
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymerization of phenylacetylene catalyzed by rhodium(I) complexes with N-functionalized N-heterocyclic carbene ligands†
Marta
Angoy
,
M. Victoria
Jiménez
 *,
Fernando J.
Lahoz
,
Eugenio
Vispe
and
Jesús J.
Pérez-Torrente
*
*,
Fernando J.
Lahoz
,
Eugenio
Vispe
and
Jesús J.
Pérez-Torrente
*
Departamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea–ISQCH, Universidad de Zaragoza–CSIC, Facultad de Ciencias, C/Pedro Cerbuna, 12, 50009 Zaragoza, Spain. E-mail: perez@unizar.es; vjimenez@unizar.es
First published on 10th February 2022
Abstract
A series of neutral [RhX(nbd)(κC-MeIm∩Z)] and cationic [Rh(nbd)(κ2C,N-MeIm∩Z)]+ (X = Cl, Br; MeIm = 3-methylimidazol-2-yliden-1-yl; ∩Z = N-functionalized wingtip; nbd = 2,5-norbornadiene) complexes featuring NHC ligands functionalized with a 1-aminopropyl, 3-dimethylaminopropyl, pyridin-2-ylmethyl, or quinolin-8-ylmethyl substituent have been prepared. These complexes efficiently catalyze the polymerization of phenylacetylene without base as a co-catalyst affording stereoregular polyphenylacetylenes of very high molar mass. Polymers of Mw up to 2 × 106 g mol−1 and moderate dispersity have been prepared with neutral chloro-complexes having aminopropyl wingtips. Catalyst precursors bearing functionalized NHC ligands with a flexible amino-alkyl wingtip are significantly more active than those having a heterocyclic substituent. These complexes are in general much more active than related compounds having N-functionalized phosphine ligands. Polymer characterization by SEC/MALS/DRI analysis has revealed the presence of a fraction of branched polymer of high molar mass in most samples obtained with catalysts having N-heterocyclic substituents at the NHC ligand. The N-donor function at the NHC ligand likely behaves as an internal base for the deprotonation of phenylacetylene to give the initiating alkynyl cationic [Rh(nbd)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-Ph)(κC-MeIm∩ZH)]+ species. However, the participation of neutral alkynyl species [Rh(nbd)(CC-Ph)(κC-MeIm∩Z)] should be considered in order to rationalize the notable catalytic activity of some neutral chloro-complexes.
C-Ph)(κC-MeIm∩ZH)]+ species. However, the participation of neutral alkynyl species [Rh(nbd)(CC-Ph)(κC-MeIm∩Z)] should be considered in order to rationalize the notable catalytic activity of some neutral chloro-complexes.
Introduction
Polyphenylacetylene (PPA) and its derivatives have attracted significant attention due to the interesting properties arising from their π-conjugated main chains. These materials have shown high solubility in a variety of organic solvents and ease of processing, which has enabled a wide range of applications in different fields. The rational choice of substituted acetylenes has allowed access to functional polymers with electronic and photoelectronic applications, stimuli-responsive materials, helical chirality or permanent microporosity.1,2 In addition, a wide range of supramolecular assemblies derived from PPAs including fibers, nanoparticles, nanotubes, gels, liquid crystals and composites have been prepared.3 PPAs are usually synthesized by transition-metal-catalyzed polymerization of substituted phenylacetylene (PA) monomers. Polymerization by early-transition-metal catalysts generally proceed through a metathesis mechanism and require the presence of cocatalysts whereas molecular complexes based on late-transition-metals such as rhodium, iridium, ruthenium and palladium, efficiently polymerize PA derivatives through a coordination–insertion mechanism.4 Rhodium(I) catalysts are particularly attractive due to their low oxophilicity, high activity and high tolerance to many of the heteroatoms in alkyne functional monomers thus facilitating access to a large number of interesting materials under mild conditions.5 In addition, rhodium catalysts provide highly stereoregular PPAs with cis-transoidal configuration, in some cases in a living manner.6 The broad applicability of rhodium catalysts is evidenced by their ability to perform the stitching polymerization of non-conjugated alkylacetylenes or 1,5-hexadienes to afford unprecedented π-conjugated polymers with ladder-type repeating units.7In recent years, there have been significant advances in the design of rhodium(I) catalysts for the controlled polymerization of alkyne-based monomers.8 In particular, well-defined Rh–vinyl9 and Rh–aryl10 complexes enable the (co)polymerization of PA derivatives to afford highly stereoregular (co)polymers with narrow molecular-weight distributions and very high initiation efficiencies. On the other hand, it is well known that many rhodium complexes in combination with an external base, such as triethylamine or 4-dimethylaminopyridine, catalyze the polymerization of PA derivatives very often in a non-controlled way.11 Our research group has developed a complementary strategy by using functionalized phosphine ligands of hemilabile character as internal base for the design of efficient PA polymerization rhodium catalysts.
We have shown that cationic rhodium(I) complexes [Rh(diene){Ph2P(CH2)nZ}]+ (n = 2 or 3; Z = OMe, NMe2) efficiently catalyze PA polymerization leading to very high-molecular-weight stereoregular PPAs with a cis-transoidal configuration and moderate dispersity.12,13 Reactivity studies on the catalyst precursor [Rh(cod){Ph2P(CH2)3NMe2}]+ revealed that the -NMe2 group behaves as an internal base for the PA deprotonation to afford the alkynyl species [Rh(CC-Ph)(cod){Ph2P-(CH2)3NHMe2}]+ which actually is the initiating species likely involved in the generation of stable rhodium–vinyl species responsible for the propagation step.12 Interestingly, characterization of the polymers by size exclusion chromatography, multiangle light scattering (SEC-MALS), or asymmetric flow field flow fractionation (A4F-MALS), showed that some PPA samples contained branched PPA of high molecular weight.13
The emergence of N-heterocyclic carbenes (NHCs) in the last two decades has spurred the development of organometallic catalysis gradually displacing the typical phosphine and amine-type ligands. Their strong σ-donor character results in very strong metal–NHC bonds, making the catalysts more robust. In addition, the easy modulation of NHC ligand substituents allows access to a variety of topologies with steric and electronic properties tailored to the specific requirements of individual catalytic transformations.14 In this context, we identified the potential of functionalized NHC ligands for the design of efficient PA polymerization rhodium catalysts.
The application of Rh(I)–NHC species as polymerization initiators is much less widespread than that of catalysts based on phosphine ligands (Chart 1). Buchmeiser et al. described in 2005 the first active Rh(I)–NHC complexes in the polymerization of PA. The neutral and cationic complexes based on the 1,3-dimesityl-3,4,5,6-tetrahydropyrimidin-2-ylidene ligand exhibited moderate activity, although the cationic complex produced highly stereoregular PPA in the presence of water with Mw up to 1.38 × 105 g mol−1.15 Trzeciak et al. reported on the catalytic activity of [RhX(cod)(bmim)] (bmim = 1-butyl-3-methylimidazol-2-ylidene, X = Cl, Br, I) complexes. Although these complexes showed low activity in dichloromethane, good activity was observed in ionic liquids affording cis-PPAs with Mw not exceeding 4.7 × 104 g mol−1.16 Zwitterionic rhodium complexes consisting of a malonate-based NHCs ligand featuring an endocyclic anionic moiety were reported by Lavigne, César et al. These compounds catalyzed the polymerization of PA affording PPAs with Mn up to 3.0 × 104 g mol−1 with a cis content up to 80%.17 Shibahara, Murai et al. reported the synthesis of rhodium complexes derived from the 1-phenyl-imidazo[1,5-a]pyridin-3-ylidene ligand. The cationic complex showed higher catalytic activity than the neutral complex with almost complete conversion in 1 h to afford a PPA of Mn 1.5 × 104 g mol−1 with moderate dispersity and a 90% cis content.18 On the other hand, Son and co-workers have recently reported a heterogeneous catalyst comprising of mesoionic carbene rhodium(I) species supported on a microporous organic polymer. This catalyst has shown good recyclability affording stereoregular PPAs with Mw up to 5.0 × 104 g mol−1.19
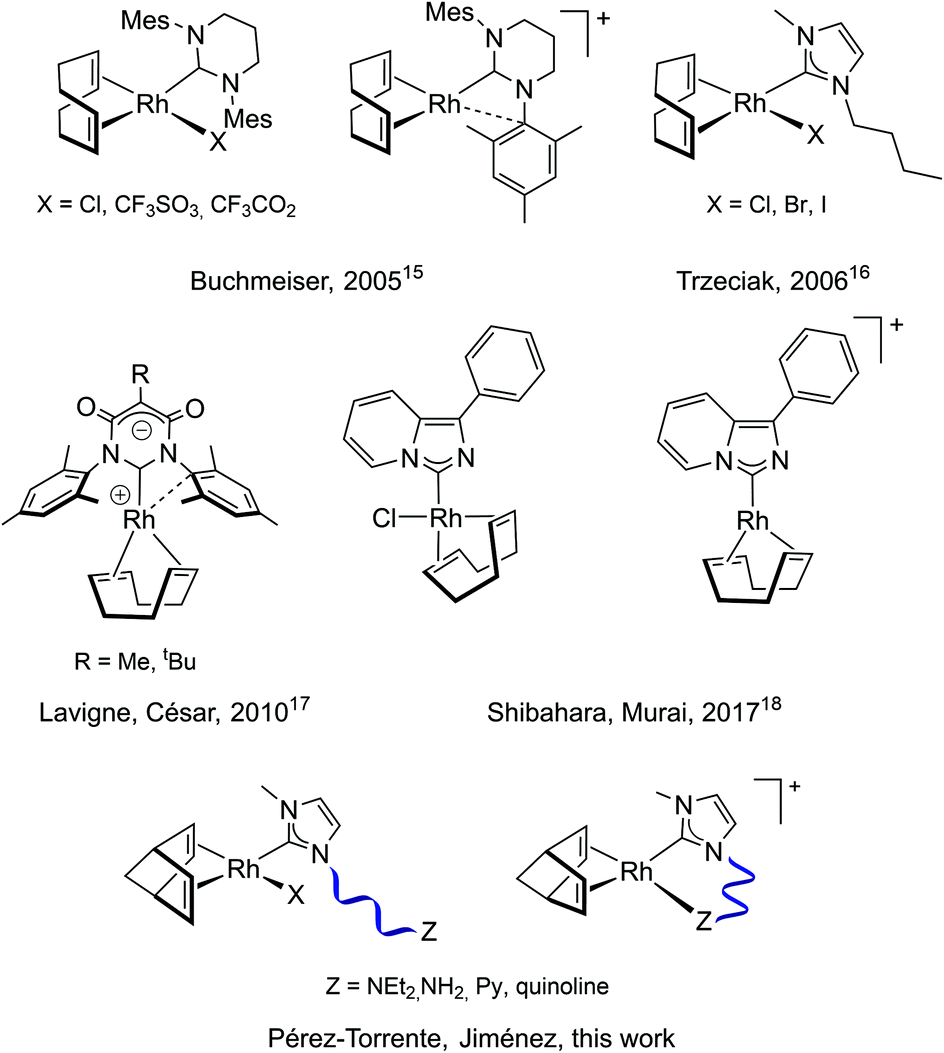 | ||
| Chart 1 Rh(I)–NHC catalysts for polymerization of phenylacetylene. | ||
We report herein on the synthesis of a series of new neutral and cationic Rh(I)–NHC complexes bearing N-functionalized NHC ligands and their application as PA polymerization catalysts. Furthermore, the observed reactivity pattern has been rationalized and compared with that of related precursors based on N-functionalized phosphine ligands.
Results and discussion
Synthesis of neutral and cationic rhodium compounds with N-functionalized NHC ligands
A series of neutral Rh(I)–NHC complexes of general formula [RhX(nbd)(κC-MeIm∩Z)] (X = Cl or Br, nbd = 2,5-norbornadiene, MeIm = 3-methylimidazol-2-yliden-1-yl, ∩Z = N-functionalized wingtip) have been prepared. In particular, Rh(I)–NHC complexes featuring flexible, such as 3-dimethylaminopropyl and 3-aminopropyl, and rigid N-functionalized wingtips, such as pyridin-2-ylmethyl and quinolin-8-ylmethyl, have been synthesized from suitable imidazolium salts following different methodologies (Chart 2). | ||
| Chart 2 Structure of Rh(I)–NHC compounds 1–9. | ||
Compound [RhCl(nbd){κC-MeIm(CH2)3NMe2}] (1) was synthesized from the hydrochloride salt [MeImH(CH2)3NMe2]Cl·HCl following a two-step procedure (Fig. 1, ii). First, monodeprotonation of the ammonium fragment with NaH followed by reaction with [Rh(μ-Cl)(nbd)]2 afforded the intermediate ion-pair [MeImH(CH2)3NMe2][RhCl2(nbd)] compound. Subsequent reaction with NaH and H2O resulted in the deprotonation of the imidazolium fragment to afford 1. Compound [RhBr(nbd){κC-MeIm(CH2)3NH2}] (2) was also prepared by double deprotonation of the hydrobromide ammonium-imidazolium salt, [MeImH(CH2)3NH2]Br·HBr, but in one single step (Fig. 1, ii). On the other hand, deprotonation of the imidazolium salt [MeImH(pyridin-2-ylmethyl)]Br by [Rh(μ-OMe)(nbd)]2 directly afforded compound [RhBr(nbd){κC-MeIm(pyridin-2-ylmethyl)}] (3) (Fig. 1, i). Finally, compound [RhCl(nbd){κC-MeIm(quinolin-8-ylmethyl)}] (4) was prepared following a different methodology involving transmetalation from the silver complex [AgBr{MeIm(quinolin-8-ylmethyl)}], generated in situ by reaction of [MeImH(quinolin-8-ylmethyl)]Br with Ag2O, to [Rh(μ-Cl)(nbd)]2. The neutral complexes were obtained as yellow or orange microcrystalline solids in moderate to good yields (45–75%).
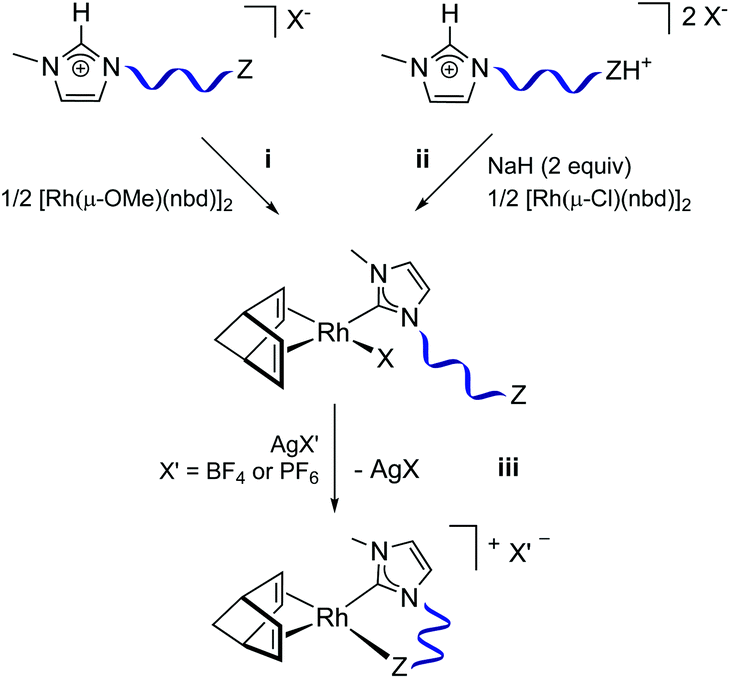 | ||
| Fig. 1 General methods for the synthesis of neutral and cationic Rh(I)–NHC complexes bearing N-functionalized NHC ligands. | ||
Compounds [RhX(nbd)(κC-MeIm∩Z)] (1–4) have been characterized by elemental analysis, mass spectrometry and NMR spectroscopy. The formation of a Rh–NHC bond in the complexes was confirmed both by the absence of the low-field signal of the NCHN acid proton of the imidazolium salts and the presence of a low field doublet or broad signal for the carbenic carbon atom (δ 185–175 ppm, JRh–C ≈ 53–58 Hz) in the 1H and 13C{1H} NMR spectra, respectively. It is remarkable that the olefinic ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH protons and carbons of the 2,5-norbornadiene (nbd) ligand in the complexes display only two resonances in the spectra. As an example, compound 1 shows two resonances at δ 4.72 and 3.47 ppm, and two doublets at δ 78.85 and 51.51 ppm (JRh–C of 5.2 and 2.7 Hz) in the 1H and 13C{1H} NMR spectra, respectively, with the more deshielded resonances corresponding to the CH bonds trans to the NHC ligand.20 In sharp contrast, related rhodium compounds featuring cod (1,5-cyclooctadiene) ligands, such as [RhCl(cod){κC-MeIm(CH2)3NMe2}]21 and [RhCl(cod){κC-MesIm(quinolin-8-ylmethyl)}],22 showed four resonances in the spectra as a consequence of the restricted rotation about the Rh–C bond of the NHC ligand due to the steric influence imparted by the bulky cod ligand. However, the small bite angle of the nbd ligand compared to cod allows the free rotation about the Rh–NHC bond thereby resulting in an effective plane of symmetry in the molecules which is responsible for the simplicity of the NMR spectra. It should be noted that this phenomenon had been previously observed by James et al. in the series of compounds [RhCl(diene)(IPr)] and [RhCl(diene)(IMes)] (diene = cod and nbd; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, IMes = 1,3- bis(2,4,6-trimethylphenyl)imidazol-2-ylidene).23
CH protons and carbons of the 2,5-norbornadiene (nbd) ligand in the complexes display only two resonances in the spectra. As an example, compound 1 shows two resonances at δ 4.72 and 3.47 ppm, and two doublets at δ 78.85 and 51.51 ppm (JRh–C of 5.2 and 2.7 Hz) in the 1H and 13C{1H} NMR spectra, respectively, with the more deshielded resonances corresponding to the CH bonds trans to the NHC ligand.20 In sharp contrast, related rhodium compounds featuring cod (1,5-cyclooctadiene) ligands, such as [RhCl(cod){κC-MeIm(CH2)3NMe2}]21 and [RhCl(cod){κC-MesIm(quinolin-8-ylmethyl)}],22 showed four resonances in the spectra as a consequence of the restricted rotation about the Rh–C bond of the NHC ligand due to the steric influence imparted by the bulky cod ligand. However, the small bite angle of the nbd ligand compared to cod allows the free rotation about the Rh–NHC bond thereby resulting in an effective plane of symmetry in the molecules which is responsible for the simplicity of the NMR spectra. It should be noted that this phenomenon had been previously observed by James et al. in the series of compounds [RhCl(diene)(IPr)] and [RhCl(diene)(IMes)] (diene = cod and nbd; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, IMes = 1,3- bis(2,4,6-trimethylphenyl)imidazol-2-ylidene).23
The abstraction of the halide ligand in complexes [RhX(nbd)(κ2C,N-MeIm∩Z)] by reaction with soluble silver salts, such as AgBF4 or AgPF6, provided access to cationic complexes having the N-donor function coordinated to the rhodium center (Chart 2). Complexes [Rh(nbd){κ2C,N-MeIm(CH2)3NMe2}]BF4 (5) and [Rh(nbd){κ2C,N-MeIm(quinolin-8-ylmethyl)}]PF6 (8) were synthesized following this methodology (Fig. 1, iii). Unexpectedly, abstraction of the bromido ligand in 3 gave the solvato compound [Rh(nbd){κC-MeIm(pyridin-2-ylmethyl)}(OCMe2)]BF4 (7). On the other hand, compound [Rh(nbd){κ2C,N-MeIm(CH2)3NH2}]PF6 (6) was prepared directly by deprotonation of the imidazolium salt [MeImH(CH2)3NH2][PF6] by [Rh(μ-OMe)(nbd)]2 in methanol. The cationic complexes were obtained as orange-yellow microcrystalline solids in moderate yields (45–55%).
Conductivity measurements of ca. 5 × 10−4 M solutions of the complexes in acetone or methanol, 70–90 Ω−1 cm2 mol−1, are within the expected range for uni-univalent electrolytes thus confirming their ionic character. In addition, the MALDI-Tof mass spectra of the compounds showed the molecular ion at the expected m/z ratio. The structure of the compounds is derived from the κ2C,N coordination of the N-functionalized NHC ligands to the fragment [Rh(nbd)]+ that result in mononuclear square-planar complexes, which was confirmed by the determination of the molecular structure of compound 8 by X-ray diffraction.
A view of the crystal structure of the cation [Rh(nbd){κ2C,N-MeIm(quinolin-8-ylmethyl)}]+ of 8 is shown in Fig. 2 along with selected bond lengths and angles. The ligand 1-(quinolin-8-ylmethyl)-3-methyl-imidazol-2-ylidene exhibits a κ2C,N coordination mode, nicely fitting a cis disposition at the metal centre [N(1)–Rh(1)–C(18) 93.13(8)°].24 The planes containing either the NHC or the quinoline fragments intersect the coordination plane at 61.47(7)° and 37.48(5)°, respectively. In addition, the mentioned planes intersect each other at 70.66(6)°. The Rh(1)–C(18) bond distance [2.018 (2) Å] falls within the range generally observed for Rh(I)–NHC compounds.25 On the other hand, the Rh–N(1) bond distance [2.184(2) Å] is slightly longer than that found in the related compound [Rh(cod){κ2C,N-MesIm(quinolin-8-ylmethyl)}]+ [2.168(18) Å].22 The remaining coordination sites are occupied by 2,5-norbonadiene rendering a distorted square planar coordination. As a result of the stronger trans influence of NHC vs. quinoline, the distance Rh(1)–ct(1) [2.103(3) Å] is longer than the distance Rh(1)–ct(2) [1.976(3) Å] as well as the bond length C(1)–C(2) [1.373(4) Å] is shorter than C(4)–C(5) [1.399(4) Å].
 | ||
| Fig. 2 ORTEP view of the cation of [Rh(nbd){κ2C,N-MeIm(quinolin-8-ylmethyl)}]+ in 8. Hydrogen atoms have been omitted for clarity and ellipsoids are at 50% probability. Selected bond lengths (Å) and angles (°) are Rh–N(1) 2.184(2), Rh–C(18) 2.018(2), Rh–C(1) 2.205(3), Rh–C(2) 2.220(3), Rh–ct(1) 2.103(3), Rh–C(4) 2.106(3), Rh–C(5) 2.087(3), Rh–ct(2) 1.976(3), C(1)–C(2) 1.373(4), C(4)–C(5) 1.399(4), N(2)–C(18) 1.347(3), N(3)–C(18) 1.351(3), N(1)–Rh(1)–C(18) 93.13(8), N(1)–Rh(1)–ct(1) 100.30(9), C(18)–Rh(1)–ct(2) 96.33(10), ct(1)–Rh(1)–ct(2) 70.80(10), Rh–C(18)–N(2) 134.5(2), Rh(1)–C(18)–N(3) 120.9(1), N(2)–Rh(1)–N(3) 104.6(2), N(1)–Rh(1)–ct(2) 162.77(9), C(18)–Rh(1)–ct(1) 166.5(9). ct(1) and ct(2) are the centroids of C(1) and C(2), and of C(4) and C(5), respectively. | ||
The coordination of the N-donor function to the rhodium center in these compounds prevents the rotation of the NHC ligand about the Rh–C which results in the loss of symmetry. This fact is evidenced by the four resonances for the olefinic CH protons and carbons of the nbd ligand observed in the 1H and 13C{1H} NMR spectra. Fig. 3 show a selected region of the 1H NMR spectrum of complexes 3 and 8 for comparison. In addition, the methylene protons >CH2 of the linkers are now diastereotopic which is also reflected in the 1H NMR spectra. However, the 1H NMR data for 7, both in acetone-d6 and CD2Cl2, showed only two resonances for the CH protons of the nbd ligand and a single resonance for the >CH2 linker which agrees with the presence an uncoordinated pyridin-2-ylmethyl fragment (Fig. 3). The formulation of this compound as the solvato complex [Rh(nbd){κC-MeIm(pyridin-2-ylmethyl)}(OCMe2)]BF4 (7) is supported by the 13C{1H}-apt spectrum that shows a resonance at δ 210.0 ppm, slightly low-field shifted with respect to that of acetone-d6, which is assigned to coordinated acetone.26 The structure of 7 contrasts with that of the iridium compound [Ir(cod){κ2C,N-MeIm(pyridin-2-ylmethyl)}]+ in which the ligand maintains its bidentate κ2C,N coordination in acetone.20 It is worth mentioning that decoordination of the hemilabile ethoxy fragment of the functionalized phosphine ligand in compound [Rh(cod){κP-(4-MeC6H4)2P(CH2)3OEt}]+ in coordinating solvents has also been observed.27
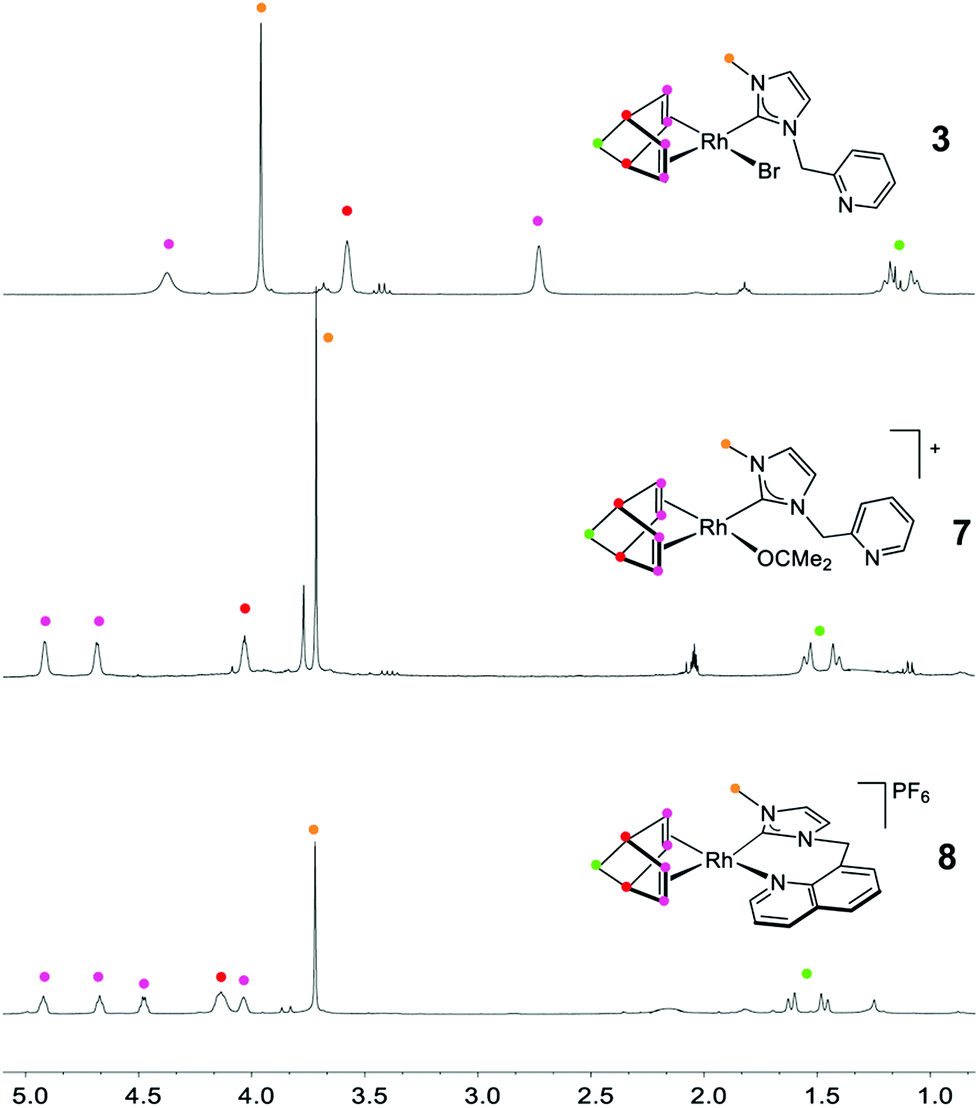 | ||
| Fig. 3 Selected region of the 1H NMR spectra of complexes 3, 7 and 8. | ||
The neutral amido complex [Rh(nbd){κ2C,N-t-BuIm(CH2)3N-t-Bu}] (9) was prepared by double deprotonation of [t-BuImH(CH2)3NH-t-Bu]Br·HBr with NaH followed by reaction with [Rh(μ-Cl)(nbd)]2 (Fig. 1, ii) and isolated as a yellow solid in moderate yield (53%) (Chart 1). The κ2-C,N coordination of the amido–NHC ligand in 9 is substantiated by the absence in the 1H NMR spectrum of the resonances of the H2 proton of the imidazolium fragment and that of the −NH2-t-Bu group present in the ammonium–imidazolium salt. Furthermore, the molecular ion observed in the MALDI-Tof mass spectrum at m/z ratio of 448.2 and conductivity measurements in acetone confirm the neutral character of the compound.
Polymerization of PA by rhodium(I) complexes with N-functionalized NHC ligands
The neutral [RhX(nbd)(κC-MeIm∩Z)] (1–4) and cationic [Rh(nbd)(κ2C,N-MeIm∩Z)]+ (5–8) complexes are efficient catalyst precursors for PA polymerization affording PPAs of very high molar mass (MM) (Table 1). PA polymerization reactions were carried out in THF at 293 K in the absence of light using a monomer-to-rhodium ratio [PA]o/[Rh] of 100. The PPAs polymers were isolated as soluble yellow-orange solids in practically quantitative yields according to the conversion values. The 1H NMR spectra showed a sharp signal at δ 5.86 ppm for the vinyl protons and six characteristic resonances in the 13C{1H} NMR spectra in CD2Cl2 which are indicative of a cis-transoidal configuration with high level of stereoregularity. In fact, a cis-content ≥95% was determined by NMR.28,29 The PPAs have been characterized by SEC/MALS/DRI that combines size-exclusion chromatography with multi-angle light scattering and refractive index detection.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | t (min) | Conv.b (%) | M w (g mol−1) | Đ | IEe (%) |
| a Reaction conditions: 293 K, [PA]o = 0.25 M, [PA]o/[Rh] = 100, in tetrahydrofuran. b Determined by GC (octane as internal standard). c Determined by SEC-MALS. d Đ = dispersity (Mw/Mn, Mn = number-average molecular weight). e Initiation efficiency, IE = Mtheor/Mn × 100; where Mtheor = [PA]o/[Rh] × MWPA × polymer yield. | ||||||
| 1 | 1 | 35 | 100 | 1.78 × 106 | 1.33 | 0.8 |
| 2 | 2 | 60 | 100 | 2.05 × 106 | 1.67 | 0.8 |
| 3 | 3 | 120 | 70 | 8.15 × 105 | 1.87 | 1.6 |
| 4 | 4 | 120 | 60 | 8.84 × 105 | 1.66 | 1.2 |
| 5 | 5 | 60 | 100 | 1.79 × 105 | 1.42 | 5.2 |
| 6 | 6 | 60 | 100 | 6.49 × 105 | 1.91 | 3.0 |
| 7 | 7 | 120 | 40 | 9.93 × 105 | 1.35 | 0.6 |
| 8 | 8 | 75 | 80 | 3.31 × 105 | 1.39 | 3.4 |
| 9 | 9 | 40 | 90 | 8.32 × 105 | 1.81 | 2.0 |
In the neutral series [RhX(nbd)(κC-MeIm∩Z)] (1–4), catalyst precursors having functionalized NHC ligands with a flexible amino-alkyl wingtip, 3-dimethylaminopropyl or 1-aminopropyl, were found to be considerably more active than those having a heterocyclic substituent (entries 1–4). Thus, catalysts 1 and 2 afforded complete PA conversion in 35 and 60 min, respectively, whereas catalysts 3 and 4 required 120 min to reach 60–70% PA conversions. The MM of the PPAs obtained with catalysts 1 and 2 is very high, weight-average molecular weights (Mw) of 1.78 × 106 and 2.05 × 106, with an initiation efficiency of 0.8% and moderate dispersities (Đ) of 1.33 and 1.67, respectively (entries 1 and 2). On the other hand, the PPA produced with catalysts 3 and 4 showed lower MM, Mw of ≈8.0 × 105, and larger Đ compared to the PA obtained with catalyst 1 (entries 3 and 4). The same trend was observed for the cationic catalysts [Rh(nbd)(κ2C,N-MeIm∩Z)]+ (5–8) with precursors 5 and 6, having a flexible amino-alkyl wingtip, as the more active catalysts in this series. Complete PA conversion was attained in 60 min affording PPAs of Mw 1.79 × 105 and 6.49 × 105, respectively, with moderate Đ (entries 5 and 6). This tendency could be associated to the bulkiness of the heterocyclic substituent compared to the amino-alkyl group at the NHC ligand that likely hampers the propagating step of the polymerization reaction. Catalyst 7 is much less active than 8 providing a PPA of higher MM, Mw of 9.93 × 105, and similar Đ (entries 7 and 8). The low PA conversion achieved with 7 points to a possible catalyst deactivation similarly as we found in rhodium catalysts featuring 2-diphenylphosphinopyridine ligands.30 Finally, the neutral amido complex [Rh(nbd){κ2C,N-t-BuIm(CH2)3N-t-Bu}] (9) showed an excellent activity reaching a 90% conversion in only 40 min to produce a PPA of Mw 8.32 × 105 and a Đ of 2.0.
It should be noted that the catalytic activity of neutral chloro-complexes compared to related cationic precursors is remarkable, particularly that of precursors having an amino-alkyl wingtip. Surprisingly, the catalytic activity of complexes 2 and 6 is comparable, although catalyst precursor 1 outperform compound 5. The excellent catalytic activity of the neutral chloro-complexes contrasts with the moderate to low catalytic activity of related complexes having unfunctionalized NHC ligands,16,18 even in the presence of an amine co-catalyst.15
Polymer characterization has been carried out by size exclusion chromatography (SEC) using multi-angle light scattering (MALS) and a refractive index (DRI) detectors, which has allowed to study the polymer morphology with regard to the presence of branching. In this context, it is worth mentioning that the analysis of the PPAs produced by related complexes [Rh(diene){κ2C,N-Ph2P(CH2)3NMe2}]+ having N-functionalized phosphine ligands showed the presence of branched polymer of high MM.13 The analysis of the PPAs obtained with the neutral catalysts evidenced the presence of branching in the samples obtained with catalysts 3 and 4, both having N-heterocyclic substituents at the NHC ligand. In contrast, catalysts 1 and 2, having N-alkyl chains as substituents, produced linear PPAs.
The light scattering and refractive index chromatograms of a PPA sample produced with catalyst [RhBr(nbd){κC-MeIm(CH2)3NH2}] (2) show a unimodal distribution of the MM (Fig. 4). The linearity of the MM representation over the elution volume range, where both MALS and DRI detectors have detectable intensity, and the linear relationship of the log–log plot of the radius of gyration (rg) vs. the molar mass (MM) in the high-molar-mass region, are characteristic of a linear polymer. In contrast, the PPA sample produced with catalyst [RhCl(nbd){κC-MeIm(quinolin-8-yl-methyl)}] (4) exhibited a very different behavior (Fig. 5). The detectable increase in MM on the high-MM region at short elution volumes suggests the presence of branched material. In addition, the log–log plot of rgvs. MM revealed an appreciable deviation from the linear behavior in the high MM region which is compatible with the presence of a small fraction of high MM branched polymer. It is important to note that the tailing intensity on the light scattering detector beyond the low-MM exclusion limit of the column set, the increase in MM with elution volume evident at long elution volumes and the quirky shape of the conformation plot in the low-MM region are a consequence of the interaction of the conjugated PPA material with the column packing. The slopes of the linear part of the conformation plots, 0.57 (2) and 0.55 (4), slightly deviate from the expected value of ca. 0.58 for a linear polymer reflecting the complex behavior of PPA in diluted solutions due to solvent-polymer and polymer-polymer interaction changes as well as σ-trans to σ-cis isomerization process.31
 | ||
| Fig. 4 (a) SEC chromatograms: light scattering detector response (90 degrees) (blue) and differential refractometer response (red), MM (molar mass) vs. elution volume plot for a PPA sample prepared with catalyst [RhBr(nbd){κC-MeIm(CH2)3NH2}] (2) in THF. (b) Log–log plot of the radius of gyration (rg) vs. MM. | ||
 | ||
| Fig. 5 (a) SEC chromatograms: light scattering detector response (90 degrees) (blue) and differential refractometer response (red), MM (molar mass) vs. elution volume plot for a PPA sample prepared with catalyst [RhCl(nbd){κC-MeIm(quinolin-8-yl-methyl)}] (4) in THF. (b) Log-log plot of the radius of gyration (rg) vs. MM. | ||
The observed trend in the neutral catalyst series is not reproduced in the cationic series. Thus, catalyst 5 afforded linear PPAs as the related neutral catalyst 1 and 2, as evidenced by the linear conformation plot. However, the PPA obtained with catalyst 6, also featuring with a flexible amino-alkyl wingtip, showed a deviation from linearity in the high-molar-mass region consistent with the presence of branched material of high MM. In the same way, catalyst 7 provided branched PPA as the related neutral catalysts 3 and 4, although catalyst 8, featuring a heterocyclic wingtip, afforded linear PPA. On the other hand, the amido complex 9 polymerize PA to give an essentially linear high molar mass PPA.
SEC chromatograms, plots of MM and rgvs. elution volume, and log–log plots of rgvs. MM for the PPA samples can be found in the ESI.†
Mechanistic considerations
The development of rhodium-based polymerization catalysts was driven by the pioneering work by Furlani and Tabata, which showed that Rh(I) complexes, such as [Rh(cod)(NN)]+ (NN = bipy, phen) in the presence of a strong base (NaOH),28 or [Rh(μ-Cl)(nbd)]2 in triethylamine as solvent,32 were efficient catalysts for the polymerization of PA. It was shown that NEt3 played a role as a cocatalyst acting as a base and facilitating the formation of the mononuclear active species [RhCl(nbd)(NEt3)].33 Interestingly, neither neutral [RhX(nbd)(κC-MeIm∩Z)] (1–4) nor cationic [RhX(nbd)(κC,N-MeIm∩Z)] (5–8) complexes require an external base to initiate the PA polymerization and, therefore, the role of the N-donor function in the NHC ligand is likely to act as an internal base for the deprotonation of PA to give an active alkynyl species, as in the case of related cationic species [Rh(cod){κ2C,P-Ph2P(CH2)3NMe2}]+ having N-functionalized phosphine ligands.12On the other hand, the initiation efficiencies calculated for both type of catalysts are very low (0.8–5.2%, Table 1). In this regard, theoretical studies by Morokuma et al.34 have demonstrated the key role of alkynyl species as PA polymerization initiators showing that the energy barrier for the PA insertion into the Rh–alkynyl bond of [Rh(nbd)(CC-Ph)(PA)] (initiation step) is almost 4 kcal mol−1 higher than the barrier for the insertion into the Rh–vinyl bond (propagation step), which explain the low initiation efficiencies observed for the Rh(NHC) catalysts (Scheme 1).
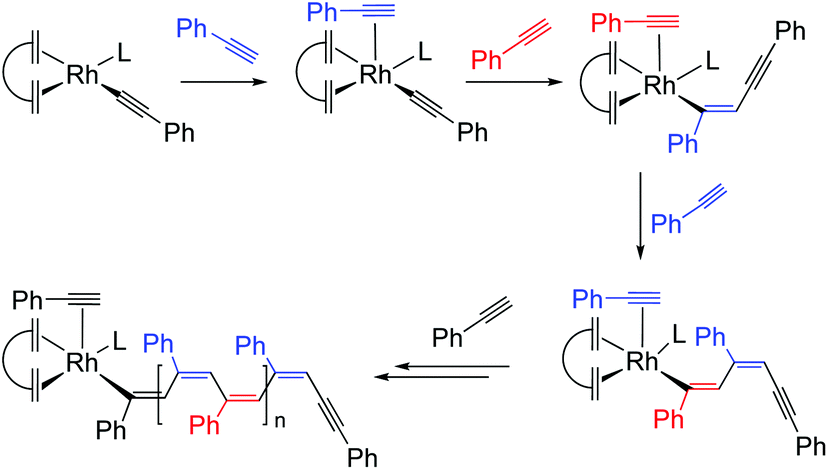 | ||
| Scheme 1 Plausible mechanism for the polymerization of PA by a generic [Rh(diene)(CC-Ph)L] species. | ||
The possible reaction pathways leading to the formation of key alkynyl initiating species from neutral or cationic complexes, as illustrate for the 3-dimethylaminopropyl-functionalized complexes 1 and 5, are shown in Scheme 2. The proposed initiating mechanism for the cationic compounds [Rh(nbd)(κ2C,N-MeIm∩Z)]+ is well documented and entails proton transfer from a η2-alkyne ligand to the N-donor atom at the wingtip, that behaves as an internal base, which results in the formation of the cationic alkynyl species [Rh(nbd)(CC-Ph)(κC-MeIm∩ZH)]+ (pathway ii). Related alkynyl species have been suggested to be the initiating species likely involved in the generation of stable rhodium–vinyl species, responsible for the propagation step, by PA insertion into the Rh–alkynyl bond.30,35
 | ||
| Scheme 2 Possible reaction pathways leading to the key Rh–alkynyl initiating species exemplified with complexes 1 and 5. | ||
This activation pattern could be also operative for the neutral complexes [RhX(nbd)(κC-MeIm∩Z)] (X = Cl or Br). After PA coordination, proton transfer to the uncoordinated N-donor function should give rise to a zwitterionic alkynyl complex, likely with a square pyramidal structure having the strong σ-donating alkynyl ligand in the apical position,36 from which ionization of the chlorido ligand may produce the same cationic species (pathway i). However, proton transfer to the reaction medium cannot be ruled out, which would give rise to a neutral alkynyl species [Rh(nbd)(CC-Ph)(κC-MeIm∩Z)] also capable of initiating the polymerization process (iii, Scheme 2). It is worth mentioning that the hydrogen bond or proton acceptor power of THF is considered to be comparable to that of methanol or monomeric water.37 In fact, mechanistic studies on PA polymerization by 2-diphenylphosphinopyridine-based rhodium(I) catalysts evidenced the formation of the neutral species [Rh(CCPh)(cod){κP-Ph2PPy}] by proton transfer to the reaction medium from the cationic [Rh(CCPh)(cod)(κP-Ph2PPyH)]+ resulting from the activation of PA.30
The efficiency of the initiation process depends on several factors: (i) the strength of the Rh–N and Rh–Cl bonds, (ii) the strain on the chelate ring that should favor the breaking of the Rh–N bond, and (iii) the basicity of the N-donor function. In general, the initiation efficiency of the cationic catalysts is greater (2–5%) than that of the neutral ones. Neutral complex 1 is significantly more active than the related cationic compound 5 and produces a polymer of greater MM as a result of a lower initiation efficiency. The different catalytic performance of both catalysts suggests the participation of distinct active species. It is reasonable to assume that the cationic [Rh(nbd)(CC-Ph){κC-MeIm(CH2)3NHMe2}]+ species is responsible for the initiation process with catalyst 5. However, we hypothesized that the chloride ion may facilitate the deprotonation of the dimethyl ammonium fragment in the cationic intermediate as an ionic pair H(thf)n+Cl− (ref. 38) leading to the efficient formation of the neutral alkynyl species [Rh(nbd)(CC-Ph){κC-MeIm(CH2)3NMe2}], which is likely the initiating species for catalyst 1.
However, this hypothesis does not hold for compounds having quinolin-8-ylmethy as wingtip since the cationic compound 8 is considerably more active than the chloro-complex 4. However, the strain in the 7-membered metallacycle likely enables the decoordination of the N-donor function while the lower basicity of the quinoline fragment facilitates proton transfer to the reaction medium (quinoline, pKa = 4.85; trimethylamine, pKa = 9.76).39 Therefore, both factors make the formation of the neutral alkynyl species through pathway ii much more favorable than through pathway i, because of the slower dissociation of the chlorido ligand compared to quinoline. Compounds 2 and 6, both having the 3-aminopropyl wingtip, have comparable activities although the initiation efficiency of 6 is greater which agrees with the observed tendency. In principle, it is likely that the Br− anion is not so effective as Cl− in promoting proton transfer to the reaction medium which, in combination with the basicity of the -NH2 group (methylamine, pKa = 10.64),39 suggest that the initiating species with both catalysts might be the cationic [Rh(nbd)(CC-Ph){κC-MeIm(CH2)3NH3}]+ species. This observation is consistent with the lower catalytic activity exhibited by 2 compared to 1. On the other hand, the poor catalytic activity of cationic precursor 7 (40% conversion in 2 h) does not allow a direct comparison with the related neutral compound 3 since both have different structures due to the coordination of acetone in 7 (Chart 2).
Catalytic performance of rhodium complexes based on N-functionalized phosphine ligands
To validate our hypothesis, we have compared the catalytic performance of complexes 1 and 5 with that of related complexes having N-functionalized phosphine ligands (Chart 3). The new neutral chloro-complexes [RhCl(nbd){κP-Ph2P(CH2)3NMe2}] (10) and [RhCl(cod){κP-Ph2P(CH2)3NMe2}] (12) have been prepared by reaction of the dinuclear compounds [Rh(μ-Cl)(diene)]2 (diene = cod, nbd) with two equiv. of Ph2P(CH2)3NMe2 (see the ESI†). The corresponding cationic compounds 11 and 13,12 and complexes 14 and 15, based on the 2-(2-(diphenylphosphino)ethyl)pyridine ligand,30 had been previously described by our research group. The catalytic performance of neutral and cationic Rh(I)–phosphine compounds 10–15 in PA polymerization is shown in Table 2. | ||
| Chart 3 Structure of Rh(I)–phosphine compounds 10–15. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | t (min) | Conv.b (%) | M w (g mol−1) | Đ | IEe (%) |
| a Reaction conditions: 293 K, [PA]o = 0.25 M, [PA]o/[Rh] = 100, in tetrahydrofuran. b Determined by GC (octane as internal standard). c Determined by SEC-MALS. d Đ = dispersity (Mw/Mn, Mn = number-average molecular weight). e Initiation efficiency, IE = Mtheor/Mn × 100; where Mtheor = [PA]o/[Rh] × MWPA × polymer yield. f Data taken from ref. 13. g Data taken from ref. 30. h Bimodal MM distribution: data for the lower mass polymer. | ||||||
| 1 | 10 | 120 | 90 | 1.49 × 106 | 1.77 | 1.2 |
| 2f | 11 | 60 | 100 | 2.18 × 106 | 2.00 | 0.9 |
| 3 | 12 | 150 | 70 | 1.30 × 105 | 1.64 | 9.0 |
| 4f | 13 | 120 | 100 | 2.38 × 105h | 1.79 | 7.7 |
| 5g | 14 | 120 | 100 | 2.04 × 106 | 1.63 | 0.9 |
| 6g | 15 | 300 | 100 | 1.66 × 106 | 1.69 | 0.5 |

Comparing data in Tables 1 and 2, it can be observed that compound 1 is much more active than 10 although both provide PPAs of similar Mw (Fig. 6). However, cationic compounds 5 and 11 have comparable activities although 11 produces a PPA of greater Mw (Fig. 7). As expected, nbd complexes 10 and 11 proved to be more active and provided higher molar mass PPAs, resulting in lower calculated initiation efficiencies than the corresponding cod complexes 12 and 13, which is consistent with the higher π-acidity of the nbd ligand compared to cod.40 However, against our initial expectations, the chloro-complexes 10 and 12 were found to be less active than the related cationic compounds 11 and 13. The rationalization of these results requires considering the very different electronic properties of the NHC and phoshine ligands. The NHC ligands are stronger σ-donor than phosphines which also have π-acceptor properties and thus, the electron density at the rhodium center in compound 1 is expected to be higher than in 10 which should facilitate the ionization of the chlorido ligand. In contrast, the neutral compound 14, featuring the 2-(2-(diphenylphosphino)ethyl)pyridine ligand, is more active than the corresponding cationic complex 15 (Table 2) which is attributed to the easy formation of the neutral active species [Rh(nbd)(CC-Ph){κP-Ph2P(CH2)2Py}] due to the efficient elimination of HCl enabled by the low basicity of the pyridine fragment (pyridine, pKa = 5.17).39
 | ||
| Fig. 6 Light scattering chromatograms (MALS) for PPA samples prepared with catalysts: [RhCl(nbd){κC-MeIm(CH2)3NMe2}] (1) (red) and [RhCl(nbd){κP-Ph2P(CH2)3NMe2}] (10) (blue). | ||
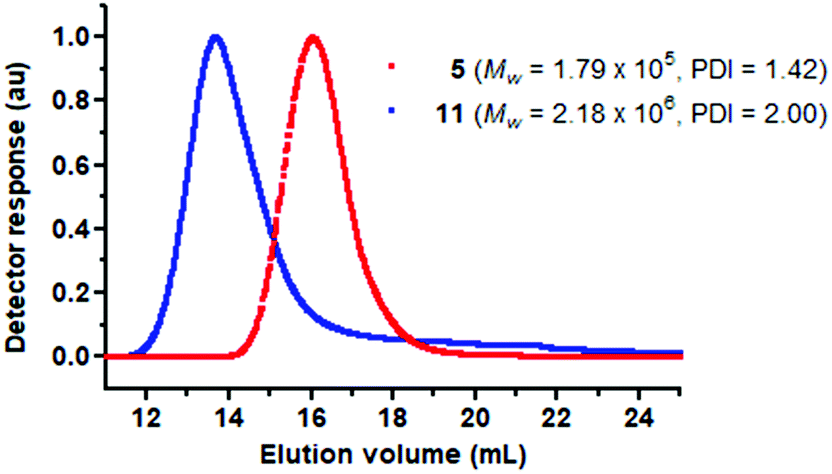 | ||
| Fig. 7 Light scattering chromatograms (MALS) for PPA samples prepared with catalysts: [Rh(nbd){κC,N-MeIm(CH2)3NMe2}][BF4] (5) (red) and [Rh(nbd){κP,N-Ph2P(CH2)3NMe2}][BF4] (11) (blue). | ||
Conclusions
A series of neutral [RhX(nbd)(κC-MeIm∩Z)] and cationic [Rh(nbd)(κ2C,N-MeIm∩Z)]+ complexes featuring NHC ligands with a N-functionalized substituent have been prepared from suitable imidazolium salts following well-established methodologies. Both types of complexes efficiently catalyze the polymerization of phenylacetylene in the absence of base affording stereoregular PPAs of very high molar mass. Catalyst precursors having functionalized NHC ligands with a flexible amino-alkyl wingtip, such as 3-dimethylaminopropyl or 1-aminopropyl, are considerably more active than those having a heterocyclic substituent, such as such as pyridin-2-ylmethyl and quinolin-8-ylmethyl. This trend is ascribed to the bulkiness of the heterocyclic substituent compared to the amino-alkyl group that likely hinders the propagating step of the polymerization reaction. Noteworthy, related compounds having N-functionalized phosphine ligands are in general much less active than the corresponding MeIm∩Z complexes. PPA polymers of weight-average molecular weights up to 2 × 106 g mol−1, with initiation efficiencies as low as 0.8%, and moderate dispersity have been prepared with the neutral chloro-complexes having aminopropyl wingtips. Analysis of the morphology of the polymers has revealed the presence of branched polymer of high molar mass in the samples obtained with neutral catalyst precursors having a heterocyclic substituent at the NHC ligand, although this trend is not reproduced in the cationic series.These catalyst precursors do not require an external base to initiate the polymerization of PA and, therefore, it is plausible that the N-donor function in the NHC ligand acts as an internal base for the deprotonation of PA to give an active alkynyl species. In the case of cationic complexes, proton transfer from a η2-alkyne ligand to the N-donor atom at the wingtip results in the formation of the cationic alkynyl intermediate [Rh(nbd)(CC-Ph)(κC-MeIm∩ZH)]+ which should actually be the initiating species. However, the formation of neutral alkynyl species [Rh(nbd)(CC-Ph)(κC-MeIm∩Z)] species by proton transfer to the reaction medium as an ionic pair H(thf)n+Cl could account for the remarkable catalytic activity of some neutral chloro-complexes compared to the related cationic precursors. In this context, the strength of the Rh–N and Rh–X bonds, the strain on the chelate ring in the cationic complexes, the electronic density at the metal center, and the basicity of the N-donor function have to be considered in order to rationalize the observed reactivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their appreciation for the financial support from the Spanish Ministerio de Ciencia e Innovación, MCIN/AEI/10.13039/501100011033, under the project PID2019-103965GB-I00, and the “Departamento de Ciencia, Universidad y Sociedad del Conocimiento del Gobierno de Aragón” (group E42_20R). The authors gratefully acknowledge Dr Vincenzo Passarelli (ISQCH) for his assistance in analyzing and discussing structural data.Notes and references
- (a) T. Masuda, Polym. Rev., 2017, 57, 1–14 CrossRef CAS; (b) A. Xu, T. Masuda and A. Zhang, Polym. Rev., 2017, 57, 138–158 CrossRef CAS; (c) R. Sakai, T. Satoh and T. Kakuchi, Polym. Rev., 2017, 57, 159–174 CrossRef CAS.
- (a) M. Goto, A. Nito, Y. Miyagi and F. Sanda, Macromol. Mater. Eng., 2019, 304, 1900275 CrossRef; (b) L. Sekerová, M. Lhotka, E. Vyskočilová, T. Faukner, E. Slováková, J. Brus, L. Červený and J. Sedláček, Chem. – Eur. J., 2018, 24, 14742–14749 CrossRef PubMed; (c) L. Liu, Y. Zang, H. Jia, T. Aoki, T. Kaneko, S. Hadano, M. Teraguchi, M. Miyata, G. Zhang and T. Namikoshi, Polym. Rev., 2017, 57, 89–118 CrossRef CAS; (d) R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, J. Am. Chem. Soc., 2016, 138, 9620–9628 CrossRef PubMed; (e) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed.
- F. Freire, E. Quiñoá and R. Riguera, Chem. Rev., 2016, 116, 1242–1271 CrossRef CAS PubMed.
- T. Masuda, F. Sanda and M. Shiotsuki, Polymerization of Acetylenes, in Comprehensive Organometallic Chemistry III, ed. D. M. P. Mingos and R. H. Crabtree, Elsevier, Amsterdam, Holland, 2007, 1st edn, vol. 11, pp. 557–593 Search PubMed.
- (a) M. Shiotsuki, F. Sanda and T. Masuda, Polym. Chem., 2011, 2, 1044–1058 RSC; (b) T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 165–180 CrossRef CAS.
- (a) T. Taniguchi, T. Yoshida, K. Echizen, K. Takayama, T. Nishimura and K. Maeda, Angew. Chem., Int. Ed., 2020, 59, 8670–8680 CrossRef CAS PubMed; (b) J. Sedláček and H. Balcar, Polym. Rev., 2017, 57, 31–51 CrossRef; (c) M. A. Casado, A. Fazal and L. A. Oro, Arabian J. Sci. Eng., 2013, 38, 1631–1646 CrossRef CAS; (d) J. Sedláček and J. Vohlídal, Collect. Czech. Chem. Commun., 2003, 68, 1745–1790 CrossRef.
- (a) S. Ikeda, Y. Hanamura, H. Tada and R. Shintani, J. Am. Chem. Soc., 2021, 143, 19559–19566 CrossRef CAS PubMed; (b) S. Ikeda and R. Shintani, Angew. Chem., Int. Ed., 2019, 58, 5734–5738 CrossRef CAS PubMed.
- N. S. L. Tan and A. B. Lowe, Angew. Chem., Int. Ed., 2020, 59, 5008–5021 CrossRef CAS PubMed.
- (a) N. S. L. Tan, P. V. Simpson, G. L. Nealon, A. N. Sobolev, P. Raiteri, M. Massi, M. I. Ogden and A. B. Lowe, Eur. J. Inorg. Chem., 2019, 592–601 CrossRef CAS; (b) N. Onishi, M. Shiotsuki, T. Masuda, N. Sano and F. Sanda, Organometallics, 2013, 32, 846–853 CrossRef CAS; (c) I. Saeed, M. Shiotsuki and T. Masuda, Macromolecules, 2006, 39, 8567–8573 CrossRef CAS; (d) M. Miyake, Y. Misumi and T. Masuda, Macromolecules, 2000, 33, 6636–6639 CrossRef CAS.
- (a) N. S. L. Tan, G. L. Nealon, G. F. Turner, S. A. Moggach, M. I. Ogden, M. Massi and A. B. Lowe, ACS Macro Lett., 2020, 9, 56–60 CrossRef; (b) N. S. L. Tan, G. L. Nealon, J. M. Lynam, A. N. Sobolev, M. R. Rowless, M. I. Ogden, M. Massi and A. B. Lowe, Dalton Trans., 2019, 48, 16437–16447 RSC.
- (a) Y. Tian, X. Li, J. Shi, B. Tonga and Y. Dong, Polym. Chem., 2017, 8, 5761–5768 RSC; (b) M. Tabata, W. Yang and K. Yokota, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1113–1120 CrossRef CAS.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, E. Vispe, F. J. Lahoz and L. A. Oro, Macromolecules, 2009, 42, 8146–8156 CrossRef.
- M. Angoy, M. I. Bartolomé, E. Vispe, P. Lebeda, M. V. Jiménez, J. J. Pérez-Torrente, S. Collins and S. Podzimek, Macromolecules, 2010, 43, 6278–6283 CrossRef CAS.
- E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- Y. Zhang, D. Wang, K. Wurst and M. R. Buchmeiser, J. Organomet. Chem., 2005, 690, 5728–5735 CrossRef CAS.
- W. Gil, T. Lis, A. M. Trzeciak and J. J. Ziółkowski, Inorg. Chim. Acta, 2006, 359, 2835–2841 CrossRef CAS.
- V. César, N. Lugan and G. Lavigne, Chem. – Eur. J., 2010, 16, 11432–11442 CrossRef PubMed.
- Y. Koto, F. Shibahara and T. Murai, Org. Biomol. Chem., 2017, 15, 1810–1820 RSC.
- K. Cho, H.-S. Yang, I.-H. Lee, S. M. Lee, H. J. Kim and S. U. Son, J. Am. Chem. Soc., 2021, 143(11), 4100–4105 CrossRef CAS PubMed.
- M. V. Jiménez, J. Fernández-Tornos, J. J. Perez-Torrente, F. J. Modrego, S. Winterle, C. Cunchillos, F. Lahoz and L. A. Oro, Organometallics, 2011, 30, 5493–5508 CrossRef.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, V. Gierz, F. J. Lahoz and L. A. Oro, Organometallics, 2008, 27, 224–234 CrossRef.
- H. M. Peng, R. D. Webster and X. Li, Organometallics, 2008, 27, 4484–4493 CrossRef CAS.
- X.-Y. Yu, B. O. Patrick and B. R. James, Organometallics, 2006, 25, 2359–2363 CrossRef CAS.
- The puckering parameters of the 7-member ring Rh(1)–N(1)–C(16)–C(15)–C(17)–N(3)–C(18): are QT = 1.089(2) Å, ϕ2 = −33.19(10)°, ϕ3 = 38.40(33)° and θ = 70.61(12)°. D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- See for example: (a) R. Azpíroz, A. Di Giuseppe, V. Passarelli, J. J. Pérez-Torrente, L. A. Oro and R. Castarlenas, Organometallics, 2018, 37, 1695–1707 CrossRef; (b) R. Azpíroz, L. Rubio-Pérez, A. Di Giuseppe, V. Passarelli, F. J. Lahoz, R. Castarlenas, J. J. Pérez-Torrente and L. A. Oro, ACS Catal., 2014, 4, 4244–4253 CrossRef.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- M. V. Jiménez, M. I. Bartolomé, J. J. Pérez-Torrente, D. Gómez, F. J. Modrego and L. A. Oro, ChemCatChem, 2013, 5, 263–276 CrossRef.
- A. Furlani, C. Napoletano, M. V. Russo, A. Camus and N. J. Marsich, Polym. Sci., Part A: Polym. Chem., 1989, 27, 75–86 CrossRef CAS.
- A. Furlani, C. Napoletano, M. V. Russo and W. J. Feast, Polym. Bull., 1986, 16, 311–317 CrossRef CAS.
- M. Angoy, M. V. Jiménez, F. J. Modrego, L. A. Oro, V. Passarelli and J. J. Pérez-Torrente, Organometallics, 2018, 37, 2778–2794 CrossRef CAS.
- C. Cametti, P. Codastefano, R. D'Amato, A. Furlani and M. Russo, Synth. Met., 2000, 114, 173–179 CrossRef CAS.
- (a) W. Yang, M. Tabata, S. Kobayashi, K. Yokota and A. Shimizu, Polym. J., 1991, 23, 1135–1138 CrossRef CAS; (b) M. Tabata, W. Yang and K. Yokota, Polym. J., 1990, 22, 1105–1107 CrossRef CAS.
- M. Tabata, T. Sone and Y. Sadahiro, Macromol. Chem. Phys., 1999, 200, 265–282 CrossRef CAS.
- Z. Ke, S. Abe, T. Ueno and K. Morokuma, J. Am. Chem. Soc., 2011, 133, 7926–7941 CrossRef CAS PubMed.
- (a) M. Angoy, M. V. Jiménez, P. García-Orduña, L. A. Oro, E. Vispe and J. J. Pérez-Torrente, Organometallics, 2019, 38, 1991–2006 CrossRef CAS; (b) Y. Tian, X. Li, J. Shi, B. Tonga and Y. Dong, Polym. Chem., 2017, 8, 5761–5768 RSC; (c) M. Shiotsuki, N. Onishi, F. Sanda and T. Masuda, Polym. J., 2011, 43, 51–57 CrossRef CAS; (d) T. Nishimura, X. X. Guo, K. Ohnishi and T. Hayashi, Adv. Synth. Catal., 2007, 349, 2669–2672 CrossRef CAS.
- (a) A. Meißner, A. Kçnig, H.-J. Drexler, R. Thede, W. Baumann and D. Heller, Chem. – Eur. J., 2014, 20, 14721–14728 CrossRef PubMed; (b) V. César, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2005, 11, 2862–2873 CrossRef PubMed.
- K. Deshmukh, S. Siddiqui and J. F. Coetzee, J. Electrochem. Soc., 1991, 138, 124–132 CrossRef.
- (a) H. Mishra, S. Enami, R. J. Nielsen, M. R. Hoffmann, W. A. Goddard and A. J. Colussi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10228–10232 CrossRef CAS PubMed; (b) V. Pilepić, C. Jakobušić, D. Vikić-Topić and C. Uršić, Tetrahedron Lett., 2006, 47, 371–375 CrossRef.
- (a) H. K. Hall, J. Am. Chem. Soc., 1957, 79, 5441–5444 CrossRef CAS; (b) R. S. Hosmane and J. F. Liebman, Struct. Chem., 2009, 20, 693–697 CrossRef CAS.
- (a) N. Onishi, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2009, 42, 4071–4076 CrossRef CAS; (b) I. Saeed, M. Shiotsuki and T. Masuda, Macromolecules, 2006, 39, 8977–8981 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of rhodium complexes, chromatograms and conformation plots of PPA samples. CCDC 1986054. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1py01650d |
| This journal is © The Royal Society of Chemistry 2022 |