
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional pH-responsive polymers containing dynamic enaminone linkages for the release of active organic amines†
Spyridon
Efstathiou
 a,
Congkai
Ma
a,
Despina
Coursari
a,
Georgios
Patias
a,
Lucas
Al-Shok
a,
Ahmed M.
Eissa
*ab and
David M.
Haddleton
*a
a,
Congkai
Ma
a,
Despina
Coursari
a,
Georgios
Patias
a,
Lucas
Al-Shok
a,
Ahmed M.
Eissa
*ab and
David M.
Haddleton
*a
aDepartment of Chemistry, University of Warwick Gibbet Hill Road, CV4 7AL Coventry, UK. E-mail: d.m.haddleton@warwick.ac.uk
bDepartment of Polymers, Chemical Industries Research Division, National Research Centre, El Bohouth St 33, Dokki, Giza 12622, Cairo, Egypt
First published on 18th March 2022
Abstract
Dynamic covalent bonds have attracted considerable attention for the development of pH-responsive polymers, however, studies using acid-cleavable enaminone linkages as a means of controlled release are limited. Herein, we report pH-sensitive benzocaine-modified poly(ethylene glycol) monomethyl ether-block-poly[2-(acetoacetoxy)ethyl methacrylate] (mPEGx-b-pAEMAy)/BNZ nanoparticles (NPs) for the aqueous controlled release of benzocaine through enaminone bond cleavage. The system is based on the commercially available monomer 2-(acetoacetoxy)ethyl methacrylate (AEMA) which contains free pendant β-ketoester functionality. Well-defined poly[2-(acetoacetoxy)ethyl methacrylate] (pAEMA) homopolymers and poly[(ethylene glycol) monomethyl ether]-block-poly[2-(acetoacetoxy)ethyl methacrylate] (mPEGx-b-pAEMAy) amphiphilic block copolymers were prepared by photoinduced Cu(II)-mediated RDRP to investigate their modification with propylamine (a model amine) and benzocaine (a primary amine containing API) through an enaminone bond. Block copolymers were prepared via two poly(ethylene glycol) monomethyl ether-2-bromo-2-phenylacetate (mPEGx-BPA, x = 43 or 113) macroinitiators synthesised by esterification which acted as the hydrophilic coronas of the ensuing NPs. The self-assembly of both mPEGx-b-pAEMAy and (mPEGx-b-pAEMAy)/BNZ was assessed in water by the direct dilution approach forming spherical NPs as characterised by dynamic light scattering (DLS) and dry-state transmission electron microscopy (TEM). Finally, the in vitro controlled release of benzocaine from mPEGx-b-pAEMAy/BNZ NPs was examined at different pH environments demonstrating faster release kinetics at lower pH with potential utility in applications with relevant chemical environments.
Introduction
Functional polymers are an important keystone of advanced technologies that continuously develop to meet the needs of today's society. At the same time, introducing new functionalities into polymers requires establishment of efficient functionalisation methodologies in order to access polymers with improved performance and properties.1,2 Of major importance are the means by which functional polymers are synthesised, with the current polymer-chemistry toolbox offering various controlled and “living” polymerisation methodologies for the synthesis of well-defined polymers and copolymers.3 Functionality can be introduced into polymers either by directly polymerising functional monomers or by post-functionalisation with both approaches being compelling depending on the availability of the starting materials and cost.4 Nevertheless, the occasionally long synthetic procedures related to the synthesis of functional monomers in conjunction with different incompatibility upon polymerisation can impart complexity.5 In that regard, post-polymerisation modification is an appealing approach, overcoming certain incompatibility issues while finding use in the functionalisation of block and random copolymers with applications in controlled stimuli-responsive materials and self-assembly.6,7Stimuli-responsive materials are functional polymers emerging for potential in a wide range of applications including controlled drug delivery,8 sensing,9 chemo-mechanical actuation,10etc.11,12 This developing class of functional polymers are designed to respond to different stimuli (e.g. temperature,13 pH,14,15 and light16) manipulating their physical or chemical properties.17 Special focus has been given to pH-sensitive nanomaterials as controlled delivery systems targeting biological sites within the body, cells and tumours based on their local pH environment.18 Introducing dynamic acid-cleavable linkages is an excellent strategy to engineer pH-sensitive nanoparticles able to release their cargo under acidic conditions.19 These dynamic covalent bonds are reversible and easily formed from fast condensation reactions,20 commonly used in the development of smart materials,21 dynamic networks,22,23 and prodrug formulations.24 Dynamic bonds such as imines,25 hydrazones,26 oximes,27etc., have found extensive use in pH-responsive drug delivery systems with specific focus on cancer therapy. However, several of these delivery systems suffer from instabilities under physiological conditions consequence of fast hydrolysis leading to deviations from optimum pH-sensitive carriers.28
Amongst the various types of dynamic chemistries, enaminones (the β-dicarbonyl adducts of enamines) have enhanced stability with respect to bonds of the same family (e.g. imines and enamines) demonstrating in parallel a dynamic nature.29 Studies investigating the hydrolysis profiles of enaminones have been previously carried out,30,31 showing a range of reaction rates that depend on the basicity of the bond and the chemical nature of the substituent groups, making enaminones promising chemical links for the development of pH-responsive nanocarriers, for example. To the best of our knowledge, the use of enaminones as acid-cleavable bonds in pH-responsive polymer nanoparticles with potential for cargo release capabilities is limited with Li et al.32 recently reporting an enaminone-based doxorubicin release copolymer system of poly(ethylene glycol) (PEG) and hexamethylenediamine (HDMA) showing potential as drug carrier for cancer therapy.
There is considerable interest in polymers bearing β-ketoester side groups which are available for post-functionalisation strategies through dynamic enaminone linkages33 and the formulation of performance materials such as vitrimers,34 dental resins,35 coatings,36 and lithography.37 A convenient way to surpass the laborious tasks of preparing monomers with reactive carbonyl moieties is the radical polymerisation of 2-(acetoacetoxy)ethyl methacrylate (AEMA); a commercially available, inexpensive, methacrylate that gives the poly[2-(acetoacetoxy)ethyl methacrylate] (pAEMA) polymer. Well-defined copolymers containing pAEMA blocks have found use as bidentate ligands for metal coordination chemistry,38,39 while the interaction of AEMA units with amines has been also proven fruitful for sensors and the development of enaminone-based dynamic organogels.40,41 Evidently, the free-radical copolymerisation of AEMA with poly(ethylene glycol) methacrylate (PEGMA) leads to biocompatible copolymers with potential applications in fluorescent bioimaging and novel polymer radioprotectors.42,43 To our knowledge there is only one reported example of the block copolymerisation of pAEMA with hydrophilic monomers with the thermally induced RAFT chain-extension of p(PEGMA) with AEMA towards polymerisation induced self-assembly (PISA) in ethanol as the solvent.44 Nevertheless, despite the wide use of pAEMA in diverse applications, the incorporation of pAEMA for release studies in aqueous media is restricted by its hydrophobicity. One example has been reported by Sanchez-Sanchez et al.45 who developed hydrophobic pAEMA containing pH-responsive single-chain nanoparticles (SCNPs) which were able to release diamines in organic media suggesting the way towards enaminones bond cleavage for controlled release applications.
Reversible-deactivation radical polymerisation techniques (RDRP) have provided access to well-defined polymers with controlled molecular weights (MWs), narrow molecular weight distributions (MWDs) and sophisticated architectures for the synthesis of macromolecules that have led to innovative materials with diverse functionalities.46–49 Among the various polymerisation types, photoinduced copper-mediated reversible-deactivation radical polymerisation (photoinduced Cu(II)-RDRP) has been established as a powerful photochemical tool for the synthesis of well-defined homopolymers, copolymers, and sequence-controlled multiblock copolymers,50–53 demonstrating fast polymerisation rates, with environmentally friendly features,54 and spatiotemporal control upon demand.
The objective of this current work was to develop pH-sensitive polymer NPs using the combination of AEMA (a β-ketoester containing monomer), and PEG to investigate the dynamic nature of enaminone bonds to allow for the aqueous acid-triggered release of primary amines. As an example, of an active pharmaceutical ingredient (API), we chose to use benzocaine, an aromatic primary amine which exhibits a topical anaesthetic effect with high permeability and low water solubility.55 Although benzocaine can be considered as a relatively safe drug that rarely causes side reactions such as hypersensitivity, it has been reported that a controlled anaesthetic effect would be beneficial, offering more lasting relief to the area in pain.56 Initially, photoinduced Cu(II)-mediated RDRP was employed to synthesize narrow disperse pAEMA homopolymers which were further post-modified with propylamine as a model amine and benzocaine as the amine of interest through enaminone linkages. Subsequently, the synthesis of well-defined amphiphilic block copolymers of poly[(ethylene glycol) monomethyl ether]-block-poly[2-(acetoacetoxy)ethyl methacrylate] (mPEGx-b-pAEMAy) is reported deriving by the chain-extension of linear in-house synthesised poly[(ethylene glycol) monomethyl ether-2-bromo-2-phenylacetate] (mPEGx-BPA, x= 43 or 113) macroinitiators with AEMA. Finally, the aqueous self-assembly of both unmodified and benzocaine-modified copolymers into spherical NPs was assessed alongside with the in vitro pH-triggered controlled release of benzocaine from drug-loaded NPs.
Results and discussion
Synthesis and characterisation of pAEMA homopolymers via photoinduced Cu(II)-RDRP
Controlled radical homo-and copolymerisations of AEMA have been previously reported using RAFT. Initial RAFT polymerisation conditions resulted in polymers with high dispersity, but optimisations of the choice of the chain transfer agent (CTA) and polymerisation temperature led to an improved control over molecular weight.33,57,58 Thermally-induced ATRP has also been previously utilised to polymerise AEMA, reaching high conversions in 40 min with a keto percent of 87% against the enol form.59 To the best of our knowledge, there is no previous report on the polymerisation of AEMA using photoinduced Cu(II)-mediated RDRP.Well-defined polymers bearing pendent β-ketoester moieties with different degrees of polymerisation (DPn) (targeting DPn, target = 20, 40, 60, and 80) were synthesised, using photo-induced Cu(II)-RDRP, taking advantage of the milder polymerisation conditions in contrast to the use of elevated temperature and the availability of spatiotemporal control.60,61 All polymerisation reactions were exposed to UV light (under a UV nail lamp or a custom-made UV box for bigger scale reactions (Fig. S1 and S2, see ESI†) using AEMA as monomer, DMSO (50 vol% to monomer) as solvent, α-methyl bromophenyl acetate (MBPA) as initiator, tris(2-(dimethylamino)ethyl)-amine (Me6TREN) as the ligand with Cu(II)Br2 as the source of copper (Scheme 1a). The photoinduced Cu-mediated polymerisations of methacrylates are more challenging than acrylates.61 Therefore, the choice of appropriate initiator and Cu-ligand complex type and molar ratio was important in order to regulate the concentration of deactivating species during polymerisation to achieve the required well-defined polymers. The MBPA initiator has been previously found effective for the polymerisation of methacrylates due to its reduced rate of initiation (ki) arising from the stabilisation of the derived radical by the aromatic ring and the relatively slow propagation rate constant of methacrylates (kp).62–64 On polymerisation, the 1H-NMR signals corresponding to the aromatic protons of MBPA initiator can be distinguished as they are well resolved, providing a method for the determination of the number average molecular weight. A reagent ratio of [Cu(II)Br2]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Me6TREN] = [0.05]:[0.36] was found to be optimal leading to very high monomer conversions (>99% as determined by 1H-NMR after 12 h) with relatively narrow dispersities achieved (Đ < 1.4) and quite symmetrical size exclusion chromatography (SEC) chromatograms showing clear shifts to higher molecular weights with an increase in the targeted degree of polymerisation (DPn, target) (Fig. 1a). Attempts to further optimise the polymerisation system were conducted by altering the Cu(II)Br2 and Me6TREN ratios as shown in Table S1.† Polymerisations led to low monomer conversions within the first 12 h, with the lowest examined ratio, [0.03]:[0.21], leading to a conversion of 1%. This was ascribed to a possible interaction between the Cu(II) and the ketoester resulting in a rate of polymerisation reduction.65,66 The 1H-NMR and SEC traces of the crude optimisation reactions (Fig. S11–S14, see ESI†) proved the existence of high monomer concentration after 12 h of polymerisation. The data from the polymerisations using the optimal reagent ratio are summarised in Table 1, where a discrepancy between the values of Mn, th and Mn, SEC was noticed due to comparison of the synthesised polymers with commercial poly(methyl methacrylate) standards. The Mn, NMR values were determined using 1H-NMR spectroscopy (Fig. S18, see ESI†) by comparing the proton integrals of the initiator –C6H5 peaks (δ = 7.24 ppm) (g + h + i, Fig. S11†) relative to the methacrylate –CH3 protons from the polymer backbone (δ = 0.43–1.10 ppm) (b, Fig. S1b†). The Mn, NMR were close to the theoretical values and the assignment of peaks in both 1H-NMR and 13C-NMR spectra confirmed the successful synthesis of pAEMA (Fig. 1b and S17†). Protons corresponding to the enol form of the β-ketoester were seen at δ = 5.02 and 11.85 ppm in the 1H-NMR spectra (Fig. S19, ESI†), showing the occurrence of keto–enol tautomerism in solution. Comparison of the peak integrals of –OCCH2CO– (δ = 3.50 ppm) and
:[Me6TREN] = [0.05]:[0.36] was found to be optimal leading to very high monomer conversions (>99% as determined by 1H-NMR after 12 h) with relatively narrow dispersities achieved (Đ < 1.4) and quite symmetrical size exclusion chromatography (SEC) chromatograms showing clear shifts to higher molecular weights with an increase in the targeted degree of polymerisation (DPn, target) (Fig. 1a). Attempts to further optimise the polymerisation system were conducted by altering the Cu(II)Br2 and Me6TREN ratios as shown in Table S1.† Polymerisations led to low monomer conversions within the first 12 h, with the lowest examined ratio, [0.03]:[0.21], leading to a conversion of 1%. This was ascribed to a possible interaction between the Cu(II) and the ketoester resulting in a rate of polymerisation reduction.65,66 The 1H-NMR and SEC traces of the crude optimisation reactions (Fig. S11–S14, see ESI†) proved the existence of high monomer concentration after 12 h of polymerisation. The data from the polymerisations using the optimal reagent ratio are summarised in Table 1, where a discrepancy between the values of Mn, th and Mn, SEC was noticed due to comparison of the synthesised polymers with commercial poly(methyl methacrylate) standards. The Mn, NMR values were determined using 1H-NMR spectroscopy (Fig. S18, see ESI†) by comparing the proton integrals of the initiator –C6H5 peaks (δ = 7.24 ppm) (g + h + i, Fig. S11†) relative to the methacrylate –CH3 protons from the polymer backbone (δ = 0.43–1.10 ppm) (b, Fig. S1b†). The Mn, NMR were close to the theoretical values and the assignment of peaks in both 1H-NMR and 13C-NMR spectra confirmed the successful synthesis of pAEMA (Fig. 1b and S17†). Protons corresponding to the enol form of the β-ketoester were seen at δ = 5.02 and 11.85 ppm in the 1H-NMR spectra (Fig. S19, ESI†), showing the occurrence of keto–enol tautomerism in solution. Comparison of the peak integrals of –OCCH2CO– (δ = 3.50 ppm) and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO– (δ = 5.02 ppm) indicated that ∼92% existed in the keto form (which is the desired functionality for post-modification) rather than the enol form. Minor peaks at δ = 3.91 and 4.76 ppm were a good indication of copolymerised 2-(hydroxy)ethyl methacrylate (HEMA) (Fig. S15, see ESI†). As previously reported,67 acetoacetate groups are susceptible to hydrolysis leading in our case to the formation of HEMA units. This was also supported by a GC-FID analysis of the AEMA monomer proving the presence of a small amount (∼7%) of HEMA available for copolymerisation (Fig. S16, see ESI†). Interestingly, the macroscopic properties of the synthesised pAEMA homopolymers could be compared to a viscoelastic material (Scheme 1b) exhibiting elasticity under an applied force.
CHCO– (δ = 5.02 ppm) indicated that ∼92% existed in the keto form (which is the desired functionality for post-modification) rather than the enol form. Minor peaks at δ = 3.91 and 4.76 ppm were a good indication of copolymerised 2-(hydroxy)ethyl methacrylate (HEMA) (Fig. S15, see ESI†). As previously reported,67 acetoacetate groups are susceptible to hydrolysis leading in our case to the formation of HEMA units. This was also supported by a GC-FID analysis of the AEMA monomer proving the presence of a small amount (∼7%) of HEMA available for copolymerisation (Fig. S16, see ESI†). Interestingly, the macroscopic properties of the synthesised pAEMA homopolymers could be compared to a viscoelastic material (Scheme 1b) exhibiting elasticity under an applied force.
 | ||
| Scheme 1 Synthetic procedure for the polymerisation of AEMA via photoinduced Cu(II)-RDRP. | ||
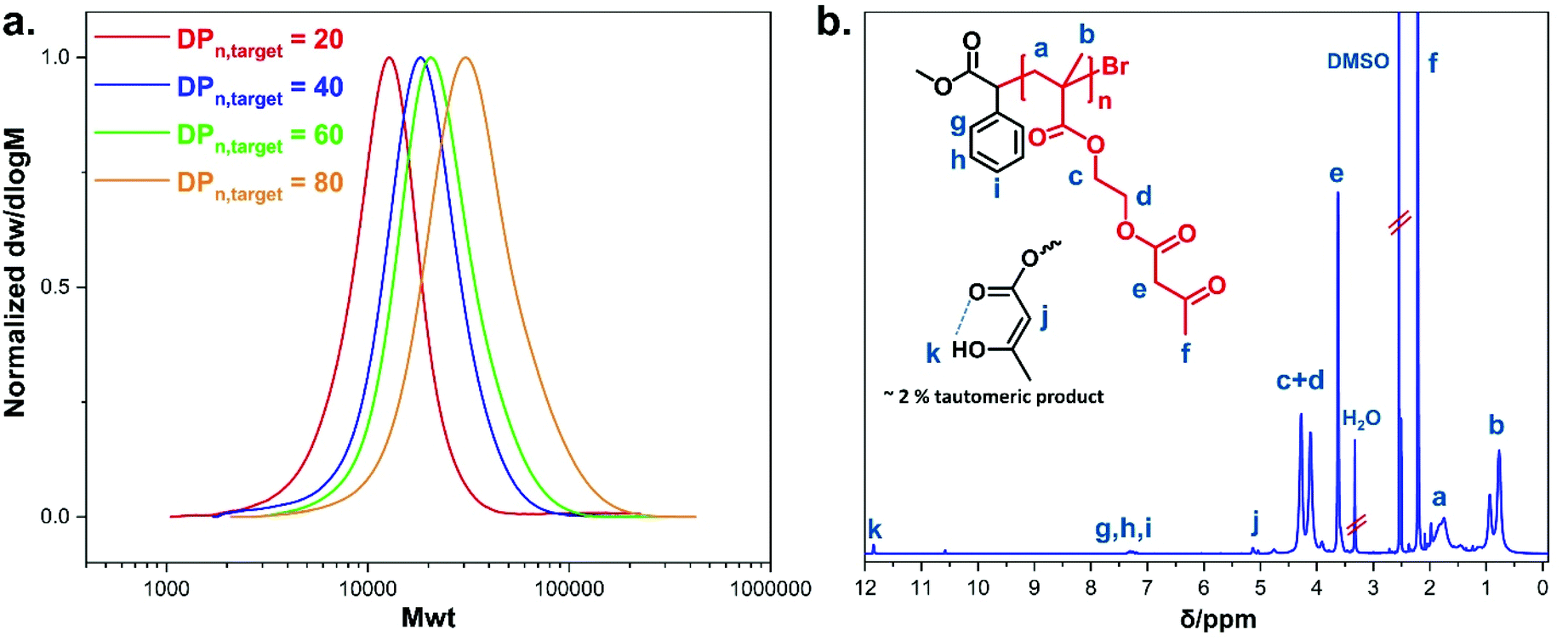 | ||
| Fig. 1 (a) Molecular weight distribution traces of pAEMA homopolymers as measured by SEC in DMF as eluent. (b) 1H-NMR (400 MHz, DMSO-d6) spectrum of pAEMA homopolymer. | ||
:MBPA and as L:Me6TREN. Polymerisation time for all reactions was 12 h
| Entry | [AEMA]:[I]:[CuII]:[L] |
M n, th (g mol−1) | M n, SEC (g mol−1) | M n, NMR (g mol−1) | Conv. (%) | Đ | DPn, NMR |
|---|---|---|---|---|---|---|---|
| a Performed on a larger scale (20 g) using an in-house UV box. | |||||||
| 1 | 20:1:0.05:0.36 |
4000 | 10300 |
4900 | >99 | 1.25 | ∼22 |
| 2 | 40:1:0.05:0.36 |
8800 | 15500 |
8400 | >99 | 1.31 | ∼38 |
| 3 | 40:1:0.05:0.36a |
8800 | 18600 |
9200 | 99 | 1.33 | ∼42 |
| 4 | 60:1:0.05:0.36 |
13000 |
19200 |
13300 |
>99 | 1.27 | ∼61 |
| 5 | 80:1:0.05:0.36 |
17200 |
27700 |
18000 |
>99 | 1.38 | ∼83 |
To gain a better insight into the photoinduced Cu(II)-mediated RDRP of AEMA in DMSO, a kinetic investigation was conducted targeting a DPn, target = 40 over 11.5 h (Fig. S22, see ESI†). Good linear first-order behaviour was noticed with an observed rate of propagation kappp = 0.43 h−1. A linear dependence between the experimental molecular weight (Mn, SEC) and % monomer conversion with relatively low dispersity (Đ) were observed throughout the reaction. Polymerisation reached 99% monomer conversion over 11.5 h with Đ = 1.33 and Mn, NMR = 9200 g mol−1 being close to the theoretical value Mn, th = 8800 g mol−1.
Post-polymerisation amine modification of pAEMA
Following the successful synthesis of well-defined pAEMA using photoinduced Cu(II)-mediated RDRP, the next objective was to explore the post-polymerisation modification of pAEMA homopolymers with amines via a condensation reaction to the formation of dynamic enaminone bonds. Initially, we chose propylamine as a small primary amine model compound (with pKa = 10.58).68 Although acid catalysis is often required to achieve full conversion of the β-ketoester to enaminone within 6 h,33 we preferred catalyst-free conditions. Addition of propylamine was conducted at room temperature (25 °C) over 24 h by mixing pAEMA42 homopolymer with a 2-fold excess of propylamine relative to the β-ketoester, yielding propylamine modified (pAEMA42/PrA) polymers. Different reaction times were also investigated with 6 h leading to low modification yields due to absence of catalyst while 72 h not offering any improvements. Thus, 24 h were found appropriate to ensure maximum modification efficiency. A colour change from transparent to amber was observed, indicating the successful formation of the enaminone as verified by FT-IR and NMR. The FT-IR of the modified polymers showed the appearance of two new strong characteristic peaks at 1600 and 1650 cm−1, corresponding to the CC stretching and N–H bending respectively (Fig. 2c). This was accompanied by a simultaneous reduction in the intensity of the CO shoulder peak at 1750 cm−1. NMR analysis revealed appearance of new peaks at δ = 8.51, 4.38 and 1.91 ppm attributed to the formation of enaminone (Fig. S25†), and complete disappearance of the carbon signals at δ = 30.4 (–CH3) and 201.5 (CO) ppm in the 13C-NMR (Fig. S26†), indicating successful reaction between propylamine and pAEMA. 2D-heteronuclear single quantum coherence (HSQC) NMR of pAEMA42/PrA, (Fig. S27†), showed appearance of two peaks at δ = 6.66 ppm and δ = 8.51 ppm which were assigned for the –NH protons of the E-isomer and the Z-isomer at a ratio of [E]:[Z]-isomer = 38:62. This ratio of isomers was in agreement with a previous report,67 where the higher percentage of Z-isomer was attributed to the formation of a stable six-membered ring as a result of hydrogen bonding interactions between the –NH and the CO groups of the enaminone. The extent of modification was determined using 1H-NMR by a comparison of integrations of the two α-protons at δ = 3.6 ppm (e, Fig. S25, see ESI†) of the β-ketoester groups before and after modification against a reference peak, in this case, δ = 2.2 ppm (f, Fig. S25, ESI†) which corresponds to the –CH3 of the ketoester group with the modification of pAEMA42/PrA being (∼93%).
 | ||
| Fig. 2 Effect of (a) molar equivalents and (b) temperature during the post-functionalisation of pAEMA42 homopolymers with benzocaine. (c) FT-IR spectra prior and after modification of pAEMA42 with propylamine (pAEMA42/PrA(93%)) and benzocaine (pAEMA61/BNZ(87%)). | ||
The focus of this work was to synthesise pH-responsive polymeric assemblies that can be used for delivery applications, thus examining the acid-triggered response of the modified polymers was important. For that purpose, modified pAEMA42/PrA(93%) homopolymers were mixed with an excess of concentrated phosphoric acid (2 eq. H3PO4, 85 wt%) to trigger hydrolysis of the enaminone and the release of the free amine. After 24 h incubation at room temperature, the solution returned to its original transparent colour. 1H-NMR spectroscopy confirmed enaminone cleavage giving back the unmodified polymer chains (Fig. S28†). The proposed mechanism for enaminone's acid triggered hydrolysis is outlined in Fig. S29.†
Following the reversible release of propylamine, benzocaine was chosen as a readily available API.69 Benzocaine has a less nucleophilic primary amine than propylamine with pKa = 2.8.70 Functionalisation of pAEMA42 with the optimised conditions for propylamine (2 fold excess of benzocaine, r.t. for 24 h) resulted in lower efficiency (50%) from 1H-NMR. Estimations of the percent of modification occurred by comparing the methacrylate –CH3 protons (δ = 0.4–1.1 ppm) against two aromatic protons of benzocaine (δ = 7.81 ppm) (c, Fig. S30†).
To further understand this system, optimisations were carried out by altering both the molar equivalents of reagents and reaction temperature. The modification of polymers containing AEMA with 4-methoxyaniline (similar reactivity to benzocaine) was achieved reaching >95% conversion with p-TsOH as catalyst and 5 eq. excess of amine.33 The initial objective was to understand how benzocaine molar equivalents affected modification efficiencies in the absence of catalyst to benzocaine modified polymers, pAEMAx/BNZ. Reactions with pAEMA42 were carried out at 25 °C at four different concentrations of benzocaine (0.5, 1, 2, and 6 eq. to the moles of acetoacetate groups), Table S1.† The recovered pAEMA42/BNZ products demonstrated an increase in the level of modification by increasing equivalents of amine, from 13% at 0.5 eq. to 55% at 6 eq. of benzocaine (Fig. 2a). In a second series of experiments, the effect of temperature was explored (Fig. 2b). Reactions at 30 °C, led to higher levels of modification efficiency with 63% and 83% conversion at 2 eq. and 6 eq. of benzocaine respectively while reaction at 40 °C caused additional improvements reaching 85% conversion with 6 eq. of benzocaine.
Structural confirmation of the pAEMAx/BNZ polymers was confirmed by FT-IR (Fig. 2c) and NMR spectroscopy (Fig. S30–S32s, see ESI†) showing formation of the enaminone. The FT-IR spectrum of pAEMA61/BNZ(87%) showed the appearance of new peaks at 1656 cm−1 corresponding to the N–H bend, 1580–1617 cm−1 corresponding to the CC stretching, and 1262 cm−1 attributed to the Ar–N bond (Fig. 2c). 1H-NMR demonstrated the successful incorporation of benzocaine as evidenced by the strong –NH signal at δ = 10.59 ppm and the aromatic signals of benzocaine at δ = 7.13 and 7.81 ppm. Interestingly, in this case, HSQC (Fig. S26†) and proton integrations showed the prevalence of the Z-isomer (94%) relative to the E-isomer which gave a weak signal peak at δ = 8.81 ppm (6%) due to possible steric hindrance effects from the aromatic group. The pAEMA61/BNZ(87%) was analysed by SEC in comparison with the unmodified pAEMA61 precursor, Fig. S33,† with a clear shift to higher molecular weight after modification, Mn, SEC = 25500 g mol−1, and little affecting on the dispersity (Đ = 1.32).
Synthesis of block copolymer via photoinduced Cu(II)-RDRP
Amphiphilic block copolymers were synthesised via photoinduced Cu(II)-RDRP using MBPA esterified PEG macroinitiators (mPEGx-BPA) as the stabilising hydrophilic blocks and pAEMA as the hydrophobic core-forming blocks, (Scheme 2). PEG was chosen due to its enhanced water solubility, stealth behaviour, antifouling properties,71 and the stability offered in liposomal and micellar formulations for drug-delivery applications.72–74 Statistical copolymers of poly(ethylene glycol methyl ether) (PEGMA) with AEMA have previously been demonstrated to have excellent biocompatibility with ∼100% viability against cells.43 | ||
| Scheme 2 Synthetic procedure for the diblock copolymers mPEGx-b-pAEMAy. | ||
Two mPEGx-BPA macroinitiators were synthesised having different molecular weights, mPEG43-BPA and mPEG113-BPA. The water-soluble macroinitiators were synthesised from mPEGx-OH (x = 43 or 113) precursors by esterification of the hydroxyl groups with α-bromophenyl acetic acid through N,N′-dicyclohexylcarbodiimide (DCC) chemistry. 1H-NMR confirmed the structure with phenyl signals (–C6H5) at δ = 7.35–7.68 ppm, the methine proton connected to the bromide and phenyl group at δ = 5.95 ppm (–C6H5CHBr) and the two protons close to the ester group at δ = 4.26 ppm alongside with additional confirmation by 13C-NMR, FT-IR, and MALDI-ToF MS (Fig. S4–S9†). The hydroxyl-terminated (–OH) macromolecular analogues were identified in the MALDI-ToF MS after the loss of the halogen atom from the terminal end group of the initiator due to potential hydrolysis during sample preparation.75,76 Esterification efficiencies were >99% after comparing the integrals of the methoxy protons of mPEG at δ = 3.25 ppm (CH3O–) with the α-bromophenyl ring protons at δ = 7.35–7.68 ppm.
The macroinitiators were chain-extended with AEMA by photoinduced Cu(II)-RDRP in DMSO using Me6TREN as ligand and Cu(II)Br2 to form the amphiphilic block copolymers mPEGx-b-pAEMAy (x = 43 or 113 and y = DPn of the block as determined by 1H-NMR spectroscopy, Fig. S20 and S21, see ESI†). MW determination by 1H-NMR occurred by comparison of the macroinitiator phenyl signals at δ = 7.10–7.35 ppm against the methyl protons at δ = 0.37–1.09 ppm. Any remaining unreacted macroinitiator was removed during precipitation. In the case of mPEG43-BPA, block copolymers of different targeted lengths were synthesised, mPEG43-b-pAEMAy, using a ratio of [Cu(II)Br2]:[Me6TREN] = [0.05]:[0.36], Table 2. Reactions reached >99% monomer conversions within 8 h forming relatively narrow dispersity block copolymers (usually Đ < 1.4) with symmetrical SEC traces shifted to higher molecular weights compared to mPEG43-BPA (Mn, SEC = 4400 g.mol−1, Đ = 1.06), Fig. 3a.
 | ||
| Fig. 3 SEC traces of diblock copolymers obtained by photo Cu(II)-RDRP in DMSO with different targeted DPn of AEMA (a) mPEG43-b-pAEMAy and (b) mPEG113-b-pAEMAy. | ||
| Blocks | [AEMA]:[I]:[CuII]:[L] |
Time (h) | Conv. (%) | M n, theor (g mol−1) |
M
n, SEC
(g mol−1) |
M n, NMR (g mol−1) | Đ |
|---|---|---|---|---|---|---|---|
|
a Determined by SEC analysis in DMF eluent and compared against PMMA narrow calibration standards.
b Calculated based on the equation: Mn, theor = MwmPEG–BPA + (Mw, AEMA × DPn, targ × Conv.)
|
|||||||
| mPEG43-b-pAEMA10 | 10:1:0.05:0.36 |
8 | >99 | 4240 | 11600 |
4240 | 1.12 |
| mPEG43-b-pAEMA25 | 20:1:0.05:0.36 |
8 | >99 | 6380 | 19000 |
7450 | 1.35 |
| mPEG43-b-pAEMA44 | 40:1:0.05:0.36 |
8 | >99 | 10660 |
22900 |
11500 |
1.24 |
| mPEG43-b-pAEMA84 | 80:1:0.05:0.36 |
8 | >99 | 19200 |
32800 |
20000 |
1.41 |
| mPEG113-b-pAEMA28 | 20:1:0.1:0.72 |
5 | 96 | 9300 | 16400 |
11200 |
1.28 |
| mPEG113-b-pAEMA52 | 60:1:0.1:0.72 |
2 | 73 | 14600 |
27700 |
16340 |
1.23 |
| mPEG113-b-pAEMA77 | 80:1:0.1:0.72 |
2.3 | 78 | 22300 |
30900 |
21690 |
1.21 |
A loss of control at high monomer conversions was noticed by chain-extending with mPEG113-BPA leading to an undesirable high molecular weight shoulder in the mass distribution. This loss was attributed to the increased viscosity of the medium causing diffusion limitations.77,78 Whereas, at high monomer conversions, the increased viscosity from the formation of long polymer chains can make the monomers and the catalytic activator and deactivator species experience diffusion limitations leading to decreased deactivation rates and uncontrolled terminations. Molecular weight shouldering was even more pronounced when targeting lower DPn blocks as the high radical concentration (higher macroinitiator concentration) increased the termination rate (kt) being second-order to the radical concentration.79 In order to circumvent this issue, chain extensions were conducted at higher ratios of [Cu(II)Br2]:[Me6TREN] = [0.1]:[0.72] which suppress termination through deactivation by Cu(II).80 Consequently, mPEG113-b-pAEMAy block copolymers were synthesised at different lengths, Table 2, with narrow and symmetrical MWDs (typically Đ < 1.3) and increased MWs compared to mPEG113-BPA (Mn, SEC = 8800 g mol−1, Đ = 1.05) based on SEC analysis (Fig. 3b).
Further kinetic studies for each copolymerisation were conducted with a first order kinetic plot observed for the chain extension of AEMA with mPEG43-BPA, (Fig. S23†). The molecular weight evolution (Mn, SEC and Mn, NMR) increased linearly with monomer conversion with decreasing dispersities reaching Đ = 1.31 at 97% conversion after 6 h with an initiator's efficiency close to 100% (as calculated from the ratio of (Mn, theor/Mn, NMR) × 100%). In the case of mPEG113-BPA (Fig. S24†), there was a linear increase in molecular weight evolution (Mn, SEC and Mn, NMR) with monomer conversion although dispersity increased at high conversions due to a high MW shoulder peak reaching a value of Đ = 1.29 at 94% conversion after 5 h with a calculated macroinitiator efficiency of 97%.81 The propagation rate values determined were similar kappp = 0.62 and 0.58 h−1 for mPEG43-BPA and mPEG113-BPA respectively and higher compared to MBPA (kappp = 0.43 h−1).
Modification of mPEGx-b-pAEMAy block copolymers with benzocaine
The modification of mPEGx-b-pAEMAy blocks with benzocaine was investigated to mPEGx-b-pAEMAy/BNZ(M%) (M%: modification percent). Attempts to react mPEG43-b-pAEMA84 with 1 eq. and 2 eq. of benzocaine at 25 °C gave lower modification efficiencies, Table S2,† with conversions of 8% and 15% respectively in contrast to 15% and 50% for pAEMA42 at the respective equivalents of benzocaine (Table S1†). Functionalisation of the mPEG43-b-pAEMA84/BNZ(15%) blocks was confirmed by 1H-NMR (complete assignment in Fig. 4a) with the appearance of the –NH signal at δ = 10.57 ppm and the aromatic protons at δ = 7.01–8.05 ppm along with FT-IR spectroscopy which additionally confirmed the desired structure, (Fig. 4c). The functionalised mPEG43-b-pAEMA84/BNZ(15%) blocks showed a clear shift to higher MW with symmetrical mass distribution as analysed by SEC, (Fig. 4b). The repeatability of this modification was assessed by functionalising mPEG43-b-pAEMA84 in triplicates using 2 eq. of benzocaine at 25 °C. The mean modification percent was determined as Mod. % = 14.0 ± 0.9% from 1H-NMR (Fig. S34†). | ||
| Fig. 4 (a) 1H-NMR (400 MHz, DMSO-d6) spectrum of functionalised mPEG43-b-pAEMA84/BNZ(15%) block copolymer. (b) Molecular weight distribution of mPEG43-b-pAEMA84 and benzocaine modified mPEG43-b-pAEMA84/BNZ(15%) block copolymers along with the UV signal as analysed by DMF SEC. (c) FT-IR comparison of mPEG43-b-pAEMA84 and benzocaine modified mPEG43-b-pAEMA84/BNZ(15%) block copolymers proving successful functionalisation. | ||
Modifications with benzocaine were extended using mPEG113-b-pAEMAy block copolymers. The reactions proved slower with low conversions even after altering solvents and temperature, (Table S2†), requiring p-TsOH as an acid catalyst (5 mol%) in DMSO as a solvent to assist solubilisation.82,83 The modification of mPEG113-b-pAEMA28 reached the highest conversion at 70% (mPEG113-b-pAEMA28/BNZ(70%)), whilst mPEG113-b-pAEMA52 reached 58% (mPEG113-b-pAEMA52/BNZ(58%)).
Thermal characterisation analysis
The polymers and macroinitiators were characterised via thermal analysis with differential scanning calorimetry (DSC). The Tg, onset and Tg, midpoint are tabulated in Table S3† and seen in Fig. 5. pAEMA homopolymers exhibited a negative glass transition temperature at Tg, onset ∼ −3 °C attributed to the flexible long pendant chains.41,84 Incorporation of benzocaine moieties reduced the chains mobility raising the Tg, onset to ∼ +31 °C and ∼ +48 °C for 55% and 92% of modification respectively. The difference in thermal properties was also seen by the physical change of the polymers from viscoelastic materials to powdery white solids following this modification (Fig. 5a). | ||
| Fig. 5 DSC thermograms of (a) pristine and modified with benzocaine pAEMA homopolymers and (b) pristine and modified with benzocaine mPEGx-b-pAEMAy block copolymers. Exo up. | ||
Both mPEGx-BPA macroinitiators did not demonstrate an observable Tg though distinct melting peaks were observed at Tm ∼ +50 °C for mPEG43-BPA and Tm ∼ +58 °C for mPEG113-BPA, (Fig. S10†). All block copolymers illustrated a single Tg and no Tm showing good compatibility of the two blocks.85,86 The presence of PEG in the block formulations led to lower Tg's compared to pAEMA homopolymers due to imparted flexibility,87 reaching a Tg, onset of ∼−27 °C for 80% PEG content in mPEG113-b-pAEMA28 blocks. The incorporation of benzocaine into the polymers raised their Tg values with them approaching the room temperature but with mPEG113-b-pAEMA28/BNZ(70%) remaining almost unchanged (Tg, onset ∼ −33 °C) compared to its block precursor mPEG113-b-pAEMA28. This was attributed to its high PEG content that rendered enough mobility and flexibility to the polymer chains thus masking any effect of the aromatic rings on the Tg.
Self-assembly studies and in vitro controlled release of benzocaine
The self-assembly behaviour of the synthesised block copolymers was investigated in water at ambient temperature. Direct dilution was the technique of choice so as to avoid the extended exposure of the polymers to aqueous media which could trigger undesirable hydrolysis.88 The NPs were evaluated by dynamic light scattering (DLS) and transition electron microscopy (TEM). mPEG43-b-pAEMAx blocks formed assemblies with an average diameter of ∼125 nm for mPEG43-b-pAEMA10 and ∼147 nm for mPEG43-b-pAEMA25 by DLS (Fig. S36†). Copolymers with longer pAEMA chains (DPn, pAEMA = 44 and 84) were insufficiently stabilised in water leading to fast precipitation or limited solubilisation even after sonication at elevated temperatures. This limitation was attributed to the high hydrophobicity of the core-forming block in conjunction with the low MW of the hydrophilic corona block. To elaborate that, assemblies of mPEG43-b-pAEMA44 were formed in methanol (diameter of ∼151 nm by DLS, Fig. S36†) showing stability in less polar media as has also been demonstrated by Zhou et al.44 with the similar pAEMA containing nanoparticles in ethanol through polymerisation induced self-assembly (PISA). Despite this, the self-assembly of benzocaine modified mPEG43-b-pAEMA10 and mPEG43-b-pAEMA25 blocks seemed to also lead to insufficient stabilization due to increased hydrophobicity after incorporating the benzocaine. The stability of the amphiphilic block copolymers was also investigated in water by NMR (Fig. S35†). As already mentioned, previous studies have shown that pAEMA units may hydrolyse to HEMA when left for several weeks at physiological pH and temperature.67 To assess that, mPEG43-b-pAEMA25 was dispersed in water for a period of two weeks and the peak integral –CH2 of HEMA at δ = 3.9 ppm was monitored. Results showed no change in the integrals proving the stability of the acetoacetate groups towards hydrolysis for a period of two weeks.An increase in the length of the hydrophilic corona PEG segment was thought to lead to more stable assemblies in water.89 The self-assembly of mPEG113-b-pAEMAy was also examined in using a direct dilution approach. This led to larger-sized micelles with average diameters of ∼74 nm and ∼116 nm for mPEG113-b-pAEMA28 and mPEG113-b-pAEMA52 respectively along with the absence of any secondary size distributions from DLS (Fig. S36†). The zeta potential values at pH 7.4 showed negative surface charges of −4.2 ± 1.2 mV for mPEG113-b-pAEMA28 and - 5.8 ± 0.2 mV for mPEG113-b-pAEMA52 in good agreement with previously reported values for PEGylated NPs (Fig. S38†).90,91 TEM showed roughly spherical NPs with the apparent mean diameters being smaller in size than from DLS due to the dry state TEM analysis. The benzocaine modified copolymer analogues, mPEG113-b-pAEMA28/BNZ(70%) and mPEG113-b-pAEMA52/BNZ(47%), demonstrated a decrease in the particle size with hydrodynamic diameters of ∼59 nm and ∼36 nm based on DLS analysis (Fig. 6a, b and Fig. S37†). TEM analysis for both copolymers agreed well with the decrease in the particle size (∼48 nm) maintaining the spherical morphologies (Fig. 6c). This decrease was attributed to the π–π stacking interactions between the aromatic rings of benzocaine,92–94 leading to a type of non-covalent “core-crosslinking” effect.95 This was reinforced after hydrolysis of mPEG113-b-pAEMA52/BNZ(47%) NPs at pH 4.4 over 2 days where a shift to higher particle sizes was detected by DLS (Fig. S39†) attributed to the escape of benzocaine from the swollen core of the NPs. In addition, slightly more negative zeta potential values were measured after modification with benzocaine, with mPEG113-b-pAEMA28/BNZ(70%) being the most stable at − 8.1 ± 1.2 mV (Fig. S38†). A similar effect was reported by Shi et al.96 whereby the π–π interactions of the aromatic groups increased both the stability and the drug loading efficiency of their micelles.
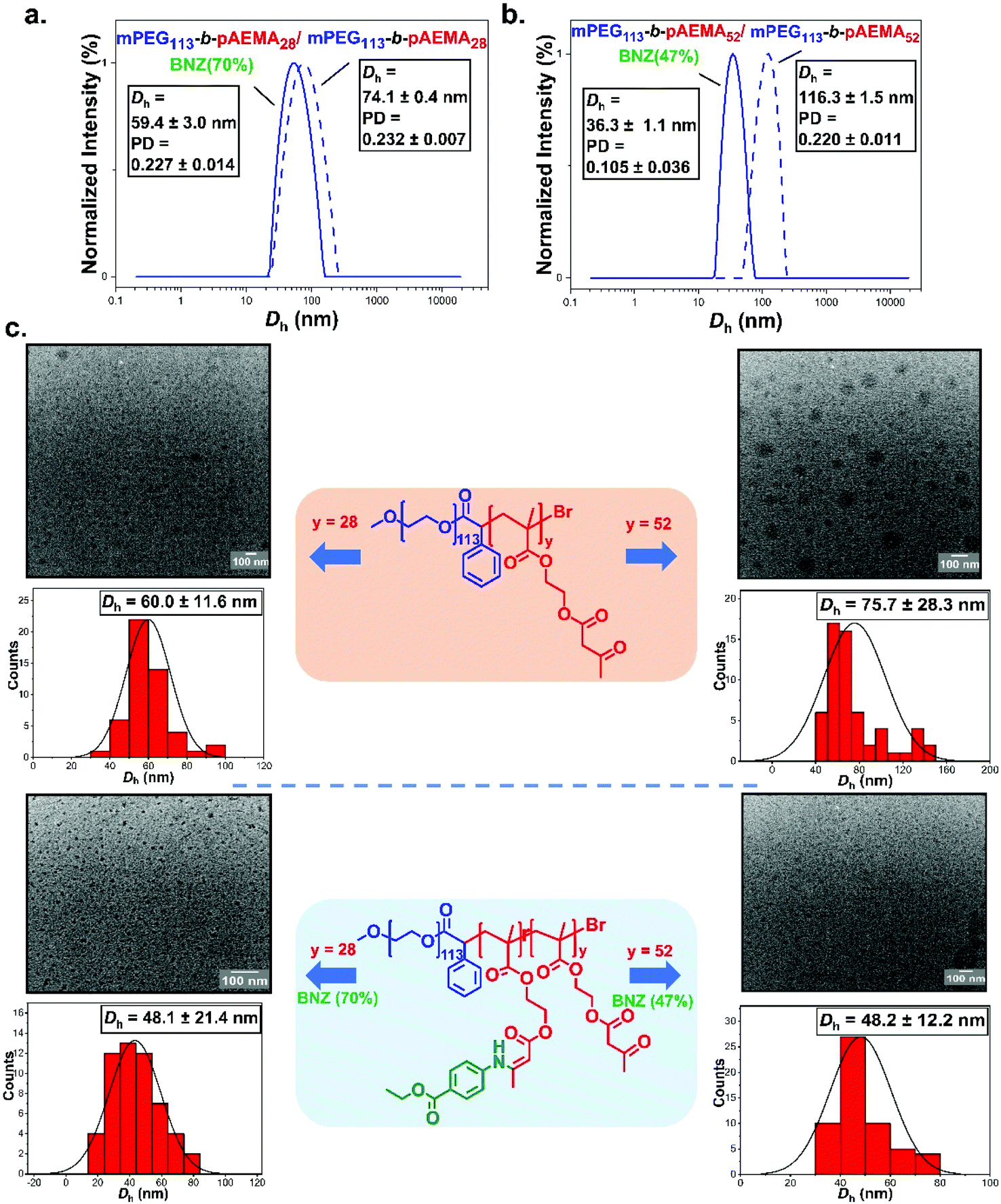 | ||
| Fig. 6 Normalised intensity-weighted size distributions comparisons obtained by DLS at 0.5 mg mL−1 between (a) mPEG113-b-pAEMA28 (dashed line) and mPEG113-b-pAEMA28/BNZ(70%) (blue line) and (b) mPEG113-b-pAEMA52 (dashed line) and mPEG113-b-pAEMA52/BNZ(47%) (blue line) along with (c) Representative dry-state TEM images of mPEG113-b-pAEMA28, mPEG113-b-pAEMA28/BNZ(70%), mPEG113-b-pAEMA52 and mPEG113-b-pAEMA52/BNZ(47%) nano-objects along with their corresponding histograms. | ||
The in vitro release of benzocaine-loaded NPs was finally studied at three different pH values (phosphate buffers at pH = 6.6, 4.4, and 3.5) using mPEG113-b-pAEMA28/BNZ(70%) NPs as an example. The selected pH values included mildly acidic and heavily acidic environments commonly reported in potential targeted delivery systems. For example, the pH of tumour cells is mildly acidic (pH 6.4–7.0), lysosomes have been reported with a pH range of pH 4.5–5.0, while the gastrointestinal tract has a gradient pH beginning with highly acidic environments in the stomach (pH < 3).97–99 The pH-responsive cumulative percent release of benzocaine with respect to time increased with decreasing pH attributed to increased hydrolysis of the enaminone bonds, (Fig. 7 and UV raw data found in Fig. S40†). In all cases a biphasic release pattern was observed,100 beginning with a burst release phase over approximately 8 hours followed by a slow steady amine release over an extended period of time which varied based on the applied pH environment. Over 6 days, a cumulative release of <10% was achieved at pH 6.6 reaching ∼36% at pH 4.4 after the same period. The highest percentage cumulative benzocaine release was seen at the lowest pH = 3.5 with a slow and continuous release profile plateauing at approximately 50%.
 | ||
| Fig. 7 (a) In vitro cumulative percent release profile of benzocaine modified mPEG113-b-pAEMA28/BNZ(70%) NPs in three different phosphate buffer solutions with pH 6.6, 4.4, and 3.5 at 37 °C. The concentration of NPs was 0.5 mg mL−1 while 2.5 mg of NPs contained 700 μg of BNZ as measured by 1H-NMR at 400 MHz using DCM as an internal standard. Quantification of BNZ was achieved by comparing with a calibration curve (Fig. S3, see ESI†). Data are presented as mean ± SD (n = 3). | ||
Thus, these pH-responsive NPs are relatively stable at physiological pH conditions with a low rate of release at pH 6.6, while demonstrating a slow release of their cargo at more acidic conditions due to the enaminone bond cleavage. The slow release profiles of these systems can be explained by the high stability of enaminone bonds which is a consequence of their tautomeric equilibria,29 in combination with a possible retardation effect due to π–π interactions between the free and conjugated to the polymer benzocaine moieties that sustain their release from the core of the NPs.101,102
Conclusion
In summary, pH-sensitive polymeric NPs containing pAEMA and PEG were prepared in water and used for the controlled release of benzocaine through enaminone bond cleavage. Optimised conditions for the photoinduced Cu(II)-RDRP of AEMA monomer are reported targeting the synthesis of well-defined pAEMA homopolymers and mPEGx-b-pAEMAy (x = 43 or 113) amphiphilic block copolymers by chain-extending mPEG-BPA synthesised macroinitiators. Polymers were further post-modified through dynamic enaminone linkage initially with propylamine, a model primary amine and with benzocaine as a model drug. Thermal characterisations via DSC analysis illustrated a variety of Tg's based on their extent of modification and the chain length of the mPEG blocks. The self-assembly of the unmodified and benzocaine modified diblocks was assessed using direct dilution in water with mPEG acting as the hydrophilic corona block and pAEMA as the hydrophobic core forming spherical micelles with diameters ranging from 30–60 nm as demonstrated by TEM and DLS. Finally, via cleavable enaminone bonds, the polymeric NPs illustrated pH dependent release characteristics as assessed by the in vitro cumulative release of benzocaine at various pH. A typical kinetic profile of a drug from NPs was observed, displaying two distinct regions; an early, rapid release followed by a slow diffusion-controlled release over an extended period.This work demonstrates that amphiphilic block copolymers of pAEMA have potential towards enaminone based controlled release applications in water and aqueous media. Since the rate-determining step of the hydrolysis of enaminones is the proton addition to the CC bond,31 different variations of amines can be tested in the future to tune the release rates and basicity of the bond depending on the desired application in the fields of biology or agriculture. In addition, we also envisage these polymers as potential crosslinkers for the fabrication of dynamic enaminone based self-healing hydrogels with potential biological interest.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Warwick Polymer Characterisation Research and Technology Platform (RTP) for all the provided characterisation equipment and more specifically Dr Daniel Lester and Dr James S. Town for their help and support and for access to equipment (Equipment funded in part by EPSRC EP/V036211/1). We are also grateful for the studentships provided by Unilever (SE), Lubrizol (GP), Syngenta (LS) as well as the University of Warwick, the EPSRC Centre of Doctoral Training in Molecular Analytical Science (EP/L015307/1) and the Electron Microscopy RTP.Notes and references
- K. Wang, K. Amin, Z. An, Z. Cai, H. Chen, H. Chen, Y. Dong, X. Feng, W. Fu, J. Gu, Y. Han, D. Hu, R. Hu, D. Huang, F. Huang, F. Huang, Y. Huang, J. Jin, X. Jin, Q. Li, T. Li, Z. Li, Z. Li, J. Liu, J. Liu, S. Liu, H. Peng, A. Qin, X. Qing, Y. Shen, J. Shi, X. Sun, B. Tong, B. Wang, H. Wang, L. Wang, S. Wang, Z. Wei, T. Xie, C. Xu, H. Xu, Z.-K. Xu, B. Yang, Y. Yu, X. Zeng, X. Zhan, G. Zhang, J. Zhang, M. Q. Zhang, X.-Z. Zhang, X. Zhang, Y. Zhang, Y. Zhang, C. Zhao, W. Zhao, Y. Zhou, Z. Zhou, J. Zhu, X. Zhu and B. Z. Tang, Mater. Chem. Front., 2020, 4, 1803–1915 RSC.
- D. F. Grishin and I. D. Grishin, Russ. Chem. Rev., 2021, 90, 231–264 CrossRef CAS.
- K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- R. Arshady, J. Macromol. Sci., Part C: Polym. Rev., 1992, 32, 101–132 CrossRef.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- M. Riedel, J. Stadermann, H. Komber, F. Simon and B. Voit, Eur. Polym. J., 2011, 47, 675–684 CrossRef CAS.
- J. Romulus, J. T. Henssler and M. Weck, Macromol, 2014, 47, 5437–5449 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- T. Shu, L. Hu, Q. Shen, L. Jiang, Q. Zhang and M. J. Serpe, J. Mater. Chem. B, 2020, 8, 7042–7061 RSC.
- L. Hu, Q. Zhang, X. Li and M. J. Serpe, Mater. Horiz., 2019, 6, 1774–1793 RSC.
- A. Bratek-Skicki, Appl. Surf. Sci. Adv., 2021, 4, 100068 CrossRef.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- E. Bellotti, M. V. Fedorchak, S. Velankar and S. R. Little, J. Mater. Chem. B, 2019, 7, 1276–1283 RSC.
- Y. Li, H. Dong, K. Wang, D. Shi, X. Zhang and R. Zhuo, Sci. China: Chem., 2010, 53, 447–457 CrossRef CAS.
- M. Wu, J. Chen, W. Huang, B. Yan, Q. Peng, J. Liu, L. Chen and H. Zeng, Biomacromolecules, 2020, 21, 2409–2420 CrossRef CAS PubMed.
- H. A. Houck, E. Blasco, F. E. Du Prez and C. Barner-Kowollik, J. Am. Chem. Soc., 2019, 141, 12329–12337 CrossRef CAS PubMed.
- D. Wang, M. D. Green, K. Chen, C. Daengngam and Y. Kotsuchibashi, Int. J. Polym. Sci., 2016, 2016, 6480259 Search PubMed.
- S. Zhuo, F. Zhang, J. Yu, X. Zhang, G. Yang and X. Liu, Molecules, 2020, 25, 5649 CrossRef CAS PubMed.
- N. Deirram, C. Zhang, S. S. Kermaniyan, A. P. R. Johnston and G. K. Such, Macromol. Rapid Commun., 2019, 40, 1800917 CrossRef PubMed.
- K. R. West and S. Otto, Curr. Drug Discovery Technol., 2005, 2, 123–160 CrossRef CAS PubMed.
- M. M. Perera and N. Ayres, Polym. Chem., 2020, 11, 1410–1423 RSC.
- S. Efstathiou, A. M. Wemyss, G. Patias, L. Al-Shok, M. Grypioti, D. Coursari, C. Ma, C. J. Atkins, A. Shegiwal, C. Wan and D. M. Haddleton, J. Mater. Chem. B, 2021, 9, 809–823 RSC.
- A. M. Wemyss, C. Ellingford, Y. Morishita, C. Bowen and C. Wan, Angew. Chem., Int. Ed., 2021, 60, 13725–13736 CrossRef CAS PubMed.
- P. Theodosis-Nobelos, D. Charalambous, C. Triantis and M. Rikkou-Kalourkoti, Curr. Drug Delivery, 2020, 17, 542–557 CrossRef PubMed.
- J. Gu, W.-P. Cheng, J. Liu, S.-Y. Lo, D. Smith, X. Qu and Z. Yang, Biomacromolecules, 2008, 9, 255–262 CrossRef CAS PubMed.
- P. Qi, X. Wu, L. Liu, H. Yu and S. Song, Front. Pharmacol., 2018, 9, 12 CrossRef PubMed.
- W. Xu, J. Ding, C. Xiao, L. Li, X. Zhuang and X. Chen, Biomater., 2015, 54, 72–86 CrossRef CAS PubMed.
- G. A. Lemieux and C. R. Bertozzi, Trends Biotechnol., 1998, 16, 506–513 CrossRef CAS PubMed.
- C. M. Kascheres, J. Braz. Chem. Soc., 2003, 14, 945–969 CrossRef CAS.
- K. Dixon and J. V. Greenhill, J. Chem. Soc., Perkin Trans. 2, 1974, 164–168 RSC.
- V. H. Naringrekar and V. J. Stella, J. Pharm. Sci., 1990, 79, 138–146 CrossRef CAS PubMed.
- D. Li, Y. Song, J. He, M. Zhang and P. Ni, ACS Biomater. Sci. Eng., 2019, 5, 2307–2315 CrossRef CAS PubMed.
- M. B. Sims, J. J. Lessard, L. Bai and B. S. Sumerlin, Macromolecules, 2018, 51, 6380–6386 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- E. K. Viljanen, S. Langer, M. Skrifvars and P. K. Vallittu, Dent. Mater., 2006, 22, 845–851 CrossRef CAS PubMed.
- I. González, J. M. Asua and J. R. Leiza, Macromol. Symp., 2006, 243, 53–62 CrossRef.
- S. S. Dinachali, M. S. M. Saifullah, R. Ganesan, E. S. Thian and C. He, Adv. Funct. Mater., 2013, 23, 2201–2211 CrossRef CAS.
- T. Krasia and H. Schlaad, in Metal-Containing and Metallosupramolecular Polymers and Materials, American Chemical Society, 2006, ch. 12, vol. 928, pp. 157–167 Search PubMed.
- H. Schlaad, T. Krasia and C. S. Patrickios, Macromolecules, 2001, 34, 7585–7588 CrossRef CAS.
- J. He, T.-Y. Zhang and G. Chen, J. Colloid Interface Sci., 2012, 373, 94–101 CrossRef CAS PubMed.
- B. Maiti, B. Ruidas and P. De, React. Funct. Polym., 2015, 93, 148–155 CrossRef CAS.
- G. Liu, Y. Zeng, T. Lv, T. Mao, Y. Wei, S. Jia, Y. Gou and L. Tao, Nat. Commun., 2020, 11, 6214 CrossRef CAS PubMed.
- G. Liu, A. Shegiwal, Y. Zeng, Y. Wei, C. Boyer, D. Haddleton and L. Tao, ACS Macro Lett., 2018, 7, 1346–1352 CrossRef CAS.
- W. Zhou, Q. Qu, W. Yu and Z. An, ACS Macro Lett., 2014, 3, 1220–1224 CrossRef CAS.
- A. Sanchez-Sanchez, D. A. Fulton and J. A. Pomposo, Chem. Commun., 2014, 50, 1871–1874 RSC.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104–137 CrossRef CAS.
- G. Lligadas, S. Grama and V. Percec, Biomacromolecules, 2017, 18, 2981–3008 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800616 CrossRef PubMed.
- Y.-M. Chuang, B. Wenn, S. Gielen, A. Ethirajan and T. Junkers, Polym. Chem., 2015, 6, 6488–6497 RSC.
- G. R. Jones, R. Whitfield, A. Anastasaki and D. M. Haddleton, J. Am. Chem. Soc., 2016, 138, 7346–7352 CrossRef CAS PubMed.
- W. Liu, Q. Yang, Y. Yang, F. Xing and P. Xiao, Ind. Eng. Chem. Res., 2021, 60, 7024–7032 CrossRef CAS.
- Y.-M. Chuang, A. Ethirajan and T. Junkers, ACS Macro Lett., 2014, 3, 732–737 CrossRef CAS.
- A. Marathianos, E. Liarou, E. Hancox, J. L. Grace, D. W. Lester and D. M. Haddleton, Green Chem., 2020, 22, 5833–5837 RSC.
- N. F. De Melo, D. R. De Araújo, R. Grillo, C. M. Moraes, A. P. De Matos, E. de Paula, A. H. Rosa and L. F. Fraceto, J. Pharm. Sci., 2012, 101, 1157–1165 CrossRef PubMed.
- J. Goscianska, A. Ejsmont, A. Kubiak, D. Ludowicz, A. Stasiłowicz and J. Cielecka-Piontek, Mater., 2021, 14, 2188 CrossRef CAS PubMed.
- P. Papaphilippou, L. Loizou, N. C. Popa, A. Han, L. Vekas, A. Odysseos and T. Krasia-Christoforou, Biomacromolecules, 2009, 10, 2662–2671 CrossRef CAS PubMed.
- Q. L. Li, X. Xiao, X. Zhang and W. Q. Zhang, Polymer, 2013, 54, 3230–3237 CrossRef CAS.
- P. Mandal, S. L. Banerjee, K. Bhattacharya and N. K. Singha, Eur. Polym. J., 2018, 103, 31–39 CrossRef CAS.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963–971 RSC.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- G. R. Jones, R. Whitfield, A. Anastasaki, N. Risangud, A. Simula, D. J. Keddie and D. M. Haddleton, Polym. Chem., 2018, 9, 2382–2388 RSC.
- N. H. Nguyen, X. Leng and V. Percec, Polym. Chem., 2013, 4, 2760–2766 RSC.
- R. Whitfield, A. Anastasaki, V. Nikolaou, G. R. Jones, N. G. Engelis, E. H. Discekici, C. Fleischmann, J. Willenbacher, C. J. Hawker and D. M. Haddleton, J. Am. Chem. Soc., 2017, 139, 1003–1010 CrossRef CAS PubMed.
- S. Kawaguchi, Coord. Chem. Rev., 1986, 70, 51–84 CrossRef CAS.
- G. Romanazzi, P. Mastrorilli, M. Latronico, M. Mali, A. Nacci and M. M. DelľAnna, Open Chem., 2018, 16, 520–534 CAS.
- S. Mohajeri, N. A. D. Burke and H. D. H. Stöver, Polym. Degrad. Stab., 2015, 114, 94–104 CrossRef CAS.
- J. Wodzinska and R. Kluger, J. Org. Chem., 2008, 73, 4753–4754 CrossRef CAS PubMed.
- S. Khair-Ul-Bariyah, M. Arshad, M. Ali, M. I. Din, A. Sharif and E. Ahmed, Mini-Rev. Med. Chem., 2020, 20, 3–11 CrossRef CAS PubMed.
- T. C. De Carvalho, F. Tosato, L. M. Souza, H. Santos, B. B. Merlo, R. S. Ortiz, R. R. T. Rodrigues, P. R. Filgueiras, H. S. França, R. Augusti, W. Romão and B. G. Vaz, Forensic Sci. Int., 2016, 262, 56–65 CrossRef CAS PubMed.
- S. Lowe, N. M. O'Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- G. S. Kwon and K. Kataoka, Adv. Drug Delivery Rev., 1995, 16, 295–309 CrossRef CAS.
- K. Kim, A. Hasneen, H.-J. Paik and T. Chang, Polym., 2013, 54, 6133–6139 CrossRef CAS.
- A. Velázquez, D. Grande and L. E. Elizalde, Polym. Bull., 2020, 1436–2449 Search PubMed.
- A. M. Rabea and S. Zhu, Polym., 2015, 7, 819–835 CAS.
- A. R. Wang and S. Zhu, Macromol., 2002, 35, 9926–9933 CrossRef CAS.
- N. Ballard and J. M. Asua, ACS Macro Lett., 2020, 9, 190–196 CrossRef CAS.
- A. Kajiwara, K. Matyjaszewski and M. Kamachi, Macromol., 1998, 31, 5695–5701 CrossRef CAS.
- A. M. Rabea and S. Zhu, Ind. Eng. Chem. Res., 2014, 53, 3472–3477 CrossRef.
- A. K. Hajare, A. R. Jagdale, G. G. Shenoy and N. Sinha, New J. Chem., 2016, 40, 4888–4890 RSC.
- J. J. Lessard, G. M. Scheutz, R. W. Hughes and B. S. Sumerlin, ACS Appl. Polym. Mater., 2020, 2, 3044–3048 CrossRef CAS.
- T. Krasia, R. Soula, H. G. Börner and H. Schlaad, Chem. Commun., 2003, 4, 538–539 RSC.
- A. I. Buzin, M. Pyda, P. Costanzo, K. Matyjaszewski and B. Wunderlich, Polym., 2002, 43, 5563–5569 CrossRef CAS.
- R. Krishnan and K. S. V. Srinivasan, J. Macromol. Sci., Part A: Pure Appl.Chem., 2005, 42, 495–508 CrossRef.
- S. H. Park, B. G. Choi, H. J. Moon, S.-H. Cho and B. Jeong, Soft Matter, 2011, 7, 6515–6521 RSC.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS.
- G. Wang, Z. Wang, B. Lee, R. Yuan, Z. Lu, J. Yan, X. Pan, Y. Song, M. R. Bockstaller and K. Matyjaszewski, Polym., 2017, 129, 57–67 CrossRef CAS.
- J. Suh, K.-L. Choy, S. K. Lai, J. S. Suk, B. C. Tang, S. Prabhu and J. Hanes, Int. J. Nanomed., 2007, 2, 735–741 CrossRef CAS PubMed.
- S. Pamujula, S. Hazari, G. Bolden, R. A. Graves, D. D. Chinta, S. Dash, V. Kishore and T. K. Mandal, J. Pharm. Pharmacol., 2011, 64, 61–67 CrossRef PubMed.
- A. J. Cruz-Cabeza, R. J. Davey, S. S. Sachithananthan, R. Smith, S. K. Tang, T. Vetter and Y. Xiao, Chem. Commun., 2017, 53, 7905–7908 RSC.
- S.-Y. Fung, H. Yang, P. Sadatmousavi, Y. Sheng, T. Mamo, R. Nazarian and P. Chen, Adv. Funct. Mater., 2011, 21, 2456–2464 CrossRef CAS.
- F. Li, Y. Zhu, B. You, D. Zhao, Q. Ruan, Y. Zeng and C. Ding, Adv. Funct. Mater., 2010, 20, 669–676 CrossRef CAS.
- M. Lin, Y. Dai, F. Xia and X. Zhang, Mater. Sci. Eng., C, 2021, 119, 111626 CrossRef CAS PubMed.
- Y. Shi, M. J. van Steenbergen, E. A. Teunissen, L. Novo, S. Gradmann, M. Baldus, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2013, 14, 1826–1837 CrossRef CAS PubMed.
- P. Qi, X. Wu, L. Liu, H. Yu and S. Song, Front. Pharmacol., 2018, 9, 1663–9812 Search PubMed.
- D. F. Evans, G. Pye, R. Bramley, A. G. Clark, T. J. Dyson and J. D. Hardcastle, Gut., 1988, 29, 1035–1041 CrossRef CAS PubMed.
- G. Hao, Z. P. Xu and L. Li, RSC Adv., 2018, 8, 22182–22192 RSC.
- J. Yoo and Y.-Y. Won, ACS Biomater. Sci. Eng., 2020, 6, 6053–6062 CrossRef CAS PubMed.
- F. Le Dévédec, L. Houdaihed and C. Allen, J. Visualized Exp., 2016, 116, 54422 Search PubMed.
- Y. Liang, X. Deng, L. Zhang, X. Peng, W. Gao, J. Cao, Z. Gu and B. He, Biomater., 2015, 71, 1–10 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Includes all synthetic procedures and additional characterisation data. See DOI: https://doi.org/10.1039/d2py00167e |
| This journal is © The Royal Society of Chemistry 2022 |