
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrain-correlated mechanochromism in different polyurethanes featuring a supramolecular mechanophore†
Hanna
Traeger
 a,
Yoshimitsu
Sagara
b,
José Augusto
Berrocal
a,
Stephen
Schrettl
*a and
Christoph
Weder
*a
a,
Yoshimitsu
Sagara
b,
José Augusto
Berrocal
a,
Stephen
Schrettl
*a and
Christoph
Weder
*a
aAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, 1700 Fribourg, Switzerland. E-mail: stephen.schrettl@unifr.ch; christoph.weder@unifr.ch
bDepartment of Materials Science and Engineering, Tokyo Institute of Technology, 2-12-1, Ookayama, Meguro-ku, Tokyo 152-8552, Japan
First published on 26th April 2022
Abstract
A previously reported, supramolecular, loop-forming mechanophore comprised of two covalently connected perylene diimide (PDI) dyes was equipped with hydroxy groups and covalently incorporated into different polyurethanes (PUs). Four PUs with different hard segments based on either isophorone diisocyanate (IDI), 4,4′-methylenebis(cyclohexyl isocyanate) (HMDI), hexamethylene diisocyanate (HDI), or methylene diphenyl diisocyanate (MDI) were investigated. The different isocyanates affect the degree of hard phase formation and crystallization, and thus bestow the materials with different mechanical properties. Except for the MDI-based polymer, whose photoluminescence is quenched, dominant excimer emission is observed for all polymers in the absence of mechanical force, which indicates that the mechanophore motif is folded into a loop with interacting PDI dyes. Upon deformation, the PDI monomer emission intensity increases to the extent that the change becomes visually discernible, suggesting that the loops unfold. The mechanophore functions well in all three matrices, but the mechanochromic response depends strongly on the polymers’ materials properties. The most pronounced response is observed for the HDI-based PU, which displays a robust hard segment formation and the highest strength and stiffness. In all materials, the mechanochromic response reliably scales with the applied strain and reversible strain sensing over multiple cycles of deformation could be shown for all PUs.
Introduction
The ability to detect force-induced bond breakage events in polymeric materials is useful to develop a fundamental understanding for failure mechanisms and to prevent catastrophic material failure.1,2 To achieve this, a broad range of mechanophores – molecules containing a mechanically cleavable bond – that can be incorporated into polymeric materials have been developed.3–9 The mechanophores are activated by application of mechanical force on the bulk material, for example by way of compression or tensile deformation, which can cause the scission of a chemical bond in the mechanophore when a threshold force is surpassed. Some mechanophores display a distinct optical (mechanochromic) response upon covalent bond scission, such as a colour change or a change of the photoluminescence properties.5,8,10 Examples of such mechanophores that have been widely used for stress sensing in polymers include spiropyrans,11–13 1,2-dioxetans,14–17 diarylbibenzofuranones,18–20 rhodamines,21–23 naphthopyrans,24–27 and oxazines.28 Since the operating principle of these motifs includes the rupture of a covalent bond, their capability of sensing stresses in the low strain regime of polymer deformation is generally limited.6 Moreover, the mechanical activation of such mechanophores is often not instantly reversible. Even in the case of mechanophores that do not undergo scissile chain cleavage, such as spiropyran,11 oxazine,28 and diarylbibenzofuranone19 derivatives, the reverse reaction is usually slow and/or requires applying external stimuli such as UV-irradiation or heating.6,29These problems can be overcome by creating mechanoresponsive motifs that operate on the basis of weak and reversible non-covalent interactions.30 Early examples of this approach include excimer-forming cyano-substituted oligo(p-phenylene vinylene) (cyano-OPV) dyes, which were first blended,31 and later covalently incorporated into the backbone of polyurethanes (PUs).32 In the stress-free state, the dye molecules aggregate and their emission is dominated by excimers. Deformation caused the disruption of the dye aggregates, leading to a clearly visible change of the photoluminescence colour to monomer emission. In the meantime, the general concept was expanded to different excimer-forming dyes, polymer systems and materials designs.30,33–35 To increase the specificity and sensitivity of mechanoresponsive motifs that rely on non-covalent interactions, some of us recently developed rotaxane mechanophores containing reversibly-separable quencher/emitter pairs36–39 and a cyclophane mechanophore bearing two excimer-forming pyrene dyes.40 The mechanical activation of these motifs is specific and instantly reversible, but their synthesis is relatively complex. Mechanoresponsive motifs in which the interactions of two chromophores are achieved in loop-like architectures41–44 can mitigate this problem. We recently introduced a loop-forming mechanophore comprised of two covalently connected, excimer-forming perylene diimide (PDI) dyes that can be accessed in three synthetic steps.45 The motif was equipped with atom transfer radical polymerization initiators, which allowed us to grow poly(methyl acrylate) (PMA) from both ends. Our investigations showed that the loops are folded in the idle state so that interactions between the PDI dyes cause the photoluminescence to be dominated by orange excimer emission. Upon deformation of the polymer the intensity of green monomer emission increases, indicating that the PDI loops unfold and the dyes disassemble. A detailed characterization of the mechanoresponsive behaviour showed that the emission colour correlates with the applied strain, and the reversible nature of the non-sacrificial mechanoresponsive motif allowed us to analyse the responses upon cyclic deformation as well as static stress-relaxation in the PMA matrix. To shed more light on its function, we now report the response of the new mechanophore motif in polymers having different mechanical properties. To achieve this, we modified reference PDI (1) and the loop-forming PDI dimer (2) with hydroxy groups and covalently incorporated the mechanophore into thermoplastic PUs with different mechanical properties (Fig. 1).
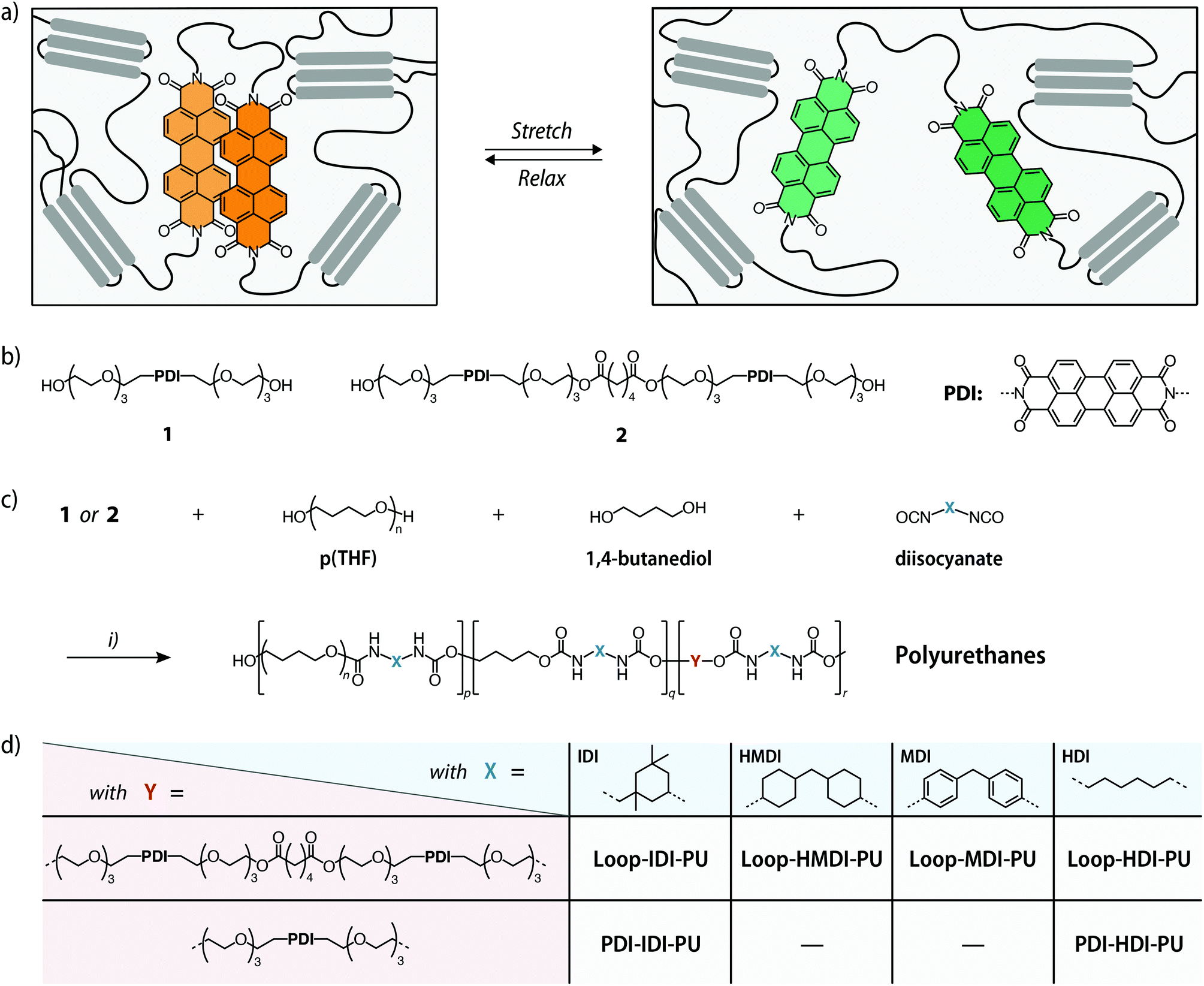 | ||
Fig. 1 (a) Schematic representation of the force-induced unfolding of the PDI-based loop-forming mechanophore in a polyurethane matrix featuring domains formed by hard and soft segments. (b) Chemical structure of the reference compound containing only one PDI dye (1) and the loop-forming mechanophore comprised of two covalently connected perylene diimide dyes (2). (c) Synthesis of the different polyurethanes (PUs) and (d) overview of the chemical structures of the polymers PDI-IDI-PU and PDI-HDI-PU with 1 and Loop-IDI-PU, Loop-HMDI-PU, Loop-HMDI-PU, and Loop-HDI-PU with 2. For PDI-IDI-PU and PDI-HDI-PU, the p![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :q:r ratio in the monomer feed was 1.0:2.2:0.002. For Loop-IDI-PU, Loop-HMDI-PU, and Loop-HDI-PU, the p:q:r ratio in the monomer feed was 1.0:2.2:0.001, while a feed ratio of 1.0:2.2:0.0005 or 1.0:2.2:0.022 was used to prepare Loop-MDI-PU with 0.03 or 0.12 wt% of 2, respectively. Reagents and conditions: (i) dibutyltin dilaurate, CHCl3, 3 h, 45 °C; then 1,4-butanediol, 48 h, 45 °C. :q:r ratio in the monomer feed was 1.0:2.2:0.002. For Loop-IDI-PU, Loop-HMDI-PU, and Loop-HDI-PU, the p:q:r ratio in the monomer feed was 1.0:2.2:0.001, while a feed ratio of 1.0:2.2:0.0005 or 1.0:2.2:0.022 was used to prepare Loop-MDI-PU with 0.03 or 0.12 wt% of 2, respectively. Reagents and conditions: (i) dibutyltin dilaurate, CHCl3, 3 h, 45 °C; then 1,4-butanediol, 48 h, 45 °C. | ||
Results and discussion
The loop-forming mechanophore comprised of two covalently connected perylene diimide (PDI) dyes and bearing hydroxy groups at each terminus (2) was synthesized in four steps (Fig. S1†).44,46 Firstly, PDI derivative 1, which also served as a reference compound containing only one PDI dye, was formed via the condensation of commercially available perylene-3,4,9,10-tetracarboxylic dianhydride and tetraethylene glycol monoamine in a microwave reaction. Subsequently, one of the hydroxy groups in 1 was protected by reaction with tert-butyldimethylsilyl chloride,46 the product was dimerized via esterification with adipic acid,44 and 2 was obtained through deprotection with tetrabutylammonium fluoride. All compounds were characterized to satisfaction by 1H-NMR, 13C-NMR, and HR-MS spectroscopy (see ESI†).Mechanophore-containing PUs with hydroxy-terminated poly(tetrahydrofuran) (p(THF)) as the soft segment and hard segments based on butanediol and either isophorone diisocyanate (IDI), 4,4′-methylenebis(cyclohexyl isocyanate) (HMDI), methylene diphenyl diisocyanate (MDI), or hexamethylene diisocyanate (HDI) were investigated with the goal to cover polymer matrices with different morphologies and mechanical properties (Fig. 1). Based on symmetry considerations, the extent of microphase separation and hard phase formation, and therewith the strength and stiffness of the PU, was expected (and observed, vide infra) to grow from IDI to HMDI and MDI to HDI.15 Thus, PUs with either 1 or 2 were prepared by employing reaction conditions based on our previous work as well as a report by Chen and coworkers.15,40 This involved reacting 1 or 2 and p(THF) (number-average molecular weight, Mn = 2000 g mol−1) for 3 h at 45 °C in CHCl3 with either IDI, HMDI, or HDI in the presence of dibutyltin dilaurate (DBTL) to form a prepolymer, which was subsequently chain extended with 1,4-butanediol for 48 h at 45 °C to yield Loop-IDI-PU, Loop-HMDI-PU, or Loop-HDI-PU. The preparation of Loop-MDI-PU was performed in the same manner at room temperature using a solvent mixture of THF and CHCl3.36 The content of 2 in the feed (with respect to the final polymer) was 0.07 wt%, corresponding to 0.035 wt% of the PDI dye for Loop-IDI-PU, Loop-HMDI-PU, and Loop-HDI-PU, while the Loop-MDI-PU samples featured a content of 2 in the polymer of 0.03 or 0.12 wt% (or 0.01 and 0.06 wt% of PDI). Reference polymers PDI-IDI-PU and PDI-HDI-PU were prepared in the same way with 0.055 wt% of 1 (or 0.03 wt% of PDI). The chemical structure of all polymers was characterized by 1H-NMR spectroscopy in either CDCl3 or THF-d8; in the case of Loop-HDI-PU and PDI-HDI-PU the addition of small amounts of trifluoroacetic acid was required to promote dissolution. The concentration of the residues of chromophore 1 and loop-forming mechanophore 2 was too low to allow detection by 1H-NMR spectroscopy. The molecular weights of Loop-IDI-PU, Loop-HMDI-PU, Loop-MDI-PU, and PDI-IDI-PU were characterized by size exclusion chromatography (SEC, Fig. S2†). The Mn of the loop-containing polymers was on the order of 30′000 g mol−1 with a Đ of 1.8 for Loop-IDI-PU and 2.1 for Loop-HMDI-PU. An Mn of 85′000 g mol−1 (Đ = 1.7) was determined for Loop-MDI-PU samples with 0.03 wt% of 2, while samples with 0.12 wt% of 2 featured an Mn of 58′000 g mol−1 (Đ = 1.9), and PDI-IDI-PU had an Mn of 133′000 g mol−1 and a Đ of 1.8. Due to the limited solubility in conventional SEC solvents, the Mn of the HDI-based PUs could not be determined. From the molecular weights of these polymers and the weight fractions of 1 and 2 one can calculate that on average only 1 of 66 Loop-IDI-PU, 1 of 77 Loop-HMDI-PU, and 1 of 10 PDI-IDI-PU macromolecules contain the respective dye motif.
To explore the extent of intramolecular versus intermolecular dye association, UV-vis absorption and fluorescence spectra of 1 and 2 were recorded in DCM (c = 5 μmol L−1) (Fig. 2a). The ratio between the A0–0 and A0–1 transition is reportedly diagnostic for the presence of ground state interactions between perylene dyes and can be used to calculate the degree of dye association.45–48 The spectra of compound 1 reveal an A0–0/A0–1 ratio of 1.62, which is in good agreement with the previously reported value for non-associated PDIs (see Table S2 and ESI† for details).44,45 The analysis of spectra of 2 yields an A0–0/A0–1 ratio of 0.82, which is assumed to correlate with an extent of dye association of up to 90%. The data, hence, suggest that the electronic ground-state interactions between the covalently linked PDI dyes promote the formation of an intramolecular loop-structure.45 The A0–0/A0–1 ratio of 2 is slightly lower than that of a recently reported perylene loop having a similar structure, which is most likely the result of the absence of bulky substituents at the termini.44,45 In agreement with these observations, the emission spectrum of 2 in DCM (c = 5 μmol L−1) shows a pronounced, broad emission band centred at ca. 623 nm that is indicative of excimer formation (Fig. 2b), as is also apparent from the orange fluorescence colour of the corresponding solutions. The magnitude of this band is much less pronounced in a corresponding solution of 1 (Fig. 2b). The UV-vis absorption spectra of the polymers Loop-IDI-PU and Loop-HMDI-PU in DCM solutions indicate that the perylene moieties in Loop-IDI-PU (A0–0/A0–1 = 0.85) and Loop-HMDI-PU (A0–0/A0–1 = 0.85) are associated to a similar extent as in 2 (Fig. 2c). Moreover, the fluorescence spectra of solutions of Loop-IDI-PU and Loop-HMDI-PU (c(2) = 7 μg mL−1) display a pronounced excimer emission (Fig. 2d), reflecting that intramolecular dye association also occurs after incorporation of 2 into the PUs. By contrast, the absorption spectrum of solutions of PDI-IDI-PU (A0–0/A0–1 = 1.74) mirror the one of 1 and similarly display no excimer emission in the fluorescence spectra (Fig. S3†). Absorption spectra of the two different Loop-MDI-PUs containing 0.03 or 0.12 wt% of 2 in THF solutions suggest strong aggregation of the PDI dyes with a A0–0/A0–1 of 0.77 and 0.61, respectively, and the corresponding fluorescence spectra display a pronounced excimer emission at 630 nm (Fig. S4†). This is likely an effect of the use of THF, which is a poor solvent for PDI dyes and promotes strong aggregation (MDI-based PUs were insoluble in DCM). Interestingly, the PDI fluorescence was completely quenched in solid samples of Loop-MDI-PU (Fig. S4†), perhaps due to an aggregation-induced quenching effect between the PDI dyes and the aromatic MDI hard segments, similar to what has been reported for naphthalene diimides.49–51 Hence, further experiments with this polymer were abandoned.
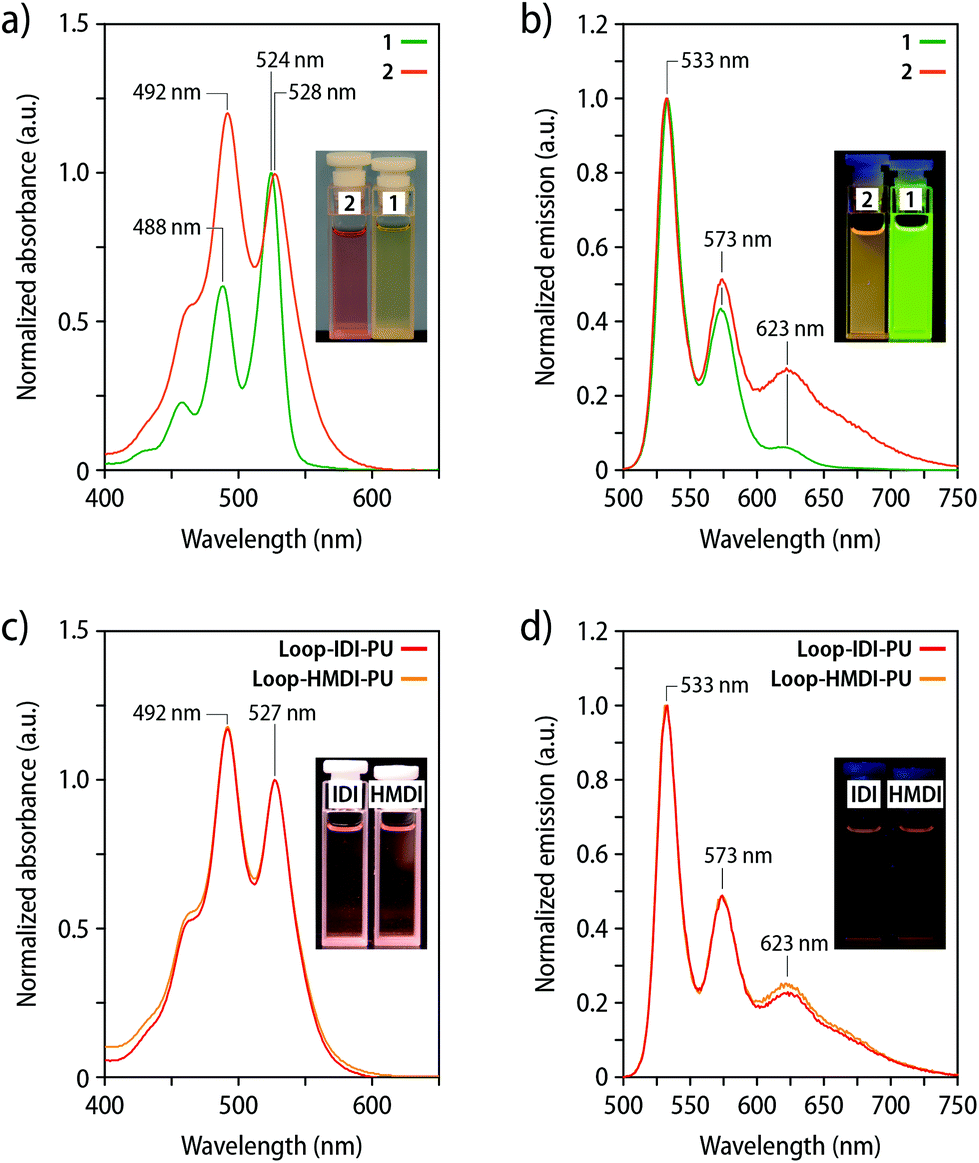 | ||
| Fig. 2 (a) UV-vis absorption spectra of 1 (green) and 2 (orange) recorded in DCM (c = 5 μmol L−1). (b) Fluorescence spectra (λex = 488 nm) of 1 (green) and 2 (orange) recorded in DCM (c = 5 μmol L−1). (c) UV-vis absorption spectra of Loop-IDI-PU and Loop-HMDI-PU recorded in DCM (c(2) = 7 μg mL−1). (d) Fluorescence spectra (λex = 488 nm) of Loop-IDI-PU and Loop-HMDI-PU recorded in DCM (c(2) = 7 μg mL−1). The insets show images of the solutions recorded (a and c) under ambient light and (b and d) under UV-light illumination (λex = 365 nm). Absorption spectra are normalized to the maximum of the A0–0 band and fluorescence spectra to 530 nm. | ||
Thermogravimetric analysis (TGA) experiments (Fig. S5†) reveal that the neat mechanophore 2, reference molecule 1, and the mechanophore-containing PUs show no appreciable weight loss at temperatures up to 230 °C. Differential scanning calorimetry (DSC) experiments show that the neat 2 is an amorphous solid, while 1 exhibits a melting transition (Tm) at ca. 40 °C (Fig. S6†). To investigate the impact of the processing history on the morphology of the various polymers, DSC traces were recorded for the as-prepared polymers, as well as for compression-molded films that were rapidly cooled to room temperature, slowly cooled to room temperature, or aged for five months. The first DSC heating trace of as-prepared Loop-IDI-PU shows only a weak, sharp endothermic peak at ca. 40 °C similar to the peak of the 5 month-old sample (Fig. S7a†), which appears to be associated with the Tm of crystalline domains formed by the p(THF) soft segments (Fig. S8†). No crystallization (Tc) can be observed in the first cooling cycle and the second heating trace is void of any transitions (Fig. S7b and c†). Indeed, no distinct features are observed in the traces of rapidly cooled and slowly cooled Loop-IDI-PU (Fig. S7a–c†) and of PDI-IDI-PU (Fig. S9a†). Taken together, the DSC data suggest that the IDI hard segments do not promote the formation of an extended hard phase, which is consistent with the non-symmetric nature of the isocyanate. This permits p(THF) crystallization, although this process is very slow or even absent in the higher molecular weight PDI-IDI-PU, so that freshly prepared films do not exhibit any substantial thermal transitions.
The DSC traces of all Loop-HMDI-PU samples show only a broad Tm between 65 °C (melt-processed samples) and 100 °C (as-prepared samples) whose magnitude is more pronounced in the as-prepared and the aged melt-processed sample than in melt-processed samples that were freshly prepared (Fig. S7d–f†). The transition is only observed in the first heating cycles, and is tentatively interpreted to be related to the melting of hard block HMDI domains.
The DSC traces of all Loop-HDI-PU samples show two reversible Tm at 4–12 °C and 145–165 °C, and the cooling scans feature pronounced Tc peaks at 88–121 °C and at ca. −25 °C (Fig. S7g–i†). The magnitude of these transitions appears to be largely independent of the sample history. Similar transitions are also observed for PDI-HDI-PU (Fig. S9d–f†). The thermal behaviour, hence, indicates that the HDI moieties in the hard segments foster a strong phase separation between hard and soft segments and fast reformation of the hard phase occurs upon cooling. With these findings in hand, we decided to carry out subsequent experiments with freshly prepared films having a uniform thickness of ca. 200 μm that were made by compression-molding (4 tons; 3 min) at 80 °C for Loop-IDI-PU and PDI-IDI-PU, 120 °C for Loop-HMDI-PU, and 140 °C for Loop-HDI-PU and PDI-HDI-PU, and slowly cooling all samples to room temperature. Optical and mechanical properties were then measured within 48 h after compression-molding to exclude possible changes in mechanical properties due to the slow crystallization of p(THF) or hard domains.
Films of the three polymers containing the loop-forming mechanophore 2 were characterized by UV-vis absorption and fluorescence spectroscopy (Fig. 3). The main absorption bands in the UV-vis spectra of Loop-IDI-PU and Loop-HMDI-PU are red-shifted relative to the bands in the solution spectra (Fig. 2c). The A0–0 and A0–1 bands are not clearly distinguishable and instead merged into a broad absorption band, preventing a reliable analysis of the extent of dye association. By contrast, the UV-vis absorption spectrum of Loop-HDI-PU shows two absorption maxima at 529 nm (A0–0) and 494 nm (A0–1, Fig. 3a). The spectrum with two maxima and a more pronounced A0–1 than A0–0 band mirrors the one reported for PMA containing a similar PDI-loop mechanophore45 and suggests the formation of some ground state aggregates. Interestingly, however, a broad excimer emission dominates the fluorescence spectra of Loop-IDI-PU (638 nm) and Loop-HMDI-PU (631 nm), while the most intense emission band of films of Loop-HDI-PU is observed at 536 nm and associated with the PDI monomer (Fig. 3b). The spectra are corroborated by the visual appearance of the films under UV-light illumination (Fig. 3b, inset). The fluorescence of Loop-IDI-PU and Loop-HMDI-PU films appears redder and less bright than that of Loop-HDI-PU, indicating different quantum yields of the folded versus the unfolded loop motif. Indeed, it has been previously reported that the fluorescence quantum yield of the unfolded PDI loops is more than three times higher than that of associated PDI dyes.44 The fact that Loop-HDI-PU displays the smallest contribution of excimer emission correlates with the rapid and robust hard-phase formation upon cooling this polymer from the melt (vide supra) and may indicate that this process kinetically traps a fraction of the loop motifs in an unfolded state, which has been previously also observed for PUs containing rotaxane mechanophores.38,39
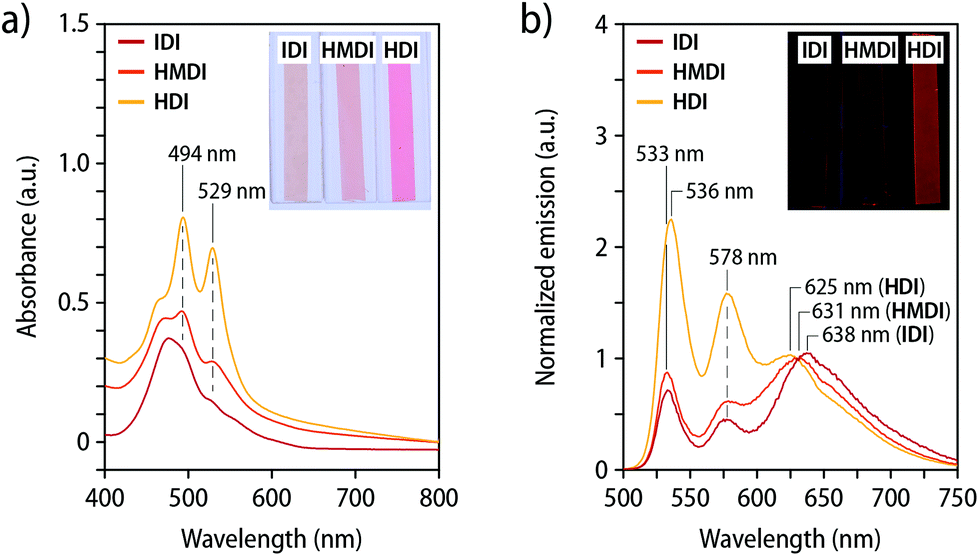 | ||
| Fig. 3 (a) UV-vis absorption and (b) fluorescence emission spectra of compression-molded films of Loop-IDI-PU, Loop-HMDI-PU, and Loop-HDI-PU and photographs of the films (insets). The fluorescence spectra were recorded with λex = 488 nm and they are normalized to the emission intensity at 630 nm. The photographs of the fluorescent films were recorded under UV-light illumination (λex = 365 nm). | ||
In order to probe to what extent intermolecular aggregation of the PDIs might be at play, absorption and emission spectra of compression-molded films of PDI-IDI-PU and PDI-HDI-PU were recorded (Fig. S3c and d†). In the absorption spectra, distinct absorption bands at 527 nm (A0–0) and 492 nm (A0–1) are visible, allowing for the determination of the A0–0/A0–1 ratio as 1.06 for PDI-IDI-PU and 1.28 for PDI-HDI-PU (Fig. S3c and Table S2†). This indicates that in PDI-IDI-PU, in which hard-phase formation is absent and the macromolecules enjoy a relatively high mobility, a small fraction of PDIs aggregate. By contrast, in PDI-HDI-PU no such aggregation can be overserved, which is consistent with the rapid assembly of the hard segments and kinetic trapping of the dyes. The emission spectra corroborate these findings (Fig. S3d†). The spectra of both polymers are governed by monomer emission, with bands centred at 535 nm in the case of PDI-IDI-PU and 540 nm in the case of PDI-HDI-PU, and a slightly more pronounced excimer emission at 630 nm for PDI-IDI-PU than for PDI-HDI-PU. Accordingly, under illumination with UV-light (365 nm), both films show green monomer emission (Fig. S3d,† inset). These findings support our hypothesis that the loop structure promotes intramolecular dye association, although the extent of association strongly depends on the polymer morphology.
It is well known that the assembly of physically or covalently incorporated, weakly interacting dye molecules in polymer matrices can vary with the temperature, and to explore this effect for the here-investigated PUs, fluorescence spectra were measured upon heating and cooling samples under well-controlled conditions (Fig. S10–S13†). Compression-molded films of Loop-IDI-PU, Loop-HMDI-PU, and Loop-HDI-PU were heated at a rate of 10 °C min−1 from 20 to 170 °C in steps of 10 °C and subsequently cooled in corresponding intervals. At each temperature, samples were equilibrated for 1 min and emission spectra were recorded (Fig. S11 and S12;†λex = 470 nm). Following previously established protocols,34,35,45 the monomer-to-excimer intensity ratio (IM/IE) was used to quantify the emission colour change and thereby the extent of dye interactions as a function of temperature (Fig. S10†). For Loop-IDI-PU and Loop-HMDI-PU, the monomer and excimer intensities were determined at 535 and 640 nm, while the intensities at 540 and 630 nm were used for Loop-HDI-PU. In samples of Loop-IDI-PU and Loop-HMDI-PU, IM/IE increases from a value of ca. 0.4, i.e., an excimer-dominated emission, to a ratio of 1.3 for Loop-IDI-PU and 1.6 for Loop-HMDI-PU (Fig. S10a and b†). The plots of IM/IEvs. temperature suggest linear regimes with different slopes that change at ca. 90 °C for Loop-IDI-PU and at ca. 70 °C for Loop-HMDI-PU. The cooling curves reveal only a small hysteresis, and after cooling samples back from 170 to 20 °C, the initial IM/IE values are almost fully recovered. In the case of Loop-HDI-PU, the initial IM/IE value (0.8) is much higher, reflecting that compression-molded samples of this polymer contain more unfolded loops. The IM/IEvs. temperature trace recorded upon heating mirrors the ones of the other PUs (Fig. S10c†), but the initial (0.8) and final (1.8) IM/IE values are higher, on account of the lower propensity of loop formation in this material. The data reflect a considerable hysteresis for samples of Loop-HDI-PU upon cooling, i.e., at each temperature, the IM/IE value is much higher than the one recorded upon heating, with a final IM/IE ratio of 1.4 at 20 °C. This effect is probably related to the crystallization of the hard segments upon step-wise cooling, as reflected by the DSC data (Fig. S7†), leading to an extensive trapping of the PDI-loop in the unfolded state and a large fraction of monomer emission (Fig. S10c†). Fluorescence spectra recorded 26 h after cooling to 20 °C show little change in the IM/IE ratio (1.5), indicating that chain rearrangement processes are, if at play, very slow (Fig. S13†). By contrast, the IM/IE ratio of a freshly hot-pressed sample was 0.73 and in this case the IM/IE ratio drops further to 0.59 when samples were stored under ambient conditions for 5 months (Fig. S14†). Thus, in Loop-HDI-PU the thermal history and ageing effects can considerably influence the extent to which the loop-motif folds.
After establishing the influence of the processing history and temperature-dependence of the monomer-to-excimer intensity ratio, the mechanochromic behaviour of freshly prepared, compression-molded films of Loop-IDI-PU, Loop-HMDI-PU, and Loop-HDI-PU was investigated. Upon uniaxial deformation to a strain of 1000–1600% no visible change of the fluorescence colour was observed for Loop-IDI-PU, whereas a clearly visible change from red to greed occurred in the case of Loop-HMDI-PU and Loop-HDI-PU (Fig. 4a). To probe the mechanochromic response more quantitatively, stress–strain curves of the different PUs were recorded with a strain rate of 50% min−1 while monitoring samples in situ by fluorescence spectroscopy measurements. Spectra were recorded at a frequency of 1 Hz and the IM/IE ratio and the applied stress were plotted as a function of the strain (Fig. 4b–d and S15†). As expected, the recorded stress–strain curves show that the mechanical properties of the PUs are strongly dependent on the nature of the hard segment. Samples of Loop-IDI-PU display a very low modulus, a yield point at ca. 50% strain, a maximum stress of ca. 0.15 MPa, failure strain of ca. 1600% (Fig. 4b).15 Beyond the yield point, the material starts to flow, which is indicated by a decrease in stress with increasing strain. Since the crystallization of the p(THF) segment is slow and the glass transition temperature (Tg) is far below ambient temperature, this material, hence, deforms at very low stresses and shows largely a viscous response, indicating that few intermolecular interactions serve as physical cross-links, presumably since the non-symmetric isophorone residues impart significant packing constrains. Whereas optical differences in the emission colour are difficult to visually discern for Loop-IDI-PU (Fig. 4a), monitoring by in situ fluorescence spectroscopy unambiguously reveals the mechanochromic response (Fig. 4e). The IM/IE ratio follows the applied strain linearly during sample deformation (Fig. 4b), with a small increase of IM/IE from 0.35 (idle state) to 0.53 (400% strain). We tentatively interpret this behaviour as associated with the low stresses required to deform Loop-IDI-PU samples and concomitantly occurring relaxation processes that may cause rapid re-aggregation of dissociated PDI dyes during deformation.
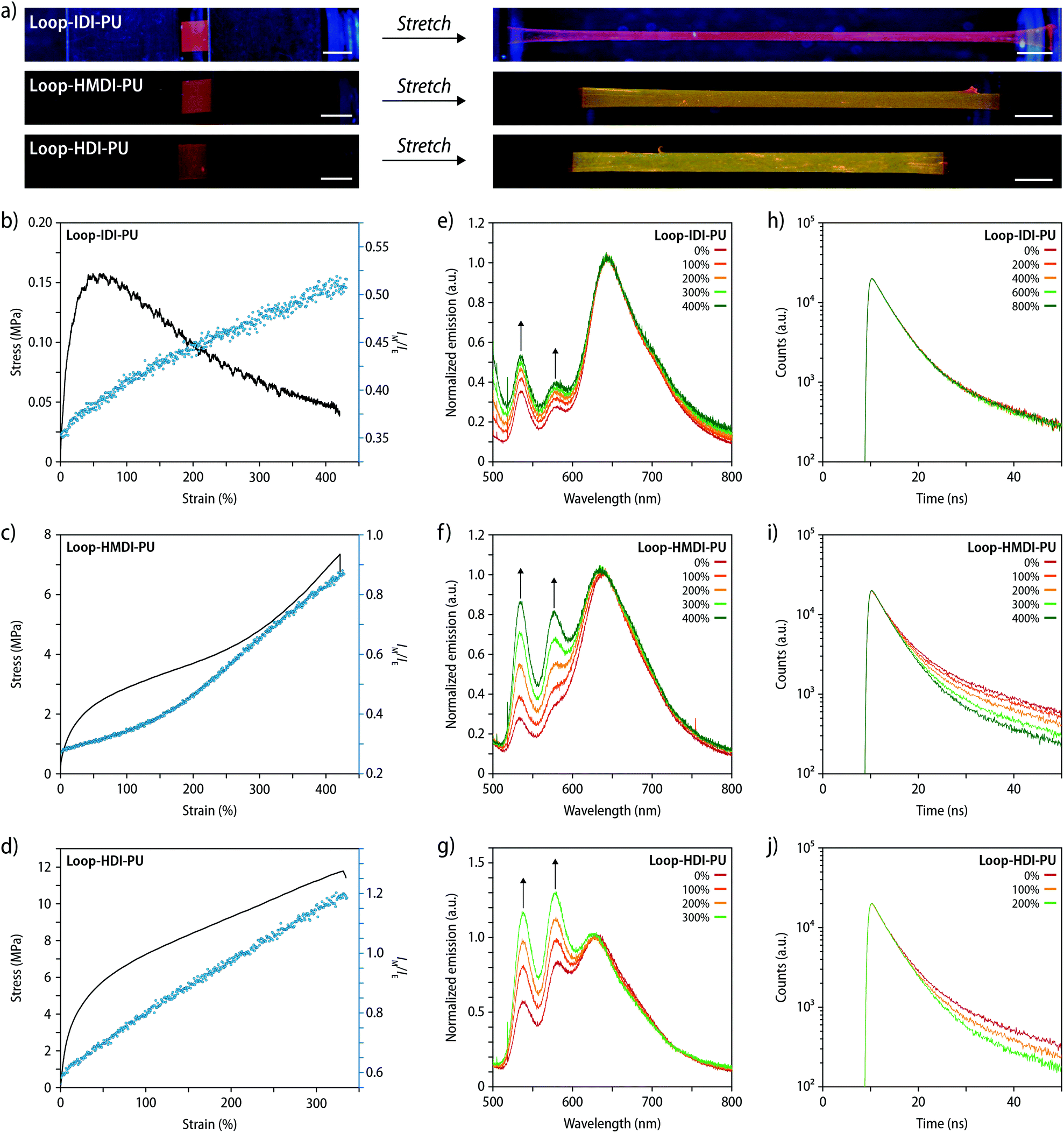 | ||
| Fig. 4 (a) Photographs of films of (top to bottom) Loop-IDI-PU, Loop-HMDI-PU, and Loop-HDI-PU in (left) the relaxed state and (right) after uniaxial deformation to strains of 1600%, 1100%, and 1000%, respectively. The pictures were taken under UV-light illumination (λex = 365 nm, scale bars = 5 mm). (b–d) Stress–strain curves (black lines, strain rate 50% min−1) and monomer/excimer intensity ratio (IM/IE, blue dots) as a function of strain for (b) Loop-IDI-PU, (c) Loop-HMDI-PU, and (d) Loop-HDI-PU. The IM/IE ratios were determined from fluorescence spectra (λex = 470 nm) of Loop-IDI-PU and Loop-HMDI-PU at 535 and 640 nm and for Loop-HDI-PU at 540 and 630 nm. (e–g) Fluorescence spectra showing the emission of (e) Loop-IDI-PU, (f) Loop-HMDI-PU, and (g) Loop-HDI-PU at different strains (λex = 470 nm). (h–j) Time-resolved fluorescence intensity decays recorded for (h) Loop-IDI-PU, (i) Loop-HMDI-PU, and (j) Loop-HDI-PU at the indicated strains at 580 nm (λex = 365 nm). | ||
The stress–strain curve of Loop-HMDI-PU displays the typical behaviour of a thermoplastic elastomer in which domains formed by hydrogen-bonded hard segments serve as physical cross-links (Fig. 4c). The data reflect a yield point at ca. 25% strain and a stress of ca. 2 MPa that is followed by a slanted rubbery plateau up to 275% strain, before a strain hardening region is observed. As already demonstrated by the pictures in Fig. 4a, the mechanochromic response of Loop-HMDI-PU is much more pronounced than that of Loop-IDI-PU. Indeed, the IM/IE ratio increases from 0.25 to 0.85 upon deformation to 400% strain (Fig. 4c and f). The plot of the IM/IE ratio against the applied strain indicates two linear regimes and a slope change as the strain hardening regime is reached. A similar response was previously observed in blends of another thermoplastic PU with a mechanochromic additive35 and related to an increase of the local forces that the mechanoresponsive motif experiences during chain alignment in the strain hardening regime. The mechanical response of Loop-HDI-PU samples is also characteristic for physically cross-linked thermoplastic elastomers, and the slightly higher stiffness and strength, relative to Loop-HMDI-PU, are indicative of an increased physical cross-linking (Fig. 4d). While high strains can be reached without observing sample failure when low strain-rates are employed (Fig. 4a), during tensile testing with a rate of 50% min−1, Loop-HDI-PU samples show no strain hardening below 325% strain, at which point samples fail (Fig. 4d and S15c†). As a result, the IM/IE ratio linearly follows the applied strain until sample failure (Fig. 4d). The relative fluorescence changes displayed by Loop-HMDI-PU with an IM/IE of 0.55 in the idle state and 1.2 at 325% strain is the highest of all PUs studied (Fig. 4d and g), which is consistent with the presence of a crystalline hard phase that is generally considered to translate into higher forces that the mechanophores experience.
To further probe how deformation affects the nature of the excited states of the mechanophore motif, lifetime measurements were conducted during uniaxial tensile deformation of films of Loop-IDI-PU, Loop-HMDI-PU, and Loop-HDI-PU (Fig. 4h–j). Emission intensity decay profiles of pristine films and samples that had been deformed to strains of 100 to 800% were recorded with excitation at 365 nm and detection at 580 nm. The recorded decay profiles were fitted to a triexponential function to separate the contributions of monomeric PDI, weekly associated dimers, and strongly associated dimers, respectively.52,53 In the case of Loop-IDI-PU, no changes were observed upon deformation (Fig. 4h), reflecting that the conformational changes of the mechanophore motif and related variations between excimer and monomer emission are minor. By contrast, the decay profiles of samples of Loop-HMDI-PU and Loop-HDI-PU display increased contributions of monomeric PDI species to the overall fluorescence lifetime (Fig. 4i and j), which increase with the applied strain and unambiguously corroborate the proposed operating mechanism.
We also explored the reversibility of the mechanochromic responses by manual stretching (Movies S1–S3†), by monitoring the fluorescence response upon repeated deformation and relaxation between strains of 0 and 200%, and by following the relaxation process after uniaxial tensile deformation to 200% strain (Fig. 5). Gratifyingly, a comparison of the stress, strain, and IM/IE values collected during these experiments (shown as a function of time) shows a high correlation for all materials. In the case of Loop-IDI-PU, a pronounced hysteresis is observed during the cyclic testing, i.e., the recovery after deformation and stress release is poor, especially in the first cycle, which is consistent with the largely viscous response of this material to deformation (vide supra). Importantly, however, also in this case the plots of IM/IEvs. time and strain vs. time are virtually identical, reflecting that the emission changes reflect the extent of strain very well (Fig. 5a). The hysteresis in Loop-HMDI-PU and Loop-HDI-PUs after the first deformation cycle is much less pronounced (Fig. 5b and c). Interestingly, the elastic recovery is better in Loop-HMDI-PU, which may be a result of irreversible rearrangements in the semi-crystalline Loop-HDI-PU after the first deformation in combination with sample buckling, which can occur since the measurement set-up does not allow to apply preload forces in tensile experiments. Throughout the subsequent deformation and relaxation cycles, the IM/IEvs. time and strain vs. time plots for Loop-HMDI-PU and Loop-HDI-PU samples mirror each other (Fig. 5b and c). Also, the fluorescence lifetime reversibly changes upon repeated deformation and relaxation (Fig. S16†). Thus, samples of Loop-HMDI-PU and Loop-HDI-PU were subjected to strains of 400 or 200%, respectively, over the course of 3 deformation/relaxation cycles and the comparison of the recorded decay profiles indicates that the changes are highly reversible. These findings suggest that dissociation and re-association of the PDI dyes within the loop motif readily occur upon mechanical deformation of the polymers.
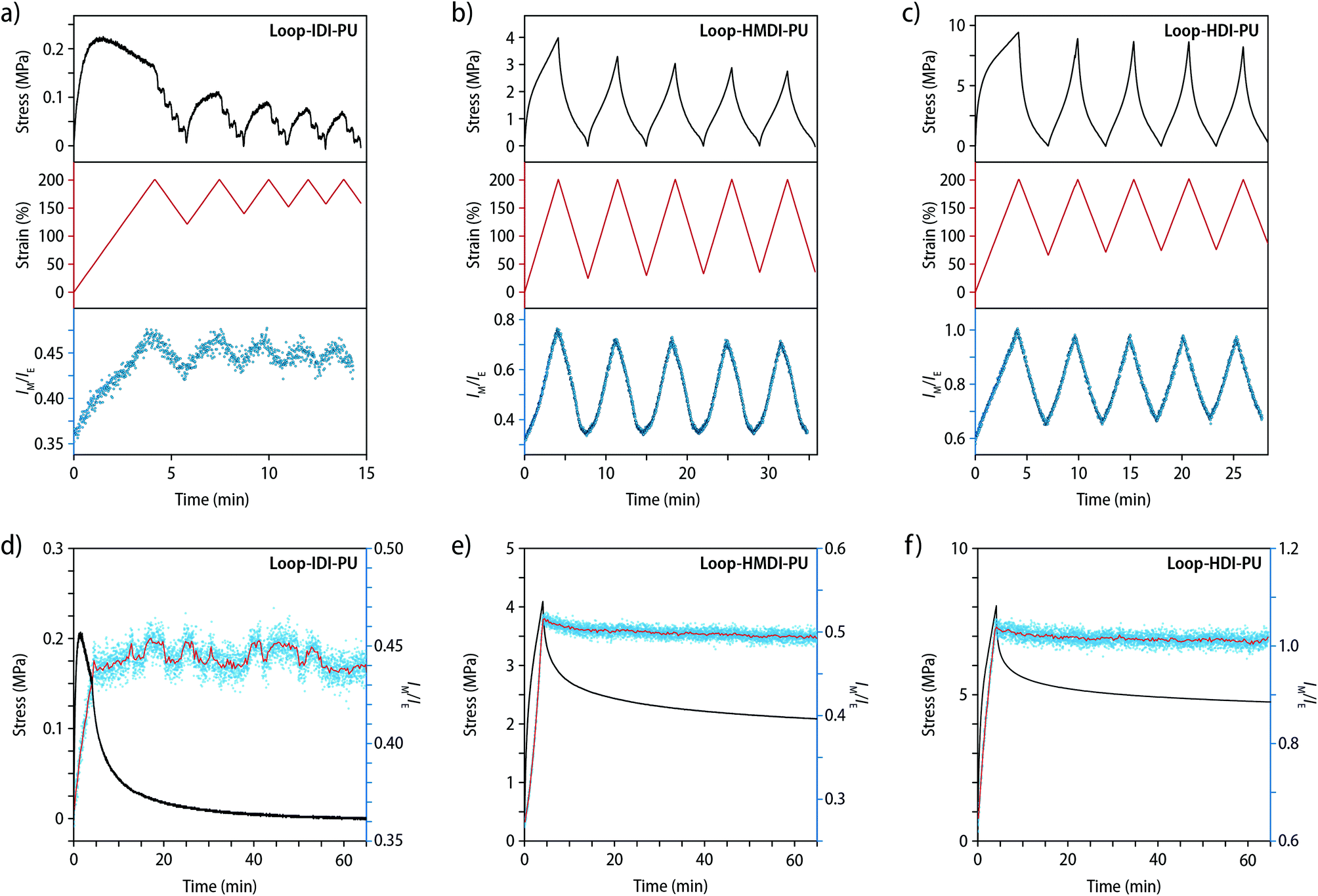 | ||
| Fig. 5 (a–c) Stress (black lines), strain (red lines), and the monomer/excimer ratio (IM/IE, blue dots) of samples of (a) Loop-IDI-PU, (b) Loop-HMDI-PU, and (c) Loop-HDI-PU recorded in cyclic deformation and relaxation experiments over 5 cycles between 0 and 200% strain (strain rate = 50% min−1). (d–f) Stress relaxation experiments showing the stress (black lines), the monomer/excimer intensity ratio (IM/IE, blue circles), and a 20-point average of the IM/IE ratio (red line) for samples of (d) Loop-IDI-PU, (e) Loop-HMDI-PU, and (f) Loop-HDI-PU over time. Films were initially stretched to 200% strain with a rate of 50% min−1. The IM/IE ratios were determined from fluorescence spectra (λex = 470 nm) of Loop-IDI-PU and Loop-HMDI-PU at 535 and 640 nm and for Loop-HDI-PU at 540 and 630 nm. | ||
Lastly, the stress relaxation behaviour of the three different PUs was probed. To this end, samples were deformed to 200% with a strain rate of 50% min−1 and held at this strain over the course of 60 min, while the stress was monitored and fluorescence spectra were recorded with a rate of 1 Hz (Fig. 5d–f and S17†). In the case of Loop-IDI-PU, samples were observed to fully relax at the end of the experiment with complete dissipation of the applied stress, again reflecting the low extent of physical cross-linking (Fig. 5d and S17†). Interestingly, the IM/IE ratio increases upon deformation in the expected manner, but in contrast to the stress, no significant changes occurred in the IM/IE ratio while the sample was kept at a strain of 200%. In the case of Loop-HMDI-PU and Loop-HDI-PUs, the extent of stress-relaxation is much smaller, reflecting again the higher density of physical cross-links in these materials (Fig. 5e and f). As for samples of Loop-IDI-PU, the IM/IE ratio during stress-relaxation of Loop-HMDI-PU and Loop-HDI-PU samples fails to reflect the observed stress decay (Fig. 5e and f and S17†), but instead mirrors the applied strain. These observations are most likely caused by the segmented built of the PUs that prevents fast relaxation processes such as chain slippage and movements compared to recently reported PMA samples featuring a related loop-forming mechanoresponsive motif.45
Conclusions
In summary, we equipped a previously established loop-forming mechanophore, comprised of two PDI dyes that are covalently connected by a short linker, with two terminal hydroxy functional groups that allowed for a covalent incorporation into the backbone of different polyurethanes. Using the mechanophore, we prepared four PUs with different hard segments and investigated their mechanochromic response. The chemical structure of the PUs is very similar, but the different hard segments impart the PUs with distinct mechanical properties on account of varying degrees of physical cross-linking, allowing us to readily correlate polymers’ properties with the responsive behaviour of the loop-forming mechanophores.In the pristine state, the photoluminescence of all polymers is dominated by excimer emission, but the monomer/excimer intensity ratio varies between the PUs and is found to be significantly influenced by the morphology and processing history. The operating principle of the mechanophore motif is based on the (un)folding of weakly associated intramolecular loops, which enables a fluorescence response even in highly dynamic polymers such as Loop-IDI-PU. Whereas changes in the emission upon deformation of such sample can be monitored, the overall response remains limited. By contrast, a pronounced response is observed for thermoplastic elastomers such as Loop-HMDI-PU and Loop-HDI-PU. In samples of the latter, the hard segment formation is robust and the highest strength and stiffness are observed. In this material, the changes in the monomer/excimer intensity ratio are most pronounced, resulting in a clearly visible fluorescence colour change. In fact, the response is much higher than in a previously reported poly(methyl acrylate) containing the same mechanophore motif.45 In the case of Loop-HMDI-PU, which displays pronounced strain hardening, a significant change in the slope of the IM/IE ratio and the applied strain was seen as the strain-hardening regime was reached, suggesting that the different deformation behaviours of the polymer are reflected in the response of the mechanoresponsive motif.
Overall, the trends of the mechanochromic responses mirror results reported by Chen and Sijbesma, who observed a 6 times higher activation of a 1,2-dioxetane mechanophore in similar PUs when compared to its use in a PMA matrix, i.e., an increased activation of mechanophores in polymers whose deformation require larger stresses.14 In comparison to the behaviour observed for similar PUs with IDI, MDI and HDI hard segments and a dioxetane mechanophore,15 the herein employed supramolecular mechanophore lacks distinct onset stresses or strains, effectively displaying a continuous response to the applied strain independent of the nature of the hard segment. However, it cannot be excluded that these differences originate from variations in the polymers’ chemical structures and topology, degrees of polymerization, and processing history.
Interestingly, the emission of the loop-forming mechanophore in the different PUs reliably reflects changes of the strain in repeated deformation/relaxation cycles, but stress relaxation processes are not translated into corresponding changes of the emission intensity ratios. For PUs with hard segments, this may be explained by a decreased reassembly of the looped aggregates upon relaxation. In other words, the data seem to suggest that while macroscopic stresses can fully relax, as in the case of Loop-IDI-PU, the individual chain segments do not necessarily return to their original conformation, perhaps on account of kinetic trapping. This effect deserves further investigation and the here reported mechanophore motif appears to be a highly suitable tool for such studies.
Author contributions
H. T., S. S., and C. W. developed the original concept for the study and designed the materials and experiments. H. T. synthesized and characterized the materials and performed the experiments with help from Y. S. and J. A. B. All authors discussed the results and contributed to the interpretation of the data. H. T., S. S., and C. W. wrote the paper. All authors contributed to editing of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge financial support through the National Center of Competence in Research Bio-Inspired Materials, a research instrument of the Swiss National Science Foundation (SNSF) as well as the Adolphe Merkle Foundation.References
- W. D. Callister and D. G. Rethwisch, Fundamentals of Materials Science and Engineering. An Integrated Approach, John Wiley & Sons, Inc, Hoboken, NJ, USA, 5th edn, 2019 Search PubMed.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- N. Deneke, M. L. Rencheck and C. S. Davis, Soft Matter, 2020, 16, 6230–6252 RSC.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- J. F. Patrick, M. J. Robb, N. R. Sottos, J. S. Moore and S. R. White, Nature, 2016, 540, 363–370 CrossRef CAS PubMed.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- R. T. O'Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Polym. Int., 2013, 62, 2–12 CrossRef CAS.
- W. H. Binder, Polymer, 2020, 202, 122639 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- T. A. Kim, B. A. Beiermann, S. R. White and N. R. Sottos, ACS Macro Lett., 2016, 5, 1312–1316 CrossRef CAS.
- C. K. Lee, B. A. Beiermann, M. N. Silberstein, J. Wang, J. S. Moore, N. R. Sottos and P. V. Braun, Macromolecules, 2013, 46, 3746–3752 CrossRef CAS.
- C. K. Lee, D. A. Davis, S. R. White, J. S. Moore, N. R. Sottos and P. V. Braun, J. Am. Chem. Soc., 2010, 132, 16107–16111 CrossRef CAS PubMed.
- Y. Chen and R. P. Sijbesma, Macromolecules, 2014, 47, 3797–3805 CrossRef CAS.
- S. Liu, Y. Yuan, J. Li, S. Sun and Y. Chen, Polym. Chem., 2020, 11, 1877–1884 RSC.
- C. Yan, F. Yang, M. Wu, Y. Yuan, F. Chen and Y. Chen, Macromolecules, 2019, 52, 9376–9382 CrossRef CAS.
- W. Yuan, Y. Yuan, F. Yang, M. Wu and Y. Chen, Macromolecules, 2018, 51, 9019–9025 CrossRef CAS.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed.
- K. Imato, T. Kanehara, S. Nojima, T. Ohishi, Y. Higaki, A. Takahara and H. Otsuka, Chem. Commun., 2016, 52, 10482–10485 RSC.
- K. Imato, T. Kanehara, T. Ohishi, M. Nishihara, H. Yajima, M. Ito, A. Takahara and H. Otsuka, ACS Macro Lett., 2015, 4, 1307–1311 CrossRef CAS.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS PubMed.
- L. Wang, W. Zhou, Q. Tang, H. Yang, Q. Zhou and X. Zhang, Polymers, 2018, 10, 994 CrossRef PubMed.
- C.-H. Wu, C.-W. Tu, J. Aimi, J. Zhang, T. Chen, C.-C. Wang and C.-F. Huang, Polym. Chem., 2020, 11, 6423–6428 RSC.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS PubMed.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed.
- B. A. Versaw, M. E. McFadden, C. C. Husic and M. J. Robb, Chem. Sci., 2020, 11, 4525–4530 RSC.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2021, 143, 7925–7929 CrossRef CAS PubMed.
- H. Qian, N. S. Purwanto, D. G. Ivanoff, A. J. Halmes, N. R. Sottos and J. S. Moore, Chem, 2021, 7, 1080–1091 CAS.
- C. Calvino, L. Neumann, C. Weder and S. Schrettl, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 640–652 CrossRef CAS.
- H. Traeger, D. J. Kiebala, C. Weder and S. Schrettl, Macromol. Rapid Commun., 2021, 42, e2000573 CrossRef PubMed.
- C. Löwe and C. Weder, Adv. Mater., 2002, 14, 1625–1629 CrossRef.
- B. R. Crenshaw and C. Weder, Macromolecules, 2006, 39, 9581–9589 CrossRef CAS.
- A. Pucci and G. Ruggeri, J. Mater. Chem., 2011, 21, 8282 RSC.
- C. Calvino, Y. Sagara, V. Buclin, A. P. Haehnel, A. Del Prado, C. Aeby, Y. C. Simon, S. Schrettl and C. Weder, Macromol. Rapid Commun., 2019, 40, e1800705 CrossRef PubMed.
- D. J. Kiebala, Z. Fan, C. Calvino, L. Fehlmann, S. Schrettl and C. Weder, Org. Mater., 2020, 02, 313–322 CrossRef CAS.
- Y. Sagara, M. Karman, E. Verde-Sesto, K. Matsuo, Y. Kim, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2018, 140, 1584–1587 CrossRef CAS PubMed.
- Y. Sagara, M. Karman, A. Seki, M. Pannipara, N. Tamaoki and C. Weder, ACS Cent. Sci., 2019, 5, 874–881 CrossRef CAS PubMed.
- T. Muramatsu, Y. Sagara, H. Traeger, N. Tamaoki and C. Weder, ACS Appl. Mater. Interfaces, 2019, 11, 24571–24576 CrossRef CAS PubMed.
- T. Muramatsu, Y. Okado, H. Traeger, S. Schrettl, N. Tamaoki, C. Weder and Y. Sagara, J. Am. Chem. Soc., 2021, 143, 9884–9892 CrossRef CAS PubMed.
- Y. Sagara, H. Traeger, J. Li, Y. Okado, S. Schrettl, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2021, 143, 5519–5525 CrossRef CAS PubMed.
- G. Creusen, R. S. Schmidt and A. Walther, ACS Macro Lett., 2021, 671–678 CrossRef CAS.
- R. Merindol, G. Delechiave, L. Heinen, L. H. Catalani and A. Walther, Nat. Commun., 2019, 10, 528 CrossRef CAS PubMed.
- K. Imato, R. Yamanaka, H. Nakajima and N. Takeda, Chem. Commun., 2020, 56, 7937–7940 RSC.
- J. Chen, A. W. Ziegler, B. Zhao, W. Wan and A. D. Q. Li, Chem. Commun., 2017, 53, 4993–4996 RSC.
- H. Traeger, Y. Sagara, D. J. Kiebala, S. Schrettl and C. Weder, Angew. Chem., Int. Ed., 2021, 60, 16191–16199 CrossRef CAS PubMed.
- W. Wang, L.-S. Li, G. Helms, H.-H. Zhou and A. D. Q. Li, J. Am. Chem. Soc., 2003, 125, 1120–1121 CrossRef CAS PubMed.
- J. J. Han, A. D. Shaller, W. Wang and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 6974–6982 CrossRef CAS PubMed.
- A. D. Q. Li, W. Wang and L.-Q. Wang, Chem. – Eur. J., 2003, 9, 4594–4601 CrossRef CAS PubMed.
- T. A. Barendt, I. Rašović, M. A. Lebedeva, G. A. Farrow, A. Auty, D. Chekulaev, I. V. Sazanovich, J. A. Weinstein, K. Porfyrakis and P. D. Beer, J. Am. Chem. Soc., 2018, 140, 1924–1936 CrossRef CAS PubMed.
- S. Alp, Ş. Erten, C. Karapire, B. Köz, A. O. Doroshenko and S. İçli, J. Photochem. Photobiol., 2000, 135, 103–110 CrossRef CAS.
- C. Kulkarni, G. Periyasamy, S. Balasubramanian and S. J. George, Phys. Chem. Chem. Phys., 2014, 16, 14661–14664 RSC.
- H. Yoo, H. W. Bahng, M. R. Wasielewski and D. Kim, Phys. Chem. Chem. Phys., 2012, 14, 2001–2007 RSC.
- H. Yoo, J. Yang, A. Yousef, M. R. Wasielewski and D. Kim, J. Am. Chem. Soc., 2010, 132, 3939–3944 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S17, Tables S1 and S2, Movies S1–S3, and a comprehensive account of all experimental details, including synthetic procedures, analytical data, and NMR spectra. See DOI: https://doi.org/10.1039/d2py00218c. The source data of this study are available from the Zenodo repository at DOI: 10.5281/zenodo.6497254. |
| This journal is © The Royal Society of Chemistry 2022 |