
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of the block copolypeptide surfactant structure on the size of polypeptide nanoparticles obtained by mini emulsion polymerisation†
Nicola
Judge
a,
Dražen
Pavlović
 a,
Evelin
Moldenhauer
b,
Paul
Clarke
c,
Ruairi
Brannigan
*ad and
Andreas
Heise
*aef
a,
Evelin
Moldenhauer
b,
Paul
Clarke
c,
Ruairi
Brannigan
*ad and
Andreas
Heise
*aef
aDepartment of Chemistry, RCSI University of Medicine and Health Sciences, Dublin 2, Ireland. E-mail: andreasheise@rcsi.ie; ruairi.brannigan@dcu.ie
bPostnova Analytics GmbH, Landsberg, Germany
cPostnova Analytics UK Ltd, Malvern, UK
dSchool of Chemical Sciences, Dublin City University, Collins Avenue, Whitehall, Dublin 9, Ireland
eScience Foundation Ireland (SFI) Centre for Research in Medical Devices (CURAM), RCSI, Dublin 2, Ireland
fAMBER, The SFI Advanced Materials and Bioengineering Research Centre, RCSI, Dublin D02, Ireland
First published on 21st April 2022
Abstract
Polypetide nanoparticles obtained by miniemulsion polymerisation of amino acid N-carboxyanhydrides (NCA) are a novel class of tuneable bio-derived functional nano materials for potential applications in nutraceutics, agriculture, and medicine. This work discloses a facile route to stable hydrophobic polypeptide nanoparticles comprising a poly(L-phenylalanine) and poly(L-leucine) core, respectively, using two amphiphilic glycosylated block copolypeptide surfactants with hydrophobic poly(L-phenylalanine) or poly(L-leucine) blocks. All surfactant/core combinations produce stable nanoparticle dispersions with average particle sizes between 160 and 220 nm. However, analyses using light scattering techniques, SEM imaging and Asymmetric Field Flow Fractionation, reveal a particle size dependence on the surfactant/core combination in that particles are reproducibly 20–30% larger if the surfactant block is identical to the amino acid polymerised in the core. It is hypothesised that this is caused by complex hydrophobic and secondary structure interactions between the surfactant and particle core. These fundamental insights will inform the future design of polypeptide nanoparticle libraries utilizing many different amino acids for example in nanomedicine.
Introduction
Degradable nanoparticles find applications in many fields such as nutraceutics,1 agriculture,2 and medicine.3 Typically degradable nanoparticles are composed of polyesters, polyamides, polysaccharides or polypeptides, whose beneficial factor is that they degrade to non-toxic, low molecular weight products which are typically easily eliminated. Conventionally, these degradable nanoparticles are formed from preformed polymers, which look to utilise hydrophobic interactions, electrostatic forces, hydrogen bonding and van der Waal forces, or some combination thereof, to maintain their integrity.4–6 The synthesis of these degradable nanoparticles typically requires a post polymerisation synthetic step and therefore, in the field of degradable nanoparticle synthesis, there has been a significant shift towards more streamlined and reproducible alternatives such as heterogeneous polymerisation systems.7 Emulsion polymerisation is an alternative method to fabricate nanoparticles which utilises emulsifying surfactants. As such, emulsion polymerisations offer enhanced control over the resulting size and morphology of nanoparticles compared to traditional particle fabrication techniques, through the selection of surfactant composition, concentration, ratio of the different phases used and the addition of co-stabilisers.8,9 However, significant amount of mass transfer between oil droplets can cause disperse size distribution. Furthermore, when considering a typical oil in water (o/w) emulsion, this methodology is unsuitable for water sensitive polymerisation procedures as often utilised in the synthesis of biodegradable polymers. Therefore, miniemulsion polymerisation has grown in popularity because of the formation of “micro-reactors” within which the polymerisation proceeds to produce the final nanoparticles.10,11 Furthermore, the utilisation of amphiphilic polymeric surfactants which self-assemble within dilute solutions and, once the monomer droplets are introduced, become kinetically irreversibly adsorbed to the oil interface, have allowed for the fabrication of particles with more precise control over size and homogeneity. This is owing to the lack of or slow exchange rate between micelles, further supressing the chance of mass transfer during polymerisation. This was demonstrated by Lu et al. when an amphiphilic poly(acrylic acid-b-styrene) was used as the surfactant within a Reversible Addition–Fragmentation chain-Transfer (RAFT) miniemulsion polymerisation to produce poly(styrene) nanocapsules.12The hydrophilic surfactant block can also be used to introduce functionality to the nanoparticles. Previously, we have applied this concept to obtain glycosylated poly(styrene) nanoparticles using a glycosylated polypeptide/polystyrene block copolymer surfactant and demonstrated selective lectin binding as well as cell uptake for intercellular oxygen sensing.13–16 In most reported cases the hydrophobic portion of the surfactant is structurally similar to the polymer produced in the oil phase to ensure complementary interfacial interaction. However, in some examples, incompatibilities between both polymer structures give rise to de-mixing phenomena caused by polymer immiscibility and repulsive molecular forces. A comprehensive study on the effect of mismatching surfactant/core structures was reported by the Heuts group where poly(styrene) and poly(methyl methacrylate) nanoparticles were synthesised using a poly(styrene) containing polymeric surfactant.17 The authors found that if the surfactant block and the core polymer are incompatible, phase separation occurs resulting in irregular particle morphologies. Moreover, Riess reported that the composition of the surfactant influenced the resulting particle size through the adsorption and intermolecular forces between the core polymer and surfactant system.18 These examples, as many others in the literature, are mostly limited to radical polymerisation techniques.19,20 Traditionally, degradable nanoparticles have been fabricated through the emulsification of pre-synthesised degradable polymers.21,22 Of the limited examples of degradable nanoparticles synthesised through miniemulsion polymerisation, almost all rely on the use of commercially available surfactants; significantly reducing the library of potential monomers available for polymerisation.23–25 Polypeptides are a class of degradable materials possessing a wide variety of inherent functionalities based on the amino acid building blocks chosen, such as amines, carboxylic acids, thiols etc. High molecular weight homo- and co-polypeptides with low molecular weight dispersities can be synthesised using the ring opening polymerisation (ROP) of amino acid-derived N-carboxyanhydride (NCA) monomers.26–28 One drawback of the ROP of NCA monomers is their moisture sensitive nature whereby anhydrous conditions are usually required for their successful controlled polymerisation. However, recently there have been reports of aqueous methods being utilised based on supressing or outpacing the hydrolysis of the NCA monomer, whilst increasing the rate of polymerisation. Song et al. utilised a biphasic DCM/aqueous system where a poly(benzyl-L-glutamate) α-helical macroinitiator was found to increase the rate of monomer consumption and allowed for the synthesis of high molecular weight polypeptides with narrow dispersities.29 More recently polymerisation induced self-assembly (PISA) has been applied within a basic aqueous buffer conditions to produce in situ polypeptide nanoparticles by ring opening polymerisation of a benzyl-L-glutamate NCA monomer.30,31 We have recently disclosed the first NCA miniemulsion technique whereby core crosslinked polypeptide nanoparticles were synthesised by a miniemulsion polymerisation utilising an amphiphilic glycopolypeptide surfactant.32 In the reported system the amphiphilic surfactant comprised an hydrophobic poly(L-phenylalanine) block, which enabled the formation of a poly(S-o-nitrobenzyl-L-cysteine) core. Subsequent UV crosslinking of the core was applied to increase the nanoparticle stability.
In this study, poly(L-phenylalanine), P(Phe), and poly(L-leucine), P(Leu), were selected as aromatic and aliphatic hydrophobic core materials, respectively. Hydrophobic polypeptides are known to assemble through secondary structure interactions such as α-helices and β-sheets,33 which was expected to strongly contribute to the stability of the polypeptide nanoparticles. Unlike for acrylic or styrenic surfactant/core systems, there is no knowledge available how the surfactant/core compatibility affects the formation and characteristics of polypeptide nanoparticles in an NCA miniemulsion polymerisation. We hypothesised that compatibility effects at the surfactant/core interface as well as secondary structure interactions could be a critical factor in this process. Here we present a fundamental study into the tuneability of sizes the system affords based on the differences in core polypeptides and the nature of the hydrophobic portion of the surfactant.
Results and discussion
Surfactant synthesis
Two amphiphilic surfactants were synthesised containing lactobionic acid modified poly(L-lysine) as the hydrophilic block and PLeu or PPhe as the hydrophobic block (Fig. 1). Firstly a poly(ε-carbobenzyloxy-L-lysine) (poly(Z-L-lysine)) block 1 was synthesised through the allylamine initiated ROP of the respective NCA targeting a degree of polymerisation (DP) of 50. Upon full monomer consumption, as monitored by ATR-FTIR spectroscopy through the disappearance of the NCA anhydride peaks (1850 cm−1 and 1790 cm−1), the second NCA (Phe or Leu) was added aiming at a DP of 10 to yield block copolymers 2. It was necessary to grow the second block, especially for phenylalanine, in an excess volume of DMF in order to prevent precipitation due to reduced solubility as the chain grows. Subsequently, the lysine block was deprotected (3) and reacted with lactobionic acid (30% glycosylation targeted) to enhance hydrophilicity and yield the desired block copolypeptides. Size Exclusion Chromatography (SEC) displayed monomodal traces (Đ ∼1.1) with the expected molecular weight shift for both surfactant variations throughout the synthetic stages (Fig. 2 and Fig. S1†). All signals for 1H NMR were in good agreement with the block copolypeptide structures (Fig. 2 and Fig. S2†) and the degree of polymerisation was calculated for the protected polypeptide through the ratio of the methine proton of the initiator allyamine (5.6 ppm) to the methylene protons of the polypeptide protecting group of P(Z-L-Lys) (5.2 ppm) and the methylene group of P(L-Phe) (2.8 ppm) or the isobutyl group of P(L-Leu) (0.8 ppm) (Fig. S3 and S4†).13 Most significant, the spectra show the disappearance of the P(Lys) protecting group signal at 5.3 ppm (green box, Fig. 2) and at 7.3 ppm (purple box) highlighting a successful deprotection as well as the appearance of lactobionic acid signals at 4–5 ppm (blue box) confirming the glycosylation of the block copolypeptide surfactants. All characterisation data are summarised in Table S1.† In the following, the surfactants will be denoted S-Phe for the phenylalanine block copolypeptide and S-Leu for the leucine containing block copolypeptide, respectively. | ||
| Fig. 1 Concept of NCA miniemulsion polymerisation by varying polypeptide segments in the surfactant and the polypeptide core. Synthesis of surfactant variations S-Phe and S-Leu. (a) ZLL NCA, allyamine, DMF, 0 °C, vacuum, (b) L-Phe NCA or L-Leu NCA, DMF, 0 °C, vacuum, (c) TFA, HBr (33% in acetic acid), 25 °C (d) lactobionic acid, sulfo-NHS, EDC, 0.1 mM, MES buffer, 25 °C. Miniemulsion nanoparticle synthesis: surfactant (S-Phe/S-Leu) DI water solution in ice bath, NCA (C-Phe or C-Leu) in DCM solution added under sonication (c = 100, A = 70%) and sonicated for 15 min before trimethylamine addition. | ||
 | ||
| Fig. 2 1H NMR spectra (400 MHz, CF3OOD) and SEC traces (HFiP, RI detection) of the three stages of synthesis of S-Leu copolypeptide surfactant. Full 1H NMR peak assignment can be found in ESI Fig. S3 and S4.† | ||
Mini-emulsion polymerization
For the miniemulsion process to form polypeptide NPs, an aqueous solution of the surfactant (Fig. 3A), was sonicated whilst a dichloromethane solution of Phe and Leu monomers of core forming polypeptides (denoted as C-Phe and C-Leu) was added forming stable emulsions (Fig. 3B). For all four surfactant/monomer combinations (S-Leu/C-Leu, S-Leu/C-Phe, S-Phe/C-Phe and S-Phe/C-Leu), the oil-in-water emulsions were formed successfully, as can be seen by the change in turbidity between Fig. 3A and B. Subsequently triethylamine was added to initiate the NCA polymerisation in the particle core in an open vessel. After 24 h the reaction mixture was dialysed and purified to afford a fully aqueous dispersion of particles (Fig. 3C). No precipitation was observed during any of these steps or after dialysis signifying high stability of the core forming homopolypeptide within the core of the nanoparticles. | ||
| Fig. 3 Images of (A) the surfactant solution S-Phe in water, (B) solution after emulsification by sonication (15 min, c = 100, A = 70%) with Phe NCA in dichloromethane and (C) the dialysis purified nanoparticle solution after core polymerisation (C-Phe). | ||
Spectroscopically the presence of the core homopolypeptide and the surfactant in the nanoparticles after dialysis was confirmed by comparative 1H NMR spectra which were recorded in d-TFA as a common solvent for surfactant and core polypeptide. Fig. 4 depicts the example of the S-Leu/C-Phe combination. The presence of diagnostic aromatic signals of P(Phe) at 7–7.5 ppm and of the S-Leu –CH3 at 0.8 ppm confirms the successful core polymerisation as well as that the surfactant remains adsorbed onto the particle surface after purification. Similar results were obtained for S-Phe/C-Leu nanoparticles (Fig. S5†) but was not conducted for the variations where the core and surfactant contained the same amino acid as no difference can be seen.
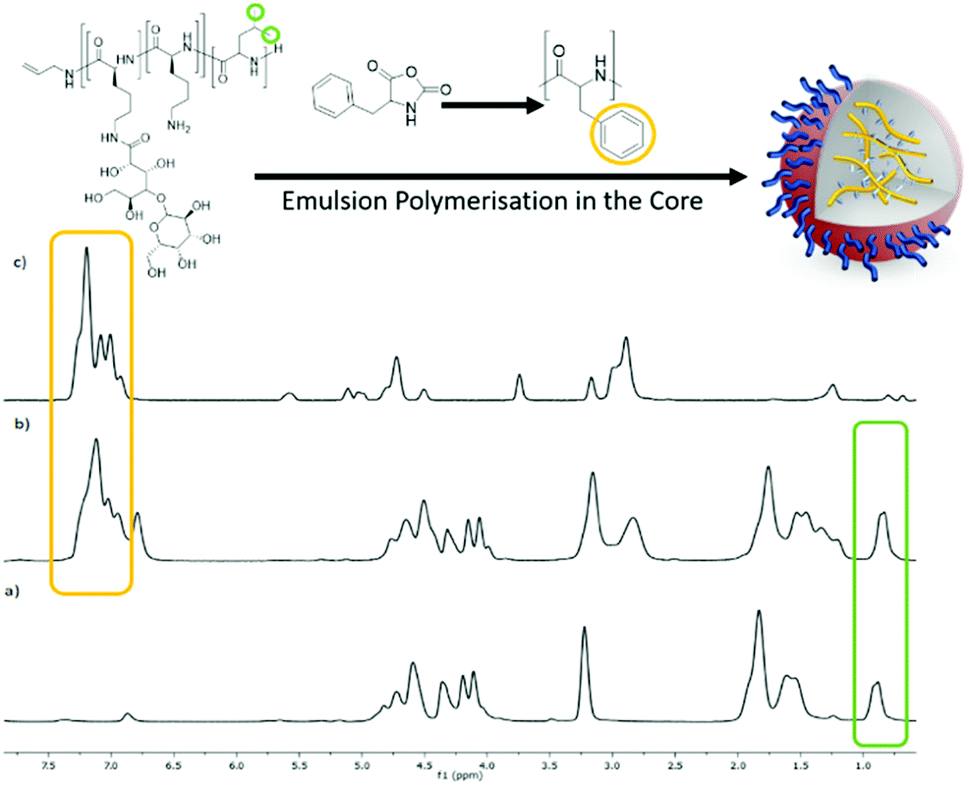 | ||
| Fig. 4 1H NMR (400 MHz, CF3COOD) spectra of (a) the surfactant S-Leu, (b) the nanoparticle obtained from the core polymerisation of Phe NCA using the S-Leu surfactant, S-Leu/C-Phe and (c) polyPhe. | ||
DLS was first used to track the emulsion droplet size for the initial 24 hours during which the polymerisation occurs. The results for all four surfactant/core combinations (n = 3) are plotted in Fig. 5 and summarised in Table S2.† It was found that the Z-average diameters are consistent for each time point, which is indicative of a stable emulsion for the duration of the polymerisation, as seen by the narrow standard deviations. Moreover, monomodal DLS traces and correlograms (Fig. S6†) achieved for each time point highlight the robustness of the data. It can therefore be said that the system is highly reproducible with a minimal batch to batch variation in z-average size when measured by DLS. Also the particles stability against dilution was eluded to through the lack of size change before and after purification by dialysis. Therefore we have successfully synthesised four variations of polypeptide NPs using a miniemulsion set up, during which the tracking of the polymerisation presented statistical differences. This eludes to the potential forces present within the core that allows for the nanoparticles to be formed in situ.
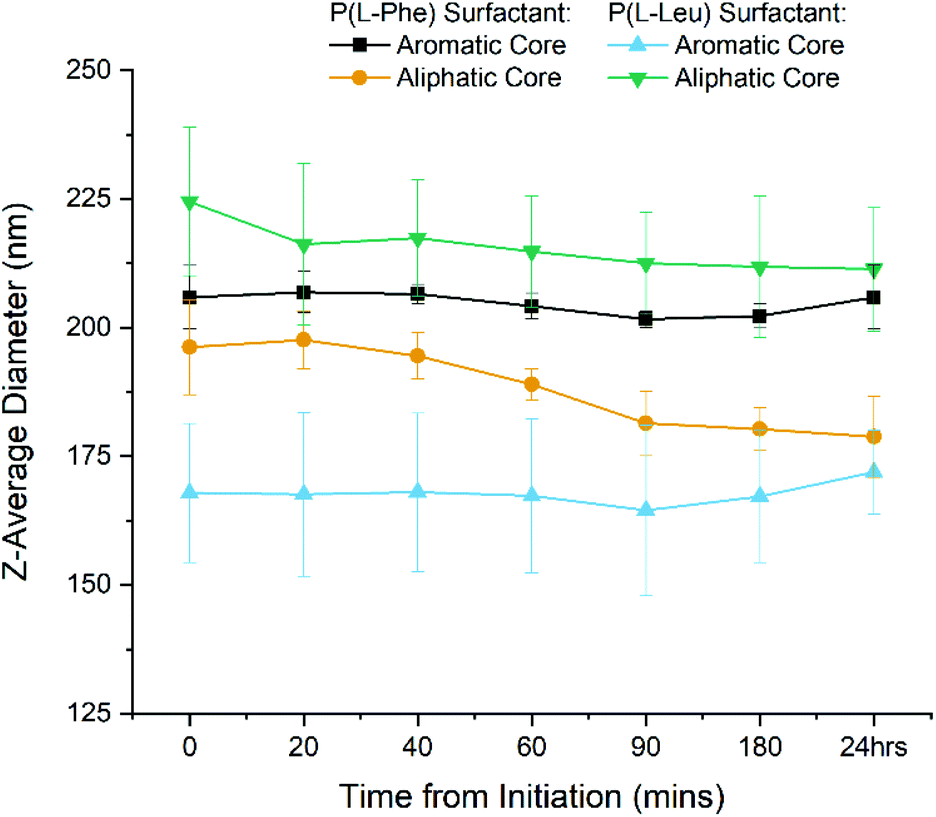 | ||
| Fig. 5 DLS Z-average diameter of the 4 different emulsion nanoparticle variations during the 24 h miniemulsion polymerisation. Samples taken at different time points directly from the miniemulsion reaction. Error bars represent standard deviation (n = 3). | ||
Nanoparticle characterisation
For all further studies the nanoparticles were collected after 24 h, purified by 72 h dialysis against water and characterised. As suggested by Kim et al.,34 to avoid any bias by a single method, the polypeptide NP's were systematically analysed by Dynamic Light Scattering (DLS), Nanoparticle Tracking Analysis (NTA), Transmission Electron Microscopy (TEM) and preliminary asymmetric flow field flow fractionation (AF4). Comparing the final DLS sizes, statistically significant differences between all four different combinations were found (Table 1, Fig. S7, S8 and Tables S3, S4†). When the surfactant polypeptide matches the core polypeptide (e.g. S-Phe/C-Phe and S-Leu/C-Leu), the resulting sizes are significantly larger with z-average diameters of 203.8 ± 8.8 nm and 224.2 ± 16.0 nm, respectively. Conversely, smaller particles are obtained when the two structures are different such as in S-Phe/C-Leu, 164.4 ± 17.5 nm, and S-Leu/C-Phe, 173.7 ± 6.5 nm. The DLS data were corroborated by NTA analysis. While both methods are light scattering techniques, the resulting data places emphasis on different factors. DLS Z-average reported values places a greater emphasis on the intensity average compared to NTA which focuses on the number average and so in general reports smaller diameters.34 However the raw number average size as measured by DLS can be compared to that of the NTA (Table S6†).| Surfactant/core | DLS | NTA | |
|---|---|---|---|
| Size [nm] | Disp. | Size [nm] | |
| S-Phe/C-Phe | 203.8 ± 8.8 | 0.19 ± 0.02 | 150.5 ± 10.9 |
| S-Phe/C-Leu | 164.4 ± 17.5 | 0.21 ± 0.02 | 134.0 ± 10.7 |
| S-Leu/C-Phe | 173.7 ± 6.5 | 0.18 ± 0.02 | 123.5 ± 2.1 |
| S-Leu/C-Leu | 224.2 ± 16.0 | 0.18 ± 0.03 | 180.9 ± 7.8 |
From the data obtained by both light scattering methods it is evident that the same trend is seen despite the technique used (Table 1 and Fig. S12†). This is a strong indication that depending on the surfactant/core composition the intermolecular forces and compatibility factors between surfactant and final core polypeptide dictate the final size. If the core polypeptide is structurally matched by the surfactant, larger particles are obtained, while in case of a mismatch, average particles sizes are smaller. As this effect is independent of the type of the core polypeptide it must be caused by a core-surfactant interaction. The exact nature of this interaction is somewhat speculative. It could be the result of polymer incompatibility leading to a phase separation between surfactant and core as seen with acrylic systems.17 However, considering the strong secondary structure interactions between polypeptides, it is conceivable that these play a significant role in this process in that surfactants capable of matching the core secondary structure are incorporated into the NP bulk assembly creating a more homogeneous interface.
Finding evidence for secondary structure interactions in this complex system is challenging. Owing to their lack of solubility, homo P(Phe) and P(Leu) are not widely studied in the literature. However, early FTIR studies on the polymerisation of L-Phe NCA and L-Leu NCA when initiated with trimethylamine, as is done here for the nanoparticle core, P(Leu) exhibited up to 95% α-helix content35,36 compared to P(Phe) which displayed a predominant β-sheet structure depending on the solvent used.37 The high propensity of P(Leu) for helical structures was ascribed to the more freely rotatable aliphatic side chain compared to the bulky aromatic side chain of P(Phe), which therefore assumes a predominantly β-sheet structure. Consequently, the two hydrophobic polypeptides differ not only in their side chain structure (aromatic vs. aliphatic) but also in their hypothetical secondary structures within the core. This eludes to the potential forces present within the core that allows for the nanoparticles to be formed and stabilised through a combination of hydrophobic and secondary structure interactions. The hydrophobic surfactant blocks have the same propensity for the respective secondary structures although it has been shown that precipitation during purification can somewhat improve the helical content of P(Phe) containing polypeptides as it resolves their thermodynamic instability as sheets.37 Therefore it can be hypothesised that in the variations containing C-Phe the core is more likely to contain a higher percentage of β-sheet structures compared to those with a C-Leu core, which is in agreement with the expectation. It can be speculated that if the surfactant block is of the same nature and can adopt the same secondary structure as the core, it integrates into the core arrangement resulting in larger nanoparticle sizes, such as that seen in the S-Leu/C-Leu or S-Phe/C-Phe compositions (Fig. 6).
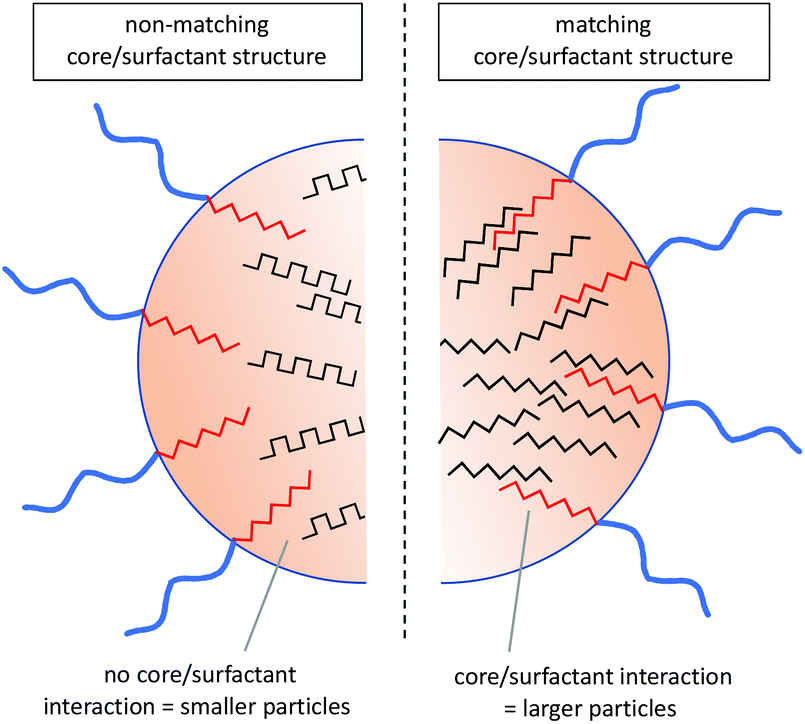 | ||
| Fig. 6 Hypothetical interactions within the core of the nanoparticles based on amino acid and theoretical secondary structure adopted. | ||
Subsequently, the stability of the NPs was further investigated upon dilution in PBS buffer compared to the standard in H2O that was used throughout (Table S5 and Fig. S9†). Using DLS the z-average size and zeta potential was taken for the four nanoparticle compositions. It was found that the zeta potential in an aqueous dispersion was roughly between 40 and 50 mV which is an indication of a stable suspension of particles.38 When measured in a 10-fold dilution in 10 mM PBS solution the measured sizes decreased slightly across all the nanoparticles but the trend was maintained. Therefore, from the light scattering sizing results, the same size trends were seen across both techniques and across the three different media used for measurements. Also the zeta potential values obtained indicate a high level of stability, theorized to be caused by the charge present in the pendent lysine of both surfactants and the presence of the bulky disaccharide group providing steric repulsion.
TEM images were taken after staining with a 1% phosphotungstic acid solution (Fig. 7 and Fig. S10†). Samples containing the S-Phe surfactant provided sufficient contrast and were also imaged unstained (Fig. S11†). While the diameters seen by the TEM images are smaller than those reported by NTA and DLS as the particles are in a dry state, all images confirm the spherical and uniform shape of all nanoparticle combinations. When comparing the P(Leu) surfactant samples (Fig. 7c and d) the same size trend is observed as such that the aromatic core nanoparticles composed of unmatched core/surfactant moieties have a smaller diameter than those with an L-Leu core. These images also allow us to presume spherical morphology in the solvated state and so support the sizes obtained by light scattering techniques as these both presume spherical morphology.
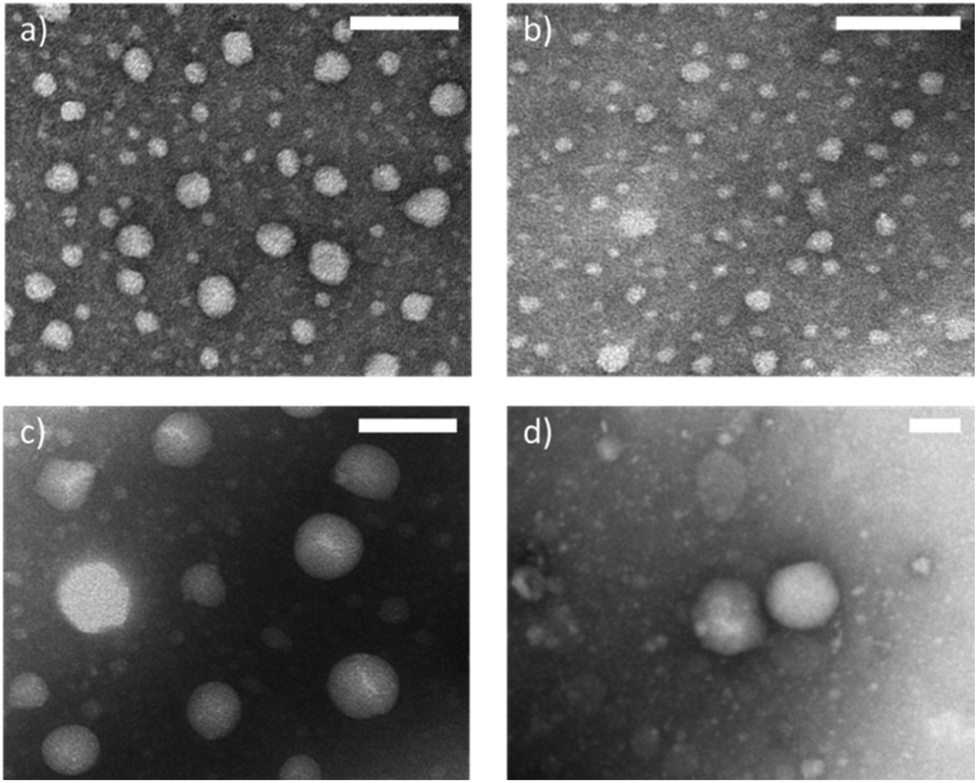 | ||
| Fig. 7 Selected TEM images of the nanoparticles of the 4 different variations. When S-Phe was used to produce a C-Phe (a) and C-Leu (b) and when S-Leu was used to produce a C-Phe (c) and a C-Leu (d). Scale bar represents 100 nm for each image respectively. | ||
Finally selected particles were also characterised by preliminary AF4. During this technique the particles are separated based on differences in dynamic diffusion when subjected to an asymmetric flow over a porous membrane; producing a nearly monodisperse size fraction which can then be analysed by online detectors including UV-Vis spectrophotometry, multi angle light scattering (MALS) and DLS.39,40 MALS was used to determine the radius of gyration (Rg) and ‘online’ DLS was used to determine the hydrodynamic radius (Rh).41 Online DLS is thought to give a more accurate Rh than that calculated by the bench top batch DLS as a measurement is taken every 3 seconds within a flow cell during separation. This eliminates the bias towards larger nanoparticles/aggregates which scatter the light more intensely and can mask the presence of smaller nanoparticles skewing the size reported.42 From the MALS and DLS elugrams, two size fractions can be identified in all samples, the lower fraction agreeing with the sizes range obtained by the static DLS and a fraction of larger particles (Table 2 and Fig. S13†). Aggregation may be hard to identify within other benchtop techniques based on the low concentration of aggregates present. For the preliminary morphology analysis, only the more populous lower size fraction was considered.
| Surfactant | Core | R g [nm] | R h [nm] | R g/Rh |
|---|---|---|---|---|
| S-Phe | C-Phe | 62 ± 6.1% | 70 ± 0.4% | 0.886 |
| C-Leu | 68 ± 1.2% | 63 ± 4.5% | 1.079 | |
| S-Leu | C-Phe | 52 ± 0.5% | 62 ± 1.2% | 0.838 |
| C-Leu | 61 ± 1.4% | 66 ± 4.8% | 0.924 |
Using the Rg and Rh values obtained from the detectors within the AF4 system, information about the morphology can be elucidated from the ratio of the two values. Rg/Rh values of roughly between 0.8 and 1 are indicative of spheres.43 More specifically, Rg/Rh value of 0.775 is indicative of a solid spherical morphology, Rg/Rh = 1 is indicative of a hollow spherical morphology and Rg/Rh >2 is indicative of an elongated structure.44 Due to the nature of the nanoparticles this can be used as an interpretation of the density or packing within the core of the nanoparticles. From Fig. 8, it can be seen that for the nanoparticles synthesised with C-Leu forming monomer Rg/Rh is closer to 1, particularly when S-Phe is used. While no explicit hollow sphere morphologies are evident from TEM, this might support the proposed less dense core morphology as opposed to the C-Phe core variants. This aids in the hypothesis that the additional π–π stacking within the aromatic core provides additional forces leading to a denser core structure.
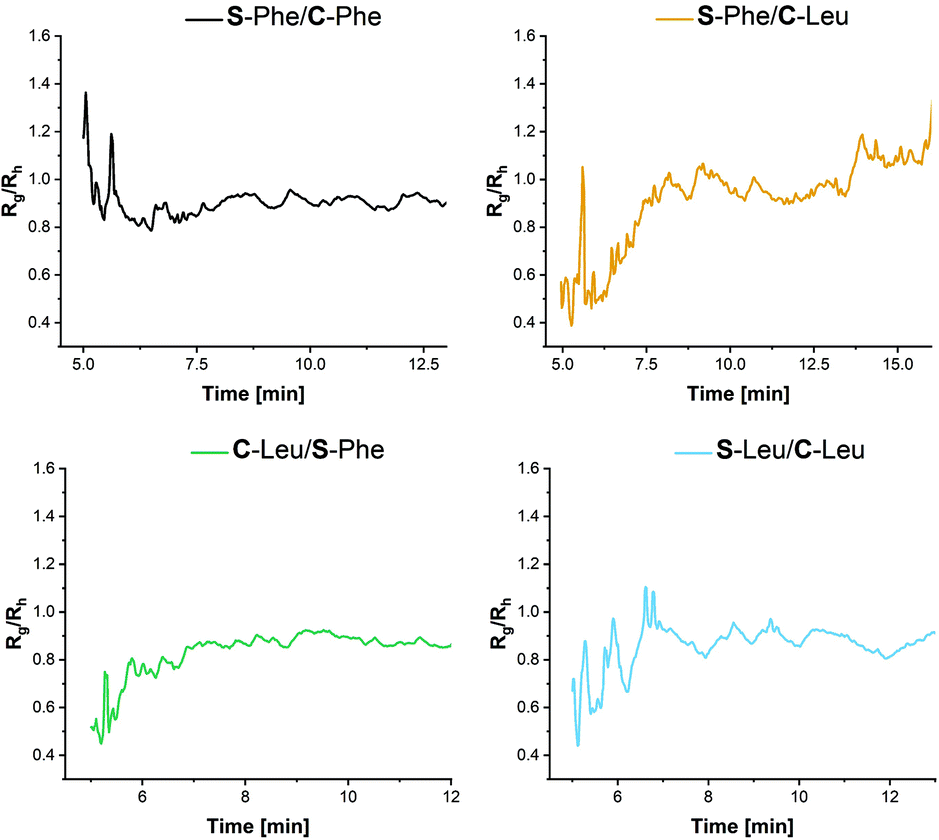 | ||
| Fig. 8 R g/Rh values for the AF4 elugrams obtained from the MALS (Rg) and DLS (Rh) to obtain information on the particle morphology for the first fraction. | ||
Conclusions
We have demonstrated the synthesis of polypeptide nanoparticles by a miniemulsion polymerisation using two different glycosylated block co-polypeptides surfactants. In this process stable aqueous dispersions of hydrophobic polypeptide nanoparticles were obtained. Depending on structural compatibility between the hydrophobic surfactant segment and the core polypeptide, nanoparticles afforded different sizes, which highlights the importance of molecular level interactions at the surfactant/core interphase in this process. We believe this fundamental understanding of polypeptide nanoparticle synthesis by emulsion polymerisation will guide the design of polypeptide nanoparticle libraries utilizing many different amino acids which can have wide applications particularly biological due to their innate biocompatibility and degradability.Experimental section
Materials
Unless otherwise noted, all reagents and chemicals were used as received without further purification. All amino acids and trifluroacetic acid were purchased from Fluorochem. Triethylamine and all solvents were purchased from Sigma Aldrich and HBr/Acetic Acid solution and Allyamine were purchased from Alfa Aesar. ε-Benzyloxycarbonyl-L-lysine (ZLLys), L-Phenylalanine (Phe) and L-Leucine (Leu) NCA was synthesized following literature procedures.45–47Experimental
P(Z-L-lysine-b-L-phenylalanine) (P(Z-L-Lys)-b-P(L-Phe)). Z-L-Lysine NCA (ZLL NCA) (9 g, 29.38 mmol) was dissolved in DMF (55 mL) and placed under vacuum at 0 °C. Allylamine (44 μL, 0.58 mmol) was dissolved in DMF (1 mL) and added in one injection. The reaction proceeded for 5 days under vacuum at 0 °C. L-Phenylalanine NCA (L-Phe NCA) (1.12 g, 5.88 mmol) was dissolved in DMF (4 mL) and added to the solution, the reaction proceeded overnight. The reaction was precipitated into diethyl ether (3 × 500 mL) and then dried under vacuum overnight. 1H NMR (400 MHz, d-TFA, δ) 7.31 (m), 5.71 (s), 5.19 (d), 4.77 (s), 4.58 (d), 3.21 (s), 2.96 (s), 2.06–1.15 (m). GPC ĐM – 1.15, Mw −16
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1.
800 g mol−1.
P(Z-L-lysine-b-L-leucine) (P(Z-L-Lys)-b-P(L-Leu)). The same procedure as previously described was used, substituting L-leucine for L-phenylalanine. 1H NMR (400 MHz, d-TFA, δ) 7.22 (m), 5.66 (s), 5.28 (d), 4.56 (d), 4.46 (s), 3.12 (s), 1.92–1.27(m), 1.16 (s), 0.83 (d). GPC ĐM – 1.07, Mw −11
900 g mol−1.
Deprotection of Z-lysine. The polypeptide (4 g, 0.32 mmol with respect to lysine repeat units) was dissolved in TFA (16 mL). HBr solution (33% in acetic acid, 3-fold excess with respect to ZLL units) was added to the solution dropwise whilst stirring in an ice bath. The reaction was left to proceed overnight and then precipitated into diethyl ether (2 × 250 mL) and then dialysed (3.5k Mw cut off) against DDI water for 3 days before being lyophilised.
P(L-Lys)-b-P(L-Phe) – 1H NMR (400 MHz, D2O, δ) 4.20 (m), 2.89 (m), 1.83–1.44 (m), 1.44–1.13 (m).
P(L-Lys)-b-P(L-Leu) – 1H NMR (400 MHz, D2O, δ) 4.30 (m), 2.99 (m), 1.75–1.43 (m) 0.92–0.86 (m).
EDC/Sulfo-NHS coupling of lactobionic acid (LBA). Lactobionic acid (1.15 g, 3.23 mmol, 15-fold excess with respect to lysine repeat units), 1-Ethyl-3-[3-dimethylaminopropyl]-carbodiimide hydrochloride (EDC) (618.2 mg, 3.23 mmol) and N-hydroxysulfosuccinimide (Sulfo-NHS) (70 mg, 0.32 mmol) were dissolved in MES buffer (7.2 mL, 10 mM, pH 4.7) and stirred for 20 minutes. This was then added to a solution of polypeptide (1.8 g, 0.22 mmol) in DDI water (14 mL). The reaction was stirred overnight and then purified by dialysis (3.5k Mw cut off) for 3 days and lyophilised.
P((L-Lys)-r-(L-Lys-LBA))-b-P(L-Phe) – 1H NMR (400 MHz, D2O, δ) 4.54 (d), 4.29 (m),4.15 (s), 4.08 (s), 3.99–3.64 (m), 3.56–3.52 (m), 2.99 (m), 1.70–1.43 (m).
P((L-Lys)-r-(L-Lys-LBA))-b-P(L-Leu) – 1H NMR (400 MHz, D2O, δ) 4.53 (d), 4.27 (m), 4.15 (s), 4.07 (s), 3.98–3.62 (m), 3.55–3.51 (m), 2.98 (m), 1.74–1.42 (m), 0.92–0.86 (m).
Mini-emulsion polymerisation. The surfactant (80 mg), was dissolved in DDI water (10 mL) and solution cooled in an ice bath for 10 minutes whilst stirring. NCA (70 mg) was dissolved in DCM (2 mL) and added to the aqueous solution dropwise while the reaction mixture was sonicated with Hielscher Ultrasonic Processor UP200 St(P = 13 W, c = 100, A = 70%) for 15 min. Triethylamine (7 μL) was added and the system allowed to stir (400 rpm) for 24 hours at room temperature. The resulting particle dispersion was dialysed (3.5k Mw cut off) against DDI for 3 days.
Author contributions
N.J.: Investigation, methodology, writing original draft, formal analysis. D.P.: Investigation. E.M.: Data formal analysis. P.C.: Resources. R.B.: Supervision, writing – review and editing. A.H.: Conceptualisation, funding acquisition, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 814236. The authors would like to thank Prof. Stefan Bon from the University of Warwick for the fruitful discussion.Notes and references
- H. Liu, R. P. Singh, Z. Zhang, X. Han, Y. Liu and L. Hu, J. Agric. Food Chem., 2021, 69, 2936 CrossRef CAS PubMed.
- S. N. Jamali, E. Assadpour and S. M. Jafari, Curr. Med. Chem., 2020, 27, 3079 CrossRef CAS PubMed.
- D. Hudson and A. Margaritis, Crit. Rev. Biotechnol., 2014, 34, 161 CrossRef CAS PubMed.
- S. Vrignaud, J.-P. Benoit and P. Saulnier, Biomaterials, 2011, 32, 8593 CrossRef CAS PubMed.
- C. P. Reis, R. J. Neufeld, A. J. Ribeiro and F. Veiga, Nanomedicine, 2006, 2, 8 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1 CrossRef CAS PubMed.
- S. C. Thickett and G. H. Teo, Polym. Chem., 2019, 10, 2906 RSC.
- M. Antonietti and K. Landfester, Prog. Polym., 2002, 27, 689 CrossRef CAS.
- Y. Reyes, S. Hamzehlou and J. R. Leiza, J. Mol. Liq., 2021, 335, 116152 CrossRef CAS.
- J. M. Asua, Miniemulsion Polymerization, in Encyclopedia of Polymeric Nanomaterials, ed. S. Kobayashi and K. Müllen, Springer, Berlin, Heidelberg, 2014, DOI:10.1007/978-3-642-36199-9_263-1.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488 CrossRef CAS PubMed.
- F. Lu, Y. Luo and B. Li, Macromol. Rapid Commun., 2007, 28, 868 CrossRef CAS.
- J. Jacobs, N. Gathergood, J. P. A. Heuts and A. Heise, Polym. Chem., 2015, 6, 4634 RSC.
- J. Jacobs, A. Byrne, N. Gathergood, T. E. Keyes, J. P. A. Heuts and A. Heise, Macromolecules, 2014, 47, 7303 CrossRef CAS.
- A. Byrne, J. Jacobs, C. S. Burke, A. Martin, A. Heise and T. E. Keyes, Analyst, 2017, 142, 3400 RSC.
- K. S. Gkika, A. Kargaard, C. S. Burke, C. Dolan, A. Heise and T. E. Keyes, RSC Chem. Biol., 2021, 2, 1520 RSC.
- A. Muñoz-Bonilla, S. I. Ali, A. del Campo, M. Fernández-García, A. M. van Herk and J. P. A. Heuts, Macromolecules, 2011, 44, 4282 CrossRef.
- G. Riess, Colloids Surf., 1999, 153, 99 CrossRef CAS.
- A. Durand and E. Marie, Adv. Colloid Interface Sci., 2009, 150, 90 CrossRef CAS PubMed.
- P. Raffa, D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Chem. Rev., 2015, 115, 8504 CrossRef CAS PubMed.
- G. Baier, A. Baki, S. Tomcin, V. Mailänder, E. Alexandrino, F. Wurm and K. Landfester, Macromol. Symp., 2014, 337, 9 CrossRef CAS.
- A. Birke, D. Huesmann, A. Kelsch, M. Weilbächer, J. Xie, M. Bros, T. Bopp, C. Becker, K. Landfester and M. Barz, Biomacromolecules, 2014, 15, 548 CrossRef CAS PubMed.
- A. Taden, M. Antonietti and K. Landfester, Macromol. Rapid Commun., 2003, 24, 512 CrossRef CAS.
- V. Chiaradia, A. E. Polloni, D. de Oliveira, J. V. de Oliveira, P. H. H. Araújo and C. Sayer, Colloid Polym. Sci., 2018, 296, 861 CrossRef CAS.
- S. Målberg, A. Finne-Wistrand and A.-C. Albertsson, Polym. J., 2010, 51, 5318 Search PubMed.
- A. R. Mazo, S. Allison-Logan, F. Karimi, N. J.-A. Chan, W. Qiu, W. Duan, N. M. O'Brien-Simpson and G. G. Qiao, Chem. Soc. Rev., 2020, 49, 4737 RSC.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528 CrossRef CAS PubMed.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373 RSC.
- Z. Song, H. Fu, J. Wang, J. Hui, T. Xue, L. A. Pacheco, H. Yan, R. Baumgartner, Z. Wang, Y. Xia, X. Wang, L. Yin, C. Chen, J. Rodríguez-López, A. L. Ferguson, Y. Lin and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10658 CrossRef CAS PubMed.
- C. Grazon, P. Salas-Ambrosio, E. Ibarboure, A. Buol, E. Garanger, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Angew. Chem., Int. Ed., 2020, 59, 622 CrossRef CAS PubMed.
- C. Grazon, P. Salas-Ambrosio, S. Antoine, E. Ibarboure, O. Sandre, A. J. Culow, B. J. Boyd, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Polym. Chem., 2021, 12, 6242 RSC.
- J. Jacobs, D. Pavlović, H. Prydderch, M.-A. Moradi, E. Ibarboure, J. P. A. Heuts, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2019, 141, 12522 CrossRef CAS PubMed.
- C. Bonduelle, Polym. Chem., 2018, 9, 1517 RSC.
- A. Kim, W. B. Ng, W. Bernt and N.-J. Cho, Sci. Rep., 2019, 9, 2639 CrossRef PubMed.
- H. E. Auer and P. Doty, Biochem, 1966, 5, 1716 CrossRef CAS PubMed.
- H. E. Auer and P. Doty, Biochem, 1966, 5, 1708 CrossRef CAS PubMed.
- H. R. Kricheldorf, D. Müller and J. Stulz, Makromol. Chem., 1983, 184, 1407 CrossRef CAS.
- D. J. Pochapski, C. Carvalho dos Santos, G. W. Leite, S. H. Pulcinelli and C. V. Santilli, Langmuir, 2021, 37, 13379 CrossRef CAS PubMed.
- A. H. Hinna, S. Hupfeld, J. Kuntsche and M. Brandl, J. Pharm. Biomed. Anal., 2016, 124, 157 CrossRef CAS PubMed.
- X. Zhang, Y. Li, S. Shen, S. Lee and H. Dou, TrAC, Trends Anal. Chem., 2018, 108, 231 CrossRef CAS.
- A. Zattoni, B. Roda, F. Borghi, V. Marassi and P. Reschiglian, J. Pharm. Biomed. Anal., 2014, 87, 53 CrossRef CAS PubMed.
- Y. Hu, R. M. Crist and J. D. Clogston, Anal. Bioanal. Chem., 2020, 412, 425 CrossRef CAS PubMed.
- D. Xie, K. Xu, R. Bai and G. Zhang, J. Phys. Chem., 2007, 111, 778 CrossRef CAS PubMed.
- R. J. Williams, A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Macromolecules, 2016, 49, 2802 CrossRef CAS PubMed.
- G. J. M. Habraken, C. E. Koning and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6883 CrossRef CAS.
- J. Sun, X. Chen, C. Deng, H. Yu and Z. Xie, Langmuir, 2007, 23, 8308 CrossRef CAS PubMed.
- Z. Y. Tian, Z. Zhang, S. Wang and H. Lu, Nat. Commun., 2021, 1, 5810 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, methods, NMR analysis, DLS data, TEM and NTA images. See DOI: https://doi.org/10.1039/d2py00331g |
| This journal is © The Royal Society of Chemistry 2022 |