
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReplacing amine by azide: dopamine azide polymerization triggered by sodium periodate†
Monika
Szukowska
ab,
Łukasz
Popenda
c,
Emerson
Coy
 c,
Claudiu
Filip
d,
Jakub
Grajewski
a,
Mateusz
Kempiński
e,
Yeonho
Kim
f and
Radosław
Mrówczyński
*ab
c,
Claudiu
Filip
d,
Jakub
Grajewski
a,
Mateusz
Kempiński
e,
Yeonho
Kim
f and
Radosław
Mrówczyński
*ab
aFaculty of Chemistry, Adam Mickiewicz University, ul. Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: radoslaw.mrowczynski@amu.edu.pl
bCenter for Advanced Technologies, ul. Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland
cNanoBioMedical Centre, Adam Mickiewicz University, ul. Wszechnicy Piastowskiej 3, 61-614, Poznań, Poland
dNational Institute for R&D of Isotopic and Molecular Technologies, 400293 Cluj, Romania
eFaculty of Physics, Adam Mickiewicz University, ul. Uniwersytetu Poznańskiego 2, 61-614 Poznań, Poland
fDepartment of Applied Chemistry, Konkuk University, Chungju 27478, Republic of Korea
First published on 9th May 2022
Abstract
Polydopamine (PDA) has been widely described for a range of biomedical and surface engineering applications. However the structure of PDA remains elusive owing to the insoluble nature of the polymer. Furthermore, the influence of the amine group present in the polydopamine and related functional polycatchols on the character and the structure of resulting functional materials remains vague. Here we perform polymerization of the dopamine analogue 4-(2-azidoethyl)benzene-1,2-diol (dopamine azide), where the amine group is switched to azide, using sodium periodate, which gives rise to particles with a diameter of up to one micrometer. The obtained particles are stable in water but, in contrast to other polycatcechol-based polymers, are soluble in organic solvents. The detailed structural investigations using various liquid and solid-state nuclear magnetic resonance (NMR) spectroscopy methods, X-ray photoelectron spectroscopy (XPS) and mass spectrometry (ESI/MALD) prove that the obtained polymeric material consists mainly of repeating monomers linked by C–C bonds of aromatic units bearing open-azidoethyl chains. Moreover, the resulting polymer shows a different morphology from polydopamine (PDA) obtained under the same polymerization conditions. Therefore, our results are an important step towards understanding the relationships between the structures of the starting catechol monomers and shed new light on the influence of the amine group on the nature of the resulting poly(catechols).
1. Introduction
PDA is considered a synthetic melanin that shares common features, in particular, with eumelanins, which are a subgroup of melanins.1 The eumelanins are naturally occurring black pigments comprising numerous cross-linked 5,6-dihydroxyindole (DHI) and 5,6-dihydroxyindole-2-carboxylic acid (DHICA) units and are characterized by structural complexity and insolubility as well as broadband absorption in UV–vis light.2–5Since PDA was reported by Lee et al. in 2007 as a substrate for independent coating materials, complete understanding of this material's character, chemistry, and formation has remained challenging.6 Different structural models of PDA have been reported (see Fig. 1). For instance, Lee proposed cation–π interactions between protonated amines of uncyclized dopamine/dopamine-quinone and π electrons from DHI and their subsequent oligomers as binding forces in PDA formation.7,8 Another model based on the pyrrole moiety incorporated in the structure was proposed by Chai et al.9 The quinhydrone model based on hydrogen bonding was proposed by Bielawski.10 Liebscher et al. proposed that PDA consists of dihydroxyindole and indoledione units with different degrees of (un)saturation covalently linked by C–C bonds between their benzene rings.11 Furthermore, they proved the presence of open-chain monomer units in the PDA structure.12
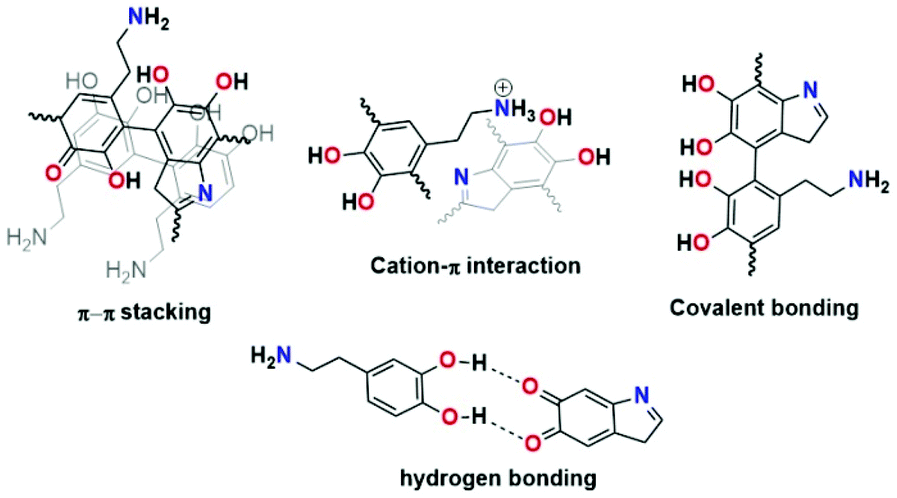 | ||
| Fig. 1 Proposed structural models of PDA. | ||
In the last few years, attention has been paid to the synthesis and structural properties of polydopamine analogues (PDANA),13–15 which can be obtained in three main different ways.13 In the first strategy, the PDA coating is subjected to post modification using available functional groups16 in the PDA with (bio)macromolecules17 bearing amine18–20 or thiols21,22 while free amino groups can be exploited in the diazotransfer reaction,12 ring-opening polymerization or sulfonation.23 The second strategy includes copolymerization of dopamine with other catecholic derivatives. The last and so far the least explored approach that can contribute to a thorough understanding of the nature of polycatechol coatings is based on chemical modification of the dopamine molecule or other catecholic molecules and their polymerization. That approach has been applied to prepare 5,6-dihydroxybenzothiophene (DHBT), the sulfur analog of the eumelanin building block 5,6-dihydroxyindole.24 In contrast to materials obtained from 5,6-dihydroxyindole resulting in a black insoluble eumelanin polymer, polymerization of DHBT yielded an amorphous solid with visible absorption and electron paramagnetic resonance properties different from those of DHI melanin. Another example of PDANA, obtained using this strategy, is polymerization of 4-(3-aminopropyl)-benzene-1,2-diol, which an additional methylene group characterizes in an aliphatic bridge at the amino group in the dopamine molecule, which gives access to faster molecular assembly and surface coating.25 Recently, Petran et al. showed oxidation of 3,4-dihydroxybenzhydrazide by ammonium persulfate, resulting in a black insoluble material.26 The resulting materials differed from PDA since the hydroxyl groups were replaced and converted to acylhydrazine, acylhydrazone, and 1,3,4-oxadiazoline bridges which prevent adhesion of the polymer. Del Campo and d'Ischia reported the oxidation of 6-nitro- and 6-chlorodopamine analogs and their influence on the resulting polymer formation.27 Experiments showed that 6-chlorodopamine in contrast to 6-nitrodopamine undergoes oxidation and forms robust polymer coatings when deposited from Tris buffer at pH 8.5. This process was less efficient in the case of 6-nitrodopamine, which required stronger oxidation conditions (NaIO4) and led to inhomogeneous films on coated substrates.
Here we report the conversion of the amino group from dopamine into the azide moiety via the diazotransfer reaction and the preparation of new poly(dopaazide) particles resulting from the oxidation of 4-(2-azidoethyl)benzene-1,2-diol (dopamine-azide) by NaIO4 in water. Because of the solubility of the resulting material in organic solvents, we performed detailed structural characterization using liquid-state NMR, mass spectrometry and solid-state NMR, as well as FTIR, XPS, DSC/TGA. In addition, the results of the theoretical DFT calculations are shown to verify the hypothesis of the possible structure of this new polymeric material. We also show differences in coatings obtained from PDA and poly(dopaazide), which shed new light on understanding of the relation between the catechol monomer and the properties of resulting polycatechol material and the role of amine groups in the PDA coating formation.
2. Experimental
2.1 Materials and methods
The reagents used in this work were dopamine hydrochloride (99%), trifluoromethanesulfonic anhydride (≥99%), zinc chloride, sodium azide, triethylamine ≥99% and sodium periodate. All of the chemical reagents were purchased from Merck, except for dopamine hydrochloride (Alfa Aesar). For all experiments, Milli-Q deionized water (resistivity 18 MΩ cm−1) was used.Scanning electron microscopy (SEM) images were recorded on a Jeol 7001 TTLS scanning electron microscope. The samples were drop-cast on a washed silica wafer and dried in a vacuum desiccator. HR-TEM images were collected in a high-resolution transmission electron (HR-TEM) microscope (JEOL ARM200F) working at 200 kV, equipped with an energy dispersive detector (EDX) for chemical composition. UV–vis spectra were recorded on a Lambda spectrophotometer 950 UV/Vis/NIR. Infrared spectra were recorded on a Jasco FT/IR-4700 Fourier transform infrared spectrometer in KBr pellets. Zeta potential measurements were carried out on a Zetasizer Nano-ZS ZEN 3600 produced by Malvern Instruments Ltd. The thermal characteristics were measured using a simultaneous TGA/DSC analyzer (SDT 650, TA Instruments, USA) in a N2 atmosphere with a heating rate of 10 °C min−1. X-ray photoelectron spectroscopy (XPS) was performed with a K-Alpha + XPS system (Thermo Fisher Scientific, UK) using a monochromated Al Kα X-ray source (hv = 1486.6 eV) with charge compensation. Contact angle measurements were performed using the SEO Contact Angle Analyzer Phoenix 300 equipped with an industrial zoom lens, Navitar TV zoom 7000, under ambient conditions (air, room temperature, atmospheric pressure). Data were acquired and analyzed using SurfaceWare software. Atomic force microscopy was performed with the Park Systems XE7 atomic force microscope. Measurements were done in non-contact mode using the PPP-NHCR cantilevers with a resonant frequency of approx. 300 kHz, to avoid possible changes to the sample surface.
All liquid-state NMR experiments were carried out at 298 K in DMSO-d6 at an approximate sample concentration of 25–30 mg ml−1. The 1H and 13C measurements were performed on an Agilent DD2 800 spectrometer, operating at resonance frequencies of 799.73 and 201.09 MHz, respectively, equipped with a 1H{13C/15N} probe. The 15N NMR spectra were recorded on either a Bruker Avance III 700 spectrometer (700.48 MHz and 70.98 MHz for 1H and 15N, respectively) equipped with a QCI-P CryoProbe or a Bruker Avance III 500 spectrometer using a PABBO probe. The 1D (1H, 13C) and 2D (1H–1H gCOSY, 1H–1H zTOCSY, multiplicity-edited 1H–13C gHSQC, 1H–13C gHMBC and 1H–15N gHMBC) experiments were recorded using standard pulse sequences and acquisition parameters. For 1H and 13C data a residual solvent signal (DMSO-d6: 2.50 ppm for 1H, 39.5 ppm for 13C) was used as an internal chemical shift reference, while 15N spectra were referenced with respect to the CH3NO2 line at 0.00 ppm.
Solid-state 13C and 15N NMR spectra of poly(dopaazide) were recorded at 125.73 and 50.66 MHz Larmor frequencies with a Bruker AVANCE III 500 MHz spectrometer operating at room temperature. Standard RAMP 13C/15N CP-MAS spectra were acquired at 14/7 kHz spinning frequencies, with 2/4 ms contact times, and proton decoupling under TPPM, by averaging 10k/300k transients with a recycle delay of 4 s. A 13C CP-MAS spectrum with a short CP contact time of 100 μs was also taken. The recorded 13C/15N NMR spectra were calibrated with respect to the CH3 line in TMS (tetramethylsilane) and the 15NO2 line in nitromethane, through an indirect procedure which uses α-glycine (176.5 ppm for the 13COOH line and −347.6 ppm for the 15NH3 line) as an external reference.
High-resolution mass spectra (HRMS) were obtained using an Impact HD mass spectrometer (Q-TOF type instrument equipped with electrospray ion source; Bruker Daltonics, Germany). The sample solutions were infused into the ESI source using a syringe pump (direct inlet) at the flow rate of 3 μL min−1. The instrument was operated under the following optimized settings: end plate voltage 500 V; capillary voltage 4.5 kV; nebulizer pressure 0.3 bar; dry gas (nitrogen) temperature 200 °C; and dry gas flow rate 4 L min−1. The spectrometer was previously calibrated with the standard tuning mixture. Matrix-assisted laser desorption ionization–time of flight (MALDI TOF) mass spectrometry was performed in reflection mode using an Ultraflex TOF/TOF (Bruker Daltonics, Germany). The thin-layer preparation method was applied. The matrix (2,5-dihydroxybenzoic acid – DHB) was dissolved at a concentration of 20 mg mL−1 in a mixture of 0.1% TFA in de-ionized water (70% v/v) and acetonitrile (30% v/v). The matrix solution was spotted onto the target and dried in air. The sample solution was deposited onto the matrix spot and dried in air in the next step.
2.2 General procedure for polymerization of 4-(2-azidoethyl)benzene-1,2-diol
4-(2-Azidoethyl)benzene-1,2-diol28 (730 mg, 4.0 mmmol) was placed in a 250 ml round-bottom flask and dissolved in water (182.5 ml) to maintain a concentration of 4 mg ml−1 followed by the addition of NaIO4 (174 mg, 0.8 mmol, 0.2 eq.). The stirring bar speed was set up at 400 rpm and was kept constant for 24 hours. The resulting precipitate was centrifuged and washed with deionized water until the color of the supernatant disappeared. Finally, the material was dispersed in water.The influence of the oxidant on the polymerization process of 4-(2-azidoethyl)benzene-1,2-diol was tested according the protocol described below.
4-(2-Azidoethyl)benzene-1,2-diol (200 mg, 1.12 mmol) was dissolved in 50 ml followed by the addition of an appropriate amount of NaIO4 (0.1 eq., 23.88 mg, 0.11 mmol; 0.2 eq., 47.77 mg, 0.22 mmol; 0.5 eq., 119.44 mg, 0.56 mmol; 1.0 eq., 238.89 mg, 1.12 mmol; 2.0 eq., 477.78 mg, 2.23 mmol). After 24 h the reaction was worked up according to the general protocol.
2.3 Synthesis of PDA triggered by different equivalents of NaIO4
Dopamine hydrochloride (200 mg, 1.05 mmol) was dissolved in 50 ml of water followed by the addition of an appropriate amount of NaIO4 (0.1 eq., 22.55 mg, 0.11 mmol; 0.2 eq., 45.11 mg, 0.21 mmol; 0.5 eq., 112.78 mg, 0.53 mmol; 1.0 eq., 225.57 mg, 1.06 mmol; 2.0 eq., 451.15 mg, 2.11 mmol). Then the reaction was continued according to the general protocol described for the polymerization of 4-(2-azidoethyl)benzene-1,2-diol.2.4 Coating protocol using 4-(2-azidoethyl)benzene-1,2-diol and dopamine
The coating reaction was carried out on three types of materials: glass, ceramics and silicon. Each material was thoroughly washed with acetone and isopropanol in an ultrasound bath and then dried under nitrogen gas before use. Then the corresponding 4-(2-azidoethyl)benzene-1,2-diol or dopamine hydrochloride was placed in a Petri dish. Subsequently, water was poured into those dishes to obtain a 4 mg ml−1 concentration and a magnetic stirrer was set at 400 rpm. After 5 minutes, the glass, ceramics and silica wafer were immersed in the solution followed by dropwise addition of 502 μl (530 μl in case of dopamine) of sodium periodate in water (20 mg ml−1), which corresponded to 0.2 eq. of NaIO4. Stirring was continued for either 24 or 48 hours, and the reaction vessels were covered with aluminum foil to protect the system against evaporation. After coating reaction substrates were taken up, rinsed with water and dried in air.3. Results and discussion
3.1 Synthesis and structural studies of poly(dopaazide)
In order to mask the amine group we synthesized the dopamine-azide derivative using a diazotrasnfer reaction according to the reported protocol28 (see the ESI† for the detailed procedure). In this reaction the diazo group (–N2) was transferred to an amine to form an azide. Further, the polymerization of the obtained (2-azidoethyl)benzene-1,2-diol was triggered by the addition of 0.2 eq. of NaIO4 in water (see Scheme 1). Upon adding the oxidant, the mixture changed color to red almost instantly, followed by its conversion to light brown in time (see Fig. 2A). In order to gain insight into the polymerization process, we followed the reaction using UV–vis spectroscopy at different time intervals. As can be seen in Fig. 2A, the spectra are characterized by three distinctive regions. The first peak is located at 250 nm, and its intensity is decreasing with the reaction's progress. This absorption band can be assigned to the absorption of aromatic rings present in the starting azide derivative of dopamine. The second region is a broad absorption in a range of 300–450 nm with a maximum located at 400 nm, which indicates the oxidation of catechol moieties and the formation of o-quinone, yielding red to orange intermediates, which is in agreement with the observed color change during the polymerization process.29,30 The intensity of this peak increases during polymerization while new intermediates are formed. The last part of the spectrum is the band between 450–650 nm with maximum absorption at 600 nm. The intensity of this band also increases along with polymerization time and comes from light scattering on particles that are forming during the polymerization.31 We further investigated the morphology of the particles using SEM. The poly(dopaazide) particles were spherical in shape and had a diameter of ∼0.7 μm (see Fig. 2B). We used HR-TEM combined with energy dispersive X-ray analysis (EDX) to investigate the composition and distribution of elements in the obtained material (see Fig. 2C). The performed analysis revealed nitrogen, carbon and oxygen atoms built in the poly(dopaazide). Interestingly, ∼3.34% by mass of all elements in the sample was iodine, which presumably comes from the NaIO4.32 This suggests that iodine forms stable complexes with the poly(dopaazide) or is covalently linked to quinones as already reported for polydopamine obtained in the polymerization process triggered by NaIO4. The FTIR spectrum of poly(dopaazide) shows several characteristic bands that were assigned to stretching vibrations of OH groups at 3500–3200 cm−1, the stretching vibration of C–H bonds from CH2 groups at 3000–2840 cm−1 and a strong peak at 2100 cm−1 resulting from stretching vibrations of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN bonds of azide moiety.
NN bonds of azide moiety.
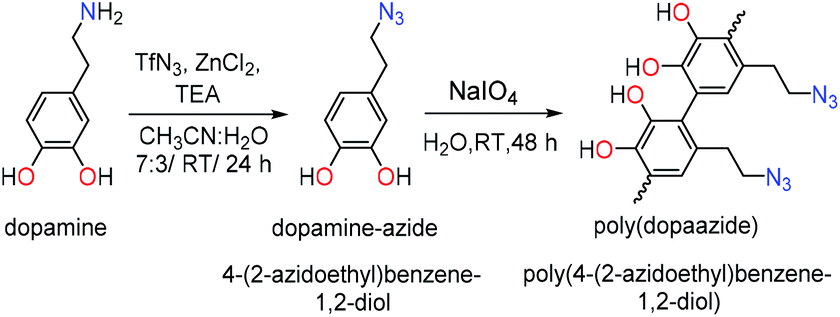 | ||
| Scheme 1 Synthetic route of polymerization of dopamine-azide to poly(dopaazide). | ||
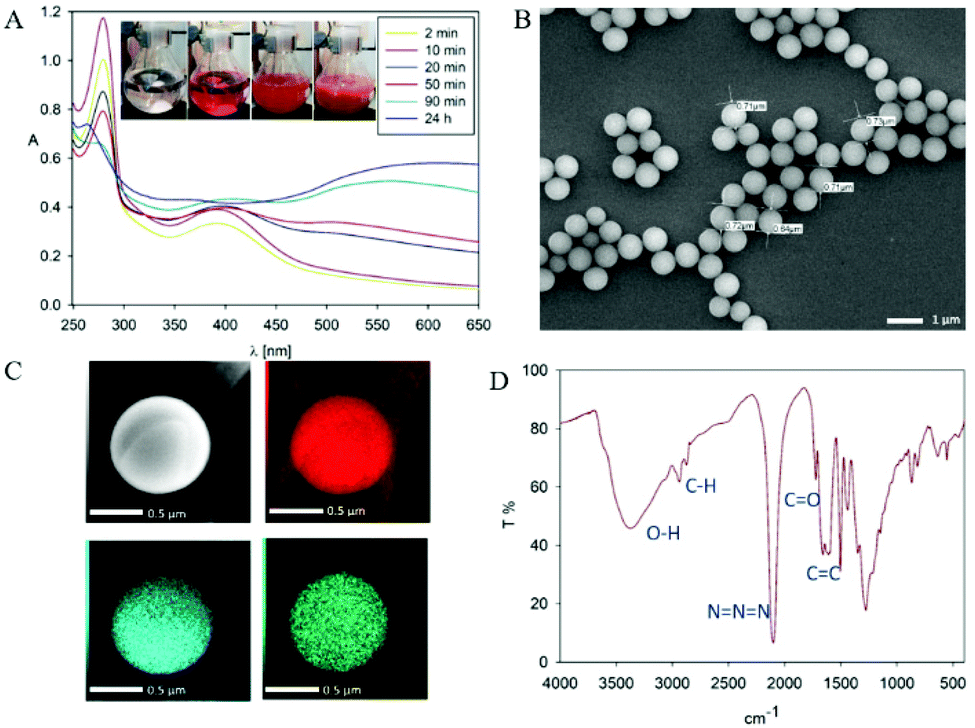 | ||
| Fig. 2 (A) The UV–Vis spectra recorded at different time intervals during the polymerization of dopamine-azide derivative (inset show the color evolution during polymerization process). (B) SEM picture of poly(dopaazide). (C) HR-TEM picture of single poly(dopaazide) particle and its elemental analysis by EDX. Carbon (red), oxygen (blue), iodine (green). (D) FTIR spectrum of poly(dopaazide). | ||
The intense signal at 1700 cm−1 was ascribed to the stretching vibration of CO groups, which pointed out the presence o-quinone in the structure of the poly(dopaazide) material (see Fig. 2D). We also verified the value of the zeta potentials of the particles in water (see the ESI Fig. S1†). Unlike PDA, which exhibits a highly negative value of zeta potential,11 the obtained particles had a value close to 0 mV. This value could suggest the low stability of the particles in suspension.33 However, they were stable in water, and we did not observe noticeable precipitation of particles for up to several days.
The results from the high-resolution XPS measurements showed two distinctive populations of the N 1S region (see Fig. 3A): one at ∼400.38 eV, which can be attributed to the amide/amine,34 and a second one at a higher binding energy of ∼404.16, attributed to azide bonds.35–37 Since we did not use primary amines in the experiments, these results suggest that the azide moiety, which is labile,38 lost nitrogen either during the polymerization process or after exposure to X-ray irradiation. The peaks of oxygen (see Fig. 3B) show a variety of components typical of organic compounds with a small but visible contribution of OC–O and high binding energies.39 The peak above is further confirmed in the oxygen deconvolution (see Fig. 3C), where a peak is observed at ∼530.7 eV which resembles that in the studies of Chehimi et al.,40,41 where C–O−/–C–O− groups were observed and attributed to a lower conjugation of the carbonyl groups in the sample. However, it can also be attributed to the iodine content of the sample, possibly iodanil hydroquinone.42
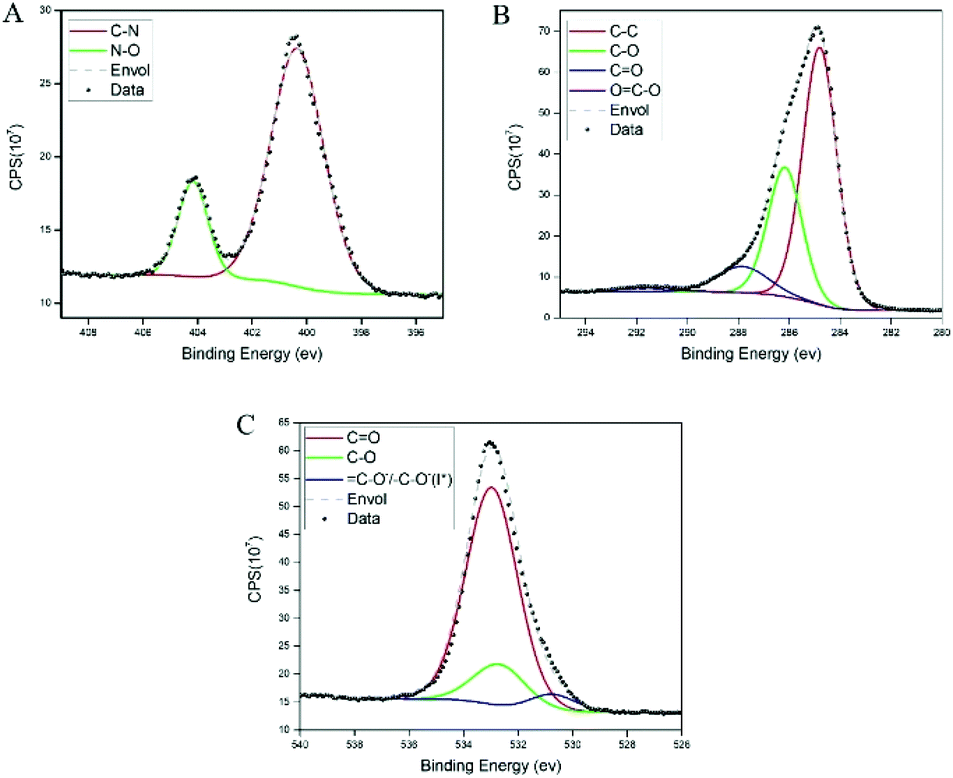 | ||
| Fig. 3 XPS spectra of poly(dopaazide) with their corresponding components. (A) The nitrogen 1s spectra. (B) Carbon 1s and (C) oxygen 1s. | ||
Then the thermal properties of poly(dopaazide) were investigated by means of STA analysis (see the ESI Fig. S2†). The TGA curve for the polymer has three regions. The first one is up to ∼120 °C, where approximately 5% weight loss is observed. This can be assigned to water evaporation from the sample. The second region ranges from 120 to 250 °C, where a ∼15% loss of mass is observed. This is assigned to the degradation of the polymer chain and azido moiety, which is thermally labile.38,43 The broad character of this change is caused by the heterogonous polymer structure. Moreover, the second process occurring in a range of 175–250 °C is exothermic, which is confirmed in DSC by two peaks at 150 and 225 °C with heat release indicating azide decomposition.44 Above 300 °C, the onset of an endothermic peak points to the decomposition of the polymer structure.
The main reason why the structure of PDA remains elusive is its insoluble character. Nevertheless, we decided to check the solubility of poly(dopaazide), which appeared to change its nature in organic solvents. For instance, the suspension of poly(dopaazide) in EtOH caused a change in color suspension from dark yellowish to dark brown (see Fig. 4A). Moreover, the UV–Vis spectrum of poly(dopaazide) in water differed significantly from the spectra obtained for poly(dopaazide) in organic solvents (see Fig. 4A). This fact may indicate the soluble nature of the poly(dopaazide) or a change in particle size (degradation) in organic solvents such as EtOH, DMSO or ACN. The SEM picture reveals that the particles significantly decreased in size to around 30–100 nm after being suspended in ethanol, which confirms the correctness of our assumptions (see Fig. 4B).
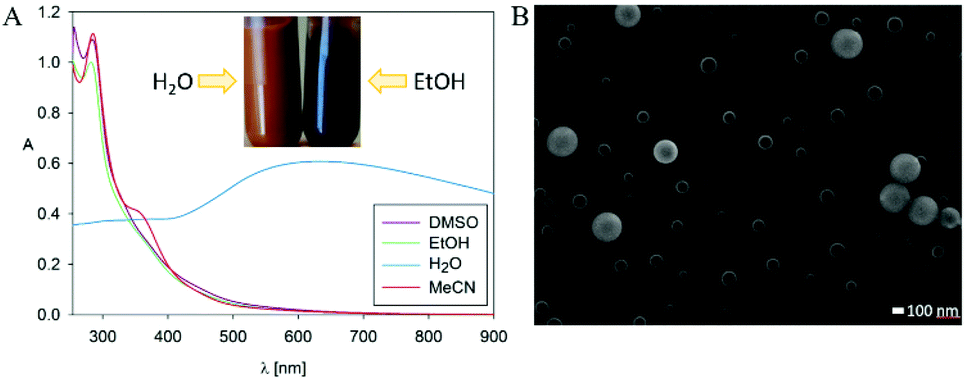 | ||
| Fig. 4 (A) UV–Vis spectra of poly(dopaazide) particles in different solvents (inset shows the colour of poly(dopaazide)) dispersion in H2O and EtOH). (B) SEM picture of poly(dopaazide) in EtOH. | ||
Since we observed a significant change in the poly(dopaazide) nature in comparison with PDA, we decided to get a deeper insight into the polymer structure, using liquid-state NMR experiments. It is worth highlighting that assignment of the signals in 2D NMR spectra of poly(dopaazide) was done on the basis of 2D spectra recorded for the monomer (see the ESI Fig. S3–S5†) The 1H NMR spectrum of poly(dopaazide) revealed a few groups of resonances (see Fig. 5A). The group of signals occurring in the range of 2.6–3.0 ppm (blue circle) was assigned to methylene protons located at the C7 position in the aliphatic chain. The adjacent group of signals at 3.3–3.7 ppm (green circle) corresponds to the protons of the methylene group at the C8 position located in the aliphatic bridge next to the azide group. The assignment of these groups of protons is also confirmed by the 1H–1H COSY spectrum (see Fig. 6), which shows strong correlations between them. Another group of signals ranging from 6 to 7 ppm in the 1H spectrum was assigned to the aromatic protons (red circle). The last set of signals appearing from 8.5 to 10 ppm was assigned to hydroxyl protons. Moreover, in the 1H spectrum we observed an additional group of protons at 4–5 ppm, which on the 1H–13C HSQC spectrum (see Fig. 7) correlates with carbon atoms at around 60 ppm and comes from additional species built in the polymer structure. The 13C NMR spectrum of poly(dopaazide) shows three signals at high chemical shift values (185 ppm, 190 ppm and 191 ppm), corresponding to carbonyl carbon atoms from the quinone present in the polymer structure (see Fig. 6). Based on the 1H–13C HSQC spectrum, as well as knowledge of the spectrum of the monomer, it can be distinguished which aromatic carbons are attached to the –OH groups (see Fig. 7). In the next step we carried out the 1H–15N HMBC experiment on the azide derivative of dopamine, where a correlation between the proton and nitrogen can be seen. The comparision of 1H-15N spectra of dopamine azide and poly(dopaazide) is presented in Fig. 8. The spectra of both compounds (dopamine-azide and poly(dopaazide)) show signals from three nitrogen atoms building azide. The chemical shift values of individual nitrogen atoms are −308, −172, and −132 ppm and are assigned to (α), (γ), and (β) nitrogen atoms, respectively. The obtained 1H–15N HMBC spectra suggest that most structures in our material are built from repetitive monomers of dopamine azide linked by an aromatic C3–C6 single bond with open-azido chains, which is in accordance with the 1H, 13C and 2D COSY results. Additionally, in the 1H NMR spectra of poly(dopaazide) most of the resonances are grouped into ranges in which we would expect signals from dimeric/trimeric or longer forms (see Fig. 5A). Further profound analysis of 1H and 13C NMR chemical shift values and cross-peaks in 2D NMR spectra confirms that these signals belong to species containing aromatic rings connected with each other through C3/C6 carbons and they are located in regions that are characteristic for dimers/trimers and tetramers. However, the ranges in which the resonances are observed tend to be slightly broader than those expected from the presence of only such structural forms in the solution.
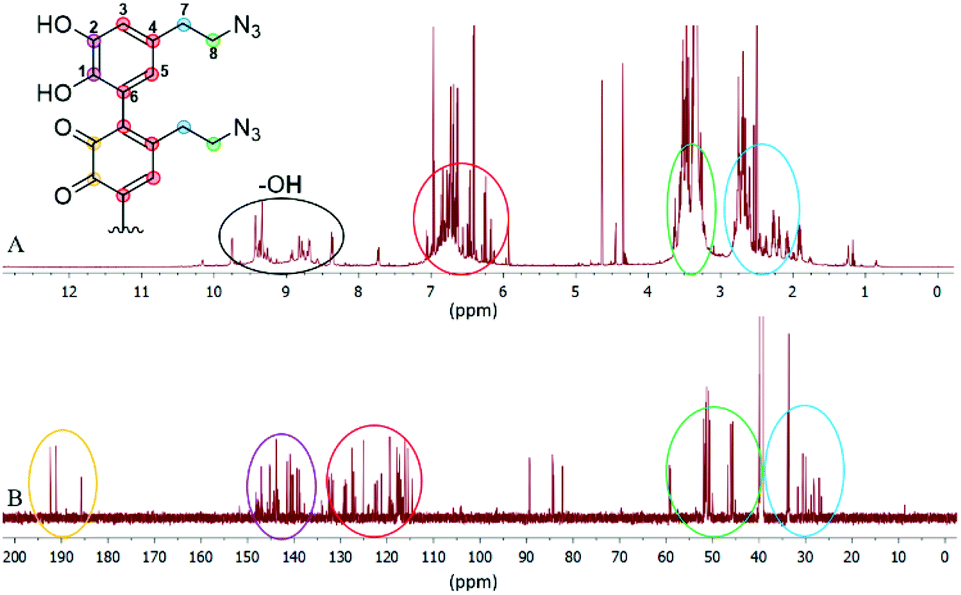 | ||
| Fig. 5 1H NMR (A) and 13C NMR (B) spectra of poly(dopaazide) in DMSO-d6. | ||
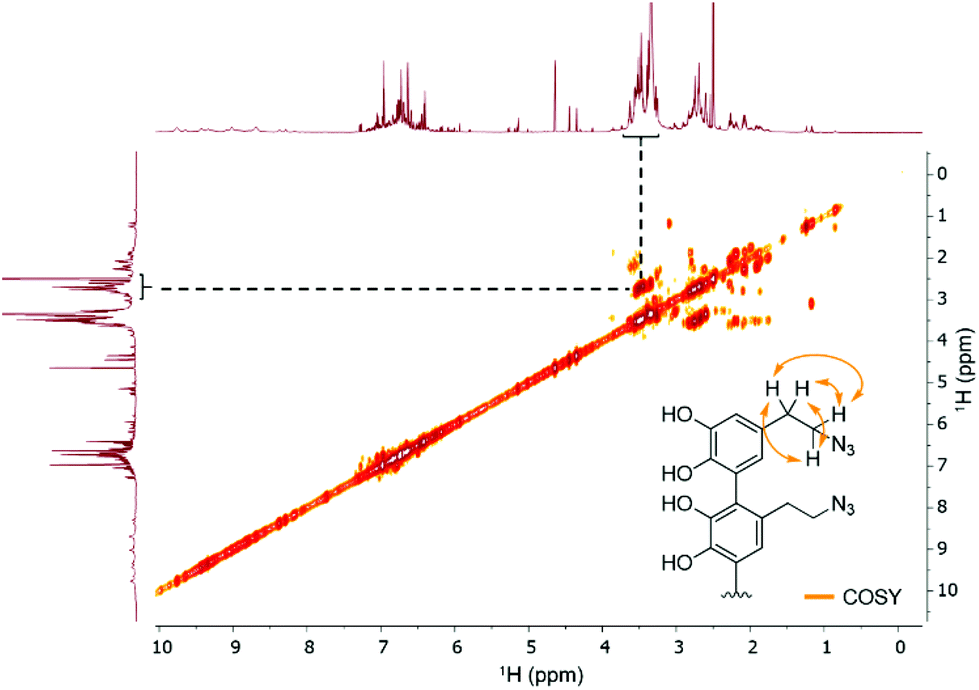 | ||
| Fig. 6 1H–1H COSY spectrum of poly(dopaazide) (DMSO-d6, 298 K). | ||
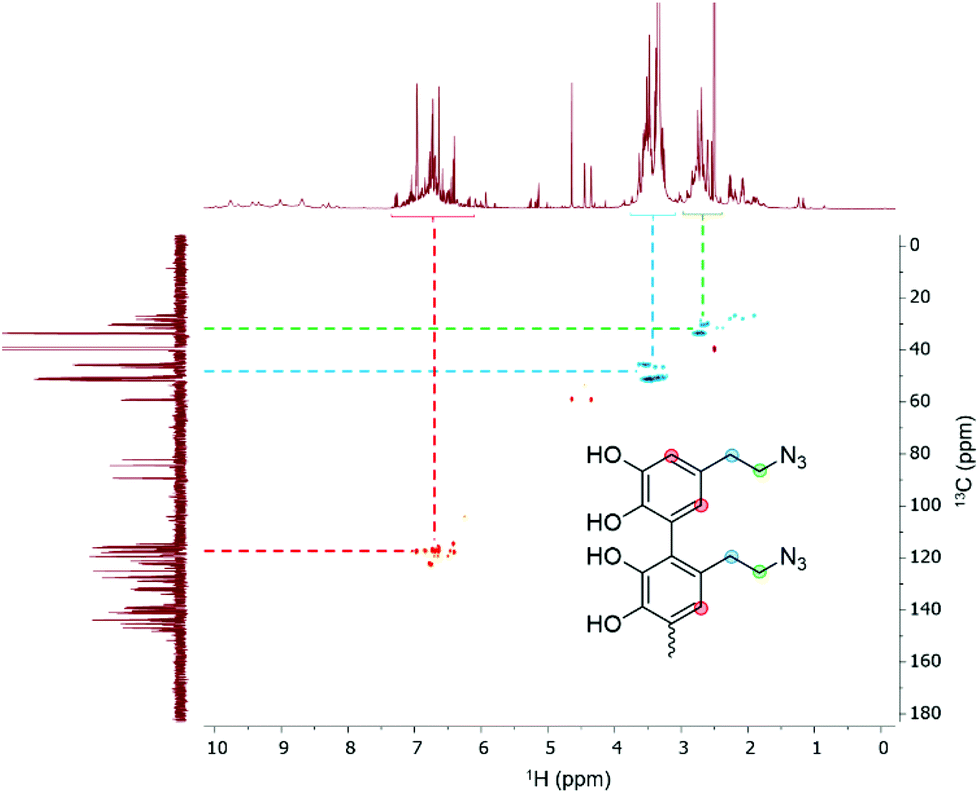 | ||
| Fig. 7 1H–13C HSQC spectrum of poly(dopaazide) (DMSO-d6, 298 K). | ||
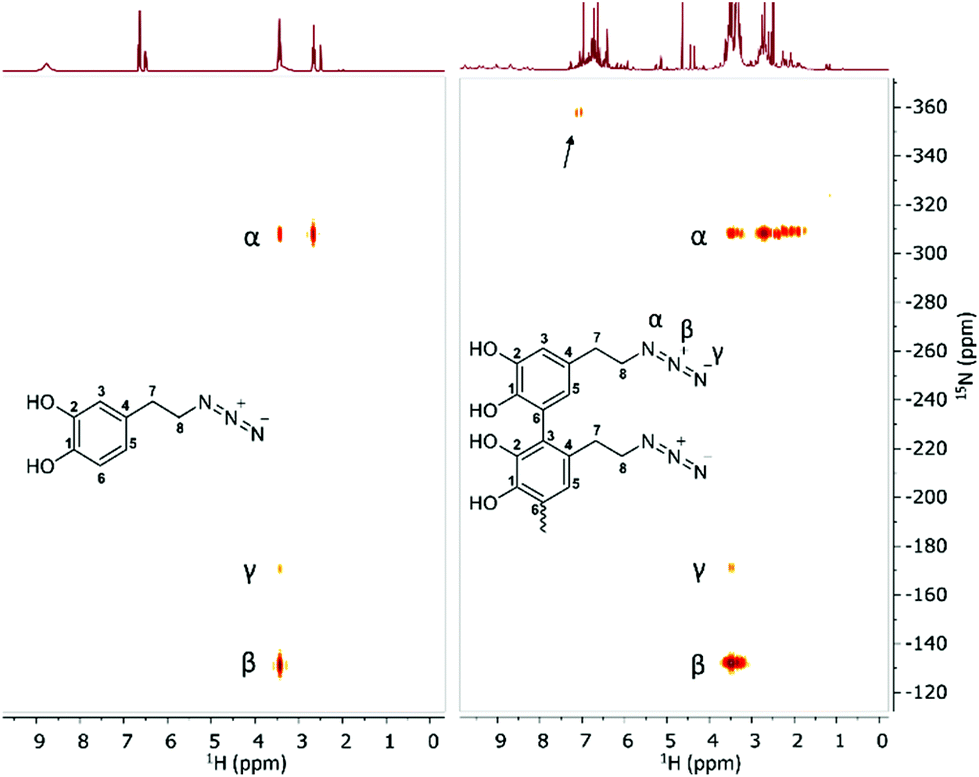 | ||
| Fig. 8 1H–15N HMBC spectrum of dopamine-azide (left) and poly(dopaazide) (right) (DMSO-d6, 298 K). The arrow indicates signals from additional species present in the sample. | ||
Indeed, the 1H–15N HMBC spectrum of poly(dopaazide) shows a nitrogen signal at −365 ppm. Moreover, in the 1H spectrum additional signals in a range of 4–5 ppm and in the 13C spectrum signals at 80–90 ppm and 190 ppm are observed, which suggest additional species. Because of the complexity of the poly(dopaazide) structure and relatively low concentration of this moiety in the sample, we could not unambiguously point out the structure of this group. We take into account the hypothesis that the additional nitrogen signal observed in the 1H–15N HMBC might come from the decomposition of azide (release of N2) during the polymerization process, leading to amine restoration and the formation of an indole-like structure built in the poly(dopaazide) structure. Also 1H and 13C experiments indicate that these forms are present to a small extent, and may contain non-aromatic rings with unsaturated bonds.
Since ssNMR is one of the most common techniques for investigating polymers’ structures, including polydopamine45 and materials obtained from the polymerization of L-dopa amides,46 we decided to use this technique to further investigate the structure of poly(dopaazide) after its suspension in ethanol. The 13C CP-MAS spectra of poly(dopaazide) were acquired with short (τCP = 100 μs) and long (τCP = 2 ms) 1H → 13C cross-polarization times, as shown in Fig. 9A. The former emphasizes protonated carbon sites, whereas in the latter the NMR lines of ipso carbons also show up with full intensity. Based on a comparison between them, spectral assignment is quite straightforward. The carbon atoms give rise to lines at 34.8 ppm (C7), 52.7 ppm (C8), 118.7 ppm (protonated C3 and C6), and 124 ppm (C4), respectively. The (C1) and (C2) carbons give rise to the 13C ss-NMR lines at 143.3 and 141 ppm, and to a broader peak centered on 131 ppm, most likely made up of severely overlapped individual NMR lines, corresponding to the C1 and the ipso C3/C6 carbon sites. The signals from C3/C6 carbons will occur in a linear 3–6 coupling scheme of the dopamine-azide monomers in the final poly(dopaazide) material.
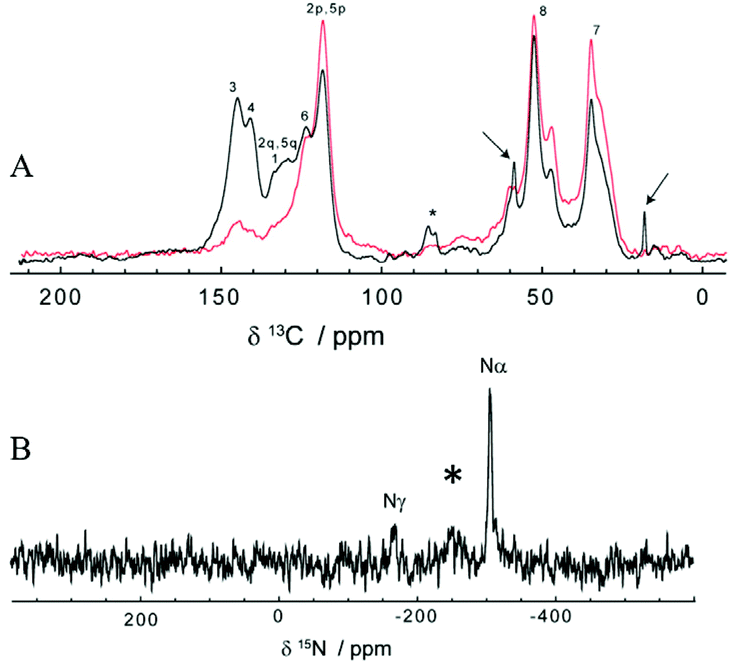 | ||
| Fig. 9 ss-NMR of poly(dopaazide). (A) Comparison between the 13C CP-MAS spectra of poly(dopaazide) recorded at short and long cross-polarization times of τCP = 100 μs (red) and τCP = 2 ms (black), respectively, (B) the 15N CP-MAS spectrum of poly(dopaazide), with assignment of the three distinct nitrogen atoms. | ||
In addition to the above discussed lines, the characteristic ethanol NMR lines, marked with arrows, are also seen in the 13C CP-MAS spectrum at 18.4 and 58.9 ppm (see Fig. 9A). This suggests that ethanol is not in a remnant liquid phase, due to incomplete drying, but its molecules are incorporated in some way in poly(dopaazide), most probably through non-covalent interactions. The existence of only weak interactions between ethanol molecules and poly(dopaazide) is supported by the following two facts: the ethanol ss-NMR lines are much narrower than those of poly(dopaazide) and they are obtained at a short cross-polarization time. Both arguments are consistent with the increased mobility of the ethanol molecules within the system. Finally, it is also worth mentioning that a small signal at about 86 ppm in the spectrum recorded at τCP = 2 ms indicates the presence of additional species, which is in agreement with peaks observed in liquid 13CNMR experiments.
Further qualitative structural analysis of poly(dopaazide) can be done from the spectra and a comparison with the results reported for polydopamine, PDA, a closely related analogue.11,45 A notable difference observed in the poly(dopaazide) 13C CP-MAS spectra in comparison with the PDA spectra is the missing NMR signal in the 100–110 ppm range, which means that cyclization of the C7/C8 carbons could not take place in poly(dopaazide), as observed for PDA. Also, the significantly narrower ss-NMR lines in the poly(dopaazide) spectra indicate less disorder in this material than in PDA. Moreover, poly(dopaazide) appears in terms of chemical disorder to be predominantly made from only one monomer type, dopamine-azide, and magnetic disorder.47 Assuming a linear 3–6 coupling of monomers, the intensity of the ipso C3/C6 carbons relative to the aromatic peak at 131 ppm, as well as to the intensities of the aliphatic C7 and C8 carbons, is indicative of short poly(dopaazide) oligomer sizes – most probably trimers. A trimer model is further supported by the fact that each aliphatic ss-NMR signal is actually split into one dominant component, which may come from the monomers at the edge, and a smaller shifted component, the contribution of the inner monomer (see Fig. 9A).
Additional structural details can be extracted from the 15N CP-MAS spectrum of poly(dopaazide), which is illustrated in Fig. 9B. Compared with the case of PDA,48 a much simpler spectrum is obtained here, with only three ss-NMR lines, of which the positions at −308 and −169 ppm agree well with the previous assignment to the distinct nitrogen sites in similar azide-containing systems,49 as well as the results from 15N liquid NMR of poly(dopaazide). Possibly due to the very poor S/N ratio, the line of the β nitrogen in the azide moiety, usually found at around −130 ppm in solution NMR spectra, is not identified here. Instead, a broad 15N ss-NMR line seems to occur at −250 ppm (marked with an asterisk in Fig. 9B) – for a detailed understanding of its origin, 15N isotopic labelling of the azide moiety would definitely be beneficial. Therefore the data from ssNMR experiments point toward the same conclusion as data from liquid NMR, that poly(dopaazide) mainly incorporates the basic dopamine-azide monomeric unit.
The solubility/degradation of poly(dopaazide) in organic solvents allowed us to use two techniques of mass spectrometry namely ESI-MS and MALDI-MS, which are frequently used to determine the structures of various polymers, including polycatechol-based materials.11,50–52 The ESI-MS spectra of poly(dopaazide) show many peaks with slightly different masses, which suggest fragmentation under the ionization conditions and make the determination of individual masses difficult. The proposed structures of interesting peaks with different degrees of saturation are presented in the ESI in Table S2.† Here we present examples of peaks and corresponding structures that unambiguously confirm the linear structure of poly(dopazazide) and prove open azidoethyl moieties in the polymer (see Fig. 10). The results obtained from ESI-MS are compatible with those obtained from ss and liquid NMR. Moreover, the obtained masses point to the incorporation of iodine and the release of nitrogen from the azide groups (see the data from MALDI-MS in the ESI in Table S2†).
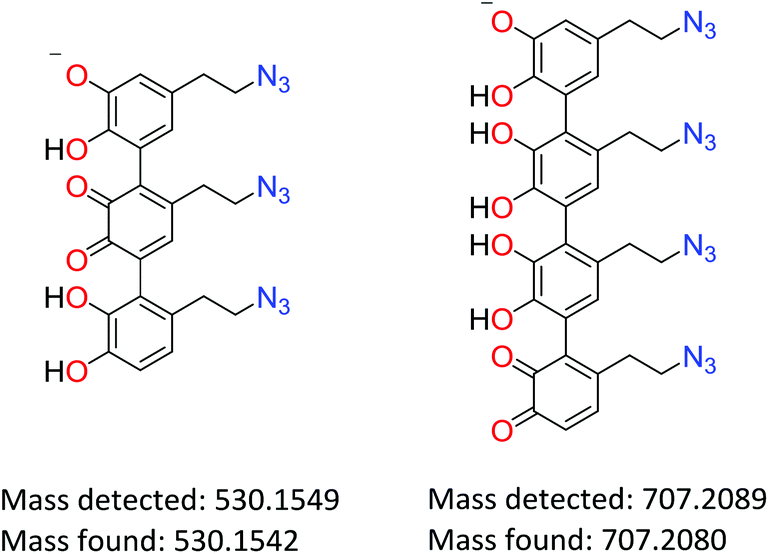 | ||
| Fig. 10 Proposed structures and masses of poly(dopaazide) building units. | ||
3.2 Influence of catchol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :oxidant ratio on the morphology of poly(dopaazide) and polydopamine particles and resulting films
:oxidant ratio on the morphology of poly(dopaazide) and polydopamine particles and resulting films
Ball and d'Ischia have shown that the ratio between NaIO4 and dopamine influences the deposition rate of PDA films and has an impact on the thickness and homogeneity of the resulting polymer.53 Therefore, we decided to vary the ratio between the dopamine azide and sodium periodate between 0.1 to 2 eq. to investigate impact of that ratio on particle morphology and the obtained coating (see the ESI Fig. S6†).
In the first experiment, we polymerized the azide derivative of dopamine with different amounts of NaIO4 and the resulting particles were collected after 24 h. The morphology of the synthesized particles was determined using SEM. In all cases particles were spherical but their sizes were slightly increasing with the amount of oxidant used. Moreover, the polydispersity of the sample was increasing. In the case of the polymerization of dopamine under the same conditions, we did not observe the formation of particles but rather the apparition of an unshaped polymer. Well-structured PDA particles were observed only when 0.5 eq. of oxidant was used (see the ESI Fig. S7†).
Further, we compared the character of poly(dopaazide) and PDA films after their deposition on glass, ceramics and silicon. The coated substrates were examined by measuring their water contact angle (CA) The results are provided in Table S1 in the ESI.† Due to the “spreading” of droplets over time, measurements were taken immediately after applying the droplet, and 1 minute and 10 minutes after droplet application. On account of the evaporation over time, there is a gradual reduction in the droplet volume, resulting in some additional reduction in the CA value. In each case, a strong change in the contact angle with time can be observed – the initial value most likely depends on the surface topography. This can be explained by a reaction with the substrate or water penetration into the layer, and probably some small contribution is also made by water evaporation. The main conclusions that have been drawn from these measurements are: the strongest CA temporal changes appeared in the case of the Si substrate samples; poly(dopaazide)/glass after 48 h of coating is more hydrophobic than poly(dopaazide)/glass after 24 h; and while PDA/glass after 48 h is more hydrophobic than PDA/glass after 24 h in the initial phase, over time the CA values converge. Then the morphology of coated surfaces was investigated by means of AFM (atomic force microscopy) and the corresponding AFM results are presented in the ESI (see the ESI Fig. S8†). Generally, samples coated with poly(dopaazide) are much rougher than those coated with PDA. The surface roughness of PDA seems to correlate with the contact angle values. In the case of poly(dopaazide), samples are coated with a homogeneous layer of material and form clusters. For poly(dopaazide)/glass after 48 h, these clusters agglomerate a bit more, and the highest cluster packing distinguishes poly(dopaazide)/Si. In the case of PDA/glass and PDA/Si, the 5 × 5 μm images show almost nothing due to the slow polymerization rate and hence very small particle sizes. The PDA/Si layer shows the most agglomerated particles compared with PDA/glass. The images of PDA show additional contrast on the surface, which might result from the existence of two different phases on the surface. Phase shift images confirm that, however, more measurements are required to actually confirm that and get more information on the origin of this effect.
3.3 DFT calculations
Information about the structures of the monomers allows a better understanding of the formation of the polymer due to the geometric predisposition of the substrates in the polymerization reaction as well as the steric repulsions arising in the oligomers. In our calculation we took into account dopamine azide both in the catecholic form and after oxidation to quinone. Detailed structures of both monomers used for comparison with the results of NMR analysis were obtained by molecular modelling using the Gaussian suite of programs.54 Initially calculations were performed using Parameterization Method 6 (PM6), then the calculated lowest energy conformers were subsequently refined using the DFT (B3LYP/631**) method. The results of structural calculations for dopamine azide in catecholic form show that this molecule adopts two conformations with energies of negligible difference. The aromatic ring is planar and the side chain adopts the expected trans conformation (see Fig. 11A). The torsion angle between the aromatic ring plane and side-chain is close to perpendicular to minimize steric repulsion between the ortho- and benzylic hydrogen atoms. The change in torsion angle between atoms C3–C4–C7–C8 from trans to gauche increases the energy of a molecule less than 0.2 kcal mol−1, suggesting almost unrestricted rotation of a side chain under the conditions of the polymerization process. In the lowest energy conformers, two neighbouring hydroxy groups attached to aromatic ring form intramolecular hydrogen bond-type interactions with the distance between the hydrogen from one hydroxy group to the next oxygen atom being 2.16 Å. Two different arrangements of this interaction originate from two forms: in one the oxygen attached to carbon 1 is a proton donor, in the other it is the oxygen attached to carbon 2.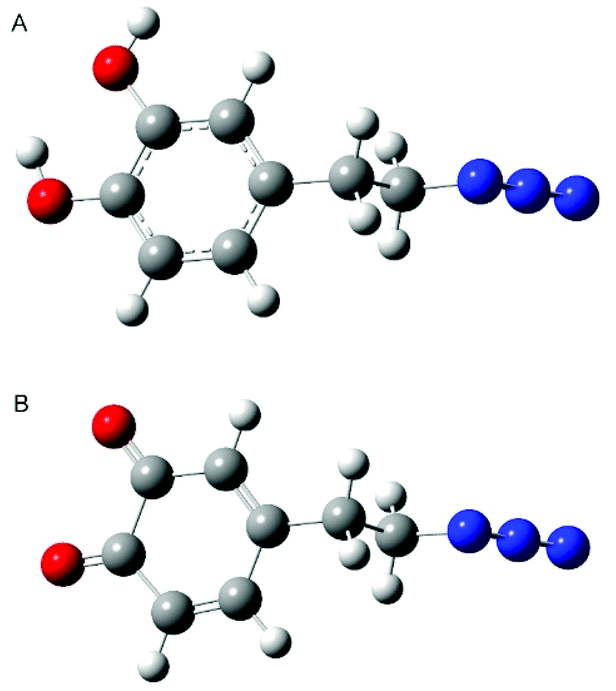 | ||
| Fig. 11 Calculated lowest energy conformers of dopamine azide (A) and dopamine azide quinone (B). Red colour, oxygen atom; blue colour, nitrogen; dark grey, carbon; light grey, hydrogen. | ||
Calculations for compound dopamine azide in the quinone form indicate the presence of only one main conformer. The oxidation of 1,2 diols caused the formation of a 1,2 diketone unit that cannot form intramolecular hydrogen bonds as its reduced form does. Oxidation of the hydroxy groups causes the ring to lose its aromaticity, but the planar structure of the ring is preserved (see Fig. 11B). The side chain of the oxidized dopamine azide also in this molecule adopts a trans conformation with a perpendicular arrangement to the ring plane.
In both molecules the negative partial charge is located on the oxygen atoms (−0.545 and −0.607 for dopamine azide and −0.406 and −0.412 for quinone dopamine azide). This suggests these atoms may interact with cations and a protic solvent such as water or methanol. The protic solvent may also interact with two molecules of dopamine azide, quinone dopamine azide or dopamine azide with quinone dopamine azide by the formation of hydrogen bonds, allowing the preorganization of substrates in the polymerization process. In the case of a dopamine azide-based polymer (or other catecholic polymers), the loss of protic solvent may cause a change in the torsion angle between two adjacent aromatic rings due to the repulsion of negatively charged carbonyl oxygen atoms, which can influence the local conformation of the polymer backbone.
4. Conclusions
In this work, we demonstrated oxidative polymerization of the 4-(2-azidoethyl)benzene-1,2-diol-azido analogue of dopamine triggered by sodium periodate as an oxidant. The obtained material resulted in spherical and smooth particles, which was confirmed by SEM/TEM analysis. The particle size can be controlled by the amount of oxidant used in the reaction, while under the same experimental conditions polydopamine formed shapeless structures. It is worth highlighting that the synthesized poly(dopaazide) polymer solubility in organic solvents made possible deep structural analysis of the polymer using liquid-state NMR. Furthermore, the performed ESI-MS, liquid and ssNMR spectroscopy, as well as the XPS and FTIR experiments, revealed that the polymer is linear and contains open ethylazido chains in the structure. In addition, the mass spectrometry experiments confirmed the presence of trimers, dimers and tetrameters building the polymer. The solubility of the polymer in organic solvents is probably caused by the lack of cross-linkage between building blocks that occurs for PDA where a free amino group is present. Thus, the replacement of the amine group by azide indicates the crucial role of NH2 moiety in tailoring properties of polycatchol materials and makes our results an important step forward in understanding the relationship between the structure of the starting catechol and the resulting polycatechol materials, opening a new path in designing polycatchol macromolecules.Author contributions
Conceptualization M. S., R. M.; formal analysis M. S., Ł. P., M. K., E. C., C. F., R. M.; funding acquisition R. M., C. F.; investigation M. S., Ł. P., M. K., Y. K., C. F., J. G., R. M.; methodology M. S., R. M., C. F., J. G.; project administration R. M.; resources R. M, C. F., J. G.; supervision R. M.; validation M. S., Ł. P., R. M., M. K., C. F.; writing – original draft M. S., R. M., C. F., Ł. P., J. G.; writing – review & editing M. S., R. M., C. F., Ł. P., J. G., E. C., M. K., Y. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support under research grant number UMO-2018/31/B/ST8/02460 from the National Science Centre is kindly acknowledged. C. F. acknowledges financial support from UEFISCDI through the project PN-III-P4-ID-PCE-2020-1463. The authors thank Zofia Gdaniec (Department of Biomolecular NMR, Institute of Bioorganic Chemistry, Polish Academy of Sciences) for giving us the opportunity to carry out NMR measurements on a Bruker Avance III 700 spectrometer. The calculations were performed at the Poznań Supercomputing and Networking Center.Notes and references
- M. L. Tran, B. J. Powell and P. Meredith, Biophys. J., 2006, 90, 743–752 CrossRef CAS PubMed.
- W. Cao, X. Zhou, N. C. Mccallum, Z. Hu, Q. Z. Ni, U. Kapoor, C. M. Heil, K. S. Cay, T. Zand, A. J. Mantanona, A. Jayaraman, A. Dhinojwala, D. D. Deheyn, M. D. Shawkey, M. D. Burkart, D. Rinehart and N. C. Gianneschi, J. Am. Chem. Soc., 2021, 143, 2622–2637 CrossRef CAS PubMed.
- E. Kaxiras, A. Tsolakidis, G. Zonios and S. Meng, Phys. Rev. Lett., 2006, 97, 218102 CrossRef PubMed.
- R. Micillo, L. Panzella, M. Iacomino, G. Prampolini, I. Cacelli, A. Ferretti, O. Crescenzi, K. Koike, A. Napolitano and M. d'Ischia, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- D. Aguilar-Ferrer, J. Szewczyk and E. Coy, Catal. Today DOI:10.1016/j.cattod.2021.08.016.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- S. Hong, Y. Wang, S. Y. Park and H. Lee, Sci. Adv., 2018, 4, 1–11 Search PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- Q. Lyu, N. Hsueh and C. L. L. Chai, Polym. Chem., 2019, 10, 5771–5777 RSC.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- J. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Haìdade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed.
- R. Mrówczyński, L. Magerusan, R. Turcu and J. Liebscher, Polym. Chem., 2014, 4, 6593–6599 RSC.
- R. Mrówczyński, R. Markiewicz and J. Liebscher, Polym. Int., 2016, 65, 1288–1299 CrossRef.
- H. A. Lee, E. Park and H. Lee, Adv. Mater., 2020, 32, 1–20 Search PubMed.
- J. Liebscher, Eur. J. Org. Chem., 2019, 1–20 Search PubMed.
- Y. Song, G. Ye, F. Wu, Z. Wang, S. Liu, M. Kopec, Z. Wang, J. Chen, J. Wang and K. Matyjaszewski, Chem. Mater., 2016, 28, 5013–5021 CrossRef CAS.
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed.
- W. Z. Qiu, G. P. Wu and Z. K. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 5902–5908 CrossRef CAS.
- S. N. Ramanan, N. Shahkaramipour, T. Tran, L. Zhu, S. R. Venna, C. K. Lim, A. Singh, P. N. Prasad and H. Lin, J. Membr. Sci., 2018, 554, 164–174 CrossRef CAS.
- B. D. McCloskey, H. B. Park, H. Ju, B. W. Rowe, D. J. Miller, B. J. Chun, K. Kin and B. D. Freeman, Polymer, 2010, 51, 3472–3485 CrossRef CAS.
- P. Xue, L. Sun, Q. Li, L. Zhang, J. Guo and Z. Xu, Colloids Surf., B, 2017, 160, 11–21 CrossRef CAS PubMed.
- P. Cao, W. W. Li, A. R. Morris, P. D. Horrocks, C. Q. Yuan and Y. Yang, R. Soc. Open Sci., 2018, 5, 172165 CrossRef PubMed.
- H. Veisi, S. Vafajoo, K. Bahrami and B. Mozafari, Catal. Lett., 2018, 2734–2745 CrossRef CAS.
- M. Iacomino, J. Mancebo-Aracil, M. Guardingo, R. Martín, G. D'Errico, M. Perfetti, P. Manini, O. Crescenzi, F. Busqué, A. Napolitano, M. d'Ischia, J. Sedó and D. Ruiz-Molina, Int. J. Mol. Sci., 2017, 18, 1–16 Search PubMed.
- J. Hong, D. G. Jwa, H. Ha, J. Kwak, M. Kim and S. M. Kang, Langmuir, 2019, 35, 6898–6904 CrossRef CAS PubMed.
- A. Petran, N. D. Hădade, C. Filip, X. Filip, A. Bende, A. Popa and J. Liebscher, Macromol. Chem. Phys., 2018, 219, 1–10 CrossRef.
- J. Cui, J. Iturri, J. Paez, Z. Shafiq, C. Serrano, M. d'Ischia and A. Del Campo, Macromol. Chem. Phys., 2014, 215, 2403–2413 CrossRef CAS.
- S. Pagoti, D. Dutta and J. Dash, Adv. Synth. Catal., 2013, 355, 3532–3538 CrossRef CAS.
- F. Ponzio, J. Barthès, J. Bour, M. Michel, P. Bertani, J. Hemmerlé, M. d'Ischia and V. Ball, Chem. Mater., 2016, 28, 4697–4705 CrossRef CAS.
- M. Bisaglia, S. Mammi and L. Bubacco, J. Biol. Chem., 2007, 282, 15597–15605 CrossRef CAS PubMed.
- X. Han, F. Tang and Z. Jin, RSC Adv., 2018, 8, 18347–18354 RSC.
- E. T. Kaiser and S. W. Weidman, J. Am. Chem. Soc., 1966, 88, 5820–5827 CrossRef.
- J. Larsson, M. Hill and A. Duffy, Annu. Trans. – Nord. Rheol. Soc., 2012, 20, 1–35 Search PubMed.
- R. J. J. Jansen and H. van Bekkum, Carbon, 1995, 33, 1021–1027 CrossRef CAS.
- G. Zorn and L. Liu, J. Phys. Chem. C, 2014, 118, 376–383 CrossRef CAS PubMed.
- A. C. Gouget-Laemmel, J. Yang, M. A. Lodhi, A. Siriwardena, D. Aureau, R. Boukherroub, J. N. Chazalviel, F. Ozanam and S. Szunerits, J. Phys. Chem. C, 2013, 117, 368–375 CrossRef CAS.
- E. D. Stenehjem, V. R. Ziatdinov, T. D. P. Stack and C. E. D. Chidsey, J. Am. Chem. Soc., 2013, 135, 1110–1116 CrossRef CAS PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- E. Coy, I. Iatsunskyi, J. C. Colmenares, Y. Kim and R. Mrówczyński, ACS Appl. Mater. Interfaces, 2021, 13, 23113–23120 CrossRef CAS PubMed.
- M. M. Chehimi and M. Delamar, J. Electron Spectrosc. Relat. Phenom., 1990, 50, 25–32 CrossRef.
- B. F. Grześkowiak, D. Maziukiewicz, A. Kozłowska, A. Kertmen, E. Coy and R. Mrówczyński, Int. J. Mol. Sci., 2021, 22, 1–16 Search PubMed.
- K. B. Yatsimirskii, V. V. Nemoskalenko, V. G. Aleshin, Y. I. Bratushko and E. P. Moiseenko, Chem. Phys. Lett., 1977, 52, 481–484 CrossRef CAS.
- S. Xie, M. Sundhoro, K. N. Houk and M. Yan, Acc. Chem. Res., 2020, 53, 937–948 CrossRef CAS PubMed.
- K. G. Malollari, P. Delparastan, C. Sobek, J. Vachhani, T. D. Fink, R. H. Zha and P. B. Messersmith, ACS Appl. Mater. Interfaces, 2019, 11, 43599–43607 CrossRef CAS PubMed.
- M. Cîrcu, C. Filip and C. F. Monica Cîrcu, Polym. Chem., 2018, 9, 3379–3387 RSC.
- A. Petran, R. Mrówczyński, C. Filip, R. Turcu and J. Liebscher, Polym. Chem., 2015, 6, 2139–2149 RSC.
- R. Mrówczyński, L. E. Coy, B. Scheibe, T. Czechowski, M. Augustyniak-Jabłokow, S. Jurga and K. Tadyszak, J. Phys. Chem. B, 2015, 119, 10341–10347 CrossRef PubMed.
- V. Proks, J. Brus, O. Pop-Georgievski, E. Večerníková, W. Wisniewski, J. Kotek, M. Urbanová, F. Rypáček, E. Vec, W. Wisniewski and M. Urbanová, Macromol. J., 2013, 214, 499–507 CAS.
- F. Joubert, O. Musa, D. R. W. Hodgson and N. R. Cameron, Polym. Chem., 2015, 6, 1567–1575 RSC.
- Y. K. Kim, R. Landis, R. W. Vachet and V. M. Rotello, ACS Appl. Mater. Interfaces, 2018, 10, 36361–36368 CrossRef CAS PubMed.
- Q. Lyu, N. Hsueh and C. L. L. Chai, Polym. Chem., 2019, 10, 5771–5777 RSC.
- J. Yang, W. Zhang, H. Zhang, M. Zhong, W. Cao, Z. Li, X. Huang, Z. Nie, J. Liu, P. Li, X. Ma and Z. Ouyang, ACS Appl. Mater. Interfaces, 2019, 11, 46140–46148 CrossRef CAS PubMed.
- F. Ponzio, J. Barthès, J. Bour, M. Michel, P. Bertani, J. Hemmerlé, M. d'Ischia and V. Ball, Chem. Mater., 2016, 28, 4697–4705 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian and J. V. Ort, Gaussian, Inc., Wallingford CT.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00293k |
| This journal is © The Royal Society of Chemistry 2022 |