed in the confinement of the miniemulsion droplets, high molecular weight polymers were produced.
Introduction
Thiol–ene polymerization is a popular synthetic platform due to its versatility and synthetic simplicity,1–4 but is often plagued by the formation of low molecular weight polymers. This limitation impacts the thermomechanical properties of the resulting polythioethers and prevents their applications where high-performance polymers are required.5–7 Because of the step-growth character of thiol–ene polymerization, high molecular weights are achieved only at nearly complete monomer conversions, which is challenging to accomplish in practice.8–10However, recent studies showed the formation of polymers with improved degrees of polymerization for thiol–ene products prepared in miniemulsion.2,11–13 Such behavior could potentially be ascribed to a confinement effect similar to that observed during free-radical polymerization carried out in nanodispersed media, which leads to higher conversion rates of the monomers, and yield polymers with higher degrees of polymerization.14,15 These observations demonstrate the need to investigate the mechanism of thiol–ene polymerization in nanoconfined systems to rationally design high molecular weight polythioethers and expand the range of applications of the resulting thiol–ene systems.
Understanding the kinetic of the thiol–ene polymerization in miniemulsion is crucial to reach high monomer conversion and produce high molecular weight polymer materials. Typically, thiol–ene polymerization reactions are studied by monitoring the conversion with 1H-NMR or FTIR spectroscopy.2,9,16 However, the detection of thiol moieties can be difficult due to intrinsic low analytical sensitivity resulting from proton exchange in NMR or weak intensity of the SH stretching in FTIR. Consequently, quantification usually relies only on the conversion of the enes alone.17,18
The study of the kinetic of the thiol–ene polymerization is further complexified by the coexistence and competition of two polymerization mechanisms.19,20 On the one hand, there is the step-growth controlled thiol–ene coupling, where a thiyl radical attacks an ene moiety producing a thiol–ene adduct. On the other hand, some commonly used enes can concomitantly undergo radical homopolymerization through a chain-growth process. Consequently, the study of a thiol–ene polymerization reaction monitored only by the detection of the conversion of the carbon–carbon double bonds could be misleading. In systems where allyl ethers are used, the reaction can be relatively well described by overlooking any chain-growth mechanism since their ene-centered radicals show greater selectivity to thiyl radicals than to homopolymerization.21,22 However, this approximation can no longer hold for monomer systems using (meth)acrylates or vinylic enes since homopolymerization can occur at a rate faster than the addition of thiol to the double bonds.16,23,24 To study such systems, the simultaneous detection of both thiol and ene functionalities could provide a more complete understanding of the reaction. In addition, it could also offer new insights into strategies to control polymerizations performed with off-stoichiometric ratios of thiols and enes, which could be used to synthesize polymers that can readily undergo post-polymerization modifications.1,25,26
It has been shown that thiol–ene polymerization performed in dispersed media yields polythioethers with high degrees of polymerization compared to thiol–ene polymerization performed in solution or in bulk.12,18 Thiol–ene polymerization, similarly to polymerization occurring in the confinement of a nanopore,27 or other reactions performed in colloidal suspensions, have been shown to take advantage of the compartmentalization of the reactants to increase the rate of conversion. During a polymerization performed in droplets created by miniemulsion, the monomers-containing droplets act as nanoreactors and become the final polymer particles.28 Typical free-radical polymerization and controlled-living radical polymerization in such dispersed systems have shown improved reaction kinetics.14,29,30 This phenomenon is primarily due to the segregation of radicals in different compartments, which reduces termination events.14,31 Most of the current understanding of the effect of confinement on polymerization reactions comes from the study of those chain-growth polymerizations,30,32–35 and the effect of confinement on step-growth polymerization remains ambiguous.36–39 However, addressing the effects of confinement on the rate and yield of step-growth reactions and harnessing those effects during thiol–ene polymerizations performed in dispersed media would be an efficient method to produce high-quality polythioethers and design new polymers.
Here we report a systematic study comparing the reaction in bulk and miniemulsion systems of thiol–ene polymerization. Monomer pairs bearing thiol and allyl/vinyl functionalities were prepared in both types of environments. The polymerization reaction was followed by Raman spectroscopy to characterize the conversion of thiols and enes during the reaction. In conjunction with the evolution of the polymer molecular weight, those results can provide insights into the thiol–ene polymerization mechanism in confined systems. Furthermore, the effect of the off-stoichiometric ratios of monomers, which would bring more flexibility in the design of new thiol–ene polymers, has been investigated. Our results pave the way for the broader use of polythioethers in applications where not only their functional versatility and reduced shrinkage stress are needed,3,40,41 but also the improved thermomechanical properties21,22 associated with higher molecular weight,42 such as in scratch-resistant coatings,7 dental restorative materials,43 fibers manufacturing,44 microfluidics scaffolds,1,45 and even in industrial-scale molding processes.46
Experimental
Materials
Diallyl adipate (DAA, 98%), divinyl adipate (DVA, 99%), 2,2′-(ethylenedioxy) diethanethiol (EDDT, 95%), hexadecane (98%), and lithium phenyl(2,4,6-trimethylbenzoyl)phosphinate (TPO-Li, 98%), were purchased from TCI Deutschland. Diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO, 97%), sodium dodecyl sulfate (SDS, 99%), acetone (99%), 4-methoxyphenol (MEHQ, 99%), 1,4-butanediol diacrylate (1,4DAc, 87%), and tetrahydrofuran (THF, 99.9%) were acquired from Sigma-Aldrich. All chemicals were used as received unless noted otherwise. DVA and 1,4DAc were purified before use with a column of aluminum oxide to remove stabilizers.Characterization
Raman spectra were collected with an Ocean Insight QEPro spectrometer, equipped with a 785 nm laser source and a coupled fiber probe. Gel permeation chromatography (GPC) was performed on an Agilent Technologies 1260 Infinity with THF as mobile phase. The GPC was calibrated with a series of 12 standards of poly(methyl methacrylate) (PMMA) with nominal weight of 1600, 1190, 675, 392, 201, 88.5, 41.4, 23.5, 12.6, 6.37, 2.2, and 0.8 kDa mol−1. The hydrodynamic diameter and size distribution of the nanoparticles in dilute suspensions were measured with a Malvern Zetasizer Nano-S90 dynamic light scattering (DLS) instrument. 1H-NMR, 13C-NMR and HSQC spectra were recorded for dried polymer samples dissolved and/or swollen in CDCl3.Preparation of monomer mixtures and emulsification
Samples for bulk polymerization were prepared by adding the photoinitiator (TPO) at a concentration of 0.1 wt% compared to the monomer mixture or di-enes and di-thiols used. Samples used during the polymerization in miniemulsion were prepared by emulsification of the monomer mixture by ultrasonication. The biphasic mixture of monomers in water contained 20 wt% of monomers to the total emulsion. The dispersed phase was composed of the monomers in an equimolar mixture of diene and dithiol monomers, containing hexadecane (4 wt% of Cmonomers) as an osmotic pressure agent to avoid Ostwald ripening and stabilize the droplets after emulsification. The continuous phase was composed of water containing SDS as a surfactant (CSDS = 0.2 wt% in water) and the photoinitiator TPO-Li (0.1 wt% of Cmonomers). The biphasic mixture was emulsified with a Branson digital sonifier SFX550 cell disruptor (70% amplitude, 2 min). Experiments with off-stoichiometry ratios employed excesses of 5 or 10% of one of the monomers both in bulk and dispersed media.UV-irradiation of the monomer mixtures
All samples were reacted in a multi-purpose photoreactor platform equipped with four 385 nm LED modules (Peschl Ultraviolet GmbH) (Fig. S1A†). In the case of bulk samples, the polymerization was carried out in a polystyrene cuvette placed in the center of the photoreactor. Aliquots were taken at 30 seconds, 1, 2, 5, 10, and 30 minutes of reaction, quenched in liquid N2, and dissolved in THF containing 200 ppm of MEHQ for GPC analysis. In the case of miniemulsion samples, the emulsion was flown through a 16 cm long tubing (Øint = 0.08 cm, V = 0.330 mL) located inside the reactor with a peristaltic pump at flow rates varying from 0.05 to 14 mL min−1. After going through the reactor (Fig. S2†), samples were collected at the outlet tubing, quenched and analyzed by GPC. The UV irradiance was varied from 4.17 mW cm−2 to 18.3 mW cm−2. Unless noted otherwise, the experiments were carried out at a constant UV irradiance of 15 mW cm−2 (Fig. S1B†).Kinetics of photopolymerization by Raman spectroscopy
The variation in the chemical composition of the samples over time was monitored using an optical fiber Raman probe. For samples prepared in bulk, the probe was placed inside the reactor in front of the reaction cuvette (Fig. S2†). For miniemulsion experiments, the samples were flown through a tube from the reactor to an external flow-through quartz cuvette placed inside a sample holder with an opening to insert the Raman probe at the optimal working distance (7 mm). To measure the extent of polymerization, Raman spectra were recorded at different intervals of time (or different flow rates) of the reaction, at a given UV-irradiance. To allow the direct comparison between bulk and miniemulsion kinetics, the reaction times in miniemulsion were calculated by converting the flow rate given by the peristaltic pump to the residence time of the monomer inside the reactor. The flow rate was varied from 0.05 mL min−1 to 7 mL min−1 corresponding to a reaction time of ca. 30 min to 30 s. OceanView spectroscopy software was used for data treatment. All the spectra were normalized to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at ca. 1735 cm−1, originating from the carbonyl present in the diene monomers.
O stretching at ca. 1735 cm−1, originating from the carbonyl present in the diene monomers.
Results and discussion
We selected a model monomer-pair composed of diallyl adipate (DAA) as the diene, and 2,2′-(ethylenedioxy) diethanethiol (EDDT) as its dithiol counterpart to study the kinetics of the thiol–ene polymerization. Raman spectroscopy was employed to detect and quantify the moieties throughout the experiments carried out in a flow-through setup (Fig. 1). The choice of this monomer system stemmed from the well-resolved peaks of the monomer couple, without any overlaps between vibrations in the thiol and ene region. Furthermore, those monomers are not absorbent at the wavelength (385 nm) used to initiate the polymerization. In addition, DAA contains two ester linkages, allowing the normalization of the spectra using the carbonyl vibration (ca. 1735 cm−1) as an internal standard (Fig. 2A). The use of Raman spectroscopy, compared for example to FTIR, enables the simultaneous analysis of both thiols and enes functionalities with sufficient accuracy to follow the conversion kinetics. Furthermore, Raman spectroscopy is well-suited for the analysis of waterborne media since the water vibrations are not a significant concern in the spectral window of interest. | ||
| Fig. 1 Scheme of the experimental setup used for the reaction and collection of the samples prepared by polymerization in miniemulsion. The miniemulsions were pumped using a peristaltic pump in a tubing going from a reservoir into the reactor and to a flow-through cuvette, allowing the measurement of the Raman spectra using a Raman probe. | ||
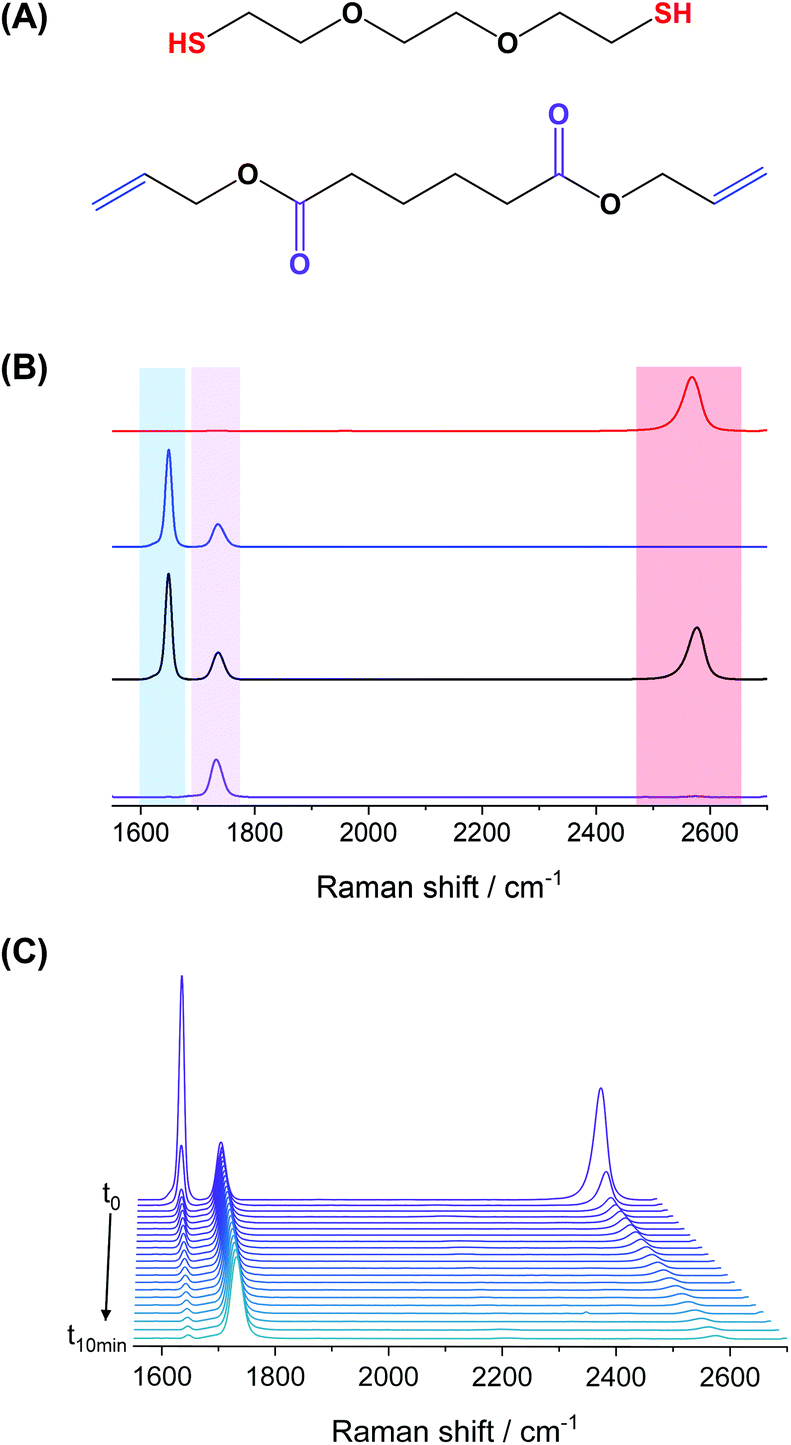 | ||
| Fig. 2 Model system used to study the kinetics of the thiol–ene polymerization. (A) Chemical structure of the monomers 2,2′-(ethylenedioxy) diethanethiol (EDDT) and diallyl adipate (DAA), and (B) Raman spectra of EDDT-DAA monomer-pair before and after polymerization in bulk. From top to bottom: EDDT (red), DAA (blue), EDDT-DAA 0% conversion – before UV irradiation (black), and EDDT-DAA 100% conversion – after UV irradiation (violet). Shaded areas highlight the vibrations of the functional groups analysed: S–H (red); CC (blue); CO (violet). (C) Evolution of the Raman spectra of the monomer mixture during a bulk polymerization from t = 0 (back) to t = 10 min (front) under UV irradiation. | ||
In addition, the use of a flow system allowed to probe a large variety of reaction conditions by conveniently changing the flow rate, using the same miniemulsion for a given set of experiments. By fixing the volume, i.e., the tubing length and diameter, of miniemulsion inside the reactor, the reaction time was calculated for each flow rate as:
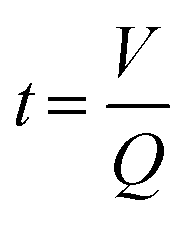 | (1) |
O stretching mode of the ester group in DAA was not involved in the reaction and remained constant. The conversion of each monomer was calculated using the variation in the integration of the thiol or ene peaks over time (Fig. 2B and C).
It is worth noting that during the polymerization in miniemulsion, the monomer droplets were stable, at least in the timeframe of the reaction. The dispersion of monomer droplets and the resulting nanoparticles suspensions were analyzed by dynamic light scattering (DLS). The size distribution measured by DLS for both the monomer droplets and the resulting nanoparticles displayed a narrow monomodal size distribution, and a moderate polymerization shrinkage was observed during the polymerization, yielding nanoparticles with an average hydrodynamic diameter of 220 nm (Fig. S3†). The size of the particles prepared with different monomer mixtures resulted in droplets and particles of similar sizes. Although phase separation between the hexadecane contained in the monomer droplets and the polythioether formed during the thiol–ene polymerization, as observed in similar systems,12 could occur, this did not influence the overall stability of the systems studied here.
Generally, the kinetics of thiol–ene photopolymerization (Fig. 3 and Fig. S4†) in bulk is chemically controlled by the interplay of the rate of reaction of the thiyl radical with the CC double bond (kP) and the rate of chain transfer from the carbon-center radical to unreacted S–H group to generate a new thiyl radical (kCT). The thiyl radicals are formed (kSH) by the reaction between a thiol and the radicals generated by the photoinitiator (kI). The chain transfer step is highly dependent on the chemical structures of the monomers involved. Typically, systems composed of thiol and allyl ethers have shown to be “chain-transfer limited”, i.e., the overall rate varies as a first-order reaction only with respect to the thiol concentration and is unaffected (zero-order) by the concentration of available CC double bonds, at least, under stoichiometric conditions.47
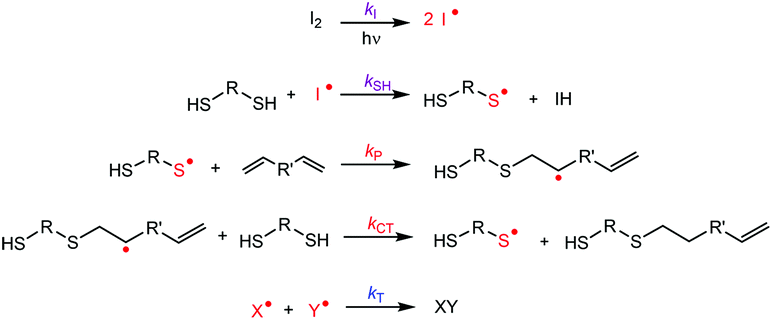 | ||
| Fig. 3 Mechanism of thiol–ene reaction polymerization of a generic system of difunctional monomers. The first step involves the generation of radical species from the UV-homolysis of the photoinitiator (kI), which then produces thiyl radicals from the reaction with thiol groups (kSH). Thiyl radicals are added to carbon–carbon double bonds (kP) in an anti-Markonikov orientation, generating carbon-centered radicals. The thiol–ene adduct is produced by a chain-transfer step (kCT), also regenerating a thiyl radical in the process. Termination (kT) happens when two radicals of the same or different species mutually quench themselves. | ||
Fig. 4 shows the kinetics of conversion of both CC ene bonds and S–H thiol bonds in bulk and in miniemulsion in the respective presence of an oil-soluble photoinitiator (TPO) and its water-soluble analog (TPO-Li), two photoinitiators displaying similar interaction with the incident light.48,49 Furthermore, the use of TPO-Li rather than TPO limited the occurrence of side reactions during the sonication process and magnified the effects of compartmentalization by segregating the initiator and the monomer in different locations. Both in bulk and miniemulsion, there is an initial rapid conversion of the functionalities, followed by a slower conversion at longer reaction times. As the conversion of the monomer increased, the molecular weight of the polymer produced increased (Fig. S5†).
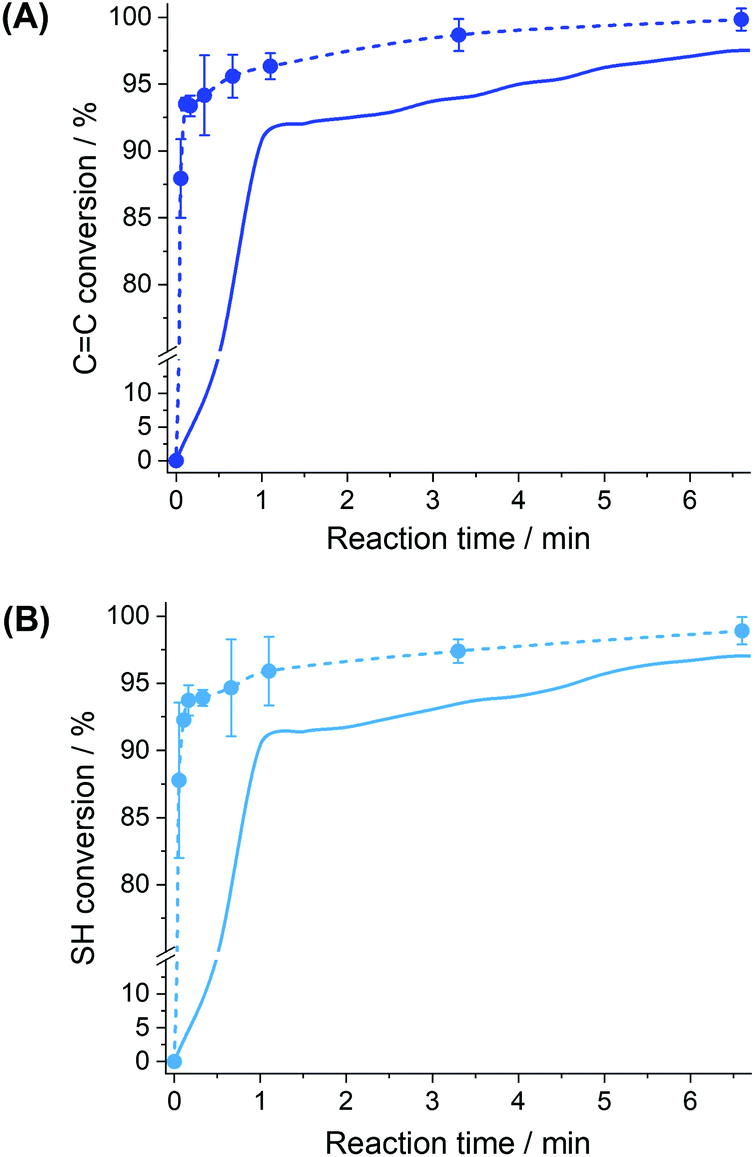 | ||
| Fig. 4 Effect of the nanoconfinement on the conversion kinetics of (A) ene and (B) thiol moieties for the EDDT-DAA monomer pair for polymerization carried out in bulk (solid line) and in miniemulsion (dashed line). The polymerizations were carried out with equimolar mixtures of monomers prepared in bulk (10 mL of monomer mixture) or in miniemulsion (2 mL of monomer mixture emulsified with 8 mL of water) irradiated with a UV-irradiance of 15 mW cm−2. | ||
Conversion profiles, such as those in Fig. 4, are characteristic of a first-order step-growth polymerizations, such as thiol–ene, where monomers are rapidly consumed to form dimers, trimers, and then oligomers, with increasing chain lengths (Fig. S5†). When high conversions of functional groups are achieved, the local concentration of reactive species is depleted, thus reducing the apparent rate of addition (Fig. S4†).
Furthermore, diffusional limitations, for example resulting from gelation, and other side reactions, could also influence the polymerization. However, these factors are not as critical as the recombination of the active sites.50 The kinetics of an ideal radical step-growth thiol–ene polymerization between dithiol and diallyl/divinyl monomers is mainly governed by its kinetics parameters, namely the ratio between propagation and chain-transfer. In reality, side reactions and bimolecular termination events between active species might also come into play and reduce the effective conversion of functional groups.51
Conversely, the conversion curves obtained for the reaction in miniemulsion show a much faster monomer conversion than the bulk reaction. Such behavior is in keeping with the increased reaction rate observed in other dispersed systems.31,32,35 This phenomenon can be ascribed to the confinement effect, provided by the formation of droplets of the monomer phase that act as individual nanoreactors, and to the mechanism of initiation of the thiol–ene reaction. This confinement can influence the polymerization reaction in different ways. The presence of the interface can force the molecules present in the droplets to adopt a reactive conformation or restrict the local movement of the reactive functionalities changing the probability of both termination and propagation occurring in the droplet. The interface also regulates the entry of the thiyl radicals in the droplets. The combination of these different factors led to the increased reaction rate observed.
Although every thiol group reacting in a thiol–ene polymerization needs to be first converted into a thiyl radical, the thiol–ene coupling is generally considered as self-sustaining, i.e., the external initiation process is essential to generate the first thiyl radical, but then following the coupling between the thiyl and the ene, a chain-transfer reaction occurs to transfer the radical from the reacted ene to an unreacted thiol present in the reaction environment. Thus, one thiyl radical entering the monomer droplet can lead to the formation of long polymer chains through the efficient propagation of the initial radical, while in bulk, the presence of a large number of thiyl and TPO-Li radicals led to competition between termination and propagation steps.
The presence of an interface between the confined and continuous spaces influences the reaction kinetics by modifying the interactions between reactants,45,52 and controlling the concentration of radicals within the droplets. Although the rate-limiting step of the thiol-allyl reaction is typically the chain transfer of the radical from the ene-radical to an unreacted thiol, the reaction rate is also affected by the amount of radical initiator used, i.e., to the concentration of thiyl radicals, as shown in Fig. 3. Experimental results (Fig. S6†) show that increasing the concentration of radicals generated by the UV-irradiation of bulk samples led, after a threshold concentration, to a reduction in the apparent polymerization rate. This phenomenon can be attributed to the recombination of free radical species produced during the degradation of the photoinitiator. Thus, the acceleration observed in the confinement of the nanodroplets can potentially be ascribed to the beneficial distribution of radical species in the system between the droplets and the continuous phase.
In the miniemulsion studied here, the radical photoinitiator was water-soluble while the monomers were confined to the hydrophobic droplets. However, the two monomers, EDDT and DAA, present different partition coefficients, with the EDDT, the di-thiol, exhibiting a higher water solubility (15 g L−1 at 25 °C).2 Consequently, EDDT can likely diffuse between the monomer droplet through the continuous aqueous phase. Furthermore, the radicals were formed during the photoconversion of the initiator TPO-Li in the continuous phase, and were unlikely to enter the monomer droplets directly due to their high hydrophilicity. The most probable reaction mechanism consists of the reaction between the TPO-Li radicals and the EDDT molecules present in the continuous phase. The reaction forms a thiyl radical, which has lower hydrophilicity than the TPO-Li radicals and can then enter the monomer droplet to initiate the polymerization. The entry of the thiyl radical into the droplets is controlled by the interface between the oil droplet and the continuous phase. Such gate control provided by the droplet interface is inexistent in bulk formulations, with all species, monomers, and radicals, being present in the medium from the onset of the irradiation and can lead to an increased termination rate. Consequently, the generation of thiyl radicals in the continuous phase of the miniemulsion and their controlled entry in the monomer droplets limited the probability of termination reaction through disproportionation or recombination in the droplets and led to the increased apparent polymerization rate observed.
The diene, DAA, was then substituted by other enes to analyze the effect of different chemical structures, more specifically electron-density and radical stability, on the polymerization kinetics. The substitution of the diene did not influence the stability, nor the size of the monomer droplets formed during miniemulsion (Fig. S3A†). The ene groups in DAA are allylic double-bonds, and their reactivity was compared to that of divinyl adipate (DVA) bearing vinylic double-bond and 1,4-butanediol diacrylate (1,4Dac) containing acrylic double bonds. Fig. 5 clearly shows that the use of different dienes yielded different polymerization.
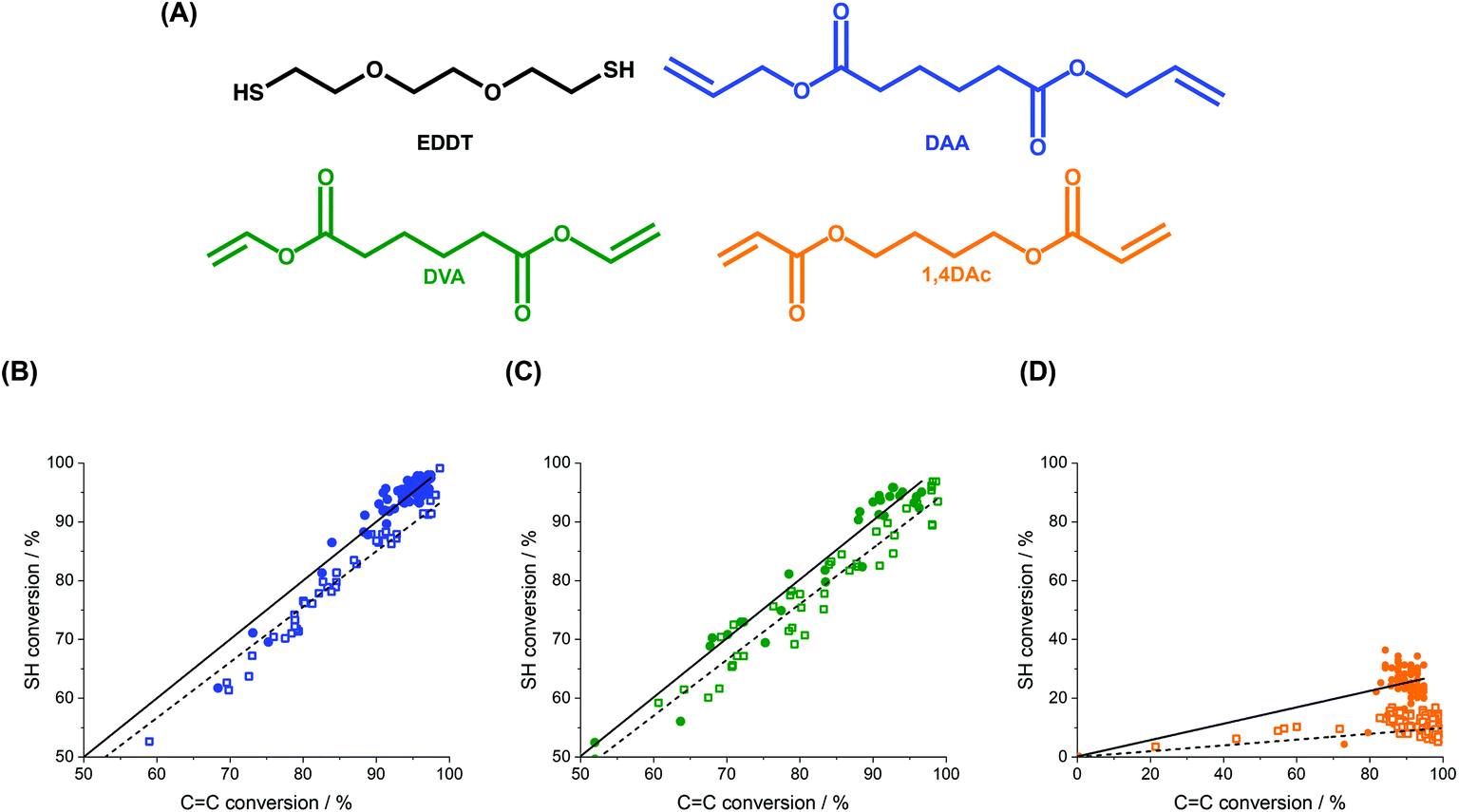 | ||
| Fig. 5 Monomer conversion during thiol–ene polymerization of different monomer pairs. (A) Chemical structures of the monomer systems. 2,2′-(ethylenedioxy)diethanethiol (EDDT) (black) was used as the di-thiol for the polymerization with diallyl adipate (DAA) (blue), divinyl adipate (DVA) (green), and 1,4-butanediol diacrylate (1,4DAc) (orange). (B-D) Comparison of the conversions of thiols and carbon–carbon double bonds in each system for polymerization carried out in miniemulsion (solid circle) and in bulk (open squares). (B) EDDT-DAA (blue), (C) EDDT-DVA (green) and (D) EDDT-1,4Dac (orange). The polymerizations were carried out with equimolar mixtures of EDDT and the respective diene prepared in bulk (10 mL of monomer mixture) or in miniemulsion (2 mL of monomer mixture emulsified with 8 mL of water) irradiated with a UV-irradiance of 15 mW cm−2. | ||
For the systems composed of DAA and DVA, the conversions of thiols and enes occurred at a similar rate. In contrast, the polymerization of 1,4Dac and EDDT led to a much faster conversion of the enes compared to the conversion of the thiols. These results suggest that, when using acrylates as the ene-component, the monomer pair did not react through a purely thiol–ene addition mechanism, but that the thiol–ene reaction competed with a free-radical chain-growth polymerization, leading to the formation of a crosslinked network, as confirmed by NMR spectroscopy (Fig. S7†). The NMR spectra of the polymers formed by the polymerization of EDDT-DAA and EDDT-DVA only show the formation of polythioethers, while the polymer obtained during the polymerization of EDDT-1,4Dac show both the formation of polythioethers and the radical homopolymerization of acrylates.
After the thiyl addition and formation of the carbon-centered radical intermediate, the thiol–ene polymerization can undergo either a chain-transfer step by abstracting a hydrogen atom from another thiol (step-growth), or can react with another alkene to undergo chain-growth homopolymerization. The fate of the reaction is defined by the relative rates of chain transfer and homopolymerization, which are in turn influenced by the chemical nature of the alkenes. Electron-poor monomers such as (meth)acrylates present high polarities and their resonance structures are stabilized by the ester moiety, which usually favors their homopolymerization. However, the chain transfer is favored in electron-rich enes, such as allyl ethers and vinyl ethers.53,54 The results presented in Fig. 5 are in keeping with the expected trends, and a mixed polymerization mechanism was observed in the mixture of monomer composed of dithiol and methacrylate-based dienes while the mixtures using diallyl and the divinyl enes polymerized primarily through thiol–ene addition.
Interestingly, Fig. 5 also shows a difference between the conversion during the polymerization in bulk and miniemulsion. In miniemulsion, the conversion of both enes and thiols occurred at an identical rate for the polymerization of DAA and DVA with EDDT. However, in bulk, the conversion of enes occurred at a moderately faster rate than the conversion of the thiols. Reactions in miniemulsion are known to reduce the incidence of side reactions,55,56 which could affect the conversion of the enes in bulk. NMR spectroscopy of the polymers obtained in bulk for EDDT-DAA and EDDT-DVA showed the presence of a limited occurrence of radical homopolymerization of the enes, which was completely absent in the polymers produced in miniemulsion (Fig. S8A and B†). The same phenomenon was also observed for the polymerization of the monomer pair EDDT-1,4Dac in miniemulsion, increasing the ratio between the conversion rate of thiol to those of enes compared to the reaction performed in bulk. Nevertheless, the conversion of enes was still highly favored in comparison to that of the thiols.
In general, the conversion of both enes and thiols occurred more rapidly in the miniemulsion nanodroplets than in the bulk (Fig. 4). This increase in the polymerization rate coefficient was observed for both the monomer pair EDDT-DAA and EDDT-DVA. Fig. 6 compares the initial rate coefficients for the polymerization of EDDT-DAA and EDDT-DVA prepared in bulk and miniemulsion. The results indicate that the polymerization rate of the reaction performed in miniemulsion increased up to 35-fold compared to the same reaction performed in bulk. This result was observed for both monomer pairs and was likely due to the lower concentration of radicals present in the monomer droplets in comparison to the bulk.
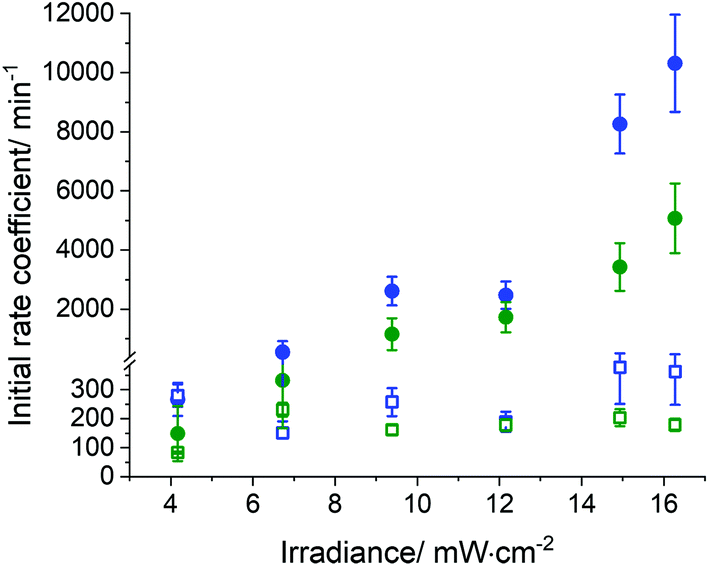 | ||
| Fig. 6 Effect of nanoconfinement in the rate of thiol–ene polymerization for EDDT-DAA (blue), and EDDT-DVA (green) performed in bulk (open squares) and miniemulsion (solid circles). The initial polymerization rate coefficients (k) were obtained from the first derivative at t = 0 of the conversion data fitted to an [CC]t = [CC]0 (1 + e−kt), where [CC]t and [CC]0 are the concentration of carbon double bonds at time t and time 0. | ||
Furthermore, the polymerization rate increased when the UV dose was increased, likely due to a faster generation of radicals from the photoinitiator, inducing the formation of the thiyl radicals, which in turn initiated the thiol–ene addition (Fig. 6 and Fig. S6†). This effect was limited for reactions performed in bulk in comparison to reactions performed in miniemulsion, but present in both. In bulk, as the local concentration of radicals increases, more termination will occur. After a threshold of irradiance or radical concentration, new species are immediately quenched, and a limited increase in the overall rate of reaction was observed (Fig. S6A†). On the other hand, when the process is performed in miniemulsion, the reaction locus is inside the nanodroplets, segregating the monomers from the radicals formed. But, even in miniemulsion, when the UV dose was increased over a certain threshold level, no net increase in the overall rate coefficient for the conversion of the ene (or thiol) was observed (Fig. 6). In the miniemulsion system, the dithiol was sparingly miscible in the continuous phase, and it can be assumed that only the thiyl radicals were able to enter the nanodroplets. The results obtained suggest that the increase in the initial reaction rate coefficient observed in miniemulsion can be ascribed to an increase in the nucleation rate of miniemulsion droplets. Increasing the number of radicals in the aqueous phase, increased the rate of termination in the aqueous phase, but also increased the mass transport of thiyl radicals from the continuous to the dispersed phase, increasing the rate of droplet nucleation and the apparent rate of conversion of the monomers. These results are in keeping with the effect observed when increasing the concentration of radical initiator during the miniemulsion of styrene, where a higher concentration of initiator in the aqueous phase led to an increase in the rate of droplet nucleation.57
We also examined the evolution of the number average molecular weight (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n) of the polymer during the reaction (Fig. 7 and Fig. S9†). The results show that as the reaction time increased, the conversion of the monomers increased, and so did the molecular weight of the polymer isolated (Fig. 7A and C) both in bulk and in miniemulsion. At high monomer conversion, after 10 minutes of polymerization, the polymers produced in miniemulsion had systematically a higher n than those obtained in bulk (Fig. S10†). Fig. 7 shows the conversion of the monomers using the consumption of the carbon–carbon double bonds. However, because of the absence of homopolymerization of the enes during the reaction of EDDT-DAA and EDDT-DVA in miniemulsion (Fig. S7 and S8†), the same trends were also observed using the consumption of the thiols groups (Fig. S9†) since the conversion of both the thiol and the enes occurred at the same rate.
n) of the polymer during the reaction (Fig. 7 and Fig. S9†). The results show that as the reaction time increased, the conversion of the monomers increased, and so did the molecular weight of the polymer isolated (Fig. 7A and C) both in bulk and in miniemulsion. At high monomer conversion, after 10 minutes of polymerization, the polymers produced in miniemulsion had systematically a higher n than those obtained in bulk (Fig. S10†). Fig. 7 shows the conversion of the monomers using the consumption of the carbon–carbon double bonds. However, because of the absence of homopolymerization of the enes during the reaction of EDDT-DAA and EDDT-DVA in miniemulsion (Fig. S7 and S8†), the same trends were also observed using the consumption of the thiols groups (Fig. S9†) since the conversion of both the thiol and the enes occurred at the same rate.
 | ||
| Fig. 7 Effect of the nanoconfinement on the average molecular weight of the polymer (n) for (A and B) EDDT-DAA (blue) and (C and D) EDDT-DVA (green). (A and C) Kinetics of the conversion of enes (left axis, black) and n of the resulting polymer (right axis, blue) in bulk (dashed line, open symbols) and in miniemulsion (solid line, closed symbols). (B and D) Influence of the monomer conversion on the n of the resulting polymers for polymerization performed in miniemulsion (closed circles) and bulk (open squares). The black curve represents a fit to eqn (2). | ||
Interestingly, n did not increase in a similar manner with the monomer conversion when the reaction was performed in bulk or in miniemulsion (Fig. 7B and D). The polymers obtained from both EDDT-DAA and EDDT-DVA in miniemulsion medium not only show higher degrees of conversions after a given reaction time, but also much larger n, even at lower monomer conversions. Typically, the variation of n, in a step-growth polymerization, such as thiol–ene polyaddition, is highly dependent on the conversion of the polymerizable groups,12,58 and high n can only be achieved at near-complete conversions, where high number average chain lengths (![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) n) can be obtained. For an ideal case, where the reactivities are equal and remain constant and in the absence of side reactions and diffusional limitations or any restrictions influencing the movement of the reactive species, the evolution of n in a step-growth polymerization can be described by:
n) can be obtained. For an ideal case, where the reactivities are equal and remain constant and in the absence of side reactions and diffusional limitations or any restrictions influencing the movement of the reactive species, the evolution of n in a step-growth polymerization can be described by:
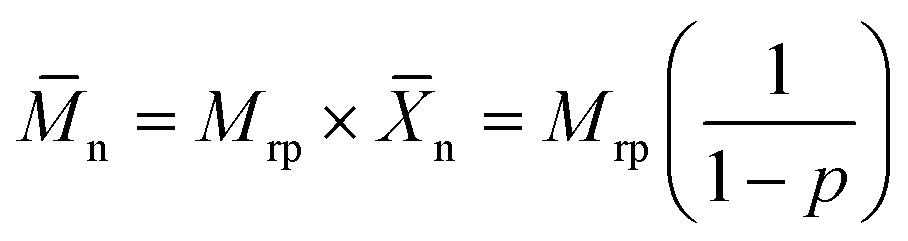 | (2) |
n is the number-average of the degree of polymerization at a monomer conversion p and Mrp is the molecular weight of the repetition unit.59
Systematically, the n obtained in bulk mostly followed the trend expected from eqn (2), although minimal deviations were observed at high conversions, likely due to the limited Raman signal from the monomer functionalities at high conversion, affecting the precise quantification of the conversion and potentially the presence of other side reactions. However, the polymers obtained by the polymerization in miniemulsion had larger n than what could be expected from eqn (2) for a given monomer conversion. The polymers synthesized during the polymerization in miniemulsion systematically displayed a larger n than what was observed for the same polymerization carried out in bulk. The maximal n observed for a given polymer produced in miniemulsion was observed earlier (both in terms of time and monomer conversion) than during the bulk polymerization, suggesting that mobility restrictions were influencing the outcome of the polymerization carried out in miniemulsion.
The molecular weight of polymers synthesized by step-growth polymerization is strongly influenced by the monomer conversion, but also the presence of impurities and stoichiometric imbalance between the reactive functionalities. Fig. 8A shows that the polymerization in miniemulsion was more tolerant to the presence of an off-stoichiometric mixture of monomers than the polymerization in bulk. In the presence of a stoichiometric excess of one of the monomers, eqn (2) can be rewritten as:
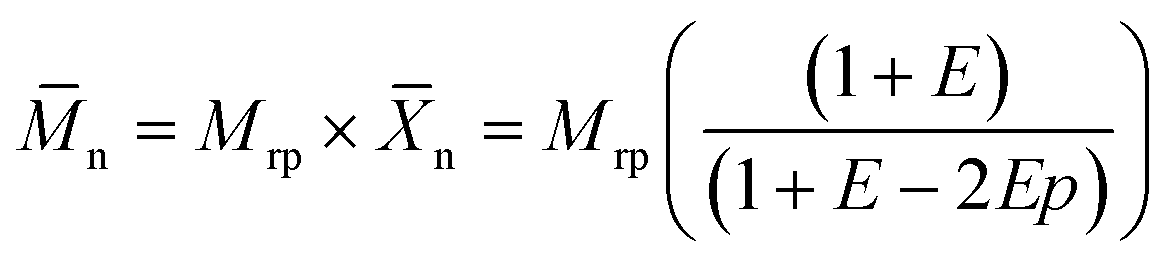 | (3) |
 | ||
| Fig. 8 Effect of excess of diene or di-thiol on the polymerization performed in miniemulsion (blue circles) and in bulk (black squares) for the EDDT-DAA monomer mixture. (A) Measured n (close symbols) and dispersity index (Đ) (open symbols). The curve represents a fit to eqn (3) for p = 98% of the limiting monomer. (B) Reaction rates for polymerization reactions performed in bulk and in miniemulsion at different stoichiometry ratios of thiol and ene-monomers. The initial polymerization rate coefficients (k) were obtained from the first derivative at t = 0 of the conversion data fitted to [CC]t = [CC]0 (1 + e−kt), where [CC]t and [CC]0 are the concentration of carbon double bonds at time t and time 0. | ||
Generally, the molecular weights obtained during the polymerization in miniemulsion were larger (15–25 kDa) than those obtained during the polymerization in bulk (4–8 kDa) (Fig. S11†). These results confirmed that the confinement effect promoted by the polymerization in a heterophase system yields high n polymers, even in off-stoichiometric conditions. Interestingly, mixtures with an excess of enes yielded polymers with higher n, and higher dispersity index (Đ), than the ones prepared with an excess of thiols, both in miniemulsion and bulk. This result could stem from side reactions occurring in the diene-rich monomer mixtures in addition to the thiol–ene polymerization. Indeed, Fig. S8† shows that while only polythioether were formed during the polymerization of the stoichiometric mixture of monomers in miniemulsion, ene–ene coupling, likely through radical homopolymerization, can be observed in the polymers produced in miniemulsion using an excess of enes. Those ene–ene coupling led to the formation of mildly crosslinked polymer samples and consequently increased the dispersity index observed for the polymers prepared with off-stoichiometric monomer mixtures. For the polymers prepared in bulk, the different monomer compositions showed evidence of enes homopolymerization (Fig. S8†).
The rate coefficient for the polymerization reaction was calculated for each stoichiometry. The rates of ene conversion showed the influence of nanoconfinement on the thiol–ene polymerization (Fig. 8B) for the different stoichiometric mixtures of dithiol (EDDT) and diene (DAA). As established previously, the monomer systems composed of EDDT-DAA undergo only thiol–ene polymerization in miniemulsion (Fig. S7 and S8†). Consequently, similar results were observed when the rate coefficients were calculated from the conversion of the thiols rather than the enes (Fig. S12†). While the rate of the reactions performed in bulk decreased as the excess of one or the other monomer increased, such a trend was absent for reactions carried out in miniemulsion. In addition to the enhancement of the polymerization rate observed for stoichiometric mixtures of monomers (Fig. 6), the effect of confinement in miniemulsion formulations prevented the decrease in the polymerization rate, even when the reaction was performed with an off-stoichiometric ratio of monomers. This result suggests that in addition to the control entry of thiyl radicals in the miniemulsion droplets, the thiol–ene step-growth polymerization performed in miniemulsion also benefited from the restricted mobility of the reactive functionalities, likely promoted by the presence of the interface of the droplets, which modified the reaction probability and influencing the rate of termination and propagation steps, regardless of the presence of 5 or 10% of stoichiometric excess of either dienes or dithiols.
Conclusions
The kinetics of the photopolymerization of thiol–ene systems in bulk and miniemulsion conditions was analyzed by Raman spectroscopy. This allowed for the simultaneous detection and quantification of both thiols and ene moieties. The analysis of the polymerization kinetics using Raman spectroscopy allowed us to detect the coexistence of different polymerization mechanisms. This situation was more pronounced in monomer pairs prone to homopolymerization, such as (meth)acrylates.Polymerizations performed in miniemulsion showed much higher reaction rates than the ones carried in bulk as a consequence of the confinement effect. Each droplet of the miniemulsion contained the two monomers and acted as nanoreactors, promoting higher conversions, and yielding polymers with higher molecular weight than what was achieved in bulk. The presence of the interface between the monomer droplet and the continuous aqueous phase regulated the entry of radicals into the nanoreactor, reducing the probability of termination.
Moreover, the reduced reaction locus appeared to create an environment that restricts the number of radicals, increasing their effective local concentration in the nanoenvironment defined by the monomer droplet. In addition, the interface could also hinder the mobility of the reactive species or induce changes in the molecule conformations, both factors influencing the rate of the propagation and termination steps. Systematically, higher molecular weights were observed in miniemulsion in comparison to the same polymerization performed in bulk. This phenomenon was even more pronounced when the polymerization was performed with non-stoichiometric mixtures of monomers. The number-average of molar masses obtained for experiments carried with non-stoichiometric ratios of thiol and ene functionalities in miniemulsion suggest that either the propagation step was promoted in the miniemulsion compared to the bulk, or the termination steps were hindered. In any case, the compartmentalization of the radicals alone is insufficient to describe the higher rate coefficient observed in the miniemulsion polymerization of the monomer mixtures prepared with an off-stoichiometric ratio.
The polymerization of thiol–ene pairs in confinement produced well-defined nanoparticles, composed of polymer with high average molecular weights, larger than what could typically be expected for step-growth polymers produced with the degree of conversion achieved. In addition, the effect of confinement seems not only to enhance the total rate of polymerization, but also to limit the effects associated with the presence of one of the monomers in stoichiometric excess. This is an interesting effect, which can be harnessed to design high molecular weight polymers with well-defined end-group that can be further used for post-polymerization functionalization with an additional thiol–ene coupling reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 765341 (Project PHOTO-EMULSION, MSCA-ITN-2017). The authors are also grateful to Ute Heinz, and Sandra Seywald for the GPC measurements, and to Cuong Minh Quoc Le for the measurements of the irradiance of the LED system. Open Access funding provided by the Max Planck Society.References
- D. Sticker, R. Geczy, U. O. Häfeli and J. P. Kutter, ACS Appl. Mater. Interfaces, 2020, 12, 10080–10095 CrossRef CAS PubMed.
- F. Jasinski, E. Lobry, B. Tarablsi, A. Chemtob, C. Croutxé-Barghorn, D. Le Nouen and A. Criqui, ACS Macro Lett., 2014, 3, 958–962 CrossRef CAS.
- E. P. Magennis, A. L. Hook, P. Williams and M. R. Alexander, ACS Appl. Mater. Interfaces, 2016, 8, 30780–30787 CrossRef CAS PubMed.
- E. D. Rodriguez, X. Luo and P. T. Mather, ACS Appl. Mater. Interfaces, 2011, 3, 152–161 CrossRef CAS.
- L. Zhang, X. Ren, Y. Zhang and K. Zhang, ACS Macro Lett., 2019, 8, 948–954 CrossRef CAS.
- T. A. Sherazi, in Encyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer, Berlin, 2015, pp. 1–2 Search PubMed.
- V. Mishra, J. Desai and K. I. Patel, J. Coat. Technol. Res., 2017, 14, 1069–1081 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- D. Love, B. Fairbanks and C. Bowman, ACS Macro Lett., 2020, 174–179 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- E. Lobry, F. Jasinski, M. Penconi, A. Chemtob, C. Croutxé-Barghorn, E. Oliveros, A. M. Braun and A. Criqui, RSC Adv., 2014, 4, 43756–43759 RSC.
- C. M. Q. Le, L. Vidal, M. Schmutz and A. Chemtob, Polym. Chem., 2021, 12, 2084–2094 RSC.
- L. Infante Teixeira, K. Landfester and H. Thérien-Aubin, Macromolecules, 2021, 54, 3659–3667 CrossRef CAS PubMed.
- P. B. Zetterlund and D. R. D'Hooge, Macromolecules, 2019, 52, 7963–7976 CrossRef CAS.
- Y. W. Marien, P. H. M. Van Steenberge, D. R. D'Hooge and G. B. Marin, Macromolecules, 2019, 52, 1408–1423 CrossRef CAS.
- B. P. Sutherland, M. Kabra and C. J. Kloxin, Polym. Chem., 2021, 12, 1562–1570 RSC.
- M. Claudino, M. Jonsson and M. Johansson, RSC Adv., 2013, 3, 11021–11034 RSC.
- F. Jasinski, A. Rannée, J. Schweitzer, D. Fischer, E. Lobry, C. Croutxé-Barghorn, M. Schmutz, D. Le Nouen, A. Criqui and A. Chemtob, Macromolecules, 2016, 49, 1143–1153 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 1540–1573 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- T. Y. Lee, Z. Smith, S. K. Reddy, N. B. Cramer and C. N. Bowman, Macromolecules, 2007, 40, 1466–1472 CrossRef CAS.
- A. F. Senyurt, H. Wei, C. E. Hoyle, S. G. Piland and T. E. Gould, Macromolecules, 2007, 40, 4901–4909 CrossRef CAS.
- A. Lungu, J. Ghitman, A. I. Cernencu, A. Serafim, N. M. Florea, E. Vasile and H. Iovu, Polymer, 2018, 145, 324–333 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Polym. Chem., 2017, 8, 6099–6105 RSC.
- Y. Liu, W. Hou, H. Sun, C. Cui, L. Zhang, Y. Jiang, Y. Wu, Y. Wang, J. Li, B. S. Sumerlin, Q. Liu and W. Tan, Chem. Sci., 2017, 8, 6182–6187 RSC.
- Q. Tian, H. Zhao and S. L. Simon, Polymer, 2020, 205, 122868 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- L. M. Forero Ramirez, J. Babin, M. Schmutz, A. Durand, J. L. Six and C. Nouvel, Eur. Polym. J., 2018, 109, 317–325 CrossRef CAS.
- P. B. Zetterlund, Polym. Chem., 2011, 2, 534–549 RSC.
- H. Tobita, Polymers, 2016, 8, 155 CrossRef PubMed.
- M. Khan, T. R. Guimarães, D. Zhou, G. Moad, S. Perrier and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1938–1946 CrossRef CAS.
- H. Y. Zhao and S. L. Simon, Polymer, 2015, 66, 173–178 CrossRef CAS.
- P. Laurino, H. F. Hernandez, J. Bräuer, K. Krüger, H. Grützmacher, K. Tauer and P. H. Seeberger, Macromol. Rapid Commun., 2012, 33, 1770–1774 CrossRef CAS PubMed.
- M. J. Monteiro, Macromolecules, 2010, 43, 1159–1168 CrossRef CAS.
- R. H. Staff, P. Rupper, I. Lieberwirth, K. Landfester and D. Crespy, Soft Matter, 2011, 7, 10219–10226 RSC.
- C. Herrmann, D. Crespy and K. Landfester, Colloid Polym. Sci., 2011, 289, 1111–1117 CrossRef CAS.
- B. Sanz, N. Ballard, A. Marcos-Fernandez, J. M. Asua and C. Mijangos, Polymer, 2018, 140, 131–139 CrossRef CAS.
- H. Zhao and S. L. Simon, Polymer, 2020, 211, 123112 CrossRef CAS.
- B. Colak, J. C. S. Da Silva, T. A. Soares and J. E. Gautrot, Bioconjugate Chem., 2016, 27, 2111–2123 CrossRef CAS PubMed.
- K. Jiang, Y. Liu, Y. Yan, S. Wang, L. Liu and W. Yang, Polym. Chem., 2017, 8, 1404–1416 RSC.
- O. Okay, S. K. Reddy and C. N. Bowman, Macromolecules, 2005, 38, 4501–4511 CrossRef CAS.
- N. B. Cramer, C. L. Couch, K. M. Schreck, J. E. Boulden, R. Wydra, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2010, 26, 799–806 CrossRef CAS.
- K. Shanmuganathan, R. K. Sankhagowit, P. Iyer and C. J. Ellison, Chem. Mater., 2011, 23, 4726–4732 CrossRef CAS.
- R. Jenjob, F. Seidi and D. Crespy, Adv. Colloid Interface Sci., 2018, 260, 24–31 CrossRef CAS PubMed.
- N. Sandström, R. Z. Shafagh, A. Vastesson, C. F. Carlborg, W. van der Wijngaart and T. Haraldsson, J. Micromech. Microeng., 2015, 25, 075002 CrossRef.
- N. B. Cramer, T. Davies, A. K. O'Brien and C. N. Bowman, Macromolecules, 2003, 36, 4631–4636 CrossRef CAS.
- T. Majima, W. Schnabel and W. Weber, Makromol. Chem., 1991, 192, 2307–2315 CrossRef CAS.
- G. W. Sluggett, C. Turro, N. J. Turro, M. W. George and I. V. Koptyug, J. Am. Chem. Soc., 1995, 117, 5148–5153 CrossRef CAS.
- D. R. D′hooge, M. F. Reyniers and G. B. Marin, Macromol. React. Eng., 2013, 7, 362–379 CrossRef.
- L. De Keer, K. I. Kilic, P. H. M. Van Steenberge, L. Daelemans, D. Kodura, H. Frisch, K. De Clerck, M. F. Reyniers, C. Barner-Kowollik, R. H. Dauskardt and D. R. D′hooge, Nat. Mater., 2021, 20, 1422–1430 CrossRef CAS PubMed.
- K. Piradashvili, E. M. Alexandrino, F. R. Wurm and K. Landfester, Chem. Rev., 2016, 116, 2141–2169 CrossRef CAS PubMed.
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Macromolecules, 2003, 36, 7964–7969 CrossRef CAS.
- F. J. Schork, Y. Luo, W. Smulders, J. P. Russum, A. Butté and K. Fontenot, Adv. Polym. Sci., 2005, 175, 129–255 CrossRef CAS.
- F. Jasinski, P. B. Zetterlund, A. M. Braun and A. Chemtob, Prog. Polym. Sci., 2018, 84, 47–88 CrossRef CAS.
- N. Bechthold and K. Landfester, Macromolecules, 2000, 33, 4682–4689 CrossRef CAS.
- J. M. Asua, Prog. Polym. Sci., 2014, 39, 1797–1826 CrossRef CAS.
- L. De Keer, P. H. M. Van Steenberge, M.-F. Reyniers and D. R. D′hooge, Polymers, 2021, 13, 2410 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization of the polymers (13C-NMR and 1H-NMR 2D-spectra), and details of kinetics. See DOI: https://doi.org/10.1039/d2py00350c |
| This journal is © The Royal Society of Chemistry 2022 |