
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Free-radical polymerization of 2-hydroxyethyl methacrylate (HEMA) supported by a high electric field†
Wenkang
Tu
abc,
Paulina
Maksym
 abd,
Kamil
Kaminski
ab,
Katarzyna
Chat
*ab and
Karolina
Adrjanowicz
*ab
abd,
Kamil
Kaminski
ab,
Katarzyna
Chat
*ab and
Karolina
Adrjanowicz
*ab
aInstitute of Physics, University of Silesia, 75 Pulku Piechoty 1, 41-500 Chorzow, Poland. E-mail: kadrjano@us.edu.pl
bSilesian Center for Education and Interdisciplinary Research (SMCEBI), 75 Pulku Piechoty 1a, 41-500 Chorzow, Poland
cCollege of Mechatronics and Control Engineering, Shenzhen University, Shenzhen 518060, China
dInstitute of Materials Engineering, University of Silesia, 75 Pulku Piechoty 1a, 41-500 Chorzow, Poland
First published on 25th April 2022
Abstract
In macromolecular science, tuning basic polymer parameters like molecular weight (Mn) or molecular weight distribution (dispersity, Đ) is an active research topic. Many prominent synthetic protocols concerning the chemical modification of a polymerization mixture (adding additional reagents) and equipment modification have been adopted for this purpose. On the other hand, less attention has been paid to studying the impact of external stimuli such as pressure, light, and spatial restrictions on the properties of resulting polymers. Here, we present a robust synthetic protocol in which a high electric field (an external factor) supports the thermally-induced free-radical polymerization (FRP) of 2-hydroxyethyl methacrylate (HEMA). The reactions were conducted with 0.1 wt% of 2′-azobisisobutyronitrile (AIBN) in the presence of high dc (direct current) electric fields with various magnitudes ranging from 0 kV cm−1 to 140 kV cm−1. By combining dielectric spectroscopy, nuclear magnetic resonance spectroscopy, exclusion chromatography, and differential scanning calorimetry, we have explored the effect of an external electric field on the progress of polymerization (via dielectric spectroscopy) and product characteristics. We found that HEMA FRP supported by a high dc voltage results in charged macromolecules (polyelectrolytes) with high conductivity ∼ 10−10 S cm−1 at a glass transition temperature, markedly reduced molecular weight of Mn ≈41–58 kg mol−1 (E > 14 kV cm−1), and low/moderate Đ ∼ 1.1–1.3, which is an unexpected finding for non-controlled free radical polymerization. In contrast, the polymer produced in the absence of an electric field was characterized by Mn ≈ 103 kg mol−1 and much higher Đ = 1.73. Therefore, we found that the electric field can be another efficient external factor such as a spatial restriction or compression that allows for fine-tuning of polymer properties.
Introduction
Nowadays, polymer chemists make great efforts to fine-tune the molecular weight (Mn) and molecular weight distribution (dispersity, Đ) of macromolecules as the key parameters that have a significant impact on the thermal rheological and morphological properties of the polymer materials.1,2 At this point, it can be noted that over the last decades, the major focus was on the development of controlled polymerization processes (CRPs), allowing polymers with the lowest possible Đ to be obtained. What is more, these methods provided a unique opportunity to produce macromolecules of well-designed compositions, topologies, and micro- and macro-structures.3,4 However, over time, it has been noticed that polymers with broad Đ are equally attractive materials with wide industrial applicability, e.g., in 3D printing technology.5 Notably, the classical free radical polymerization process (FRP) is the best for producing polymers with broad Đ; however, it is rather dedicated to synthesizing macromolecules with very high Mn.Thus, it is extremely important to search for novel solutions and efficient procedures to tune a polymer's Mn and Đ for special needs. In this context, the most commonly implemented strategies that should be mentioned include the blending of polymer batches of known Mn or Đ,6,7 some equipment solutions (cascade reactors,8 flow reactors9 or innovative computer-controlled flow reactors10), and recent prominent works concerning CRPs, where Đ can be easily tuned by varying catalyst concentrations,11–13 and mixing different initiators14 or chain transfer agents.7,15,16 It is also worth paying attention to temporally regulating chain initiation in a controlled nitroxide-mediated polymerization (NMP) as proposed by Fors et al. This method allows for precisely controlling Mn and Đ through modulation of the initiator addition rate and time.17
Nevertheless, one should not forget about equally effective synthetic protocols utilizing external factors such as light,18 compression,19,20 ultrasonic waves,21,22 spatial restriction, friction, magnetic field,23 and finally. electric field.24 Interestingly, two of these,i.e., compression or 2D hard confinement, were successfully applied to FRP and CRP, yielding polymers (or polymer-based materials) with complex architectures25,26 and superior macroscopic performances.27–29 Unfortunately, considerably less attention has been paid to implementing the electric field in the polymerization process. It seems quite surprising since the electric field is a clean, inexpensive, and straightforward physical stimulus. It provides a unique opportunity to change the field direction, intensity, and frequency at any time of the reaction. This aspect appears to be particularly interesting for polar macromolecules that can become polarized with the subsequent alignment of the molecules under the electric field. In this context, it is worth emphasizing that the strong impact of the electric field was well demonstrated in the case of the phase transformation, self-ordering,24 or self-assembling30,31 of different materials (including polymeric ones). Here, it is worth mentioning the interesting work by Cai et al. on the reversible transformation between the two-dimensional covalent organic frameworks (2D COFs) and the self-assembled molecular networks (SAMNs) of 1,3,5-tris(4-biphenyl boronic acid)benzene (TBPBA) on a graphite surface at room temperature underneath the tip of a scanning tunneling microscope (STM). The direction of the electric field generated between the STM tip and the graphite can be changed to locally control the on-surface polymerization (of SAMNs)/depolymerization (of COFs) processes.32 In turn, in the work of McFarland et al., macroscopic P3HT (poly(3-hexylthiophene)) fibers (>6 cm) were assembled by employing P3HT nanowhiskers as fundamental building units under an external dc electric field. Moreover, the assembled P3HT fibers will remain in the macroscopic form even when the electric field is switched off.33 What is more, as far as we are aware, there is only one paper by Chen et al., where chitosan, polyacrylic acid, and montmorillonite were polymerized in the presence/absence of a relatively low electric field E ≤ 15 kV cm−1. As a result, two kinds of elastomers with similar chemical compositions but different microstructures and electro-responses were obtained.34
Inspired by this work and highly motivated to check how a very high electric field could influence the polymerization pathway and Mw, and Đ of the produced polymers, we proposed and developed the bulk thermally-induced FRP protocol supported by a high external electric field of different magnitudes (up to 140 kV cm−1). Here, we focused on the FRP of 2-hydroxyethyl methacrylate (HEMA). Our choice was dictated by the fact that HEMA is an important hydrogel material (manufactured primarily on soft contact lenses).35 On the other hand, it is a pretty demanding and challenging monomer for classical batch FRP (due to the highly reactive –OH group), which generally ran with poor control and a high number of side reactions,36–38e.g., the Trommsdorf–Norrish effect. In contrast, externally-regulated HEMA FRP supported by high-pressure,39 ultra-waves,40 or UV light irradiation41 turned out to be more controlled processes reaching higher rates, HEMA consumptions and providing poly(2-(hydroxyethyl)methacrylate)s (PHEMA)s of much lower Đ. Thus, the above physical factors have become an alternative to the commonly known chemical method using polymerization of the trimethylsilyl protected monomer.42
Herein, the presented protocol allowed us to tune the Mn and Đ of PHEMA just by simple modulation in the electric field magnitude. Our specific interest was also in the detailed characteristics of PHEMAs via nuclear magnetic resonance spectroscopy (NMR), size exclusion chromatography-low-angle laser-light scattering (SEC-LALLS), differential scanning calorimetry (DSC), and broadband dielectric spectroscopy (BDS). We found that by implementing the high electric field to HEMA FRP, the charged polymers of much lower Mn and low Đ, unexpected for classical FRP processes, are produced (Mn = 41.0–58.0 kg mol−1, Đ = 1.07–1.26, dc conductivity σdc ∼ 10−10 at Tg). In contrast, the FRP of HEMA at E = 0 kV cm−1 yielded PHEMA of very high Mn ∼ 1.0 × 104 kg mol−1 and high Đ = 1.73. We envisage that our robust, environmentally friendly, and, more importantly, very simple (only a two-component polymerizing mixture) protocol could be a novel method for fine-tuning Mn and Đ of macromolecules.
Experimental section
Materials
2-(Hydroxyethyl)methacrylate (HEMA, ≥99%), (AIBN, 98%), methanol (CDA, Chempur), chloroform-d (CDCl3, 99.8 atom% D), and dimethyl sulfoxide (DMSO, ≥99.5%, for HPLC), were purchased from Sigma Aldrich. HEMA was purified by passing it over a short column of activated basic alumina (Sigma-Aldrich) to remove the inhibitor and stored at −27 °C. Other reagents were used as received.Synthetic procedures
The polymers were prepared by free-radical polymerization of a reaction mixture composed of HEMA and 0.1 wt% AIBN, which is a low initiator concentration for the classical FRP process. The reaction mixture was prepared in a Schlenk flask and degassed by three freeze–pump cycles. The polymerization mechanism involves an interplay between initiation, propagation, and termination reactions, for which more information can be found elsewhere.37 Polymerization experiments in the presence and absence of high static e-fields were carried out under isothermal conditions (T = 343 K) for a period of 3 hours and 20 min with a reaction mixture held in-between a sample cell mounted inside the dielectric apparatus used in the present study. A sketch of the polymerization experiment is presented in Scheme 1. The sample cell consists of two highly polished stainless-steel electrodes (20 mm diameter) separated by a Teflon ring of 25 μm thickness. The distance between the electrodes was fixed. High-electric field experiments require very flat and polished electrode surfaces to maintain homogeneous fields. Such conditions ensure that the field lines are parallel to each other. So in this way, there is a good description and reproducibility of the results.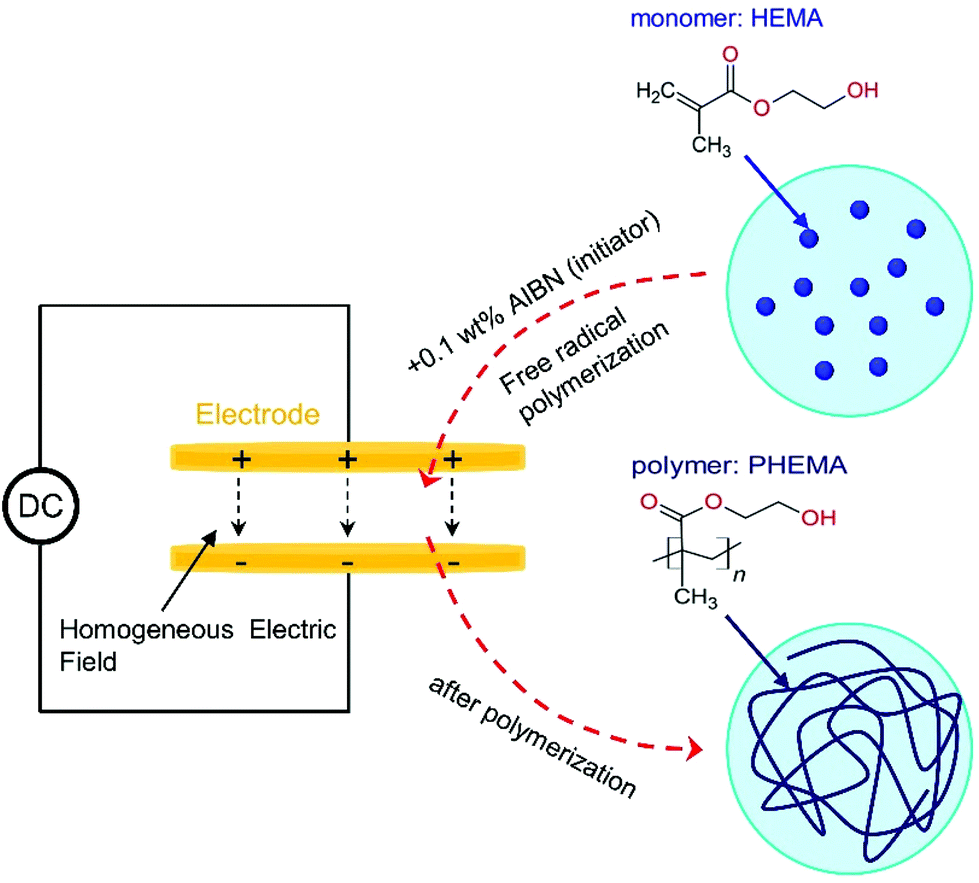 | ||
| Scheme 1 Sketch of the polymerization experiment using HEMA with a 0.1 wt% AIBN initiator carried out in-between a dielectric sample cell in the absence/presence of a high static electric field. The chemical structures of the HEMA monomer and PHEMA polymer are also shown. | ||
In each experiment, the tested polymerizable mixture was loaded into the sample cell at room temperature and then heated to T = 343 K, where the reaction can be completed within t = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s. After each polymerization experiment, the reaction mixtures were removed from the electrodes by dissolving in DMSO-d6. After performing NMR analyses, DMSO was evaporated from each sample, and recovered samples were precipitated in cold ethyl ether. Finally, the produced PHEMAs were dried to a constant mass.
000 s. After each polymerization experiment, the reaction mixtures were removed from the electrodes by dissolving in DMSO-d6. After performing NMR analyses, DMSO was evaporated from each sample, and recovered samples were precipitated in cold ethyl ether. Finally, the produced PHEMAs were dried to a constant mass.
BDS measurements
The measurements were performed using a frequency-response analyzer (Alpha Analyzer) in combination with a high-voltage booster “HVB1000”, both obtained from Novocontrol (Germany). The high voltage extension boosts the voltages of the Alpha Analyzer up to 500 V (peak voltage). In this configuration, we can measure frequencies from mHz up to 10 kHz range. A high-voltage unit combined with the dielectric spectrometer allows us not only to apply a high electric field (ac or dc) to the sample but also to measure its dielectric response at the same time. This is a very useful feature because the ongoing changes in the samples due to the presence of a high electric field can be followed. Therefore, together with the high amplitude dc electric field, a low amplitude sinusoidal field (E < 1 kV cm−1) was also applied. In this way, we were able to monitor the experiment of the isothermal polymerization progress of the tested system in real-time by following the evolutions of the real (ε′) and imaginary (ε′′) parts of complex permittivity, ε* = ε′ − iε′′ in the presence/absence of the high electric field. We have also used a dielectric spectrometer to characterize molecular dynamics and conductivity behavior in the glassy and supercooled liquid states of two PHEMA samples obtained under electric dc fields of E = 0 kV cm−1 and E = 60 kV cm−1. Before that, the polymers were purified from the residual impurities and monomer. In both cases, sample cells of the same kind were used, but with different electrode diameters, viz., 20 mm (polymerization) and 5 mm (dynamics). In all experiments, a Novocontrol Quatro system was used to control the temperature of the sample cell, which ensures the temperature stability to be within 0.1 K. In the present work, dielectric spectra for isothermal polymerization measurements were recorded within a frequency range of 0.7 Hz to 104 Hz, and those for non-isothermal measurements were within 0.09 Hz to 106 Hz. Please note that dielectric spectroscopy is an impedance technique, which means that the impedance and capacitance properties of the investigated sample are followed as a function of the reaction time. As these are closely related quantities, they provide the same set of data about the polymerization progress.NMR measurements
Proton nuclear magnetic resonance (1H NMR) spectra were recorded using a Bruker Ascend 600 spectrometer operating at 600 MHz in DMSO-d6 as a solvent. Standard experimental conditions and a standard Bruker program were used. The HEMA conversions were calculated by integrating the vinyl protons at 6.1 ppm (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, monomer) and 5.6 ppm (CH2C, monomer) against the methylene protons at 3.9 ppm (–CH2–O–CO, polymer). The structures of PHEMAs were also confirmed by 1H NMR (see the ESI, Fig. S1†).
C, monomer) and 5.6 ppm (CH2C, monomer) against the methylene protons at 3.9 ppm (–CH2–O–CO, polymer). The structures of PHEMAs were also confirmed by 1H NMR (see the ESI, Fig. S1†).
Exclusion chromatography (SEC-LALLS)
The molecular weights and dispersity indexes were determined by gel permeation chromatography (GPC) using a Viscotek GPC Max VE 2001 and a Viscotek TDA 305 triple detection instrument (refractometer, viscosimeter, and low angle laser light scattering) for data collection and OmniSec 5.12 for processing. Two D6000M general mixed bead columns were used for separation. The measurements were carried out by DMF HPLC purity (Merck) with the addition of 10 mM lithium bromide (Merck) as solvent at 323.15 K at a flow rate of 0.7 mL min−1 to reduce polymer/column interactions. The apparatus was used in a triple detection mode, and absolute molar mass (Mn and Mw) and the dispersity (Đ) were determined with a triple detection calibrated with a narrow poly(methyl methacrylate) standard – 50k PMMA (multidetection – homopolymer methods).DSC measurements
The thermodynamic properties of PHEMA obtained via purifying and drying the products under the electric fields of E = 0 kV cm−1 and E = 60 kV cm−1 were studied using a Mettler-Toledo DSC 1STARe system. The instrument has an HSS8 ceramic sensor with 120 thermocouples and a liquid nitrogen cooling station. Zinc and indium standards were used to calibrate the devices before the measurements. Samples (1.2 mg) sealed in an aluminum crucible (40 μL) were measured at a 10 K min−1 fixed heating rate. DSC thermograms were recorded in the temperature range from 243 K to 423 K. The glass transition temperatures Tg were obtained from the midpoints of the heat capacity increments. The DSC measurements for polymers obtained at 0 kV cm−1 and 60 kV cm−1 were repeated three times (for each sample) to ensure the reproducibility of the results. We have observed that the value of the glass transition temperature does not change, which means that the polymers are thermally stable even when heated up to 423 K. As a matter of fact, the literature results demonstrate that PHEMA is thermally stable up to ∼200 °C, i.e. 473 K.43 In all our experiments we were far below that limit.Results and discussion
Following the polymerization progress with the use of dielectric spectroscopy
Different techniques can be utilized to track the polymerization progress, such as Fourier transform infrared (FTIR) spectroscopy,44 Raman spectroscopy,41 NMR45 and DSC.38 Dielectric spectroscopy is also a powerful method for investigating systems that are undergoing chemical or physical changes as a consequence of chemical reaction, vitrification, crystallization, etc.46,47 In the past decade, the successful applications of dielectric measurements to in situ monitoring of tautomerization,48 crystallization of pharmaceuticals,49 small organic molecules,50 ionic liquids,51 and polymerization processes52,53 have been repeatedly demonstrated by our group. Apart from that, dielectric studies can be carried out under high pressure54 and high ac and dc electric fields.55,56 It is important to mention that the use of the dielectric technique is based on the electric dipole moment of the material being tested. As the polymerization/crystallization proceeds, the concentration of the initial material changes continuously, leading to the consecutive variation in the dipole moment distribution, which can be monitored using the dielectric technique.In the present study, we have investigated the polymerization kinetics of HEMA with 0.1 wt% AIBN as an initiator in the presence of a static electric field of different magnitudes, namely E = 0 kV cm−1, 14 kV cm−1, 28 kV cm−1, 60 kV cm−1, 100 kV cm−1 and 140 kV cm−1. The most exciting part of our work is studying phenomena that can be observed only in excessively high electric fields, which drive the dipolar material out of that linear regime. Under such high electric field conditions, the system can absorb the energy from the field. The high electric field is also known to modify thermodynamic variables such as entropy and enthalpy, resulting in structural changes that can bring the system to its new equilibrium state.55,57,58 Thus, when it comes to the chemistry of a reaction, high electric fields can significantly alter its pathway, promoting or hindering the entire reaction sequence.59
As demonstrated in Fig. 1, the BDS technique was employed to record the evolution of dielectric permittivity ε′ (panels (a) and (c)) and ε′′ (panels (b) and (d)) within the frequency range of 0.7 Hz–104 Hz under two representative e-fields, E = 0 kV cm−1 and 60 kV cm−1. In the dielectric ε′ spectra recorded at E = 0 kV cm−1 (see Fig. 1a), we can notice the electrode polarization effect (which is due to the accumulation of ions at the sample/electrode interface) at low frequencies and a kind of plateau at high frequencies. In Fig. 1b, the effects due to high dc conductivity (which is related to the charge transport) dominate the dielectric ε′′ loss spectra. As the polymerization reaction proceeds, we observed that conductivity shifts towards the lower frequencies with time. At some point, no further changes were observed in the dielectric spectra. We assign this as the endpoint of the polymerization reaction. Similar changes in the dielectric response of the polymerizing system were observed for glycidol (ring-opening polymerization) by Tarnacka et al.52
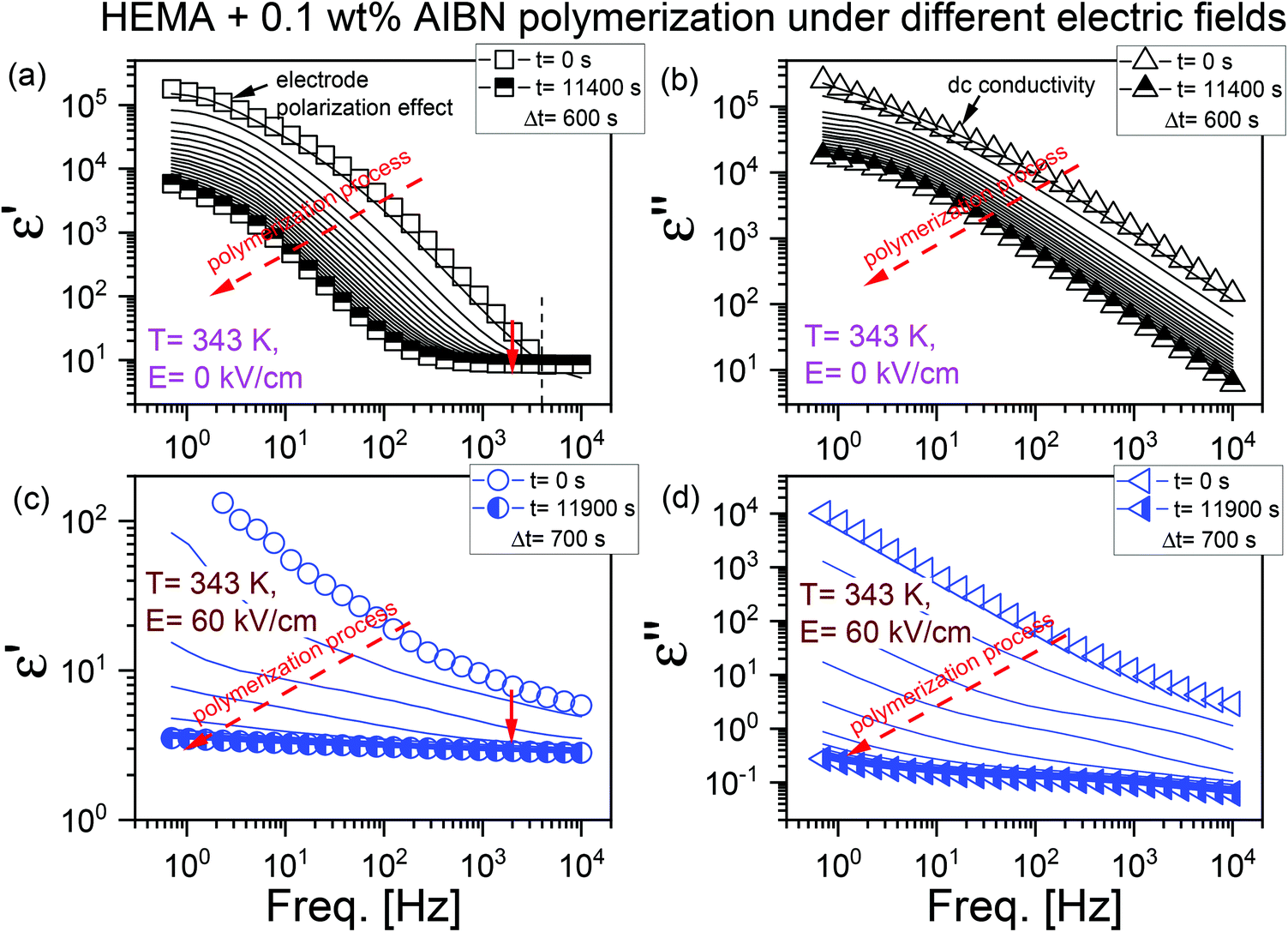 | ||
| Fig. 1 Exemplified results of the real (ε′, panels (a) and (c)) and imaginary (ε′′ panels (b) and (d)) parts of dielectric permittivity recorded upon isothermal polymerization of HEMA with 0.1 wt% AIBN at T = 343 K under static electric fields of (panel (a) and (b)) E = 0 kV cm−1 (reference) and (panel (c) and (d)) 60 kV cm−1. Dielectric spectra were obtained at different times as the reaction proceeded. The measured frequency region is within 0.7 Hz–104 Hz. | ||
As we know, electric fields of sufficiently high magnitude can affect the orientation of the dipoles in the tested system. When a dc e-field of E = 60 kV cm−1 was applied, both the dielectric ε′ and ε′′ spectra of the initial material, especially at low frequencies, differ from those observed in the case of E = 0 kV cm−1 (our reference). The electrode polarization effect becomes less pronounced at high frequencies while the conductivity is still seen. Interestingly, at the initial stages, the conductivity of the polymerizing system exposed to a high electric field was ∼10−9 S cm−1, while that at the zero-field was ∼10−4 S cm−1 (see panels (b) and (d) in Fig. 1). A decrease in dc conductivity with an increase in the reaction time is due to the formation of the polymer chains and increasing viscosity of the system. To further analyze the reaction kinetics, we have used dielectric permittivity values from this high-frequency dielectric region. Previous literature results confirm the validity of this approach to follow the polymerization progress via the dielectric technique.52,54,60,61
Analysis of the polymerization kinetics
To gain a deeper understanding of the mechanism and kinetics of the reaction, we have employed a normalized parameter, αε0, to evaluate the degree of reaction at a certain time in terms of the following equation,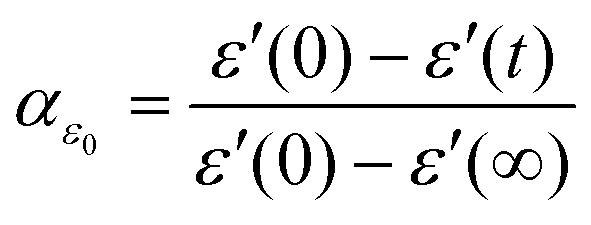 | (1) |
In this study, the NMR measurements on the polymer-contained products in each polymerization experiment provided information on the corresponding αM values. That is, conversions αM of 77%, 74%, 70%, 70%, 68% and 68%, respectively, were determined for the experiments carried out under static electric fields with different intensities E, viz., 0 kV cm−1, 14 kV cm−1, 28 kV cm−1, 60 kV cm−1, 100 kV cm−1 and 140 kV cm−1. The monomer conversion was determined from NMR measurements taken immediately after finishing high-field polymerization experiments.
With that, we can determine α via the formula, α = αε0 × αM. In Fig. 2, the evolution of monomer conversion α as a function of time under various static field conditions is shown. To analyze the overall kinetics, we have used an empirical relation, as expressed below,
| α = A0 + A·exp(−kt) | (2) |
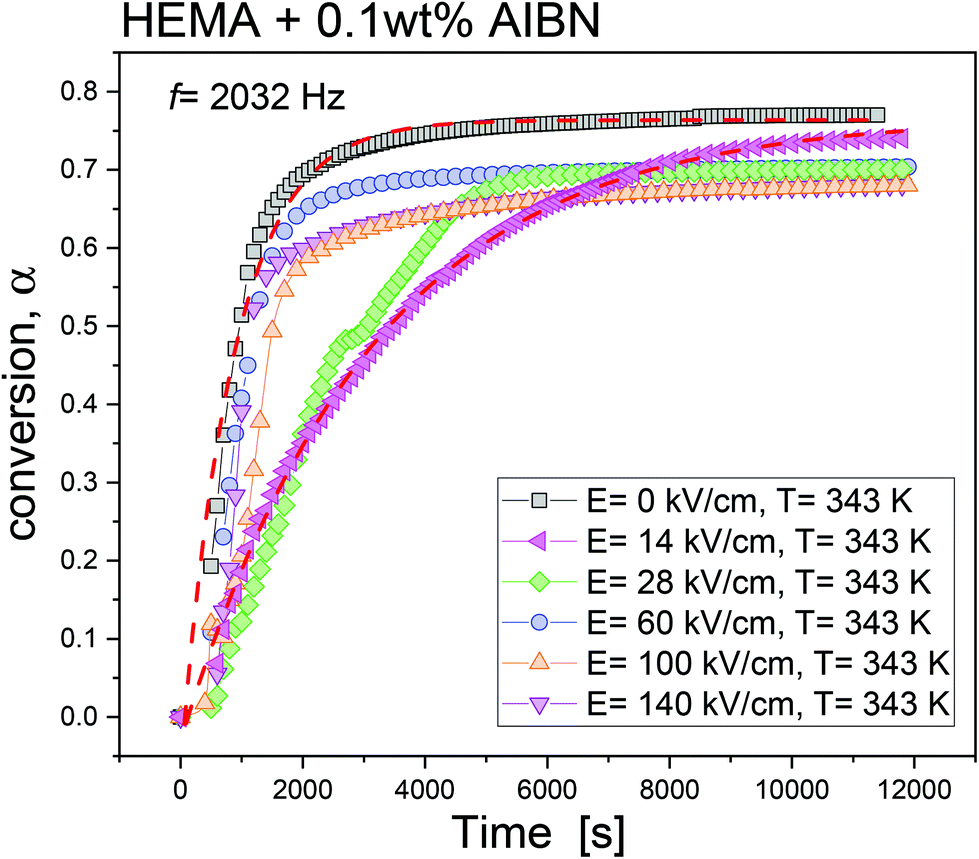 | ||
| Fig. 2 Time evolutions of monomer conversion, α, determined for the free-radical polymerizations of HEMA carried out at T = 343 K under static electric fields of various magnitudes. The data were determined at a fixed frequency of f = 2032 Hz. Two dashed lines represent the best fits to the results obtained under static electric fields of E = 0 kV cm−1 and 14 kV cm−1 in terms of an exponential function. | ||
The effect of the high electric field on the properties of obtained polymers
In Fig. 3, we show the effect of the dc field on the maximum monomer conversion αM (panel (a)), polymerization rate k (panel (b)) and molecular weight Mn together with its molecular weight distribution Mw/Mn (panel (c)). Fig. 3a shows that increasing the field intensity E will result in a reduction in the maximum monomer conversion αM. Interestingly, there is a kind of a minimum in the reaction rate at low fields, as seen in Fig. 3b. For example, at E = 14 kV cm−1, the reaction progress is sluggish relative to the zero-field case. However, as the dc field increases, the polymerization rate speeds up. When E exceeds 100 kV cm−1, the polymerization rate becomes comparable to that at E = 0 kV cm−1. In Fig. 3c, it is noticeable that PHEMA with a high molecular weight Mn ∼ 106 g mol−1 and dispersity Đ = 1.73 was synthesized in the absence of the high dc electric field. This is in agreement with the literature results. For example, Nguyen et al. have also reported the synthesis of PHEMA products with Mn ∼ 106 g mol−1 and Đ = 1.49–1.6 by single-electron transfer living radical polymerization.62 | ||
| Fig. 3 Evolutions of maximum monomer conversion αM (panel (a)), polymerization rate (panel (b)), number-average molecular weight Mn and its dispersity Đ (panel (c)) as functions of dc electric field E. Results of αM were determined based on the NMR measurements of the final products obtained upon high-field polymerization experiments. The molecular weight Mn and dispersity Đ were determined based on GPC measurements. Lines in all the panels are for understanding how the parameters evolve as the electric field magnitude is enhanced. | ||
Once polymerization of HEMA takes place in the presence of a high dc field, the obtained product is characterized by significantly reduced molecular weight (Mn = 41.0–58.0 kg mol−1) and dispersity (Đ = 1.07–1.26). As the dc field intensity increases from 14 kV cm−1 to 140 kV cm−1, a gradual decrease and an increase, respectively, are noticed for the molecular weight and dispersity index of PHEMA. When applying low dc fields within 14–60 kV cm−1, we have obtained PHEMA with Đ values of around 1.1. To the authors’ knowledge, such low dispersity has only been reported by Robinson et al. for PHEMA with Mn = 39.0 kg mol−1 and Đ = 1.09 synthesized using atom transfer radical polymerization in methanol at 293 K.63 This indicates that a high dc field could be as great as numerous chemical methods in tuning polymer properties, including their molecular weight and their distribution. On top of that, it is much simpler, “green” and does not involve complicated intermediate steps or synthesis procedures. Notably, we did not observe the ongoing crosslinking process for each experiment, and all recovered PHEMA samples were fully soluble in DMSO or DMF. The SEC-LALLS traces of all produced polymers are presented in Fig. 4. Chromatograms recorded for the sample synthesized at E = 0 kV cm−1 show a quite broad, typical peak, with no variation in the baseline at a high retention time. On the other hand, for the polymers produced at high electric fields a slightly low baseline at higher retention times was noted. Interestingly, a similar effect has been reported earlier for poly(ionic liquid)s.64 It is due to additional coulombic interactions between polymers and columns. This finding may indicate that macromolecules obtained at the high electric field are in fact charged. This supposition in some way was further confirmed by complementary dielectric investigations that revealed the higher conductivity of these macromolecules with respect to the ones synthesized under ordinary conditions.
 | ||
| Fig. 4 (a) SEC-LALLS trace of PHEMA prepared via FRP in the absence of external electric field (zero-field reference) and (b) SEC-LALLS traces of PHEMAs obtained upon FRP in the presence of dc bias, E = 140 kV cm−1. | ||
The glass transition and conductivity behavior of the obtained polymers
As illustrated in Fig. 3, relative to the zero-field case, i.e., when E = 0 kV cm−1, the application of the high electric field can result in dramatic differences in the polymerization kinetics and the macromolecular properties of the resultant polymers. It is of interest to further check the thermal and dynamic properties of the polymers obtained in the absence and presence of a dc field. Thus, we have conducted calorimetric and dielectric measurements for two PHEMA samples achieved under static electric fields of E = 0 kV cm−1 and E = 60 kV cm−1. These measurements were performed on purified and dried polymer products. The ascertained results are collected in Fig. 5. | ||
| Fig. 5 Panel (a) DSC scans recorded upon heating (10 K min−1) of two PHEMA samples obtained via polymerization under dc electric fields of E = 0 kV cm−1 and E = 60 kV cm−1. Panel (b) temperature dependences of conductivity relaxation time τM for the tested PHEMA samples. Panel (c) representative conductivity σ′ spectra collected for the tested PHEMA samples at T = 383 K. In panel (b), solid lines represent the fits to the data using the empirical VFT and Arrhenius equations. DSC and dielectric measurements were carried out after purification and drying of the obtained polymer products. | ||
The DSC thermograms depicted in Fig. 5a show that the glass transition temperature Tg values for PHEMA samples obtained at 0 kV cm−1 and 60 kV cm−1 are almost the same, namely 384 K and 383 K. These Tg values are comparable to the result of 388 ± 1.6 K for a PHEMA sample as reported in the literature.65 Subsequently, the dielectric measurements were performed to characterize the glass transition and conductivity of the tested samples. Dielectric loss ε′′ and electrical loss modulus M′′ spectra in a temperature region between 293 K and 433 K can be found in the ESI (Fig. S2 and S3†). Due to the high contribution of ionic conductivity (which results in the large rise in dielectric loss ε′′ spectra at low frequencies), the dielectric spectra can be analyzed in terms of complex modulus M*(f) and complex conductivity σ*(f) based on their equivalent correlations to ε*(f) with expressions shown below,66
 | (3) |
 | (4) |
| τ = τ0exp(Ea/kT) | (5) |
In addition to that, it is worth noting that these two synthesized PHEMA samples were characterized by high conductivity. Fig. 5c depicts the conductivity σ′ spectra for the studied PHEMA samples at their Tg. The spectra are very similar and yield a conductivity value of σdc ∼ 10−10 S cm−1, which is relatively high and comparable to the property of some PolyILs, for instance, the polymerized imidazolium-based protic ionic liquid [HSO3–BVIm][OTf].72 It should be noted here that a relatively low conductivity characterizes most of the polymer materials (σdc ∼ 10−18–10−20 S cm−1 at the glass transition temperature). Therefore, we found it very interesting that PHEMA samples reveal such an exceptionally high conductivity. As we suppose, it might originate from a proton hopping within the hydrogen-bonding network, as each monomer unit contains a hydroxyl group capable of forming an OH⋯O bonding. A similar finding was also reported for hyperbranched polymers, e.g., the family of bis-MPA polyesters (σdc ∼ 10−12–10−13 S cm−1 at Tg). In that case, the most significant contribution to the total ionic conductivity was ascribed to proton transfer along the extensive hydrogen-bonding networks.73 The exceptionally high conductivity of the synthesized PHEMA samples makes it a promising material for further development of solid-state polymer-based electrolytes for various applications. Such polymer matrices can then be further doped with ions/salts to give the building blocks of the hybrid material with high ionic conductivity, improved mechanical properties, good thermal stability, and safety.
Based on the results of our preliminary studies, we suppose that the effect of a high electric field on polymerization might be related to the dipole moment of the single building block unit. It is well known that the higher the dipole moment of the molecule, the greater its propensity to orient with the direction of the external field. Hence, with increasing the value of the dipole moment of the monomer, there should be much better control of the polymer chain alignment in such a high-field polymerization experiment. In addition to HEMA, we have also performed FRP (free radical polymerization) in the presence of a high electric field for MMA (methyl methacrylate). MMA has a very similar chemical structure to HEMA, except that it is less polar. The dipole moment of HEMA is 3.55 D, while for MMA it is ∼1.6–1.9 D.74 Field-assisted FRP of both monomers (with AIBN as a radical source) results in obtaining polymers with reduced molecular weight and low dispersity. However, the preliminary results of our study demonstrate that for MMA, we were not able to decrease Đ below ∼1.5, even using similar field magnitudes as for HEMA (∼150 kV cm−1).75
Conclusions
This study explores the effects of high electric fields on AIBN-induced (0.1 wt%) HEMA polymerization kinetics and properties of produced hydroxyl-functionalized polymers. In this work, isothermal polymerizations were conducted at T = 343 K within a period of t = 12000 s under various static fields with the magnitudes E ranging from 0 kV cm−1 to 140 kV cm−1. The dielectric technique was used to monitor the progression of the reaction in real-time. Subsequently, we employed NMR to confirm the expected structures of the polymer products obtained in each experiment and to evaluate the maximum monomer conversion αM. Based on the dielectric and NMR results, we can ascertain a general idea of the time evolution of monomer conversion α under each polymerization condition. Subsequently, the fits to the time evolution of monomer conversion α in terms of an exponential function yield the polymerization rate k. As expected, HEMA polymerization in the absence of the high dc field gave PHEMA with both high Mn ∼ 106 g mol−1 and dispersity Đ = 1.73 (=77% within t = 12000 s). Interestingly, the addition of high dc fields resulted in reduced monomer conversion α = 68–74%. More interestingly, the produced polymers were characterized by much lower Mn = 41.0–58.0 kg mol−1 and unusually low for an uncontrolled FRP process with dispersity Đ = 1.07–1.26. When the dc field increases from 0 kV cm−1 to 140 kV cm−1, the polymerization rate k decreases firstly and then increases. Next, we studied the glass transition and conductivity properties for PHEMA samples obtained after purification and drying of the polymer products achieved under static e-fields of E = 0 kV cm−1 and E = 60 kV cm−1. Surprisingly, the tested samples possess the same Tg value of 383 K and high conductivity σdc ∼ 10−10 S cm−1 at Tg. Since conductivity values of the synthesized PHEMA samples are comparable (at Tg) with those of polymerized ionic liquids, they emerge as very promising building blocks for developing polymer-based composite solid electrolytes. In addition to ionic conductivity characteristics for liquid electrolytes, such materials possess improved mechanical properties and thermal stability, and are very safe.
As proposed by us, the synthetic protocol supported by a high electric field turned out to be facile and robust in producing functionalized polymers (even as challenging as HEMA for FRP). We showed that by simple manipulation of the magnitude of the high electric field, the polymer Mn and Đ values could be adjusted. As we supposed, electric fields of sufficiently high magnitudes introduce some degree of alignment and orientation of the active groups and therefore generate charged polymers with preserved structures. However, further investigations are required to develop a general method to influence the reaction kinetics and mechanisms, which will benefit the development of more sophisticated techniques for preparing products with desirable properties. Thus, polymerization in the presence of high electric fields emerges as a topic worth much more attention.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
WT, KC and KA are grateful for the financial support from the National Science Centre within the framework of the SONATA BIS project (Grant No. 2017/26/E/ST3/00077).References
- T. Junkers, Macromol. Chem. Phys., 2020, 221, 1–6 CrossRef.
- D. T. Gentekos, R. J. Sifri and B. P. Fors, Nat. Rev. Mater., 2019, 4, 761–774 CrossRef.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1–22 CrossRef PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- R. Whitfield, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2021, 60, 19383–19388 CrossRef CAS PubMed.
- R. Whitfield, N. P. Truong, D. Messmer, K. Parkatzidis, M. Rolland and A. Anastasaki, Chem. Sci., 2019, 10, 8724–8734 RSC.
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef PubMed.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS.
- D. J. Walsh, D. A. Schinski, R. A. Schneider and D. Guironnet, Nat. Commun., 2020, 11, 3094 CrossRef CAS PubMed.
- R. Whitfield, K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2019, 58, 13323–13328 CrossRef CAS PubMed.
- M. Rolland, N. P. Truong, R. Whitfield and A. Anastasaki, ACS Macro Lett., 2020, 9, 459–463 CrossRef CAS.
- T. Shimizu, N. P. Truong, R. Whitfield and A. Anastasaki, ACS Polym. Au, 2021, 1, 187–195 CrossRef CAS PubMed.
- K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Polym. Chem., 2021, 12, 5583–5588 RSC.
- K. Parkatzidis, N. P. Truong, M. N. Antonopoulou, R. Whitfield, D. Konkolewicz and A. Anastasaki, Polym. Chem., 2020, 11, 4968–4972 RSC.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1340–1352 CAS.
- D. T. Gentekos, L. N. Dupuis and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 1848–1851 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- A. Dzienia, P. Maksym, M. Tarnacka, I. Grudzka-Flak, S. Golba, A. Zięba, K. Kaminski and M. Paluch, Green Chem., 2017, 19, 3618–3627 RSC.
- P. Maksym, M. Tarnacka, A. Dzienia, K. Erfurt, A. Brzęczek-Szafran, A. Chrobok, A. Zięba, K. Kaminski and M. Paluch, Polymer, 2018, 140, 158–166 CrossRef CAS.
- M. Kubo, T. Sone, M. Ohata and T. Tsukada, Ultrason. Sonochem., 2018, 49, 310–315 CrossRef CAS PubMed.
- T. G. McKenzie, F. Karimi, M. Ashokkumar and G. G. Qiao, Chem. – Eur. J., 2019, 25, 5372–5388 CrossRef CAS PubMed.
- L. Lv, W. Wu, G. Zou and Q. Zhang, Polym. Chem., 2013, 4, 908 RSC.
- B. Liu, H. Zhang, T. Ma, L. Bai, J. Lin and D. Lu, J. Mater. Chem. C, 2020, 8, 6503–6512 RSC.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- N. Murakami, N. Wakabayashi, R. Matsushima, A. Kishida and Y. Igarashi, J. Mech. Behav. Biomed. Mater., 2013, 20, 98–104 CrossRef CAS PubMed.
- J. Zheng, L. Ma, M. Gan, J. Yan, Z. Li, X. Shen and J. Zhang, J. Appl. Polym. Sci., 2014, 131(13), 40467 CrossRef.
- Z. Wang, Z. Wang, X. Pan, L. Fu, S. Lathwal, M. Olszewski, J. Yan, A. E. Enciso, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 275–280 CrossRef CAS.
- M. A. Kamarudin, A. A. Khan, E. Tan, G. Rughoobur, S. M. Said, M. M. Qasim and T. D. Wilkinson, RSC Adv., 2017, 7, 31989–31996 RSC.
- T. Ma, N. Song, B. Liu, J. Ren, H. Zhang and D. Lu, J. Phys. Chem. C, 2019, 123, 13993–14002 CrossRef CAS.
- Z.-F. Cai, G. Zhan, L. Daukiya, S. Eyley, W. Thielemans, K. Severin and S. De Feyter, J. Am. Chem. Soc., 2019, 141, 11404–11408 CrossRef CAS PubMed.
- F. M. McFarland, X. Liu, S. Zhang, K. Tang, N. K. Kreis, X. Gu and S. Guo, Polymer, 2018, 151, 56–64 CrossRef CAS.
- J. Chen, L. Gao, X. Han, T. Chen, J. Luo, K. Liu, Z. Gao and W. Zhang, Mater. Chem. Phys., 2016, 169, 105–112 CrossRef CAS.
- N. López-López, I. Muñoz Resta, R. de Llanos, J. F. Miravet, M. Mikhaylov, M. N. Sokolov, S. Ballesta, I. García-Luque and F. Galindo, ACS Biomater. Sci. Eng., 2020, 6, 6995–7003 CrossRef PubMed.
- J.-P. Montheard, M. Chatzopoulos and D. Chappard, J. Macromol. Sci. C., 1992, 32, 1–34 CrossRef.
- G. Mabilleau, C. Cincu, M. F. Baslé and D. Chappard, J. Raman Spectrosc., 2008, 39, 767–771 CrossRef CAS.
- D. Achilias and P. Siafaka, Processes, 2017, 5, 21 CrossRef.
- E. Evlyukhin, L. Museur, M. Traore, C. Perruchot, A. Zerr and A. Kanaev, Sci. Rep., 2016, 5, 18244 CrossRef PubMed.
- M. Kubo, T. Kondo, H. Matsui, N. Shibasaki-Kitakawa and T. Yonemoto, Ultrason. Sonochem., 2018, 40, 736–741 CrossRef CAS PubMed.
- T. T. H. Luu, Z. Jia, A. Kanaev and L. Museur, J. Phys. Chem. B, 2020, 124, 6857–6866 CrossRef CAS PubMed.
- K. L. Beers, S. Boo, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1999, 32, 5772–5776 CrossRef CAS.
- K. Demirelli, M. Coşkun and E. Kaya, Polym. Degrad. Stab., 2001, 72, 75–80 CrossRef CAS.
- H. Deng, Z. Shen, L. Li, H. Yin and J. Chen, J. Appl. Polym. Sci., 2014, 131(15), 40503 CrossRef.
- J. B. McLeary, F. M. Calitz, J. M. McKenzie, M. P. Tonge, R. D. Sanderson and B. Klumperman, Macromolecules, 2005, 38, 3151–3161 CrossRef CAS.
- J. Mijovic and S. Andjelic, Macromolecules, 1995, 28, 2787–2796 CrossRef CAS.
- J. Mijovic, Dielectric Spectroscopy of Reactive Network-Forming Polymers, 2003 Search PubMed.
- Z. Wojnarowska, P. Wlodarczyk, K. Kaminski, K. Grzybowska, L. Hawelek and M. Paluch, J. Chem. Phys., 2010, 133, 094507 CrossRef CAS PubMed.
- K. Adrjanowicz, K. Kaminski, Z. Wojnarowska, M. Dulski, L. Hawelek, S. Pawlus, M. Paluch and W. Sawicki, J. Phys. Chem. B, 2010, 114, 6579–6593 CrossRef CAS PubMed.
- K. Adrjanowicz, G. Szklarz, K. Koperwas and M. Paluch, Phys. Chem. Chem. Phys., 2017, 19, 14366–14375 RSC.
- W. Tu, G. Szklarz, K. Adrjanowicz, K. Grzybowska, J. Knapik-kowalczuk and M. Paluch, J. Phys. Chem. C, 2019, 123, 12623–12637 CrossRef CAS.
- M. Tarnacka, T. Flak, M. Dulski, S. Pawlus, K. Adrjanowicz, A. Swinarew, K. Kaminski and M. Paluch, Polymer, 2014, 55, 1984–1990 CrossRef CAS.
- P. Maksym, M. Tarnacka, A. Dzienia, K. Matuszek, A. Chrobok, K. Kaminski and M. Paluch, Macromolecules, 2017, 50, 3262–3272 CrossRef CAS.
- M. Tarnacka, M. Wikarek, S. Pawlus, K. Kaminski and M. Paluch, RSC Adv., 2015, 5, 105934–105942 RSC.
- D. M. Duarte, R. Richert and K. Adrjanowicz, J. Phys. Chem. Lett., 2020, 11, 3975–3979 CrossRef CAS PubMed.
- K. Adrjanowicz, M. Paluch and R. Richert, Phys. Chem. Chem. Phys., 2018, 20, 925–931 RSC.
- R. Richert, J. Phys.: Condens. Matter, 2017, 29, 363001 CrossRef PubMed.
- P. Lunkenheimer, M. Michl, T. Bauer and A. Loidl, Eur. Phys. J.: Spec. Top., 2017, 226, 3157–3183 Search PubMed.
- H. J. Kreuzer, Surf. Interface Anal., 2004, 36, 372–379 CrossRef CAS.
- M. Tarnacka, M. Dulski, S. Starzonek, K. Adrjanowicz, E. U. Mapesa, K. Kaminski and M. Paluch, Polymer, 2015, 68, 253–261 CrossRef CAS.
- M. Tarnacka, A. Chrobok, K. Matuszek, D. Neugebauer, R. Bielas, S. Golba, K. Wolnica, M. Dulski, K. Kaminski and M. Paluch, Polym. Chem., 2016, 7, 6363–6374 RSC.
- N. H. Nguyen, X. Leng and V. Percec, Polym. Chem., 2013, 4, 2760 RSC.
- K. L. Robinson, M. A. Khan, M. V. de Paz Báñez, X. S. Wang and S. P. Armes, Macromolecules, 2001, 34, 3155–3158 CrossRef CAS.
- P. Maksym, M. Tarnacka, R. Bernat, A. Dzienia, A. Szelwicka, B. Hachuła, A. Chrobok, M. Paluch and K. Kamiński, Polym. Chem., 2021, 12, 4418–4427 RSC.
- D. S. Jones, C. P. McCoy, G. P. Andrews, R. M. McCrory and S. P. Gorman, Mol. Pharm., 2015, 12, 2928–2936 CrossRef CAS PubMed.
- F. Kremer and A. Schönhals, Broadband Dielectric Spectroscopy, Springer Berlin Heidelberg, 2003 Search PubMed.
- Z. Wojnarowska, K. J. Paluch, E. Shoifet, C. Schick, L. Tajber, J. Knapik, P. Wlodarczyk, K. Grzybowska, S. Hensel-Bielowka, S. P. Verevkin and M. Paluch, J. Am. Chem. Soc., 2015, 137, 1157–1164 CrossRef CAS PubMed.
- M. Paluch, Dielectric Properties of Ionic Liquids, Springer, 2016 Search PubMed.
- Z. Wojnarowska, H. Feng, Y. Fu, S. Cheng, B. Carroll, R. Kumar, V. N. Novikov, A. M. Kisliuk, T. Saito, N.-G. Kang, J. W. Mays, A. P. Sokolov and V. Bocharova, Macromolecules, 2017, 50, 6710–6721 CrossRef CAS.
- F. Fan, W. Wang, A. P. Holt, H. Feng, D. Uhrig, X. Lu, T. Hong, Y. Wang, N.-G. Kang, J. Mays and A. P. Sokolov, Macromolecules, 2016, 49, 4557–4570 CrossRef CAS.
- Z. Wojnarowska, C. M. Roland, A. Swiety-Pospiech, K. Grzybowska and M. Paluch, Phys. Rev. Lett., 2012, 108, 015701 CrossRef CAS PubMed.
- Z. Wojnarowska, J. Knapik, M. Díaz, A. Ortiz, I. Ortiz and M. Paluch, Macromolecules, 2014, 47, 4056–4065 CrossRef CAS.
- K. Adrjanowicz, K. Kaminski, M. Dulski, M. Jasiurkowska-Delaporte, K. Kolodziejczyk, M. Jarek, G. Bartkowiak, L. Hawelek, S. Jurga and M. Paluch, Macromolecules, 2014, 47, 5798–5807 CrossRef CAS.
- O. Karahan, D. Avci and V. Aväyente, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3058–3068 CrossRef CAS.
- K. Chat, P. Maksym, K. Kaminski and K. Adrjanowicz, Chemical Communications, 2022 10.1039/D2CC01186G.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and SEC-LALS chromatograms of the obtained polymers. Dielectric loss ε′′ (a) and electrical loss modulus M′′ (b) spectra of a PHEMA sample obtained through the FRP of HEMA at E = 0 kV cm−1 in a temperature region between 293 K and 433 K; Dielectric loss ε′′ (a) and electrical loss modulus M′′ (b) spectra of a PHEMA sample obtained through the FRP of HEMA at E = 60 kV cm−1 in a temperature region between 293 K and 433 K. See DOI: https://doi.org/10.1039/d2py00320a |
| This journal is © The Royal Society of Chemistry 2022 |