
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metallocene catalysts for the ring-opening co-polymerisation of epoxides and cyclic anhydrides†
Matthew S.
Shaw
a,
Morgan R.
Bates
a,
Matthew D.
Jones
 b and
Benjamin D.
Ward
*a
b and
Benjamin D.
Ward
*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: WardBD@Cardiff.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 16th May 2022
Abstract
The ring-opening co-polymerisation (ROCOP) of epoxides and cyclic anhydrides is a versatile route to new polyesters. The vast number of monomers that are readily available means that an effectively limitless number of unique polymeric materials can be prepared using a common synthetic route, thereby paving the way to polymers that are more recyclable and could be used in applications that currently lie exclusively within the remit of polyolefins. Metallocene complexes [Cp2MCl2] (M = Ti, Zr, Hf, Cp = η5-C5H5) are known to be excellent pre-catalysts for olefin polymerisation and yet have not been reported for ROCOP, despite their supreme pedigree in the polymerisation realm. Herein, we report the first application of [Cp2MCl2] as catalysts for the ROCOP of epoxides and anhydrides and show that they are effective catalysts for this reaction. The catalytic performances are good with standard epoxides such as cyclohexene oxide but are excellent with more challenging sterically demanding limonene oxide, and this therefore represents a highly effective catalyst system for the preparation of bio-derived recyclable polymers.
Introduction
We live in a plastic-hungry society where polymeric materials pervade almost every facet of daily life. Global polymer production is dominated by polyethylene, polypropylene, and polyvinyl chloride, which together make up 59% of global polymer demand.1 These materials do not readily degrade in the environment, meaning their improper disposal has led to extensive pollution of aquatic environments.2–4 The ever-increasing use of plastics and a heightened awareness of their environmental footprint means that chemists must develop novel polymers with enhanced properties to those in common use, whilst simultaneously incorporating recyclability at the point of synthesis. Attention often turns to polyesters since they are hydrolysable, making them both recyclable through depolymerisation as well as potentially biodegradable, thereby alleviating the end-of-life issues that plague many plastics.5–8 Traditionally, polyesters are synthesised by step-growth polycondensation, which requires high energy input, necessitates the removal of by-products, and offers poor control of molecular weight and polymer microstructure.9,10 However, a chain-growth polymerisation involving the catalytic ring-opening copolymerisation (ROCOP) of cyclic anhydrides and epoxides (Scheme 1) is an appealing alternative, given it can be used to selectively access alternating copolymers of high and tuneable molecular weight.11–13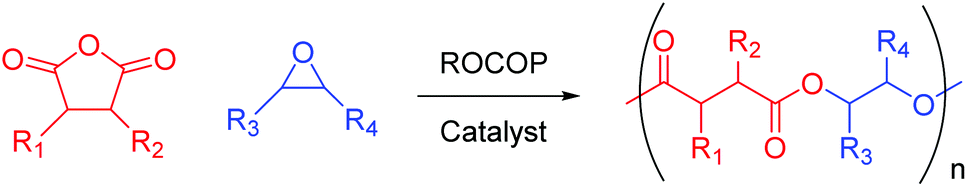 | ||
| Scheme 1 The ring-opening copolymerisation (ROCOP) of cyclic anhydrides and epoxides to produce polyesters. | ||
Epoxides and cyclic anhydrides are widely available feedstocks, some of which can be obtained from renewable sources, meaning the properties of the polymer can be tuned for many applications simply by exchanging the monomers.14–22 Furthermore, the functional group tolerance of ROCOP readily allows for post-polymerisation modifications to further enhance the material's attributes.23–26 This contrasts with the more widely investigated ring-opening polymerisation (ROP) of lactones, where a relatively small selection of suitable monomers restricts the potential physical properties of the resultant polyesters, as well as the scope for subsequent modification.27
Many different catalysts have been used to effect epoxide–anhydride ROCOP, including organocatalysts,28–30 and complexes of aluminium,31–33 cobalt,10,12,34 chromium,35,36 iron,16,17 magnesium,27,37,38 and zinc.39–41 However, there are only limited reports of the Group 4 metals (Ti, Zr, Hf) being used in this transformation; perhaps surprising given there are multiple studies detailing their efficacy in the ROP of cyclic esters42–46 and the ROCOP of epoxides and CO2.47–55 In 1999, Endo and co-workers reported the use of titanium alkoxide catalysts in the ROCOP of oxetane and anhydrides, whilst heterogeneous zirconium phosphate materials with intercalated amines have also been demonstrated to catalyse the ROCOP of glycidyl phenyl ether and hexahydro-4-methylphthalic anhydride.56–59 Furthermore, Chakraborty, Chand and colleagues have detailed the use of 8-hydroxyquinolinate based ligands complexed to titanium and zirconium in the copolymerisation of maleic anhydride and tert-butyl glycidyl ether.60 More recently, the Williams group have reported a bis(acetate) Zr(IV) catalyst for the terpolymerisation of anhydrides, tetrahydrofurans and epoxides or oxetanes to produce poly(esters-alt-ethers).61
When considering the Group 4 metals, the mind immediately turns to cyclopentadienyl (η5-C5H5−, Cp) based metallocene complexes, and in turn the polymerisation of olefins. Interestingly, despite [Cp2MCl2] (M = Ti, Zr) being used to produce cyclic carbonate from CO2 and propylene oxide, and the Ti(III) species [Cp2TiCl] promoting radical-initiated ROCOP,62–65 [Cp2MCl2] (M = Ti, Zr, Hf) have not been probed for their efficacy in epoxide–anhydride ROCOP. On the whole, investigations into metallocenes have slowed somewhat since the turn of the century in part due to an exhaustive patent literature in olefin polymerisation, yet they represent a new class (in the context of epoxide–anhydride ROCOP) of readily synthesisable catalysts.66 Herein, we describe the use of three commercially available Group 4 metallocene dichloride complexes ([Cp2MCl2], M = Ti, Zr, Hf) in conjunction with the nucleophilic co-catalyst 4-(dimethylamino)pyridine (DMAP) in the ring-opening copolymerisation of epoxides and cyclic anhydrides. This includes the large-scale synthesis of partially bio-derived polyesters from limonene oxide and (to our knowledge) the first example of hafnium used in epoxide–anhydride ROCOP.
Results and discussion
Starting from a simple 2 × 2 matrix of epoxides and cyclic anhydrides, the three metallocene complexes [Cp2MCl2] (M = Ti, Zr, Hf) were investigated for their use in ROCOP. DMAP was used as a co-catalyst to allow differentiation of initiation by external co-catalyst and chlorides present in the precatalyst, see MALDI-ToF data below. Substrates examined include cyclohexene oxide (CHO) and 4-vinylcyclohexene oxide (VCHO), as well as phthalic anhydride (PA) and tetrachlorophthalic anhydride (TCPA). VCHO is of interest as its vinyl functionality allows for post-polymerisation modification through hydroboration and thiol–ene reactions, whilst TCPA is used in the production of flame retardant materials.67–70 The polymerisation data for PA are presented in Table 1. The addition of metallocene complex increases anhydride conversion significantly for both CHO (entries 1–3) and VCHO (entries 5–7), in comparison to when DMAP alone is used over the same time period (entries 4 and 8). This activity increase is most prominent for [Cp2TiCl2] (entries 1 and 5), followed by [Cp2ZrCl2] (entries 2 and 6) and [Cp2HfCl2] (entries 3 and 7), respectively. Interestingly, an identical trend is seen for the selectivity towards alternating (AB)n microstructure (determined from ester vs. ether regions in the 1H NMR spectra of each polymer), with [Cp2TiCl2] slightly more selective than [Cp2ZrCl2], which in turn is far more selective than [Cp2HfCl2]. Whilst the selectivities remain high for titanium and zirconium, copolymers with near perfect alternating selectivity were observed in the absence of metallocene; with the region of the 1H NMR spectrum corresponding to ether linkages (arising from homopolymerisation of epoxide) often containing no discernible resonances. In terms of molecular weight, both [Cp2TiCl2] and [Cp2ZrCl2] produced polymers with a significantly higher molecular weight (far more than if one adjusts for the extra mass of the vinyl group and differences in conversion) for VCHO, indicating a reduced propensity for chain transfer processes when compared to CHO. This, however, does not extend to [Cp2HfCl2], where the molecular weights were similar regardless of the epoxide used. The highest molecular weights were observed in the absence of metallic catalyst, reflecting the relatively small number of propagating chains, a result of the reduced number of initiating groups (chlorides and DMAP) in the reactions. The polyesters generally possessed relatively low dispersities (Đ) and were similar across both epoxides; aside from [Cp2HfCl2], which gave the highest dispersities (1.61, entry 3) for the CHO–PA copolymer. In general, the conversions and molecular weights seen are of similar order to commonly employed Schiff-base-ligated metal complexes in toluene, albeit at different reaction temperatures and catalyst loadings.71 Polymerisations were also conducted “in bulk” using neat CHO as the solvent (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2000:400 [Cp2TiCl2]:[DMAP]:[CHO]:[PA], 80 °C) which yielded the expected pseudo zero-order rate plot (Fig. S1†), albeit producing polymers with lower (75%) ester selectivity.
:2:2000:400 [Cp2TiCl2]:[DMAP]:[CHO]:[PA], 80 °C) which yielded the expected pseudo zero-order rate plot (Fig. S1†), albeit producing polymers with lower (75%) ester selectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Epoxide | M | Conv.b/% | Esterc/% | M n /kDa | Đ , |
| a Average of at least two runs. Conditions: 1 eq. = 6.4 μmol (1 eq.) metallocene (unless otherwise stated), 2 eq. DMAP, 400 eq. PA, 400 eq. epoxide, 1 mL toluene, 80 °C for 21 hours. b Determined from 1H NMR spectra of the reaction mixtures. c Determined from 1H NMR spectra of the resultant polymers. d Determined by GPC (viscometry detection against polystyrene standards). e Đ = Mw/Mn. | ||||||
| 1 | CHO | Ti | 74 | 89 | 16.8 | 1.24 |
| 2 | Zr | 61 | 83 | 17.7 | 1.39 | |
| 3 | Hf | 55 | 66 | 14.4 | 1.61 | |
| 4 | None | 32 | >95 | 19.1 | 1.22 | |
| 5 | VCHO | Ti | 74 | 85 | 37.0 | 1.20 |
| 6 | Zr | 66 | 80 | 31.4 | 1.47 | |
| 7 | Hf | 57 | 52 | 17.7 | 1.44 | |
| 8 | None | 35 | 94 | 60.2 | 1.33 | |
In addition, polymerisations of CHO and PA conducted without DMAP showed low (ca. 30%) ester selectivity.
The analogous reactions using TCPA are described in Table 2. Due to the reduced solubility of TCPA, the amounts of all reagents aside from the solvent were halved (molar ratios of [metallocene]:[DMAP]:[epoxide]:[anhydride] remained 1:2:400:400). Despite this however, reaction times decreased dramatically, with near quantitative conversion of CHO observed after 14 hours using both [Cp2ZrCl2] and [Cp2HfCl2] (entries 2 and 3). Given that the rate determining step of this reaction remains epoxide opening, this increase in activity could be attributed to the electron-withdrawing chloro substituents stabilising the carboxylate anion in non-polar media, better facilitating its de-coordination and subsequent reaction with metal-bound epoxide. In another departure from copolymerisations of PA discussed previously, reactions using [Cp2TiCl2] (entries 1 and 5) gave lower conversions than both the zirconium and hafnium congeners (entries 2, 3 and 6, 7). This reversal in activity could be attributed to steric congestion around the metal centres, where the larger zirconium and hafnium can more easily accommodate the significant extra volume occupied by the chloro-substituted polymer chains. Concurrently, the selectivity of reactions involving [Cp2HfCl2] and CHO–TCPA increased relative to CHO–PA (entry 3), although hafnium remained the least selective of the three metals.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Epoxide | t/h | M | Conv.b/% | Esterc/% | M n /kDa | Đ , |
| a Average of at least three runs. Conditions: 1 eq. = 3.2 μmol (1 eq.) metallocene (unless otherwise stated), 2 eq. DMAP, 400 eq. TCPA, 400 eq. epoxide, 1 mL toluene, 80 °C. b Determined from 1H NMR spectra of the reaction mixtures. c Determined from 1H NMR spectra of the resultant polymers. d Determined by GPC (viscometry detection against polystyrene standards). e Đ = Mw/Mn. | |||||||
| 1 | CHO | 14 | Ti | 88 | 94 | 11.2 | 1.23 |
| 2 | Zr | 99 | 89 | 20.3 | 1.10 | ||
| 3 | Hf | 98 | 81 | 15.3 | 1.39 | ||
| 4 | None | 77 | 95 | 11.8 | 1.30 | ||
| 5 | VCHO | 15 | Ti | 85 | >95 | 18.5 | 1.15 |
| 6 | Zr | >99 | 82 | 19.2 | 1.28 | ||
| 7 | Hf | >99 | 65 | 27.8 | 1.39 | ||
| 8 | None | 76 | 90 | 28.5 | 1.21 | ||
Also noteworthy is the detection of polyether resonances when no metallocene is added, in particular for VCHO where the addition of [Cp2TiCl2] actually increased ester selectivity (entries 5 and 8). Despite the molecular weights being similar for both TCPA and PA in absolute terms (with the exception of VCHO with [Cp2HfCl2], entry 7), the increased mass of the monomer means this translates to fewer repeat units. This is likely due to chain transfer processes, specifically the increased favourability of transesterification at a more electrophilic ester carbonyl group.
MALDI-ToF mass spectrometry of the polymers was used to ascertain whether the observed polyether resonances in the 1H NMR spectra of the polymers were incorporated into predominantly polyester chains, or distinct polyether chains produced by a separate polymerisation process. A representative MALDI-ToF spectrum of the CHO–PA polymer produced by [Cp2TiCl2] is displayed in Fig. 1, with the repeating series corresponding to the CHO–PA unit of 246 Da indicated by different colours. All observed series in positive mode were initiated by DMAP, highlighting its importance as a nucleophilic cocatalyst, with these findings consistent with previously reported MALDI-ToF spectra for polyesters produced by an aluminium bis(methyl) pre-catalyst and DMAP.72
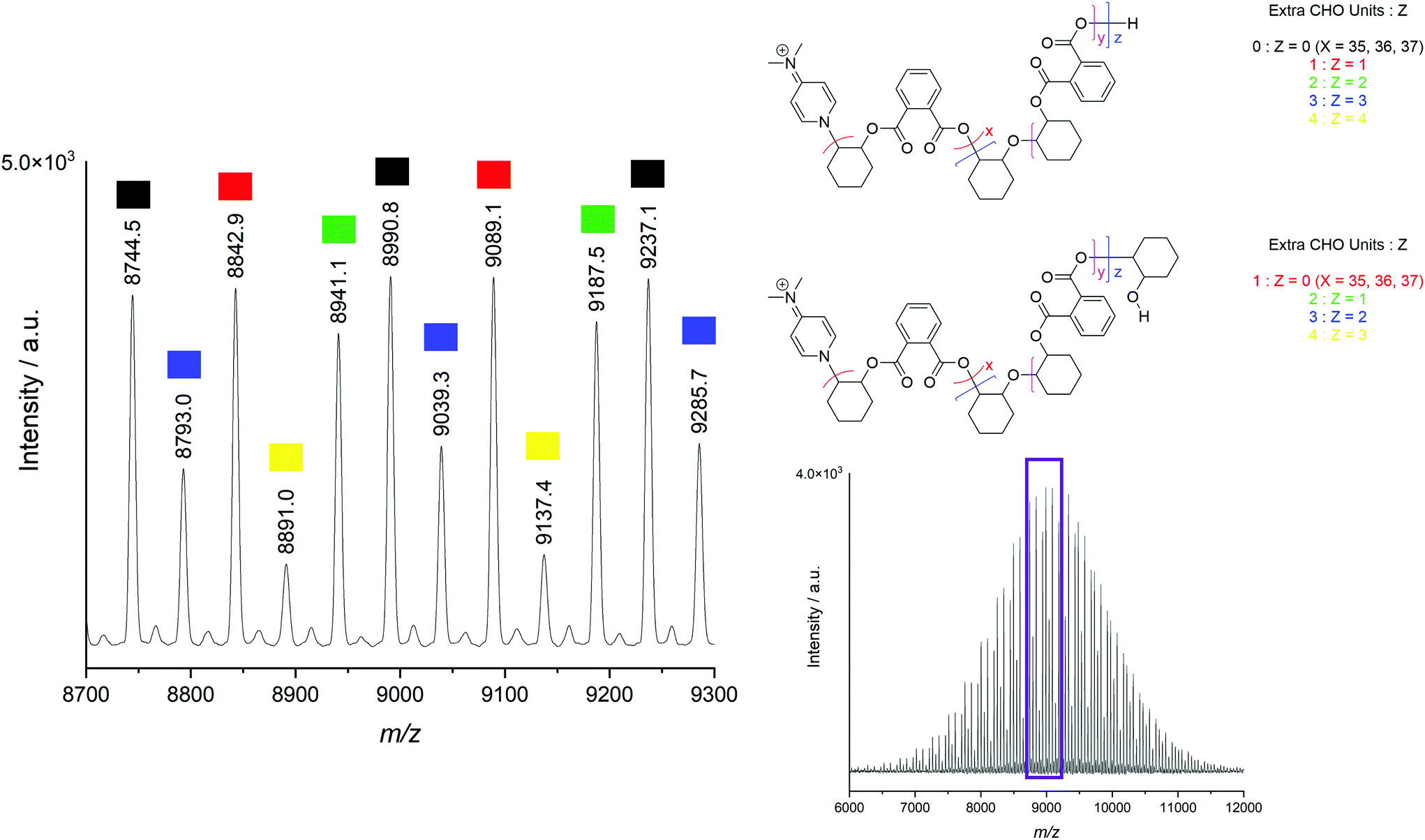 | ||
| Fig. 1 Positive mode MALDI-ToF spectrum of the CHO-PA copolymer produced by [Cp2TiCl2] (entry 1, Table 1). Also shown is an expanded section with five series indicated, each with 0–4 (N) additional CHO units in the polymer chain, corresponding to either N polyether linkages or N − 1 polyether linkages and a protonated epoxide end group. | ||
The appearance of several series corresponding to various “extra” CHO units (up to 4 across the polymer chain) indicate that there are polyether linkages within the chains, as well as potentially as end groups. These possibilities are indistinguishably by mass and therefore both are shown in Fig. 1. Similarly, MALDI series containing up to 8 extra epoxide units have been observed for a polymer with higher ether content (20% vs. 11%, Fig. S22†). These findings are consistent with previous mechanistic work where ester and ether producing catalytic cycles are thought to originate from a single catalytic species.61 Despite this, under identical reaction conditions in the absence of anhydride, [Cp2ZrCl2] did not produce poly(CHO), with this highlighting the intrinsic differences in rate of reaction between the monomers.
Mechanistic studies: stoichiometric reactions
Although significant increases in conversion followed the introduction of all three metallocenes, there remained no direct evidence explaining how they initiate ROCOP reactions. Therefore, experiments were performed to detect the generation of catalytic intermediates and follow the propagation of nascent polymer chains. [Cp2ZrCl2] reacts with propylene oxide in the absence of co-catalyst to give two species, likely corresponding to a mono- and bis(alkoxide) (Scheme 2). Addition of further propylene oxide pushed conversion to a major product with 1H NMR signals in a 10:2:4:6 integration ratio; COSY experiments confirmed the mutual coupling of the signal at δH = 4.15 with the two resonances at δH = 3.34 and 1.16 (Fig. S2†). This is consistent with a bis(alkoxide) complex and confirms the feasibility of reaction of the metallocene pre-catalysts with epoxides. Furthermore, calculation of NMR shielding tensors in Gaussian 09 yield predicted chemical shifts for the alkoxide protons that are compatible with those observed experimentally, suggesting the assigned regiochemistry in Scheme 2 (i.e. chloride attack at the least hindered carbon of propylene oxide) is the major product [DFT predicted δH = 4.25 (2H), 3.35 (4H), 1.27 (6H), observed δH = 4.15 (2H), 3.34 (4H), 1.16 (6H)].73,74 Indeed, a similar prediction on the isomer resulting from attack of the most hindered position gave δH = 4.24 (2H), 4.22 (4H), 1.45 (6H); a far poorer match to the observed spectrum (Fig. S3†).
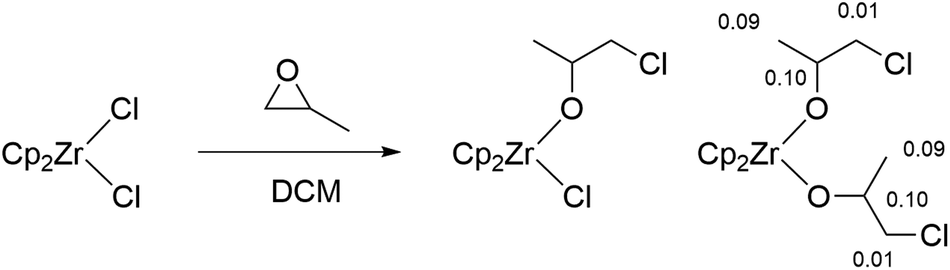 | ||
| Scheme 2 Synthesis of mono- and bis(alkoxide) complexes from [Cp2ZrCl2] and propylene oxide. Note the regiochemistry of the ring-opening at the least hindered carbon of propylene oxide. Also shown are the differences between the DFT calculated 1H NMR chemical shifts and the experimentally observed values. | ||
To experimentally confirm the regiochemistry, analogous reactions with epichlorohydrin were conducted, which gave a 1:4:6:4:1 quintet (J = 5.3 Hz) resulting from reaction at the least hindered position (Fig. S4†). This coupling to four equivalent protons is not possible if attack occurs at the most hindered carbon, where one would expect a triplet of triplets for the single proton environment.
Similar experiments, this time with two equivalents of DMAP and [Cp2ZrCl2] in a precise 2:1 ratio gave rise to new metallocene species once two equivalents of CHO were added (at room temperature, Fig. S5†). This, in conjunction with the above discussion shows that the ring-opening of metal-bound epoxide by external attack of dissociated chloride or DMAP at a zirconium centre are viable initiation pathways. Once heat was applied (Fig. S6†), the new metallocene peaks became more intense, with signals beginning to appear in the ether region of the 1H NMR spectrum, again likely attributed to metal alkoxide complexes. More succinctly, resonances corresponding to DMAP-opened alkoxides (as indicated by COSY experiments) highlight the feasibility of metal-free initiation, albeit the ratio of these signals to those of unreacted DMAP (δH = 8.21, 6.47, 2.98) is far lower than the equivalent ratio for metallocene species, where the original [Cp2ZrCl2] is completely consumed. This mirrors the previously discussed experimental data, in that DMAP is an effective catalyst, but its initiation is far slower than when used in combination with metallocenes, as reflected by the lower conversions in polymerisation reactions when only DMAP was used. Addition of two equivalents of PA led to new resonances comparable to signals in the spectrum of the CHO–PA copolymer, both in the ester and aromatic regions (Fig. S6†). No heating or any appreciable timeframe was required to see complete conversion of PA, showing the facile nature of anhydride opening compared to that of epoxide.
Following the addition of two more equivalents of CHO, the reaction mixture was analysed by electrospray ionisation mass spectrometry (ESI-MS), with ring-opened products initiated by DMAP apparent in positive mode (Fig. 2, full spectra shown in Fig. S8 and S9†). A chloride-initiated CHO–PA unit was seen in negative mode spectrum, alongside an unsaturated fragment of m/z = 245.09 Da, presumably forming within the mass spectrometer after a loss of HCl.
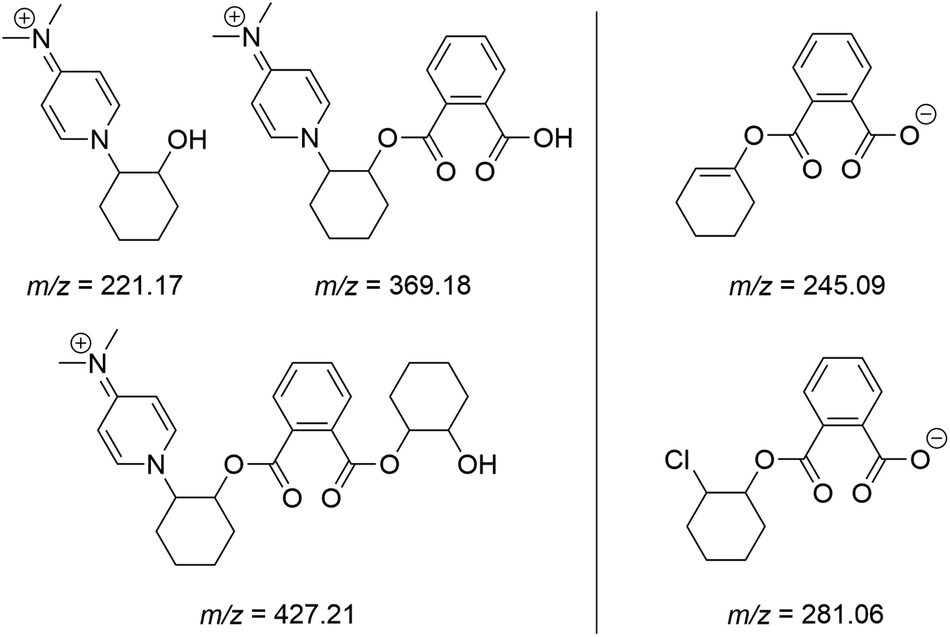 | ||
| Fig. 2 Ions detected by ESI-MS in both positive (L) and negative mode (R) in a 1:2:4:2 reaction of [Cp2ZrCl2]:DMAP:CHO:PA conducted in CDCl3. | ||
A summary of the proposed initiation modes is shown in Scheme 3, which details how both DMAP and chloride initiated chains can arise, with the prevalence of both types of initiation consistent with both the stoichiometric reactions and MALDI-ToF spectra of the polymers.
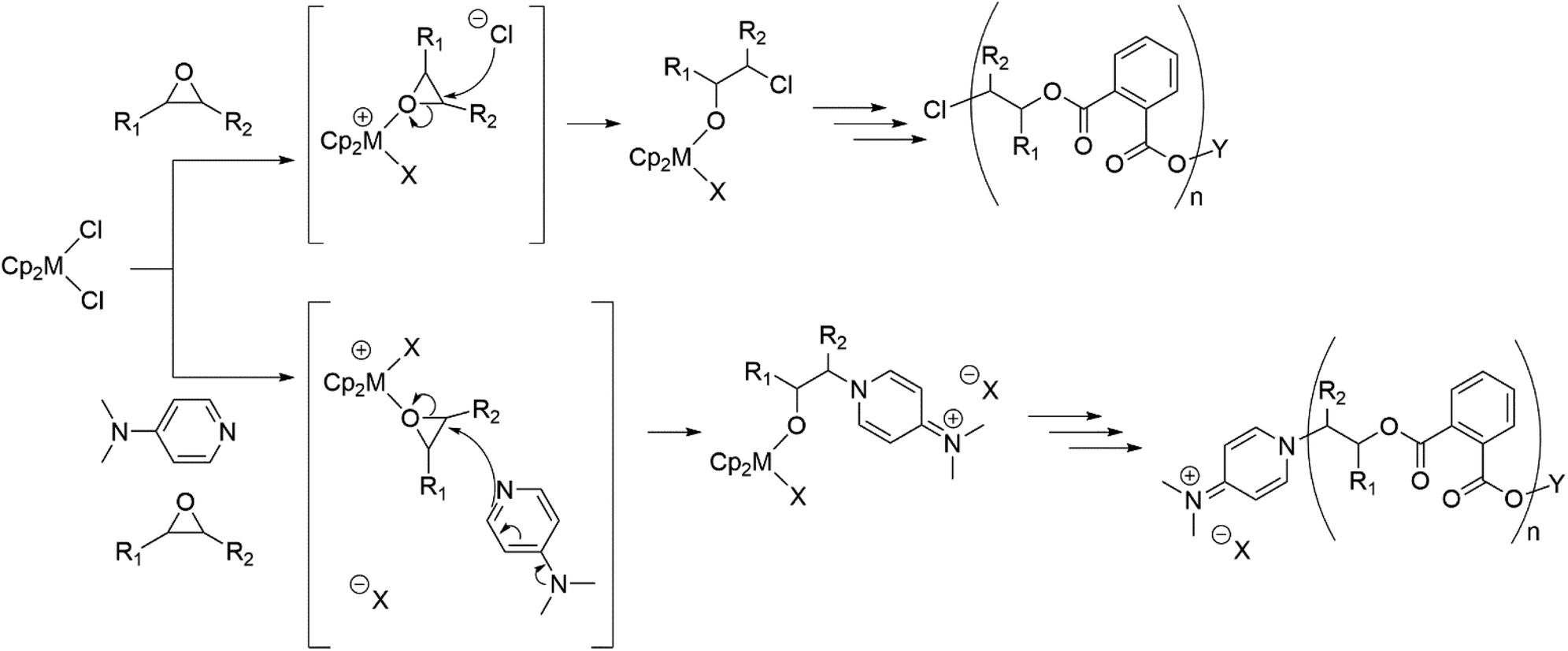 | ||
| Scheme 3 Proposed initiation routes to generate both chloride initiated polymer chains (observed in stoichiometric studies) and DMAP initiated polymer chains (observed in stoichiometric studies and MALDI-ToF of polymers) for the ROCOP of a generic epoxide and PA. For reaction with the first chloride of the pre-catalyst, X = Cl. For reaction of the second chloride, X = carboxylate. Note that DMAP initiated chains are zwitterionic and so X may represent the carboxylate terminus of the same or different polymer chain, as well as chloride. Y = chain terminus. | ||
Mechanistic studies: density functional theory calculations
Further understanding of the propagation mechanism of the metallocene-mediated polymerisation was obtained using DFT calculations (Scheme 4). Previous calculations performed by Cramer, Coates, Tolman and co-workers have demonstrated a feasible propagation cycle for an aluminium Salph catalyst; our experimental observations are consistent with this previously reported mechanism, which was therefore used as the starting point for our calculations, examining [Cp2Ti(OAc)2] as an on-cycle catalyst for the copolymerisation of ethylene oxide (EO) and succinic anhydride (SA).31 Since experimentally, the rate-limiting step is epoxide opening (affording an alkoxide) and anhydride addition (affording a carboxylate) is fast, the resting state of the catalyst is expected to contain two carboxylate-terminated polymer chains, which we model as acetate. The initiation steps (i.e. reaction of chloride pre-catalysts) examined by stoichiometric reactions were not studied here. EO and SA were chosen as substrates both for computational simplicity and to eliminate a large volume of conformational ambiguity leading to difficulty in isolating true minima for each intermediate. Therefore, the energies are expected be attenuated by the steric and electronic parameters of the experimental substrates, as well as changeable ring strain.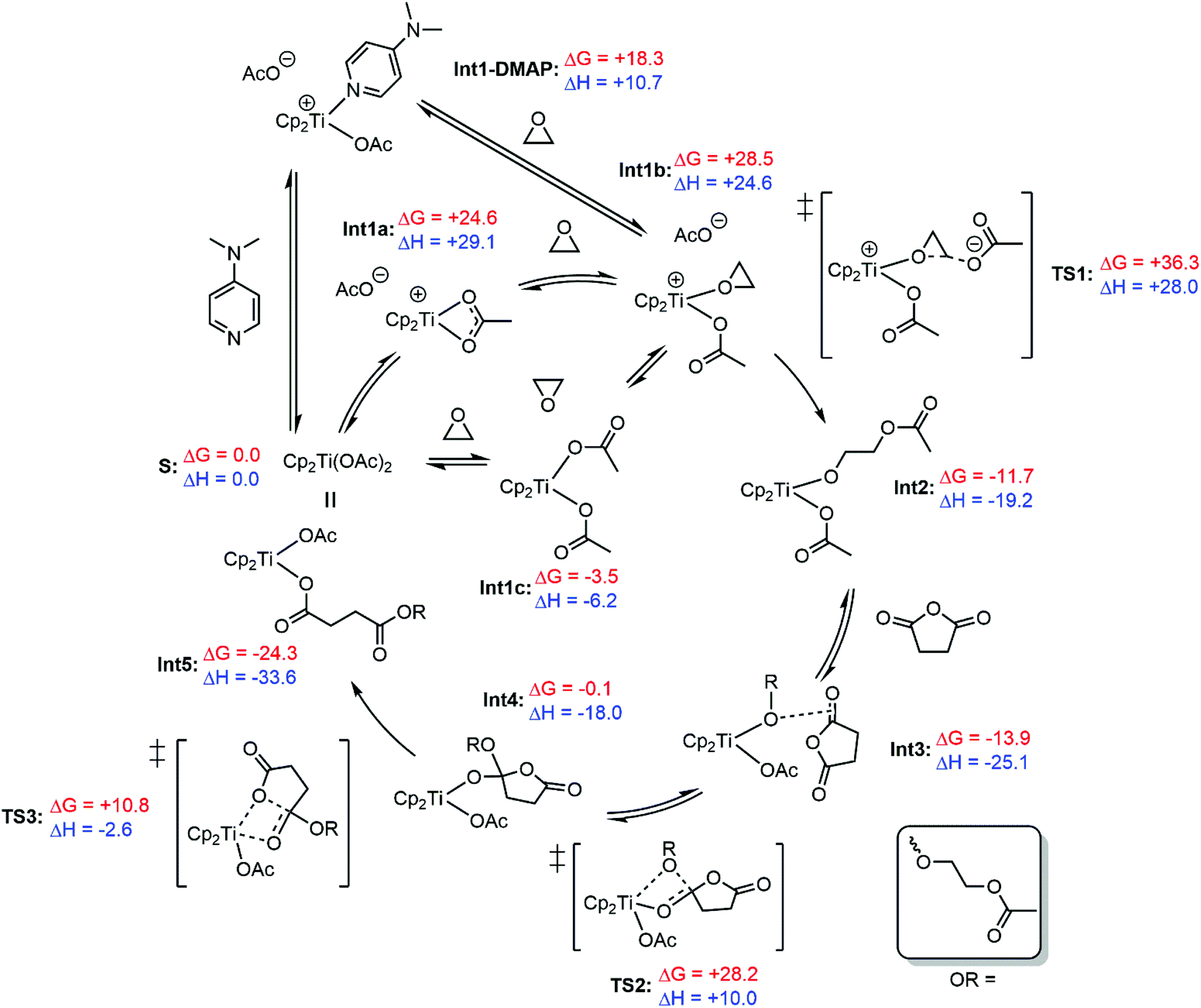 | ||
| Scheme 4 Energetically feasible propagation cycle for the polymerisation of ethylene oxide and succinic anhydride with a [Cp2Ti(OAc)2] on-cycle catalyst. Calculated with Gaussian 09 at the M06/def2-TZVP level of theory (implicit toluene solvent).73,74,77–79 Free energies and enthalpies are quoted in kcal mol−1, and have been temperature corrected (70 °C) and concentration corrected using the Sackur–Tetrode equation, applied via the GoodVibes software package.80 | ||
The proximity of the labile sites in the [Cp2Ti] intermediates allows for two different routes for anhydride opening: (a) insertion of a metal-alkoxide into a non-coordinating anhydride (as described by Cramer, Coates and Tolman) along with spontaneous ring-opening, or (b) migratory insertion of a metal-alkoxide into a pre-coordinated anhydride, similar to a route often invoked in the ring-opening polymerisation (ROP) of lactones.40,75,76 However, no minima corresponding to anhydride pre-coordination could be found for this system, and so the energetically feasible propagation cycle displayed in Scheme 4 features the initial insertion without pre-coordination to the metal, and is followed by a distinct ring-opening step.
There is a choice of pathways to Int1b, the precursor to epoxide opening. Either association of ethylene oxide (though not coordination, no such minima could be found) to [Cp2Ti(OAc)2] to give Int1c, or the formation of an ion pair resulting from the dissociation of one acetate ligand (Int1a). In addition to its role as a nucleophilic cocatalyst (as indicated by MALDI-ToF and stoichiometric reactions), it is possible to imagine a role for DMAP in the propagation cycle; as a strong donor one may expect coordination to the metal centre, helping to facilitate the de-coordination of polymer chains necessary for the binding and reaction of addition substrate. Although Int1-DMAP is energetically accessible (+18.3 kcal mol−1), attempts to observe metal–DMAP complexes by 1H NMR spectroscopy proved fruitless, although this does not necessarily preclude a potentially transient existence.
The subsequent epoxide opening transition state (TS1) is significantly higher in energy than anhydride insertion (36.3 kcal mol−1vs. 28.2 kcal mol−1), which corroborates experimental findings that epoxide opening is the rate limiting step. The product of this opening, Int2, is significantly lower in energy than Int1b. As with ethylene oxide, no minima corresponding to Int2 with an additional coordinated anhydride could be found, and the equivalent complex with a decoordinated acetate was found to be significantly (≈40 kcal mol−1) higher in free energy than Int3. Quantum Theory of Atoms in Molecules (QTAIM) (Fig. S32†) and Natural Bonding Orbital (NBO) analyses highlighted weak interactions between the anhydride protons and acetate oxygens, as well as a bond critical point between the alkoxide oxygen and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the anhydride. Although weak, this interaction may be significant, as it mirrors the reaction coordinate in TS2 for anhydride insertion. Finally, the ring-opening of the anhydride is completed with the relatively low energy TS3, remaking a bis(carboxylate) complex analogous to the starting point [Cp2TiOAc2].
O bond of the anhydride. Although weak, this interaction may be significant, as it mirrors the reaction coordinate in TS2 for anhydride insertion. Finally, the ring-opening of the anhydride is completed with the relatively low energy TS3, remaking a bis(carboxylate) complex analogous to the starting point [Cp2TiOAc2].
As previously discussed in the context of the MALDI-ToF spectra of synthesised polymers, many of these carboxylate-terminated polymer chains will be DMAP initiated, and thereby zwitterionic. In this sense, DMAP may perform a role analogous to that described for [PPN]+ counterions in previous computational studies.31
Overall, the cycle is both exothermic (ΔH < 0) and spontaneous (ΔG < 0), whilst the lower energy barriers to anhydride insertion/opening compared to epoxide opening are consistent with the lack of heating required for anhydride consumption in the stoichiometric reactions described previously.
Copolymerisation of limonene oxide
A key advantage of ROCOP is the wide variety of available substrates, meaning unsaturated natural products such as pinene or limonene can be epoxidised and used to produce partially renewable polyesters. Therefore, we decided to extend our investigations to (+)-limonene-1,2-epoxide (mixture of cis/trans isomers) (LO), which required harsher conditions to polymerise given the increased level of substitution about the epoxide functionality. The results of its copolymerisation with PA at 100 °C with all three Group 4 metallocenes are detailed in Table 3 where, similarly to the results for TCPA above, both [Cp2ZrCl2] and [Cp2HfCl2] (entries 2 and 3) led to the largest increases in conversion in comparison to control reactions (entry 4).|
|
|||||
|---|---|---|---|---|---|
| Entry | Cp2MCl2 | Conversion PAb/% | Esterc/% | M n /kDa | Đ , |
| a Average of at least three runs. Conditions: 1 eq. = 6.4 μmol (1 eq.) metallocene (unless otherwise stated), 2 eq. DMAP, 400 eq. PA, 400 eq. epoxide, 1 mL toluene, 80 °C for 29 hours. b Determined from 1H NMR spectra of the reaction mixtures. c Determined from 1H NMR spectra of the resultant polymers. d Determined by GPC (viscometry detection against polystyrene standards). e Đ = Mw/Mn. | |||||
| 1 | Ti | 58 | >95 | 6.4 | 1.51 |
| 2 | Zr | 75 | >95 | 5.8 | 1.52 |
| 3 | Hf | 78 | >95 | 5.1 | 1.59 |
| 4 | None | 34 | >95 | 14.6 | 1.27 |
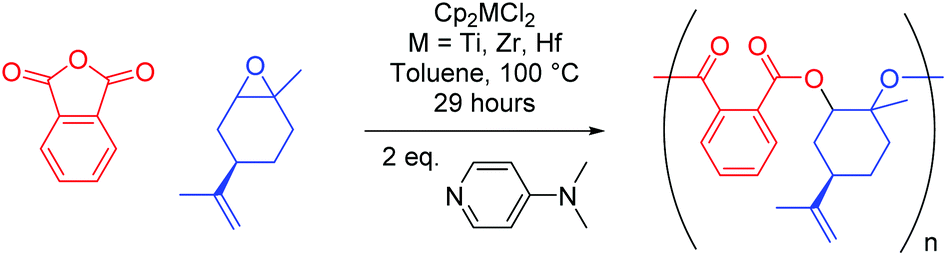
Again, this could be attributed to the greater ionic radii of zirconium and hafnium facilitating reaction with a sterically demanding monomer. Indeed, the slow rate of reaction with LO has in turn greatly reduced the rate of epoxide homopolymerisation, as seen through the complete absence of ether linkages detected in any of the polymers. Unsurprisingly, molecular weights are relatively high when only DMAP is used, and in general molecular weights are decreased compared to copolymers of CHO and VCHO with PA (Table 1). In conjunction with this are the observed increases in Đ (aside from CHO–PA [Cp2HfCl2], entry 3 Table 1, which does not significantly change when moving from CHO or VCHO to LO), suggesting that chain transfer is far more favourable with LO at higher reaction temperatures.
Although comparisons to previously reported catalytic systems are tentative given significant differences in both reaction conditions and catalyst loading, the performance of [Cp2ZrCl2] is somewhat comparable to known examples. For example, [(salophan)Al–Cl] and [(salophan)Cr–Cl] pre-catalysts have produced 24 and 28% conversion after 5 hours in toluene respectively at a higher catalyst loading and elevated temperature (110 °C),14 whilst an aminotriphenolate iron complex gave 46% conversion (84% in THF) in 24 hours at a higher concentration but far reduced temperature (65 °C) with [PPN]Cl as a co-catalyst.17
In contrast to the high ether selectivity seen in the absence of DMAP for CHO and VCHO, polymerisations performed using [Cp2ZrCl2] and LO–PA produced oligomers (Mn = 950 Da, PDI = 1.5, ca. 3–4 LO–PA units) with no discernible ether linkages in the 1H NMR spectrum. Although these reactions occurred with far lower activity (52% conversion of PA in 72 h) than those conducted employing DMAP as a cocatalyst, there is clearly potential for further catalyst development to produce a metallocene-like pre-catalyst which is both highly active and selective without the need for a co-catalyst.
Despite extensive research in the field, epoxide–anhydride ROCOP is in its infancy as a technology, and as such there are few examples of multi-gram scale polymer syntheses using ROCOP. The ability to tolerate scale-up is clearly an important factor in process development, whilst also bridging the gap between catalytic chemistry and materials science. Therefore, given its favourable characteristics, a large-scale synthesis (identical monomer feed ratios and temperature used, see Experimental section for full details) of the LO–PA copolymer was performed with [Cp2ZrCl2], yielding 218 g (57%) of perfectly alternating polyester when run to 81% anhydride conversion. On this scale, distilling epoxides from CaH2 and subliming anhydrides becomes impractical, and so a special consideration to whether the catalyst system is still effective under necessarily less stringently controlled reaction conditions is required. The observed drop in Mn from 5.8 to 4.1 kg mol−1 without using these techniques is unsurprising since trace amounts of water/diacid can facilitate chain transfer. Pleasingly however, the Đ of the large-scale polymer decreased to 1.36 (vs. 1.52 on the small scale), meaning the reaction remains well controlled even without extensive purification of the monomers. This clearly demonstrates the utility of ROCOP in producing larger quantities of polymer, and investigations into the material properties of these polymers are ongoing in our laboratory.
Conclusions
In summary, we have demonstrated the use of simple, commercially available metallocene complexes of the Group 4 metals in the ROCOP of several epoxides and cyclic anhydrides, including the bio-derived limonene oxide. This was extended to a large scale synthesis, demonstrating the reaction is robust enough to be successful without extensive drying and degassing techniques typically used to purify epoxides. Mechanistic insights were also gained from stoichiometric reactions and DFT calculations.The polymerisation performance was somewhat modest in comparison to using DMAP alone with CHO or VCHO, yet for more challenging substrates there is more a value to be had in their usage. The catalysts probed in this study have not been optimised through traditional ligand screening, yet open the gateway to a whole plethora of complexes based upon the metallocene ligand framework for use in ROCOP. This includes ansa and half metallocenes including so-called “constrained geometry catalysts”, all of which have all been widely investigated for olefin polymerisation and have approached commercialisation in several cases.81–83
Data availability
The data supporting the results presented in this article are freely available via the Cardiff University Data Catalogue at https://dx.doi.org/10.17035/d.2022.0152098705Author contributions
MSS and MRB performed the experimental and analytical work. MSS and BDW performed the computational work, MDJ and BDW supervised and guided the project and assisted with data interpretation, all authors contributed to the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the EPSRC (studentship to MSS, EP/L016443/1). Access to the Advanced Research Computing facility at Cardiff University (ARCCA) is gratefully acknowledged.Notes and references
- C. Wang, Y. Liu, W. Q. Chen, B. Zhu, S. Qu and M. Xu, J. Ind. Ecol., 2021, 25, 1300–1317 CrossRef.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- J. M. D. Souza, F. M. Windsor, D. Santillo and S. J. Ormerod, Glob. Change Biol., 2020, 26, 3846–3857 CrossRef PubMed.
- T. Stößer and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 CrossRef PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K. Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- L. Feng, Y. Liu, J. Hao, X. Li, C. Xiong and X. Deng, Macromol. Chem. Phys., 2011, 212, 2626–2632 CrossRef CAS.
- A. M. Diciccio, J. M. Longo, G. G. Rodríguez-Calero and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 7107–7113 CrossRef CAS PubMed.
- Y. Hiranoi and K. Nakano, Beilstein J. Org. Chem., 2018, 14, 2779–2788 CrossRef CAS PubMed.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1985, 107, 1358–1364 CrossRef CAS.
- J. Li, B.-H. Ren, Z.-Q. Wan, S.-Y. Chen, Y. Liu, W.-M. Ren and X.-B. Lu, J. Am. Chem. Soc., 2019, 141, 8937–8942 CrossRef CAS PubMed.
- Y. Zhou, C. Hu, T. Zhang, X. Xu, R. Duan, Y. Luo, Z. Sun, X. Pang and X. Chen, Macromolecules, 2019, 52, 3462–3470 CrossRef CAS.
- E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS.
- N. J. Van Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef CAS PubMed.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- L. Peña Carrodeguas, C. Martín and A. W. Kleij, Macromolecules, 2017, 50, 5337–5345 CrossRef.
- R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed.
- B. Liu, J. Chen, N. Liu, H. Ding, X. Wu, B. Dai and I. Kim, Green Chem., 2020, 22, 5742–5750 RSC.
- N. J. Van Zee, M. J. Sanford and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 2755–2761 CrossRef CAS PubMed.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- H. Zhang, G. Zhou, M. Jiang, H. Zhang, H. Wang, Y. Wu and R. Wang, Macromolecules, 2020, 53, 5475–5486 CrossRef CAS.
- Y. Chen, J. A. Wilson, S. R. Petersen, D. Luong, S. Sallam, J. Mao, C. Wesdemiotis and M. L. Becker, Angew. Chem., Int. Ed., 2018, 57, 12759–12764 CrossRef CAS PubMed.
- R. Baumgartner, Z. Song, Y. Zhang and J. Cheng, Polym. Chem., 2015, 6, 3586–3590 RSC.
- M. J. Sanford, N. J. Van Zee and G. W. Coates, Chem. Sci., 2018, 9, 134–142 RSC.
- H. Ding, X. Wu, B. Han, N. Liu, B. Liu and I. Kim, Polymer, 2020, 213, 123199 CrossRef.
- A. Virachotikul, N. Laiwattanapaisarn, P. Wongmahasirikun, P. Piromjitpong, K. Chainok and K. Phomphrai, Inorg. Chem., 2020, 59, 8983–8994 CrossRef CAS PubMed.
- B. Han, L. Zhang, B. Liu, X. Dong, I. Kim, Z. Duan and P. Theato, Macromolecules, 2015, 48, 3431–3437 CrossRef CAS.
- C.-M. Chen, X. Xu, H.-Y. Ji, B. Wang, L. Pan, Y. Luo and Y.-S. Li, Macromolecules, 2021, 54, 713–724 CrossRef CAS.
- S. Zhu, Y. Wang, W. Ding, X. Zhou, Y. Liao and X. Xie, Polym. Chem., 2020, 11, 1691–1695 RSC.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
- A. J. Gaston, G. Navickaite, G. S. Nichol, M. P. Shaver and J. A. Garden, Eur. Polym. J., 2019, 119, 507–513 CrossRef CAS.
- J. Li, B.-H. Ren, S.-Y. Chen, G.-H. He, Y. Liu, W.-M. Ren, H. Zhou and X.-B. Lu, ACS Catal., 2019, 9, 1915–1922 CrossRef CAS.
- M. Hatazawa, R. Takahashi, J. Deng, H. Houjou and K. Nozaki, Macromolecules, 2017, 50, 7895–7900 CrossRef CAS.
- H. K. Ryu, D. Y. Bae, H. Lim, E. Lee and K. S. Son, Polym. Chem., 2020, 11, 3756–3761 RSC.
- Y. Liu, J.-Z. Guo, H.-W. Lu, H.-B. Wang and X.-B. Lu, Macromolecules, 2018, 51, 771–778 CrossRef CAS.
- D. Wannipurage, T. S. Hollingsworth, F. Santulli, M. Cozzolino, M. Lamberti, S. Groysman and M. Mazzeo, Dalton Trans., 2020, 49, 2715–2723 RSC.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- S. Abbina, V. K. Chidara and G. Du, ChemCatChem, 2017, 9, 1343–1348 CrossRef CAS.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS PubMed.
- S. Impemba, G. Roviello, S. Milione and C. Capacchione, Inorg. Chem., 2021, 60, 7561–7572 CrossRef CAS PubMed.
- T. Xing, T. J. Prior, K. Chen and C. Redshaw, Dalton Trans., 2021, 50, 4396–4407 RSC.
- Z. Sun, Y. Zhao, O. Santoro, M. R. J. Elsegood, E. V. Bedwell, K. Zahra, A. Walton and C. Redshaw, Catal. Sci. Technol., 2020, 10, 1619–1639 RSC.
- A. G. Morozov, T. V. Martemyanova, V. A. Dodonov, O. V. Kazarina and I. L. Fedushkin, Eur. J. Inorg. Chem., 2019, 4198–4204 CrossRef CAS.
- M. D. Jones, L. Brady, P. McKeown, A. Buchard, P. M. Schäfer, L. H. Thomas, M. F. Mahon, T. J. Woodman and J. P. Lowe, Chem. Sci., 2015, 6, 5034–5039 RSC.
- T. Xing, Z.-Y. Wang, Y.-C. Sun, Z.-H. He, K. Wang, Z.-T. Liu, M. R. J. Elsegood, E. V. Bedwell and C. Redshaw, J. Appl. Polym. Sci., 2021, 138, e50513 CrossRef.
- S. K. Raman, A. C. Deacy, L. P. Carrodeguas, N. V. Reis, R. W. F. Kerr, A. Phanopoulos, S. Morton, M. G. Davidson and C. K. Williams, Organometallics, 2020, 39, 1619–1627 CrossRef CAS PubMed.
- L. Suresh, R. Lalrempuia, J. B. Ekeli, F. Gillis-D'Hamers, K. W. Törnroos, V. R. Jensen and E. Le Roux, Molecules, 2020, 25, 4364 CrossRef CAS PubMed.
- R. Lalrempuia, J. Underhaug, K. W. Törnroos and E. Le Roux, Chem. Commun., 2019, 55, 7227–7230 RSC.
- R. Lalrempuia, F. Breivik, K. W. Törnroos and E. Le Roux, Dalton Trans., 2017, 46, 8065–8076 RSC.
- J. A. Garden, A. J. P. White and C. K. Williams, Dalton Trans., 2017, 46, 2532–2541 RSC.
- C. C. Quadri, R. Lalrempuia, J. Hessevik, K. W. Törnroos and E. Le Roux, Organometallics, 2017, 36, 4477–4489 CrossRef CAS.
- M. Mandal, D. Chakraborty and V. Ramkumar, RSC Adv., 2015, 5, 28536–28553 RSC.
- M. Mandal, J. Organomet. Chem., 2020, 907, 121067 CrossRef CAS.
- D. Takeuchi, T. Aida and T. Endo, Macromol. Rapid Commun., 1999, 20, 646–649 CrossRef CAS.
- O. Shimomura, T. Nishisako, S. Yamaguchi, J. Ichihara, M. Kirino, A. Ohtaka and R. Nomura, J. Mol. Catal. A: Chem., 2016, 411, 230–238 CrossRef CAS.
- O. Shimomura, S. Sasaki, K. Kume, A. Ohtaka and R. Nomura, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2557–2561 CrossRef CAS.
- O. Shimomura, K. Tokizane, T. Nishisako, S. Yamaguchi, J. Ichihara, M. Kirino, A. Ohtaka and R. Nomura, Catalysts, 2017, 7, 172 CrossRef.
- S. Pappuru, D. Chakraborty, V. Ramkumar and D. K. Chand, Polymer, 2017, 123, 267–281 CrossRef CAS.
- R. W. F. Kerr and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 6882–6893 CrossRef CAS PubMed.
- S. H. Kim, D. Ahn, Y. Y. Kang, M. Kim, K.-S. Lee, J. Lee, M. H. Park and Y. Kim, Eur. J. Inorg. Chem., 2014, 5107–5112 CrossRef CAS.
- D. Bai, H. Jing, Q. Liu, Q. Zhu and X. Zhao, Catal. Commun., 2009, 11, 155–157 CrossRef CAS.
- D. Bai, G. Nian, G. Wang and Z. Wang, Appl. Organomet. Chem., 2013, 27, 184–187 CrossRef CAS.
- A. D. Asandei, Y. Chen and B. P. Olivieri,Polymeric Materials: Science & Engineering, American Chemical Society, Atlanta, Georgia, USA, 2006, vol. 94, pp. 461–462 Search PubMed.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS PubMed.
- N. Yi, T. T. D. Chen, J. Unruangsri, Y. Zhu and C. K. Williams, Chem. Sci., 2019, 10, 9974–9980 RSC.
- G. Si, L. Zhang, B. Han, Z. Duan, B. Li, J. Dong, X. Li and B. Liu, Polym. Chem., 2015, 6, 6372–6377 RSC.
- J. P. Agrawal and K. S. Kulkarni, J. Appl. Polym. Sci., 1986, 32, 5203–5214 CrossRef CAS.
- V. Shahedifar, A. Masoud Rezadoust and I. Amiri Amraei, Propellants, Explos., Pyrotech., 2016, 41, 321–326 CrossRef CAS.
- E. Hosseini Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef.
- F. Isnard, F. Santulli, M. Cozzolino, M. Lamberti, C. Pellecchia and M. Mazzeo, Catal. Sci. Technol., 2019, 9, 3090–3098 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- H. E. Dyer, S. Huijser, N. Susperregui, F. Bonnet, A. D. Schwarz, R. Duchateau, L. Maron and P. Mountford, Organometallics, 2010, 29, 3602–3621 CrossRef CAS.
- J. Wei, M. N. Riffel and P. L. Diaconescu, Macromolecules, 2017, 50, 1847–1861 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardois and R. Paton, GoodVibes: automated thermochemistry for heterogeneous computational chemistry data (version 3.1.0), F1000Research, 2020, 9, 291 Search PubMed.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS PubMed.
- J. Klosin, P. P. Fontaine and R. Figueroa, Acc. Chem. Res., 2015, 48, 2004–2016 CrossRef CAS PubMed.
- W. Kaminsky, J. Chem. Soc., Dalton Trans., 1998, 1413–1418 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00335j |
| This journal is © The Royal Society of Chemistry 2022 |