
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterisation of polyamides based on 2,5-furandicarboxylic acid as a sustainable building block for engineering plastics†
Muhammad
Kamran
 *ab,
Matthew G.
Davidson
*ab,
Sicco
de Vos
c,
Vasilios
Tsanaktsis
c and
Bahar
Yeniad
c
*ab,
Matthew G.
Davidson
*ab,
Sicco
de Vos
c,
Vasilios
Tsanaktsis
c and
Bahar
Yeniad
c
aCentre for Sustainable and Circular Technologies, University of Bath, Claverton Down, BA2 7AY, UK. E-mail: m.kamran@bath.ac.uk; m.g.davidson@bath.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, BA2 7AY, UK
cCorbion Biochem B.V., Arkelsedijk 46, 4206 AC, Gorinchem, The Netherlands
First published on 16th May 2022
Abstract
Polyphthalamides (PPAs) are promising engineering thermoplastics employed in several demanding applications. At present, most of the commercially available PPAs are based on non-renewable petroleum derived resources. Herein, we investigated the synthesis of a semi-aromatic polyamide, poly(hexamethylene furanamide) (PA6F), based on a bio-based monomer, 2,5-furandicarboxylic acid (FDCA), using melt polycondesation in the presence of two catalysts: Ti-isopropoxide (TIPT) and Ti-citrate (TIC). Initial experimentation and optimisation conducted in a thin-film reactor led to PA6F having number-average molecular weight (Mn) of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 at very low catalyst loading (400 ppm TIPT). The reaction was scaled-up to further assess the catalytic activity of these catalysts. Both catalysts displayed strong selectivity for the ester amidation reaction compared to the uncatalysed system, resulting in PA6Fs with improved molecular weights and glass transition temperatures. Incorporating a slight excess (4.5 mol%) of hexamethylenediamine (HMDA) in the feed, resulted in further enhancement in the glass transition temperature; an increase of up to 10 °C was observed. The polymer structure and properties were extensively investigated with the help of a range of analytical techniques. The resultant polymer showed high glass transition temperature (130 °C) and elastic modulus (3.5 GPa), and comparable thermal stability to its structural analogue poly(hexamethylene terephthalamide) (PA6T). For the uncatalysed system, MALDI-ToF mass spectrometry revealed a series of methylated chain ends at both oligomerisation and polycondensation steps, inhibiting molecular weight growth.
000 g mol−1 at very low catalyst loading (400 ppm TIPT). The reaction was scaled-up to further assess the catalytic activity of these catalysts. Both catalysts displayed strong selectivity for the ester amidation reaction compared to the uncatalysed system, resulting in PA6Fs with improved molecular weights and glass transition temperatures. Incorporating a slight excess (4.5 mol%) of hexamethylenediamine (HMDA) in the feed, resulted in further enhancement in the glass transition temperature; an increase of up to 10 °C was observed. The polymer structure and properties were extensively investigated with the help of a range of analytical techniques. The resultant polymer showed high glass transition temperature (130 °C) and elastic modulus (3.5 GPa), and comparable thermal stability to its structural analogue poly(hexamethylene terephthalamide) (PA6T). For the uncatalysed system, MALDI-ToF mass spectrometry revealed a series of methylated chain ends at both oligomerisation and polycondensation steps, inhibiting molecular weight growth.
Introduction
Polyamide (PA) is a vital member of the engineering plastics family with an estimated market size of 30 billion USD in 2020.1 A recent market study has projected a CAGR (compound annual growth rate) of 6.2% for the global polyamide market which is estimated to reach 53 billion USD by 2028.2 Polyamides in general are characterised by their superior thermal, solvent and abrasion resistance and gas barrier properties along with excellent mechanical strength.3,4 Due to these highly desirable properties, polyamides are utilised in fibre production, electrical, electronics, automotive, household goods and food packaging applications.1 However, a major drawback of aliphatic PAs such as poly(butylene adipamide) (PA46), poly(hexamethylene adipamide) (PA66, nylon 66), and poly(caprolactam) (PA6, nylon 6) is their moisture susceptibility. Moisture acts as a plasticiser, resulting in mechanical strength deprivation as well as poor dimensional stability.5 Introducing aromatic rings into the polymer backbone not only reduces moisture uptake, it also provides added benefits of higher glass transition temperature (Tg), melting temperature (Tm) and better mechanical property retention at higher temperatures. This leads to the production of semi or all-aromatic (aramid) polyamides, depending on the structure of diacid and diamine units utilised for the synthesis. Semi-aromatic polyamides, also termed as polyphthalamides (PPAs) display excellent chemical resistance, dimensional stability and high temperature performance; with the additional advantage of being melt process-able, which is not possible for aramids.6–9 For these reasons, PPAs have attracted a great deal of commercial attention and to date, several different grades and chemistries are available from various producers for use as high-performance engineering plastics.3,10 At present, poly(decamethylene terephthalamide) (PA10T), which utilises C10 diamine (1,10-diaminodecane) derived from castor oil, is the only (partially) bio-based PPA that is commercially available.11,12 To further expand the bio-based options available for PPAs, renewable replacements for the aromatic monomers, terephthalic acid (TPA) and isophthalic acid (IPA) are needed. 2,5-furandicarboxylic acid (FDCA) is an excellent candidate for this role due to its structural similarities with TPA13 and has been considered as one of the most valuable bio-based building block that can be converted into high value chemicals and materials.14,15 FDCA can be produced by the oxidation of 5-(hydroxymethyl) furfural (HMF) derived from a number of renewable carbohydrates based resources.16–19The initial studies on furan-based polyamides (FPAs) were mainly concern on their synthesis and limited data was available on the properties of the resultant polymers.20–24 However, decarboxylation of the FDCA-based ammonium salt was identified as a major side reaction resulting in low molecular weight polymers.13,20–22 Cousin and co-workers synthesised furan-based polyamides and its co-polyamides with IPA using the ammonium salt route.25 Again, strong decarboxylation was reported in the system where only FDCA and hexamethylenediamine (HMDA) salt was subjected to polycondensation. The undesired decarboxylation resulted in a low molecular weight product (Mn ∼2400 g mol−1) having a Tg of 95 °C. In another study, Cao et al. also reported decarboxylation during synthesis of polyamides starting from FDCA and a 1,10-diaminodecane derived salt.12 Recently, T. Shen et al. have synthesised FPAs with improved molecular weights (Mn ∼10000 g mol−1, relative to polymethyl methacrylate standards) via the ammonium salt route.26
To circumvent this decarboxylation, alternative synthetic methodologies were investigated by various groups, primarily using solvents-based techniques. Luo and co-workers have published a study on the direct polycondensation of FDCA with various aromatic diamines.27,28 After reaction optimisation, the group was able to synthesise furanic-aromatic polyamides in high molecular weights (Mn ∼40–80 kg mol−1). Interfacial polymerisation was employed by Cureton et al. to prepare high molecular weight FPAs with various aliphatic and aromatic diamines.29 In a series of patents, a process for preparing FPAs starting from dimethyl 2,5-furandicarboxylate (DMFDC) has been disclosed.30,31 In this process, synthesis was partially carried out in solution. However, the poly(hexamethylene furanamide) (PA6F) obtained in the example had a Mn ∼7000 g mol−1. Enzymatic polymerisation was performed by Jiang and co-workers to prepare FPAs.8,9 In their work, DMFDC was reacted with various aliphatic diamines in toluene or diphenyl ether.
Melt polycondensation of DMFDC in the absence of solvent has also been explored by some researchers. Grosshardt et al. synthesised FPAs by melt polycondensation of DMFDC and various diamines in the presence of a butyltin chloride dihydroxide catalyst.32 However, their results showed low molecular weight products despite using higher catalyst concentrations (0.13–0.20 mol%). A process for preparing FPAs through melt condensation of DMFDC and HMDA utilising catalyst was disclosed in a patent.33 Again, higher catalyst concentration and longer polycondensation time (up to 6 hours) were employed in the examples. No data for molecular weights on as-synthesised polymer samples was provided. Results from the best example showed Mn of 11600 g mol−1 after solvent purification.
In spite of these extensive efforts, existing synthetic approaches to produce FPAs have limited application since they are unable to produce polymers with a reasonable molecular weight through a scalable process. Ammonium salt-based approach is prone to decarboxylation, while solvent-based routes are not feasible for industrial scale-up. Moreover, the commonly used solvents for these polymerisations are expensive, challenging to recycle and are derived from petroleum resources, thus contributing towards the carbon footprint of the product. Melt polycondensation of DMFDC stands out as an attractive process from industrial scale-up potential, however, attempts until now have only resulted in low molecular weight polymers. Herein, due to the renewed interest in bio-based semi-aromatic polyamides, we have addressed the key challenges associated with developing a scalable and sustainable process for FPAs production. We report the melt polycondensation of DMFDC and HMDA using highly active and environmentally benign titanium-based catalyst systems to yield higher molecular weight furan-based polyamide PA6F.
Results and discussion
Synthesis of dimethyl furan-2,5-dicarboxylate (DMFDC)
Dimethyl furan-2,5-dicarboxylate (DMFDC) was prepared by esterification of furan-2,5-dicarboxylic acid (FDCA) with methanol in the presence of an acid (H2SO4). The structure of DMFDC was confirmed by 1H NMR and 13C NMR (Fig. S1, ESI†). The furanic protons at 7.44 ppm and methyl (–CH3) protons at 3.87 ppm were found to be in the expected 1:3 ratio. Minor peaks around furan and methyl protons are satellite peaks. Importantly, no traces of FDCA were found to be present as confirmed by absence of carboxylic acid protons at a chemical shift of around 13.5 ppm.
Synthesis and characterisation of bio-based furanic polyamide poly(hexamethylene furanamide) (PA6F)
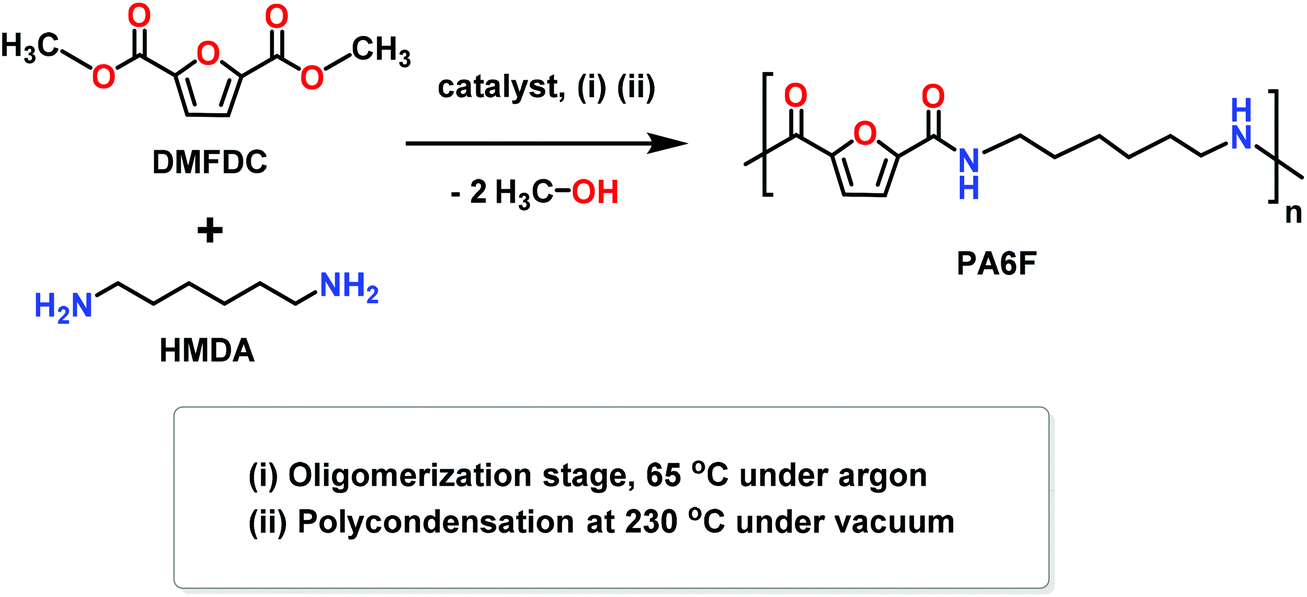 | ||
| Scheme 1 Synthesis of furan-based polyamide PA6F using melt polycondensation. | ||
Initially, the melt phase polycondensation of DMFDC and HMDA in stoichiometric amounts was carried out in a small scale (250 mg feed) thin-film reactor.38 Thin-film reactors are considered effective in countering mass transfer limitations and to facilitate diffusion and removal of volatiles, which are challenging tasks to achieve in a high viscosity polymer system. In our study, the main objective of utilising the thin-film reactor was to test and further develop the melt polycondensation of FPAs, screen catalysts and perform initial optimisation of the reaction (see section 4, ESI†).
As mentioned earlier, Grosshardt et al. had synthesised various furan-based polyamides by catalytic melt polycondensation technique.32 However, the resultant polymers showed low molecular weights. In their work, the oligomerisation was conducted at a higher temperature (140 °C), which may have caused significant N-methylation of the macromolecules, a side-reaction that limits molecular weight growth (section 4, ESI†).30 For our two-step synthesis procedure, the first step of oligomerisation was conducted at 65 °C under an argon atmosphere to minimize the N-methylation of the chain ends. 65 °C was chosen to promote the removal of some of the methanol produced in the initial phase of the reaction. Whereas, polycondensation was carried out at 230 °C under reduced pressure. Preliminary catalyst screening revealed that titanium(IV) isopropoxide (TIPT) and ultra-pure titanium(IV) citrate (TIC) showed promising results. Fig. 1 represents the inherent viscosities and glass transition temperature (Tg) of PA6F polyamides synthesised at different loadings of TIPT (see also Fig. S8 ESI†). A gradual increase in both inherent viscosity and Tg is evident with catalyst incorporation compared to uncatalysed reaction. These results were also corroborated with molecular weight data obtained by GPC analysis (Table S4 ESI†). Thus, a bio-based, semi-aromatice polyamide with a relatively high molecular weight (Mn = 14000 g mol−1, Mw = 43000 g mol−1) was achieved at 400 ppm TIPT, following a simple and environmentally benign catalytic approach. During the review of this paper, S. Xie et al. published a study on the synthesis of poly(decamethylene furanamide) (PA10F) by melt polycondensation using organocatalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) also having a relatively high molecular weight (Mn up to 29000 g mol−1, relative to polymethyl methacrylate calibration).39
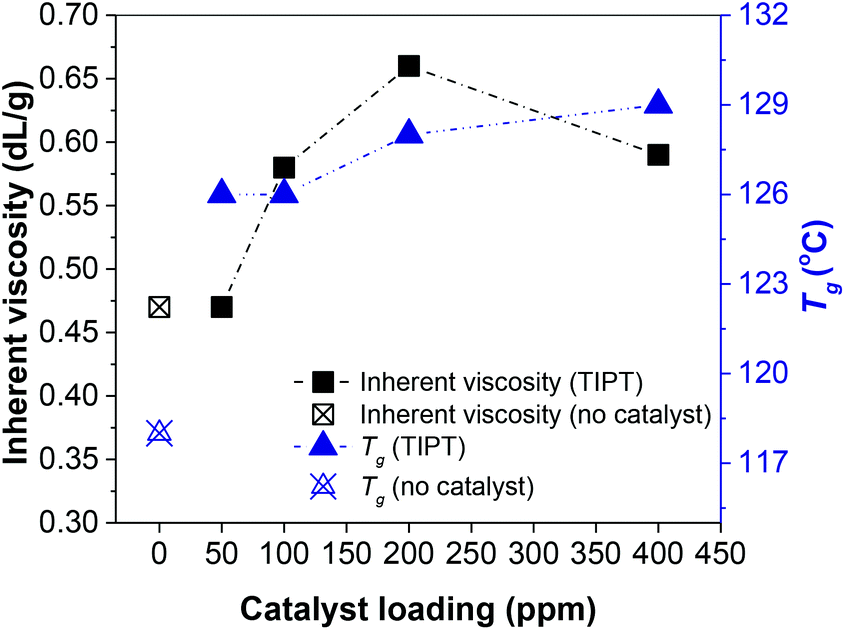 | ||
| Fig. 1 Inherent viscosity and glass transition temperatures (Tg) for PA6Fs synthesised using thin-film reactor. | ||
The chemical structure of PA6F was confirmed with 1H and {1H}13C NMR experiments. As a representative example, Fig. 2(A) depicts 1H and 13C NMR spectra for PA6F in DMSO-d6 with corresponding assignments. Chemical shifts were assigned with assistance from 2D experiments (not shown here). From 1H NMR, the characteristic amide proton at 8.51 ppm is distinguishable due to strong deshielding effect of the neighbouring nitrogen atom.
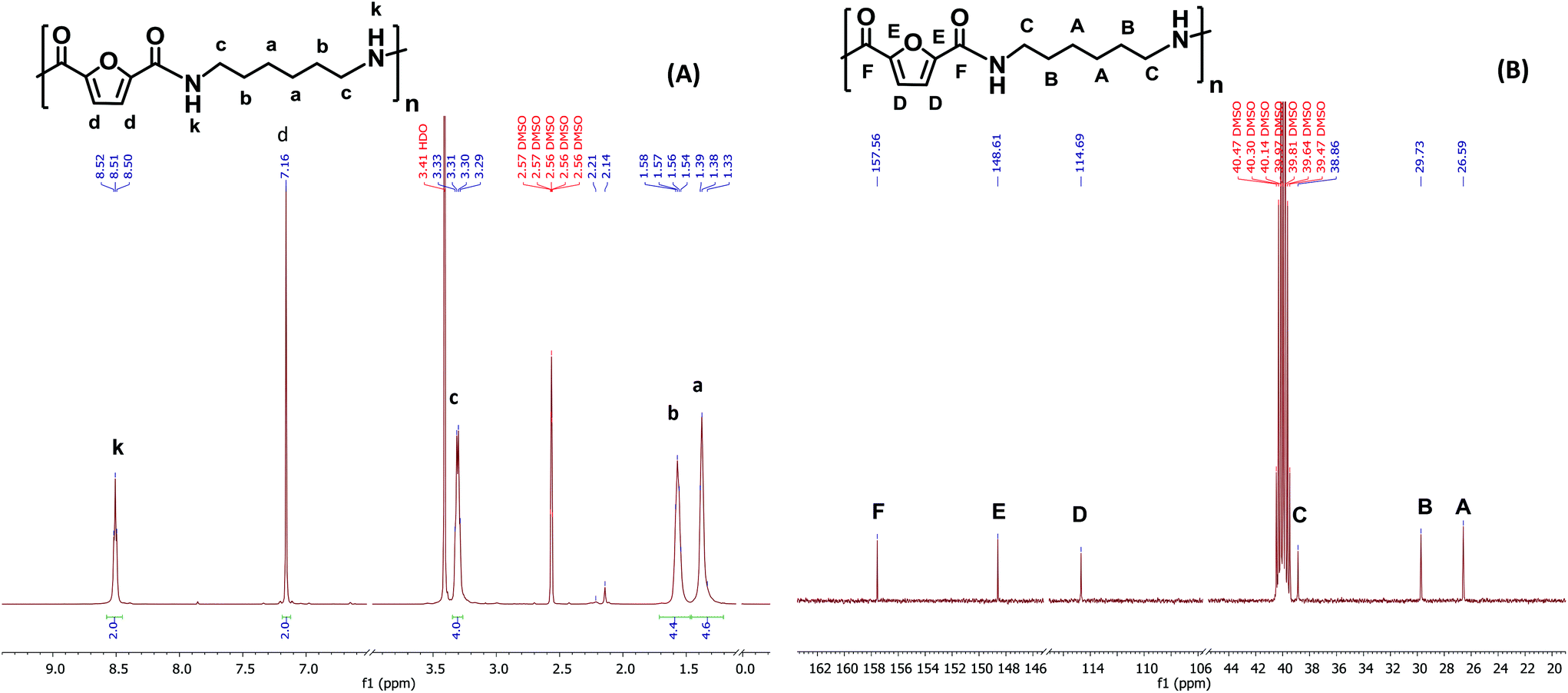 | ||
| Fig. 2 Representative NMR spectra (A) 1H NMR and (B) {1H}13C NMR, recorded for a PA6F polymer sample in DMSO-d6 at 298 K. | ||
For the oligomerisation step, the reaction contents were stirred at 65 °C for a period of time until the reaction resulted in a white solid agglomerate after subsequent increase in the viscosity. Beyond this point, it was not possible to continue stirring at the set temperature. The progress of the reaction was monitored by recording the time needed to form the solid agglomerate and the amount of methanol collected through distillation, calculated as a percentage of the theoretical amount to be liberated when reaction goes to completion. Fig. 3 represents performance comparison of both catalysts during the oligomer formation. It is evident that both catalysts showed faster kinetics to oligomerisation compared to uncatalysed control reaction, TIC in particular was much quicker. Overall, almost similar amounts of methanol were recovered after the agglomerate formation, suggesting comparable conversions during this stage, irrespective of the catalyst addition. The degree of polymerisation (DP), calculated using 1H NMR after oligomerisation, was also found to be in the close range of 10 to 13 units (Fig. S9 and Table S5 ESI†).
 | ||
| Fig. 3 Oligomerisation step conducted in the presence of both TIPT and TIC at different loadings and uncatalysed reaction. Time needed to reach solid agglomerate formation (blue pattern bars) and amount of methanol distilled off expressed as percentage of theoretical amount to be liberated when reaction goes to completion (red squares). | ||
Fig. 4(A) depicts torque versus time data recorded over the course of the polycondensation step for these samples. It is clear that the uncatalysed reaction failed to show any viscosity increase after two hours of polycondensation. On the other hand, samples where catalysts were incorporated, showed torque increase over time. For TIPT, an induction period of approximately 40 minutes was observed, after which the torque increase was significant. In the case of TIC, virtually no induction period was observed and increase in torque was evident from the start of the polycondensation reaction. Moreover, it is also apparent that TIC resulted in higher torque values at similar loadings compare to TIPT (Fig. 4B). These results also support our earlier observations in the oligomerisation step, where TIC showed faster kinetics.
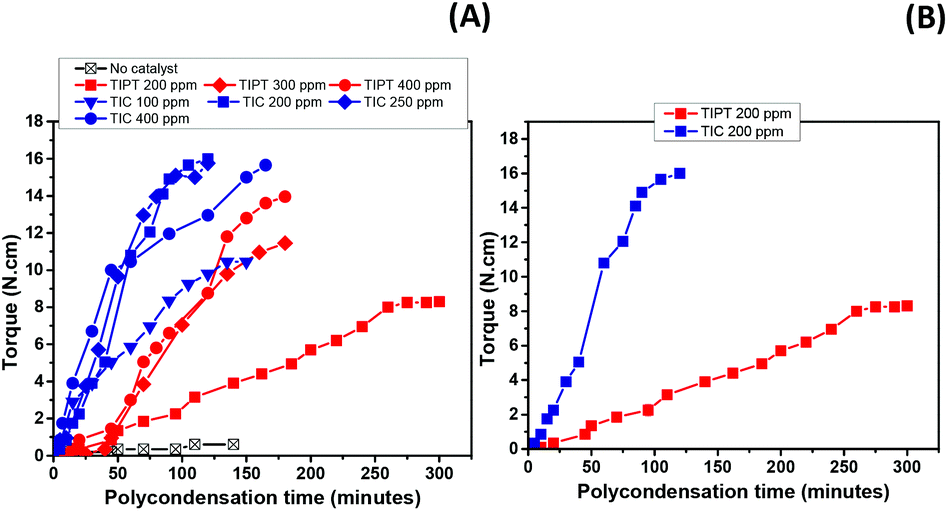 | ||
| Fig. 4 (A) Toque values recorded during polycondensation reactions in the melt employing different loadings for both catalysts (TIPT and TIC) and (B) Torque comparison at similar loadings for TIPT and TIC. | ||
The faster kinetics and absence of an induction period in the TIC case, as observed in our reaction system, lies in the fact that TIC is produced using ultra-pure titanium(IV) isopropoxide which limits the formation of disadvantageous titanium-oxo species.40
During DSC analyses of PA6Fs (Fig. S10–S11 ESI†), no melting endotherms were observed on the second heating curves together with an absence of crystallisation exotherms on the cooling or heating scans. It has been proposed that the presence of a furan ring in polyamides reduces favourable interchain hydrogen bonding interactions between amide and carbonyl moieties.28,41 Interchain hydrogen bonding is considered vital for the crystallisation and improved thermal properties of polyamides in general. The weaker interchain hydrogen bonding in the kinked PA6F structure could result in lower, in addition to slower crystallisation behaviour, which could explain the absence of melting endotherms in our DSC analyses. Moreover, on the first heating scan, a broad Tg and a less pronounced melting endotherm was observed for some samples (e.g., Fig. 5). We suggest this behaviour is due to annealing affects caused by the thermal history of the sample.
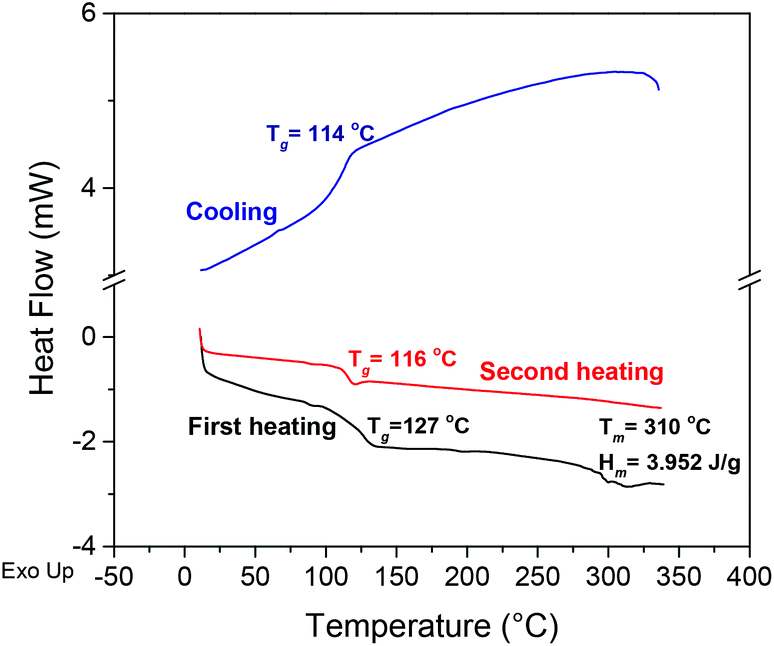 | ||
| Fig. 5 DSC thermograms for PA6F sample synthesised with TIPT 200 ppm (Table 1, entry 2). | ||
Detailed DSC and GPC data for above samples is presented in Table 1. As evident, Tg of the uncatalysed sample is lower than all other samples where catalyst was incorporated. It was not possible to determine the absolute molecular weight of the uncatalysed sample due to very low light-scattering signals in the GPC analysis, hampering the peak integration.
| Entry | Cat. | Catalyst loadinga (ppm) | As-synthesised polymer analysis | Polymer analysis after purification | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T g (°C) | M n (kg mol−1) | M w (kg mol−1) | Đ | T g (°C) | M n (kg mol−1) | M w (kg mol−1) | Isolated yieldd (%) | |||
| a Calculated based on weight of metal (Ti) in the catalyst relative to the weight of DMFDC in the feed. b Measured on the second heating curve of the DSC thermogram, no melting endotherm was detected in any of these samples. c Performed using HFIP as mobile phase and evaluated using triple detection method to obtain absolute molecular weights. d Calculated from the purification step. e Sample displayed low intensity light scattering signals, hampering absolute molecular weight determination. f For this run, polycondensation was conducted for 5 hours under similar conditions as entry 4. | ||||||||||
| 1 | No cat. | 0 | 110 | —e | 131 | 8 | 11 | 36 | ||
| 2 | TIPT | 200 | 116 | 5 | 10 | 1.94 | 128 | 12 | 16 | 73 |
| 3 | TIPT | 300 | 121 | 6 | 10 | 1.67 | 130 | 13 | 17 | 64 |
| 4 | TIPT | 400 | 121 | 8 | 12 | 1.51 | 130 | 14 | 18 | 71 |
| 5 | TIPT | 400 | 119 | 8 | 13 | 1.62 | 130 | 14 | 18 | 66 |
| 6 | TIC | 100 | 118 | 5 | 10 | 1.92 | 130 | 13 | 16 | 66 |
| 7 | TIC | 200 | 116 | 6 | 12 | 1.98 | 130 | 13 | 19 | 70 |
| 8 | TIC | 250 | 116 | 7 | 12 | 1.71 | 130 | 12 | 16 | 77 |
| 9 | TIC | 400 | 117 | 6 | 12 | 1.92 | 130 | 14 | 20 | 72 |
It is apparent from these results that catalyst addition has a considerable impact in improving the molecular weight of PA6F under our conditions. TIPT at 400 ppm loading appears to give the best results. For TIC, lower concentration of catalyst was preferred, as at higher loadings, low molecular weight dark coloured polymer was produced. Indeed, longer polycondensation time did not show any significant impact on the end product (Table 1, entries 4 and 5). Moreover, when glass reactor samples were analysed after subjecting them to a solvent purification step, both Tg and molecular weights were improved. The yield of purified polymer in the case of uncatalysed sample was significantly lower compared to catalysed reactions.
It is clear from data presented in Table 1 and those for the thin-film reactor (Table S4 ESI†), that molecular weights achieved with glass reactor were on lower side at similar synthesis conditions. This could be attributed to the effective diffusion and volatile (methanol) removal from the system in the case of polymer thin film.
Reactions employing excess diamine (HMDA)
Reaction stoichiometry plays a pivotal role in step-growth polymerisation to attain high molecular weight polymer. We were interested to study the effect of reaction stoichiometry on the polymer molecular weight and properties. In particular, the reactions employing an excess of diamine monomer. Experiments were conducted to investigate the effect of utilising excess HMDA in our system. TIPT at 400 ppm and TIC 100 ppm were chosen as optimum catalyst loadings, whereas the amount of diamine was varied in 2, 4.5 and 10 mol% excess relative to the DMFDC in the feed. Results are compared with those without and with excess diamine (4.5 mol% excess) but with no catalyst. The torque versus time behaviour, representing the viscosity of the melt during polycondensation step, is depicted in Fig. 6, while DSC and GPC analysis results of these samples are presented in Table 2.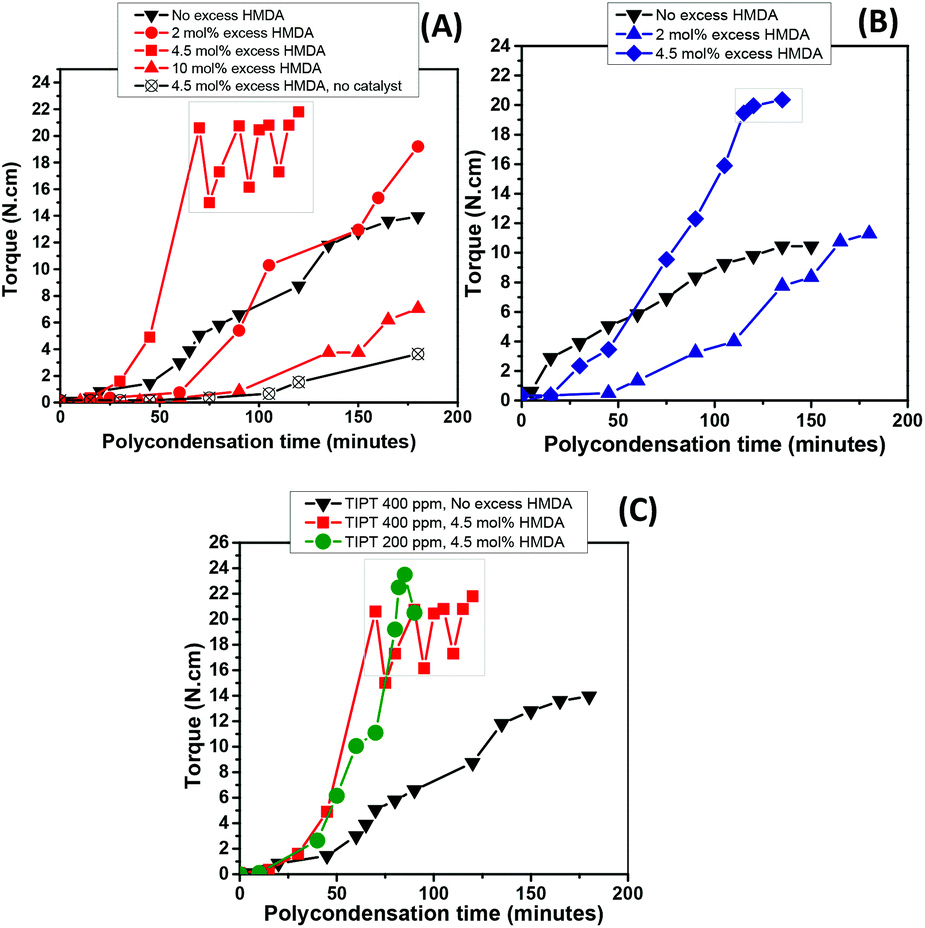 | ||
| Fig. 6 Torque versus time plots for the polycondensation step of PA6F synthesised with an excess HMDA using different catalyst loadings (A) TIPT 400 ppm (B) TIC 100 ppm and (C) TIPT 400 ppm and 200 ppm. | ||
| Entry | Cat. | HMDA excess (mol%) | T g (°C) | M n (kg mol−1) | M w (kg mol−1) | Đ |
|---|---|---|---|---|---|---|
|
a Measured on the second heating curve of the DSC thermogram.
b Performed using HFIP as mobile phase and evaluated using triple detection method to obtain absolute molecular weights.
c Monomers in stoichiometric amounts (DMFDC:HMDA = 1:1).
d Reaction performed at TIPT loading of 200 ppm.
e Polycondensation temperature 230 °C for 1 h then 240 °C for 0.5 h.
f Polycondensation temperature 240 °C.
|
||||||
| 1 | No cat. | 4.5 | 119 | 5 | 11 | 2.20 |
| 2 | TIPT | —c | 121 | 8 | 12 | 1.51 |
| 3 | TIPT | 2 | 125 | 9 | 17 | 1.90 |
| 4 | TIPT | 4.5 | 130 | 9 | 23 | 2.56 |
| 5 | TIPT | 10 | 131 | 8 | 17 | 2.18 |
| 7 | TIPTd | 4.5 | 128 | 8 | 17 | 2.14 |
| 8e | TIPT | 4.5 | 132 | 11 | 85 | 7.60 |
| 9f | TIPT | —c | 120 | 9 | 14 | 1.61 |
| 10 | TIC | —c | 118 | 5 | 10 | 1.92 |
| 11 | TIC | 2 | 123 | 8 | 13 | 1.69 |
| 12 | TIC | 4.5 | 132 | 9 | 24 | 2.65 |
It is clear from Fig. 6(A) and (B) that a 2 mol% excess HMDA does not affect the polycondensation rate significantly and final torque values achieved with both catalysts were almost comparable to those without any excess HMDA. However, at 4.5 mol% excess, a dramatic increase in the melt viscosity was observed as evident by the rapid torque increase. The reaction reached the maximum torque limit of our equipment (20 N cm) in approximately 70 and 120 minutes with TIPT (400 ppm) and TIC (100 ppm), respectively. After which, either reaction temperature was increased by 5–10 °C, or stirrer rpm was reduced to bring the torque value within the limits (enclosed within the boxes in Fig. 6). Due to very high torque response within such a short time, it was possible to reduce the TIPT catalyst concentration from 400 to 200 ppm (Fig. 6C) without significantly changing the torque profile. However, 10 mol% excess HMDA run did not reach high torque values even after 180 minutes.
The effect of introducing excess diamine was also evident in the polymer properties (Table 2). An increase in glass transition temperatures (Tg) was noticeable with increase in molar excess of HMDA. The increase in Tg was less pronounced with 2 mol% excess, while higher increase (approximately 10 °C) was observed in the case of 4.5 mol% and 10 mol%. Again, no melting endotherm was observed for any of these samples. Overall, samples synthesised with excess HMDA displayed broader molecular weight distribution, as represented by slightly higher dispersities (Đ) in contrast to the runs conducted with stoichiometric amounts (Table 1). This phenomenon can be explained by the fact that incorporation of excess diamine may result in more amino terminated chain ends which can combine with polymer chains containing terminal ester groups (see MALDI-ToF analysis), accounting for some of the molecular weight improvement observed. However, the amino end-groups are also prone to initiate transamidation in the macromolecules under these conditions, resulting in chain scission, therefore, producing polymer with broader molecular weight distribution.42,43 Moreover, the reaction performed at 4.5 mol% excess using higher polycondensation temperature (Table 2, entry 8) in contrast to the stoichiometric run (Table 2, entry 9), showed higher dispersity. This could be attributed to the chain branching and subsequent crosslinking phenomenon happening as a result of the condensation reaction between amino end-groups, also observed by Puglisi et al. while studying the PA66 thermal gelation at high temepratures.44 This is also consistent with the presence of an insoluble polymer fraction observed when sample from this run was dissolved in HFIP for GPC analysis.
As mentioned previously, torque increase over time was not witnessed in the case of 10 mol% excess diamine in the feed. Interestingly, the run with 10 mol% excess, despite having marked differences in molecular weight with uncatalysed sample (Table 2, entry 1), ended up showing similar torque development during the polycondensation (Fig. 6A). The fact that this observation was only apparent at 10 mol% diamine excess, suggests that some residual fraction of the unreacted diamine may still be present in the system which may have reduce the melt viscosity by disrupting the interchain hydrogen bonding interactions.
Polymers synthesised using stoichiometric quantities of monomers in the feed showed slight yellowish colouration, but those without catalyst and excess diamine had a strong orange hue (Table S6 ESI†). Further investigation into the underlying phenomenon for this discoloration was beyond the scope of this study. A plausible explanation could be the thermo-oxidative degradation of polymer under these conditions resulting in the formation of azomethine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) intermediates, which undergo a series of consecutive condensation reactions, resulting in sequences with conjugated double bonds which generate UV/Vis active chromophores.4,45 Since this process is facilitated by the presence of amino end-groups which help produce more azomethines, this explains more discolouration observed with increasing molar excess of diamine.
N) intermediates, which undergo a series of consecutive condensation reactions, resulting in sequences with conjugated double bonds which generate UV/Vis active chromophores.4,45 Since this process is facilitated by the presence of amino end-groups which help produce more azomethines, this explains more discolouration observed with increasing molar excess of diamine.
MALDI-ToF mass spectrometry analysis
MALDI-ToF analyses were conducted to study the end-groups distribution in the PA6F oligomer and polymer samples. Fig. 7(A) and (B) shows MALDI spectra recorded for the polymer synthesised without any catalyst and with 400 ppm TIPT respectively (also Fig. S12 ESI†). Whereas, Tables S7 and S8 ESI† list data after the interpretation of MALDI peaks. Both samples showed several distinctive end-groups series in addition to the expected amino and ester terminated end-groups. In the uncatalysed sample (Fig. 7A and Table S7†), significant methylation of terminal amino groups was observed, as evident from end-groups shown in series B, C, H and I. Moreover, some decarboxylation of the end-groups also took place (series A). Both of these side reactions have already been cited as the main reason for lower molecular weight of FPA's. In contrast, catalysed sample (Fig. 7B and Table S8†) showed only three less-abundant series of methylated end-groups (C, D and G).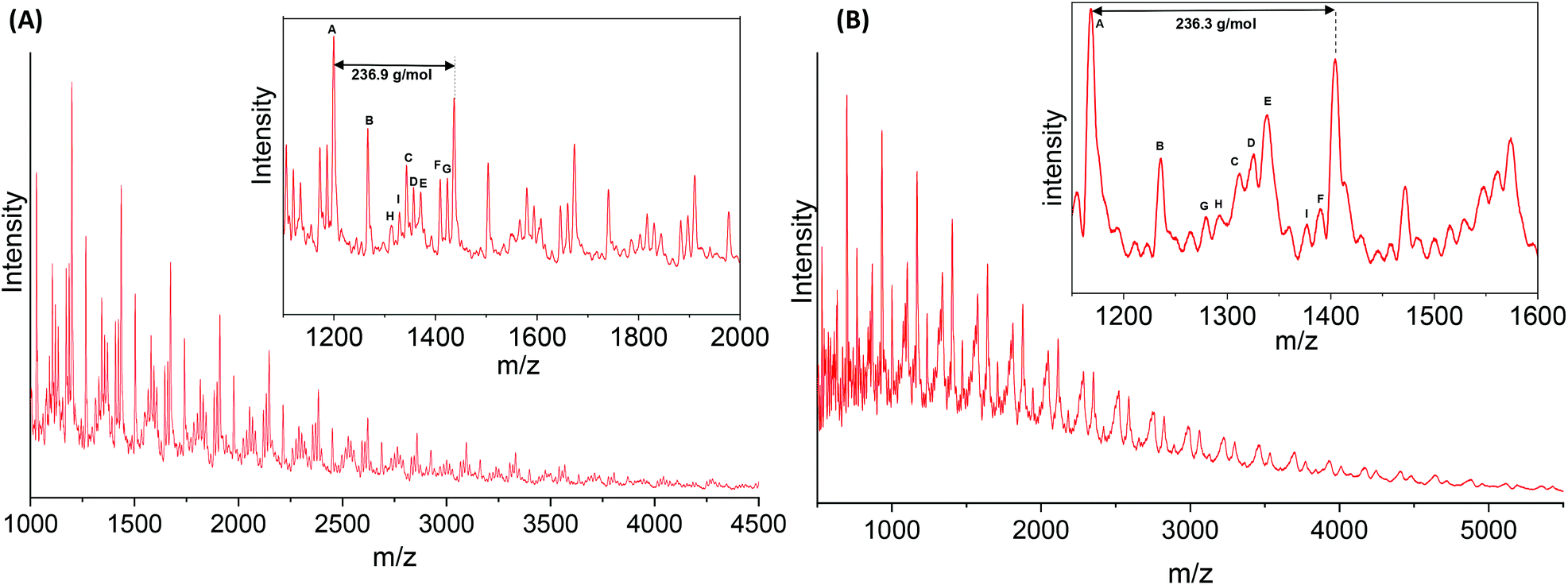 | ||
| Fig. 7 MALDI-ToF MS spectra for PA6F polyamides synthesised (A) without any catalyst (Table 1, entry 1) and (B) using TIPT 400 ppm catalyst (Table 1, entry 4). | ||
Both samples also showed chains terminated with acid end-groups. Acid end-groups may generate as a result of the hydrolysis of esters,9 possibly due to the presence of residual water. Another possible route to acid formation is the N-methylation of amino end-groups which also results in carboxylic acid as a by-product (section 4 ESI†). As the furanic carboxylic acid terminal group is known to have lower thermal stability, existence of acid end-groups could also explain the decarboxylated end-group seen in both samples.12,25 Interestingly, the uncatalysed oligomer sample analysed at the end of the first step also showed a significantly greater proportion of methylated end-groups in comparison to TIPT catalysed oligomer (Fig. S13, S14 and Tables S9, S10 ESI†). This reflects the selectivity of TIPT catalyst towards amidation over competing methylation reaction,46 leading to higher molecular weight PA6F. However, more detailed mechanistic studies are required to provide conclusive evidence on catalytic activity and selectivity of TIPT and the side reactions taking place.
Dynamic mechanical analysis (DMA)
As mentioned earlier during DSC analysis, no melting endotherm or crystallisation exotherm was observed on the second heating scan or on the cooling scan for any of the synthesised PA6F samples. A typical behaviour indicating predominantly amorphous characteristics. To further assess the morphology of PA6F, we conducted DMA on compression moulded films of PA6F. For this purpose, polymer having a reasonably high molecular weight was chosen (Mw = 23 kg mol−1) due to the excellent film forming properties (Fig. S15†). The films were subjected to different heat treatments prior to DMA to investigate the effect of thermal history on the thermomechanical behaviour. One of the films was quenched in ice-cold water soon after the compression moulding process. This film was subsequently dried under ambient conditions. To facilitate the mobility of the amorphous phase, the second film was subjected to an isothermal annealing process just above the Tg of the polymer, i.e., at 150 °C for 15 hours. A third film, termed as “as-pressed”, was analysed without any heat treatment. All films were dried at a temperature below the Tg (100 °C) in vacuo prior to DMA measurements to remove any absorbed moisture.From Fig. 8(A), no variation in storage modulus (E′) was observed for any of the films below 120 °C, suggesting good stability and elasticity retention at temperatures below the measured Tg by DSC (130 °C). However, on further increase in the temperature, a significant drop in E′ was observed for all three samples without any apparent signs of recovery, a characteristic behaviour of glassy polymers. This change in E′ was accompanied by an increase in the tan delta, which showed a single peak maximum for the quenched film, whereas for the other two samples, two broad overlapping peaks were observed. The annealed sample showed a slightly higher softening point then the other two films. However, this increase is relatively insignificant. Further characterisation of these three films was conducted using wide-angle X-ray diffraction (WAXD).
 | ||
| Fig. 8 DMA of compression moulded PA6F film subjected to different heat treatments after the moulding (A) log storage modulus and (B) tan delta, as a function of temperature. Polymer used had a Mn = 9 kg mol−1 and Mw = 23 kg mol−1. | ||
Wide-angle X-ray diffraction (WAXD)
WAXD measurements were conducted on the same films used for the DMA. No distinct diffraction peaks were observed. Instead, all samples exhibited a broad amorphous halo signal at around 2θ ∼21° (Fig. 9). This corroborates our earlier results that PA6F demonstrates a perturbed crystallisation tendency and displays predominantly amorphous characteristics under the tested conditions.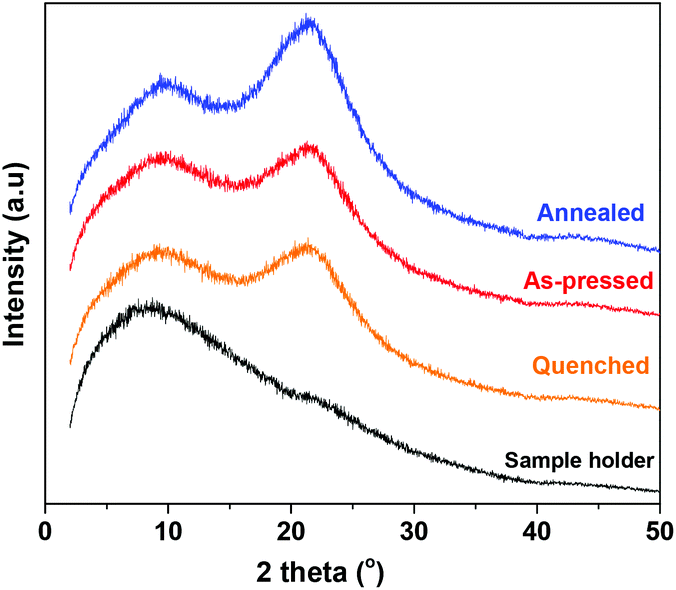 | ||
| Fig. 9 WAXD pattern of compression moulded PA6F films subjected to different heat treatments. Polymer used had a Mn = 9 kg mol−1 and Mw = 23 kg mol−1. | ||
Thermogravimetric analysis (TGA)
To evaluate the thermal stability of PA6F polyamides synthesised in this study, TGA was conducted on selected samples. Fig. 10(A) and (B) represents the TGA curves and the derivative weight loss respectively, for samples without catalyst, with 400 ppm TIPT catalyst and with 400 ppm TIPT and 4.5 mol% excess diamine in the feed. It can be observed that the uncatalysed sample showed a two-step decomposition, a minor weight loss step at around 288 °C followed by a major decomposition step aligned with other two polymers. Irrespective of the sample tested, the major decomposition step had an onset degradation temperature of 367 °C, whereas temperature for maximum rate of decomposition was recorded to be 434 °C. The early degradation step in the uncatalysed sample could be associated with the differences in end-group composition compared to the catalysed samples as observed earlier during MALDI-ToF analysis.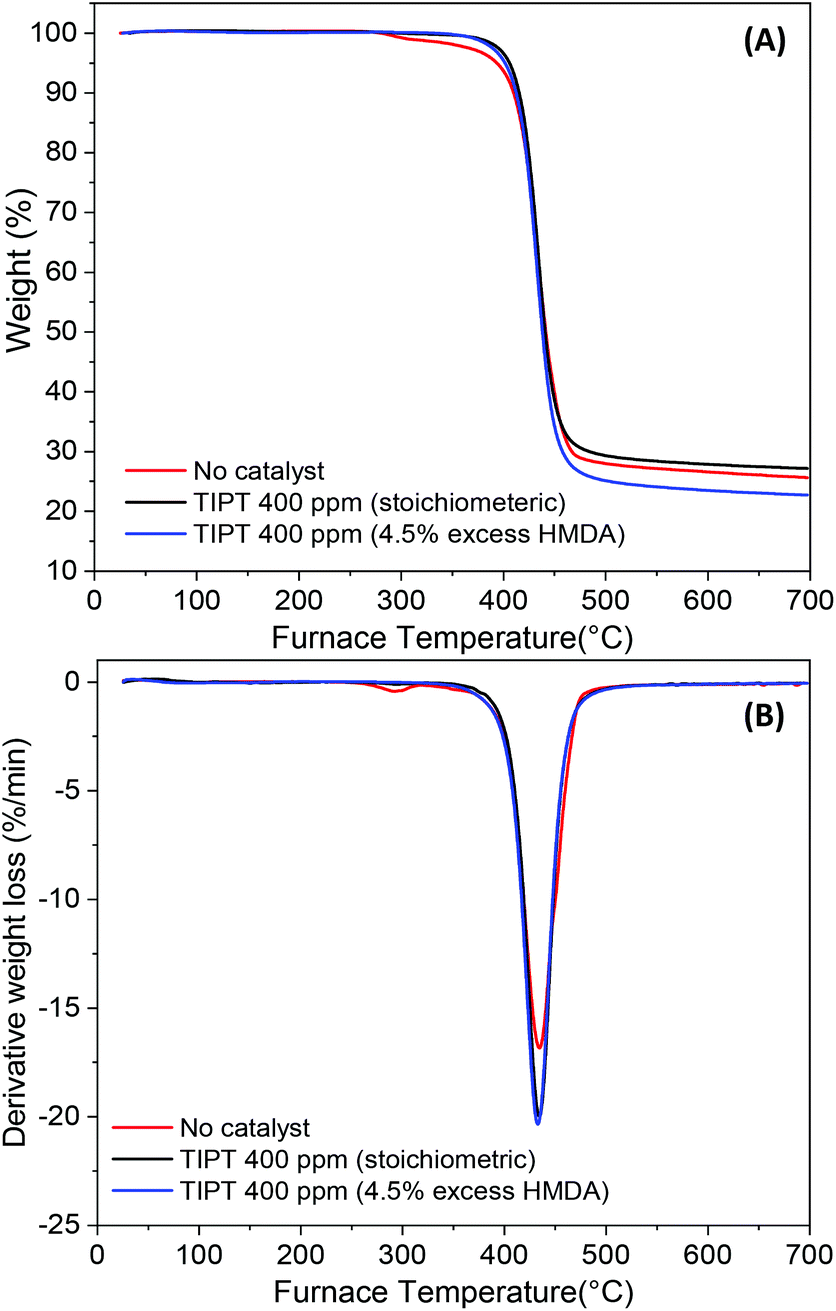 | ||
| Fig. 10 TGA analysis of PA6F polyamides (A) TGA curves and (B) derivative weight loss. | ||
PA6F's petrochemical analogue PA6T has a high melting temperature (371 °C), which makes thermal processing without degradation, an enormous challenge. Thus, PA6T is often modified during synthesis by copolymerisation with other acid/lactam/diamine monomers to reduce its melting temperature to about 280–320 °C range and enhance processability. These modifications to the polymer backbone can adversely affect some of the unique properties offered by PPAs, for instance their low moisture uptake. In contrast, besides being derived from renewable furan-based monomer, PA6F shows comparable thermal stability (Table 3) but a much wider thermal processing window, thus facilitating thermal processing of the polymer without significant degradation.
| Synthesis technique | M n (kg mol−1) | M w (kg mol−1) | T g (°C) | T m (°C) | Degradation temperatures (°C) | PA6X [Ref.] | ||
|---|---|---|---|---|---|---|---|---|
| MP = Melt polymerisation, ASR = Ammonium salt route, EP = Enzymatic polymerisation, IP = Interfacial polymerisation.a Measured through GPC using HFIP as mobile phase.b Recorded on DSC second heating scan.c Not detected following DSC second heating scan.d Onset of degradation temperature.e Temperature for 50% weight loss (Td-50%).f Temperature at maximum rate of degradation (Td-max).g Measured following cooling scan of DSC.h Temperature for 5% weight loss (Td-5%).i Measured following first heating scan in DSC.j Measured on SEC using DMSO/LiBr as mobile phase.k Determine via SEC using DMF/LiCl as mobile phase.l Not mentioned in the study.m Measured using DTA (differential thermal analysis).n Method not described. | ||||||||
| MP | 9a | 23a | 130b | —c | 367 d | 434 e | 433 f | PA6F [this work] |
| MP | 5.2a | 13.8a | 110b | —c | 350–450 | PA6F32 | ||
| ASP | 2.4a | 7.2a | 95g | —c | 380 h | 435 e | PA6F25 | |
| EP | 13.4j | 20.6j | 119b | 162i | 322 h | 443 f | PA6F9 | |
| IP | 30k | 52k | 86 | 287 h | 355 f | PA6F29 | ||
| IP | —l | —l | 125m | 371 m | 350 n | PA6T3,47 | ||
Mechanical properties
Uniaxial tensile testing was performed on compression moulded PA6F samples to assess the mechanical properties. Table 4 summarises the test results (stress–strain curves are depicted in Fig. S16 ESI†). All specimens tested showed elastic deformation and brittle fracture with low elongation at break, a typical behaviour displayed by semi-aromatic polyamides. A tensile modulus of 3.5 ± 0.7 GPa was recorded for the PA6F which is comparable to that reported for poly(hexamethylene terephthalamide-co-caprolactam) (PA6T/6) (Table 4). PA6F showed a marginally lower tensile strength of 58.8 ± 7.4 MPa compared to 72.5 MPa observed for PA6T/6. Overall, these excellent mechanical properties indicate strong potential of the furan-based semi-aromatic polyamide PA6F as a high-performance bio-based alternative to petroleum derived analogues.| Polymer | E y (GPa) | σ u (MPa) | σ b (MPa) | ε b (%) |
|---|---|---|---|---|
| a Elastic modulus, calculated within linear 0.1–0.5% strain region. b Ultimate tensile strength. c Tensile strength at break. d Elongation at break. e Polymer used had a Mn = 9 kg mol−1 and Mw = 23 kg mol−1. | ||||
| PA6Fe (this work) | 3.5 ± 0.7 | 60.9 ± 10.1 | 58.8 ± 7.4 | 3.4 ± 0.12 |
| PA6T/648 | 3.8 | — | 72.5 | 2.7 |
| PA10T49 | 1.5 | — | 63.0 | 10.0 |
Conclusions
A systematic study towards the synthesis of a high molecular weight furan-based polyamide poly(hexamethylene furanamide) (PA6F) has been conducted. The PA6F was produced in the melt using two titanium-based catalysts, Ti-isopropoxide (TIPT) and Ti-citrate (TIC) by utilising very low catalyst concentrations. The initial investigations of catalyst activities in a thin-film reactor showed that at 400 ppm TIPT, PA6F having Mn = 14000 g mol–1 and Mw = 43000 g mol−1, and a Tg of 130 °C could be synthesised following a simple two step synthesis approach.
Studying the reaction on a multigram scale revealed that both selected catalysts showed higher activity towards amidation compared to uncatalysed reactions, leading to polymers with improved molecular weight and thermal properties. TIC in particular, showed faster oligomerisation and polycondensation rates.
Moreover, investigation into reaction stoichiometry revealed that slight excess of HMDA (4.5 mol%) resulted in considerable enhancement in the glass transition temperatures over stoichiometric reactions carried out under similar conditions. Following these conditions, we were able to produce PA6F having a molecular weight of Mn = 9000 g mol−1 and Mw = 23000 g mol−1 within 2 hours of polycondensation.
The polymer structure and properties were characterised using a wide range of analytical techniques. DSC analysis showed a predominantly amorphous nature of the PA6F polymer. This was further confirmed with DMA and WAXD measurements. PA6F showed comparable thermal stability to its petroleum-based counterpart in TGA measurements, with major degradation temperature (Td-max) around 434 °C. PA6F also showed outstanding mechanical properties and displayed an elastic modulus in the similar range as PA6T/6. MALDI-ToF mass spectrometry analysis revealed significant methylation of end-groups in the uncatalysed sample, both at the oligomerisation and polycondensation steps. This side reaction may be regarded as a primary reason for inhibiting chain growth of PA6F macromolecules in the absence of catalyst, thus resulting in a low molecular weight product.
These results demonstrate that PA6F synthesised using a renewable furanic monomer and a benign catalyst system has the potential to be utilised as a promising engineering thermoplastic polymer. This could lead the foundation for directing the research in this area towards more environmentally sustainable materials and processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement no. 665992 and from Corbion Biochem B.V. (PhD studentship for MK). UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC grant EP/R027129/1. The authors gratefully acknowledge the contributions of Taco Visser, Jessica Kraak and Stefan van Berkel from Corbion for their assistance in GPC analysis and for their helpful discussions.References
- M. Pervaiz, M. Faruq, M. Jawaid and M. Sain, Curr. Org. Synth., 2016, 14, 146–155 CrossRef.
- Bloomberg, Polyamide Market Size Worth $53.32 Billion By 2028 | CAGR: 6.2%, Grand View Research, Inc., 2021, https://www.bloomberg.com/press-releases/2021-10-12/polyamide-market-size-worth-53-32-billion-by-2028-cagr-6-2-grand-view-research-inc, (accessed 20 January 2022) Search PubMed.
- C. Zhang, e-Polym., 2018, 18, 373–408 CAS.
- S. V. Levchik, E. D. Weil and M. Lewin, Polym. Int., 1999, 48, 532–557 CrossRef CAS.
- M. Hewel, US Pat., 20080274355A1, 2008 Search PubMed.
- J. M. García, F. C. García, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623–686 CrossRef.
- D. Glasscock, G. Kozielski and M. Martens, DuPont Eng. Polym., 2008, 1–9 Search PubMed.
- Y. Jiang, D. Maniar, A. J. J. Woortman, G. O. R. Alberda Van Ekenstein and K. Loos, Biomacromolecules, 2015, 16, 3674–3685 CrossRef CAS PubMed.
- Y. Jiang, D. Maniar, A. J. J. Woortman and K. Loos, RSC Adv., 2016, 6, 67941–67953 RSC.
- J. K. Fink, in High Performance Polymers, William Andrew Publishing, 2nd edn, 2014, pp. 281–299 Search PubMed.
- Y. Gao, T. Yang, Y. Xia and Y. He, Polymer, 2021, 237, 124385 CrossRef CAS.
- M. Cao, C. Zhang, B. He, M. Huang and S. Jiang, Macromol. Res., 2017, 25, 722–729 CrossRef CAS.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass Volume I: Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U.S.D.O. Energy, 2004 Search PubMed.
- Q. Hou, X. Qi, M. Zhen, H. Qian, Y. Nie, C. Bai, S. Zhang, X. Bai and M. Ju, Green Chem., 2021, 23, 119–231 RSC.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- L. Hu, L. Lin, Z. Wu, S. Zhou and S. Liu, Renewable Sustainable Energy Rev., 2017, 74, 230–257 CrossRef CAS.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- V. H. Hopff and A. Krieger, Makromol. Chem., 1961, 47, 93–113 CrossRef.
- H. Hopff and A. Krieger, Helv. Chim. Acta, 1961, 44, 1058–1063 CrossRef CAS.
- P. M. Heertjes and G. J. Kok, Delft Prog. Rep., Ser. A, 1974, 1, 59–63 CAS.
- A. Mitiakoudis, A. Gandini and H. Cheradame, Polym. Commun., 1985, 26, 246–249 CAS.
- A. Mitiakoudis and A. Gandini, Macromolecules, 1991, 24, 830–835 CrossRef CAS.
- T. Cousin, J. Galy, A. Rousseau and J. Dupuy, J. Appl. Polym. Sci., 2018, 135, 45901 CrossRef.
- T. Shen, B. Zhang, Y. Wang, P. Yang, M. Li, R. Hu, K. Guo, K. Chen, N. Zhu, L. Wang, C. Zhu and H. Ying, Chem. Eng. J., 2022, 437, 135361 CrossRef CAS.
- W. Huang, X. Hu, J. Zhai, N. Zhu and K. Guo, Mater. Today Sustainability, 2020, 10, 100049 CrossRef.
- K. Luo, Y. Wang, J. Yu, J. Zhu and Z. Hu, RSC Adv., 2016, 6, 87013–87020 RSC.
- L. S. T. Cureton, E. Napadensky, C. Annunziato and J. J. La Scala, J. Appl. Polym. Sci., 2017, 134, 1–12 CrossRef.
- A. Duursma, R. Aberson, D. D. Smith, J. Flores, M. A. Dam and G. J. M. Gruter, US Pat., 9938376B2, 2018 Search PubMed.
- D. D. Smith, J. Flores. R. Aberson, M. A. Dam, A. Duursma and G. J. M. Gruter, US Pat., 9951181B2, 2018 Search PubMed.
- U. Fehrenbacher, O. Grosshardt, K. Kowollik, B. Tübke, N. Dingenouts and M. Wilhelm, Chem. Ing. Tech., 2009, 81, 1829–1835 CrossRef CAS.
- S. Percec and S. N. Bair, WO Pat., 2017106405A1, 2017 Search PubMed.
- H. Lundberg, F. Tinnis and H. Adolfsson, Chem. – Eur. J., 2012, 18, 3822–3826 CrossRef CAS PubMed.
- C. L. Allen, A. R. Chhatwal and J. M. J. Williams, Chem. Commun., 2012, 48, 666–668 RSC.
- H. Lundberg, F. Tinnis and H. Adolfsson, Synlett, 2012, 23, 2201–2204 CrossRef CAS.
- C. Han, J. P. Lee, E. Lobkovsky and J. A. Porco, J. Am. Chem. Soc., 2005, 127, 10039–10044 CrossRef CAS PubMed.
- G.-J. M. Gruter, L. Sipos and M. Adrianus Dam, Comb. Chem. High Throughput Screening, 2012, 15, 180–188 CrossRef CAS PubMed.
- S. Xie, J. Yang, X. Wang and J. Yang, Eur. Polym. J., 2022, 162, 110932 CrossRef CAS.
- M. Schoennagel and A. T. Cooper, US Pat., 2019, 10,442,824 B2, 2019 Search PubMed.
- C. H. R. M. Wilsens, Y. S. Deshmukh, B. A. J. Noordover and S. Rastogi, Macromolecules, 2014, 47, 6196–6206 CrossRef CAS.
- A. Jeyakumar, PhD Thesis, Technische Universiteit Eindhoven, 2012, 176.
- S. Cakir, M. Nieuwenhuizen, P. G. A. Janssen, R. Rulkens and C. E. Koning, Polymer, 2012, 53, 5242–5250 CrossRef CAS.
- C. Puglisi, F. Samperi, S. Di Giorgi and G. Montaudo, Polym. Degrad. Stab., 2002, 78, 369–378 CrossRef CAS.
- S. Bandi, S. Mehta and D. A. Schiraldi, Polym. Degrad. Stab., 2005, 88, 341–348 CrossRef CAS.
- J. Malluche, G. P. Hellmann, M. Hewel and H.-J. Liedloff, Polym. Eng. Sci., 2007, 47, 1589–1599 CrossRef CAS.
- P. W. Morgan and S. L. Kwolek, Macromolecules, 1975, 8, 104–111 CrossRef CAS.
- J. Huang, X. Tong, J. Yang, Z. Wang, M. Zhang, X. Wang and J. Yang, J. Polym. Res., 2020, 27, 138 CrossRef CAS.
- M. Li and T. J. Dingemans, Polymer, 2017, 108, 372–382 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00189f |
| This journal is © The Royal Society of Chemistry 2022 |