
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of radical copolymerization of the (oxa)norbornene end-group of RAFT-prepared macromonomers on bottlebrush copolymer synthesis via ROMP†
Mohamed
Naguib
ab,
Kate L.
Nixon
ac and
Daniel J.
Keddie
 *a
*a
aSchool of Sciences, Faculty of Science and Engineering, University of Wolverhampton, Wulfruna Street, Wolverhampton, WV1 1LY, UK. E-mail: d.keddie@wlv.ac.uk
bPolymer and Pigments Department, National Research Centre, Cairo, 12622, Egypt
cSchool of Life, Health and Chemical Sciences, The Open University, Walton Hall, Milton Keynes, MK7 6AA, UK
First published on 2nd February 2022
Abstract
Bottlebrush polymers are attractive for use in a variety of different applications. Here we report the synthesis of two novel trithiocarbonate RAFT agents bearing either an oxanorbornenyl or norbornenyl moiety for bottlebrush synthesis via ROMP grafting-through polymerization. RAFT polymerization kinetics was evaluated as a function of the monomer type, number-average degree of polymerization (Xn) and RAFT agent structure. The correlation between the oxa/norbornenyl moiety and the type of RAFT monomer (methyl acrylate, n-butyl acrylate, and styrene) has been investigated. The reactivity of the oxa/norbornenyl group of the RAFT agent towards the radical propagating species during RAFT polymerization influences the molar mass, molar mass distribution and the residual olefinic end-group functionality of the resulting polymeric macromonomers. The RAFT synthesized macromonomers (MMs) are subjected to “grafting-through” ROMP using a Grubbs 3rd generation catalyst, resulting in bottlebrush polymers. The ‘defects’ in the MM structures have been found to be responsible for the higher amount of MM residue after the ROMP process and hence affect the microstructures of the synthesized bottlebrush polymers.
Introduction
Due to their outstanding and tunable properties, molecular bottlebrush polymers have attracted great interest in the last few years.1–3 Molecular bottlebrushes consist of long linear polymeric backbones densely grafted with short side-chains. Based on their well-defined structures and fascinating properties they can be functionalized and utilized in many applications such as coatings, lubrication, nanomedicine, drug delivery, and photonics.4–6There are three main approaches for the preparation of bottlebrush polymers: (1) ‘grafting-to’ – attachment of side chains to the polymeric backbone; (2) ‘grafting-from’ – polymerization of monomers from the backbone; and (3) ‘grafting-through’ – polymerization of macromonomers.7 Each method has its own advantages and limitations. Generally, these well-defined bottlebrushes are synthesised via a combination of two or more polymerization techniques.8,9
The grafting-through strategy is the polymerization of macromonomers (MMs) that contain polymerizable end-groups. The grafting density and dispersity play a crucial role in the performance of these bottlebrush polymers. Unlike the other two strategies, the grafting-through technique ensures high grafting density as well as low brush dispersity and hence extraordinary properties and high efficacy toward applications. Often, ring-opening metathesis polymerization (ROMP), using Grubbs-type Ru catalysts, has been utilized for the grafting-through technique due to its rapid polymerization rates. In addition, the molar mass of MMs and the reactivity of polymerizable moieties are key factors in the grafting-through strategy.1,10
For the preparation of functional MMs with tailored molecular properties (i.e. targeted molar mass, low dispersity) reversible deactivation radical polymerization (RDRP) techniques are highly attractive. Reversible addition–fragmentation chain transfer (RAFT) polymerization is arguably the most versatile RDRP method as it has a superior tolerance for a wide range of functional groups;11 the RAFT technique is compatible with non-ionic, cationic, and anionic monomers. Through careful selection of the RAFT agent and reaction conditions, MMs amenable for polymerization by “grafting-through” ROMP can be prepared.1,10,12
In the broader scientific literature exo-norbornenes are the cyclic olefins (monomers) of choice for ROMP; their rapid ring opening metathesis kinetics and low incidence of chain transfer render their polymerization ‘living’.13 An added advantage of norbornenes is their thermal stability. While being less commonly used, exo-oxanorbornenes (i.e. oxygen-bridged analogues of norbornenes) are also widely reported in ROMP.7
In the context of functional RAFT agent synthesis, exo-oxanorbornene derivatives are quite attractive starting materials as they are relatively inexpensive. In contrast, exo-norbornenes tend to be quite expensive and are often prepared ‘in-house’ by laborious isomerization methods from the cheaper endo-isomers. Whilst this suggests oxanorbornenes as an attractive alternative to norbornenes, for the synthesis of ‘ROMP-able’ RAFT agents, their main drawback is their thermal lability; they can readily undergo retro-Diels–Alder reactions extruding furan.14
Herein, we directly compare the utility of oxanorbornene- and norbornene-based trithiocarbonate RAFT agents for the preparation of bottlebrush polymers via sequential RAFT/ROMP. The influence of the identities of the RAFT polymerizable monomer (i.e. methyl acrylate (MA), n-butyl acrylate (BA), and styrene (St)) and the strained olefin end-group (i.e. oxanorbornenyl, norbornenyl) on macromonomer (MM) synthesis is investigated in detail. While both norbornene1,12,15–17 and oxanorbornene5,18 end-groups have been used for the synthesis of polymers via a sequential RAFT/ROMP strategy (or other RDRP/ROMP methods) in the past, the incidence and effect of radical propagation to the end-group has largely been ignored and a direct comparison is lacking; here we seek to remedy these points. Additionally, the effect of the resultant MM structure on the subsequent ROMP grafting-through polymerization is also investigated.
Experimental
Materials
2-Bromopropionyl bromide, carbon disulfide (CS2), chlorobenzene, dodecanethiol, ethanolamine, n-heptane, neutral alumina Brockmann activity I (70–230 mesh), cis-5-norbornene-endo-2,3-dicarboxylic anhydride, potassium tert-butoxide, pyridine, triethylamine (TEA) and all solvents were purchased from Fisher Scientific. Solvents were of analytical reagent grade unless otherwise stated. exo-3,6-Epoxy-1,2,3,6-tetrahydrophthalic anhydride 1, a Grubbs 3rd generation catalyst (G3) (dichloro[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene](benzylidene)bis(3-bromopyridine) ruthenium(II)), and anhydrous sodium sulfate (Na2SO4) were obtained from Sigma-Aldrich. Methyl acrylate (MA), n-butyl acrylate (BA) and styrene (St) were purchased from Sigma-Aldrich and freed from inhibitors by passage through neutral alumina activity I (70–230 mesh). 2,2′-Azobis[2-methylpropionitrile] (AIBN) was purchased from Acros and purified by recrystallization twice from methanol prior to use. All NMR solvents were produced by Cambridge Isotope Laboratories and obtained through Goss Scientific. Silica gel (ZEO prep 60 HYD 40–63 μm) was obtained from Apollo Scientific. Hydrochloric acid was purchased from Better Equipped.Characterization
Nuclear magnetic resonance (NMR) spectra were recorded on a Jeol 400 MHz spectrometer at room temperature. 1H and 13C NMR spectra were internally referenced to the residual solvent.19 Size exclusion chromatography (SEC) was conducted on an EcoSEC-HLC 8320GPC system with a dual flow RI detector and a TSKgel Super HZM-N 3 μm (4.6 × 150 mm) column. THF was used as the eluent at a flow rate of 0.35 mL min−1 at 40 °C and low dispersity polystyrene standards were used for the calibration.Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH). These data are in agreement with those of Kötteritzsch et al.20
CH); 13C NMR (50 MHz, CDCl3) δ 21.5, 37.6, 39.9, 47.5, 62.0, 80.9, 136.6, 170.0, 176.0. These data are in agreement with those of Runge and Bowden.21
CH); 13C NMR (50 MHz, CDCl3) δ 14.2, 16.8, 22.8, 27.9, 29.0, 29.2, 29.4, 29.5, 29.6, 29.7, 29.7, 32.0, 37.4, 37.7, 47.6, 47.6, 47.8, 62.0, 77.3, 136.6, 171.0, 176.0, 222.1.
CH).These data are in agreement with those of Matson and Grubbs.23
CH);13C NMR (50 MHz, CDCl3) δ 41.4, 42.9, 45.3, 48.0, 60.3, 137.9, 178.9. These data are in agreement with those of Matson and Grubbs.23
CH); 13C NMR (50 MHz, CDCl3) δ 20.4, 36.2, 38.6, 41.8, 44.2, 46.8, 61.2, 136.7, 168.8, 176.7.
CH). These data are in agreement with those of Kötteritzsch et al.20
CH); 13C NMR (50 MHz, CDCl3) δ 21.5, 37.6, 39.9, 47.5, 62.0, 80.9, 136.6, 170.0, 176.0. These data are in agreement with those of Runge and Bowden.21
CH); 13C NMR (50 MHz, CDCl3) δ 14.2, 16.8, 22.8, 27.9, 29.0, 29.2, 29.4, 29.5, 29.6, 29.7, 29.7, 32.0, 37.4, 37.7, 47.6, 47.6, 47.8, 62.0, 77.3, 136.6, 171.0, 176.0, 222.1.
CH).These data are in agreement with those of Matson and Grubbs.23
CH);13C NMR (50 MHz, CDCl3) δ 41.4, 42.9, 45.3, 48.0, 60.3, 137.9, 178.9. These data are in agreement with those of Matson and Grubbs.23
CH); 13C NMR (50 MHz, CDCl3) δ 20.4, 36.2, 38.6, 41.8, 44.2, 46.8, 61.2, 136.7, 168.8, 176.7.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2)). Removal of the solvent under reduced pressure gave the norbornene-functional RAFT agent 8-Nb as a yellow solid (6.1 g, 11.3 mmol, 55%); 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J 6.8 Hz, 3H, CH3), 1.20–1.30 (m, 16H, alkyl CH2), 1.29 (d, J 9.7 Hz, 1H, CHH), 1.37 (m, 2H, SCH2CH2CH2), 1.51 (m, 1H, CHH), 1.56 (d, J 7.3 Hz, 3H, CHCH3), 1.66 (p, J 7.3 Hz, 2H, SCH2CH2), 2.69 (d, J 1.2 Hz, 2H, 2 × endo-CH), 3.28 (m, 2H, 2 × bridgehead-CH), 3.33 (t, J 7.3 Hz, 2H, SCH2), 3.76 (m, 2H, NCH2), 4.38 (m, 2H, OCH2), 4.80 (q, J 7.3, 1H, CHCH3), 6.28 (t, J 1.8 Hz, 2H, 2 × CH); 13C NMR (50 MHz, CDCl3) δ 14.2, 16.8, 22.8, 28.0, 29.0, 29.2, 29.4, 29.6, 29.7, 32.0, 37.4, 37.5, 43.0, 45.4, 47.6, 48.0, 62.1, 137.9, 170.9, 177.8, 221.9.
:2)). Removal of the solvent under reduced pressure gave the norbornene-functional RAFT agent 8-Nb as a yellow solid (6.1 g, 11.3 mmol, 55%); 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J 6.8 Hz, 3H, CH3), 1.20–1.30 (m, 16H, alkyl CH2), 1.29 (d, J 9.7 Hz, 1H, CHH), 1.37 (m, 2H, SCH2CH2CH2), 1.51 (m, 1H, CHH), 1.56 (d, J 7.3 Hz, 3H, CHCH3), 1.66 (p, J 7.3 Hz, 2H, SCH2CH2), 2.69 (d, J 1.2 Hz, 2H, 2 × endo-CH), 3.28 (m, 2H, 2 × bridgehead-CH), 3.33 (t, J 7.3 Hz, 2H, SCH2), 3.76 (m, 2H, NCH2), 4.38 (m, 2H, OCH2), 4.80 (q, J 7.3, 1H, CHCH3), 6.28 (t, J 1.8 Hz, 2H, 2 × CH); 13C NMR (50 MHz, CDCl3) δ 14.2, 16.8, 22.8, 28.0, 29.0, 29.2, 29.4, 29.6, 29.7, 32.0, 37.4, 37.5, 43.0, 45.4, 47.6, 48.0, 62.1, 137.9, 170.9, 177.8, 221.9.
RAFT polymerization
:1:0.1 or 50:1:0.1 were mixed in a 25 mL round bottomed flask (RBF), and the resulting solution was degassed by sparging with nitrogen for 30 min. The solution polymerization was initiated by increasing the temperature to 60 °C. For kinetic studies, an aliquot of the reaction mixture (0.3 mL) was taken at predetermined times and quenched by rapid cooling in liquid nitrogen. The polymer was recovered by precipitation three times in a methanol/water solution.
:1:0.1 or 50:1:0.1 were mixed in a 25 mL RBF, and the resulting solution was degassed by sparging with nitrogen for 30 min. The polymerization was initiated by increasing the temperature to 65 °C. For kinetic studies, an aliquot of the reaction mixture (0.3 mL) was taken at predetermined times and quenched by rapid cooling in liquid nitrogen. The polymer was recovered by precipitation three times in methanol.
Grafting through polymerization via ring opening metathesis polymerization
:RAFT:I = 50:1:0.1) and the polymerization was quenched after a certain time to obtain the desirable molar mass.
Results and discussion
RAFT agent design and synthesis
To probe the differences in the performance between oxanorbornene and norbornene strained olefinic groups in both (a) RAFT-based macromonomer (MM) synthesis and (b) ROMP-based bottlebrush polymer synthesis via grafting-through polymerization, two RAFT agents were prepared; these were the oxanorbornenyl RAFT agent 7-ONb and the norbornenyl RAFT agent 8-Nb (see Scheme 1). Briefly, the alcohol functional imides 3 and 4 were obtained in moderate to high yields from the relevant exo-anhydrides 1 and 2 by treatment with ethanolamine as per published literature procedures.20,23 The subsequent reaction of alcohols 3 and 4 with 2-bromopropionyl bromide adapted from the procedure of Keddie et al.24 delivered the alkylating agents 5 and 6. The RAFT agents 7-ONb and 8-Nb were isolated in moderate yields by following standard RAFT agent syntheses,25i.e. by alkylation of the carbodithioate salt derived from dodecanethiol and CS2.26 | ||
| Scheme 1 Synthesis of (i) alkylating agents 5 and 6 and (ii) RAFT agents 7-ONb and 8-Nb. | ||
Kinetic analysis of RAFT polymerization
Three monosubstituted monomers of interest, namely, methyl acrylate (MA), n-butyl acrylate (BA), and styrene (St), were selected for the preparation of macromonomers via RAFT polymerization (see Scheme 2). Importantly, these monomers allow us to probe the effect of electronics and/or sterics in the preparation of (oxa)norbornenyl-functional macromonomers. | ||
| Scheme 2 RAFT polymerization of MA, BA and St using RAFT agents 7-ONb and 8-Nb. | ||
MA was the first monomer investigated. Initially, we targeted a number-average degree of polymerization (Xn) of 50 (i.e. [MA]:[RAFT] = 50:1), using the RAFT agents 7-ONb (see Table 1, entry 1, and Fig. 1(a and b)) or 8-Nb (see Table 1, entry 2, and Fig. 1(c and d)). From the SEC data, it can be clearly observed that the oxanorbornene RAFT agent 7-ONb delivered polymers of higher molar masses and higher dispersity (see Table 1, entry 1, and Fig. 1(b)) than those of the analogous norbornene RAFT agent 8-Nb (see Table 1, entry 2, and Fig. 1(d)). Of note, the high percentage “livingness” (L%)27,28 calculated from kinetic factors indicates that the high molar mass shoulder(s) observed in the SEC traces are due to ‘branching’, formed via the reaction of the olefinic RAFT end-groups, rather than chain-coupling via termination by combination (see Table 1). The degree of branching (DB%), quantification of the presence of branched polymers (i.e. polymer dimers, trimers, etc.) calculated by either NMR analysis of olefinic RAFT end-group consumption‡ or SEC deconvolution, was significantly higher for 7-ONb than for 8-Nb (see Table 1 entries 1 and 2). This is also clearly evidenced by kinetic analysis of the rate of olefinic end-group consumption during polymerization; the oxanorbornene end-group is consumed to a greater extent than that of norbornene (see Table 1, entries 1 and 2, and Fig. 1(a) and (c)). Clearly the propensity for the cross-propagation of the poly(methyl acrylate) propagating radical (PMA˙) to the olefinic polymer end-group is higher for 7-ONb based systems than those that use 8-Nb. We postulate that this is likely due to a retro Diels–Alder extrusion of furan from 7-ONb derived chain-ends during the reaction to produce a more reactive maleimide end-group,§ which can then undergo rapid copolymerization with PMA˙ (see Scheme 3(a) and (b)). Indeed estimations of indicative copolymerization reactivity ratios, using the Alfrey-Price Q–e system,32,33 indicate a significantly larger preference for PMA˙ to cross-propagate to a maleimide than to a norbornene.¶,|| When targeting a higher chain length of Xn = 200 (i.e. [MA]:[RAFT] = 200:1) almost the same trends were observed as those for the Xn = 50 examples, with the oxanobornene-based materials displaying a higher number-average molar mass (Mn), molar mass dispersity (Đ) and degree of branching (DB%) (see Table S1, entries 1 and 2, and Fig. S1(a) and (b)†).
 | ||
| Fig. 1 Pseudo-first order kinetics plots (a and c) for the conversion of MA (red circles) and the (oxa)norbornene end-group (black squares), and SEC chromatograms illustrating the evolution of the molar mass distribution with the reaction time (b and d) for polymerizations of MA with the RAFT agents 7-ONb (a and b) or 8-Nb (c and d). All polymerizations were performed with [MA]:[RAFT] = 50:1. | ||
 | ||
| Scheme 3 (a) Proposed formation of a maleimide chain-end via a retro Diels–Alder reaction, and (b) cross-propagation of the propagating polymer species to the maleimide chain-end leading to branching, during RAFT polymerization reactions using the oxanorbornene RAFT agent 7-ONb. Pn, Pm = polymer chains; M = monomer. | ||
| Entrya | Monomer | [M] (mol L−1) | RAFT agent | T (°C) | Time (h) | M conv.b (%) |
M
n
c (calc) |
M n | Đ | L% (ω-end)e | DB% (NMR)b,f | DB % (SEC)f,g |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a [M]0:[RAFT]0:[AIBN]0 = 50:1:0.1.
b Calculated from1H NMR.
c
M
n
(calc) = ([M]0 – [M]t)/([RAFT]0) × MWmonomer + MWRAFT.
d SEC THF eluent, T = 40 °C (data reported in polystyrene equivalents).
e
L% = ([CTA]0/([CTA]0 + df × [I]0 × 1 − ekdt) × 100%,27,28 where f is the initiator efficiency ( =0.7),29d is the number of chains formed by radical–radical termination ( =1),30 and kd = 9.67 × 10–6 s−1at 60 °C (ref. 31) or kd = 1.95 × 10–5 s−1at 65 °C (calculated from Arrhenius parameters).31
f DB% = percentage degree of branching.
g calculated following the deconvolution of SEC chromatograms.
|
||||||||||||
| 1 | MA | 6.79 | 7-ONb | 60 | 0.5 | 32 | 1920 | 2200 | 1.19 | 99.9 | 7 | 7.3 |
| 1.5 | 69 | 3510 | 4500 | 1.37 | 99.6 | 24 | 24.3 | |||||
| 2.5 | 80 | 3980 | 5900 | 1.49 | 99.4 | 37 | 26.5 | |||||
| 3.5 | 85 | 4200 | 7000 | 1.64 | 99.2 | 42 | 32.7 | |||||
| 5 | 92 | 4500 | 7900 | 1.80 | 98.9 | 47 | 35.4 | |||||
| 2 | MA | 6.79 | 8-Nb | 60 | 0.5 | 19 | 1360 | 1100 | 1.12 | 99.9 | 4 | 0 |
| 1.5 | 57 | 2990 | 3200 | 1.13 | 99.6 | 10 | 4.1 | |||||
| 2.5 | 76 | 3810 | 4400 | 1.17 | 99.4 | 13 | 9.7 | |||||
| 3.5 | 85 | 4200 | 5000 | 1.22 | 99.2 | 15 | 14.2 | |||||
| 5 | 90 | 4410 | 5500 | 1.25 | 98.9 | 15 | 19.2 | |||||
| 3 | BA | 4.68 | 7-ONb | 60 | 0.5 | 34 | 2720 | 2900 | 1.19 | 99.9 | 17 | 3.4 |
| 1.5 | 68 | 4900 | 6300 | 1.33 | 99.6 | 33 | 35.3 | |||||
| 2.5 | 78 | 5540 | 7800 | 1.43 | 99.4 | 40 | 41.6 | |||||
| 3.5 | 84 | 5920 | 8900 | 1.51 | 99.2 | 45 | 41.6 | |||||
| 4.5 | 87 | 6120 | 9400 | 1.60 | 99.0 | 47 | 39.2 | |||||
| 6 | 90 | 6310 | 10300 |
1.69 | 98.7 | 49 | 44.5 | |||||
| 4 | BA | 4.68 | 8-Nb | 60 | 0.5 | 23 | 2010 | 2100 | 1.14 | 99.9 | 5 | 0 |
| 1.5 | 63 | 4580 | 5000 | 1.16 | 99.6 | 7 | 5.5 | |||||
| 2.5 | 77 | 5470 | 6200 | 1.19 | 99.4 | 10 | 9.9 | |||||
| 3.5 | 83 | 5860 | 6900 | 1.22 | 99.2 | 16 | 13.4 | |||||
| 4.5 | 87 | 6120 | 7400 | 1.24 | 99.0 | 18 | 15.5 | |||||
| 6 | 92 | 6440 | 7900 | 1.28 | 98.7 | 21 | 20.0 | |||||
| 5 | St | 8.73 | 7-ONb | 65 | 2 | 9 | 1010 | — | — | 99.1 | 7 | — |
| 4 | 12 | 1160 | 1500 | 1.13 | 98.3 | 9 | 5.0 | |||||
| 7 | 31 | 2150 | 2200 | 1.18 | 97.4 | 15 | 9.4 | |||||
| 10 | 42 | 2730 | 2900 | 1.20 | 96.6 | 20 | 14.2 | |||||
| 22.5 | 68 | 4080 | 4800 | 1.33 | 94.7 | 34 | 27.9 | |||||
| 6 | St | 8.73 | 8-Nb | 65 | 2 | 11 | 1110 | — | — | 99.1 | 4 | — |
| 4 | 21 | 1630 | 1400 | 1.09 | 98.3 | 5 | 0 | |||||
| 7 | 36 | 2410 | 2000 | 1.10 | 97.4 | 8 | 0 | |||||
| 10 | 47 | 2990 | 2500 | 1.09 | 96.6 | 11 | 0 | |||||
| 22.5 | 82 | 4810 | 4200 | 1.09 | 94.7 | 17 | 1.2 | |||||
To probe the effect of the acrylate ester chain length on the RAFT system, BA was the next monomer investigated, targeting Xn = 50. Unsurprisingly, the additional sterics from the n-butyl ester of the monomer made a little difference to the reactivity of the poly(n-butyl acrylate) propagating species (PBA˙) towards the different end-groups of 7-ONb and 8-Nb when compared to the PMA˙ systems. PBA prepared in the presence of oxanorbornene 7-ONb displayed significantly higher Mn, Đ and DB% values than the PBA samples prepared in the presence of norbornene 8-Nb (see Table 1, entries 3 and 4, and Fig. 2). Again, analogous outcomes were observed when targeting PBA of Xn = 200 (see Table S1, entries 3 and 4, and Fig. S1(c) and (d)†).
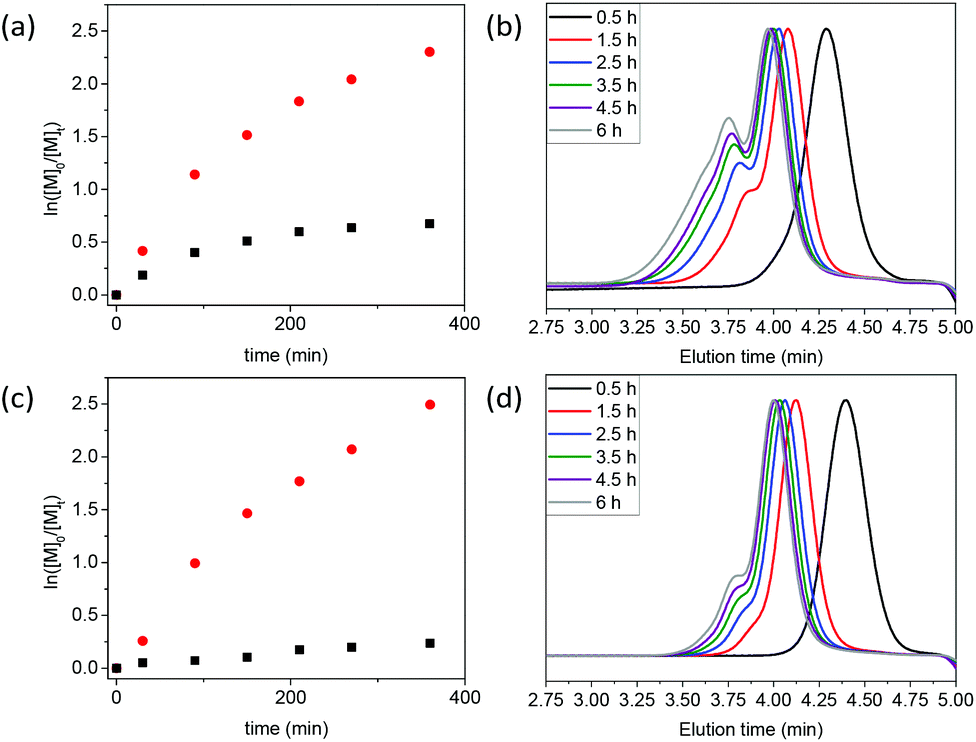 | ||
| Fig. 2 Pseudo-first order kinetics plots (a and c) for the conversion of BA (red circles) and the (oxa)norbornene end-group (black squares), and SEC chromatograms illustrating the evolution of the molar mass distribution with the reaction time (b and d) for polymerizations of BA with the RAFT agents 7-ONb (a and b) or 8-Nb (c and d). All polymerizations were performed with [BA]:[RAFT] = 50:1. | ||
The final monomer investigated was St, again initially targeting Xn = 50 (i.e. [St]:[RAFT] = 50:1). As with the previous examples discussed above, for St polymerization the oxanorbornene RAFT agent 7-ONb led to significantly higher Mn, Đ and DB% values than the norbornene RAFT agent 8-Nb (see Table 1, entries 5 and 6, and Fig. 3). Interestingly, for the polymerization of St controlled with the norbornene RAFT agent 8-Nb minimal branching (DB% = 1.2%) was observed, particularly when compared to the acrylate systems (cf. DB% ∼20% for MA and BA at ∼90% monomer conversion). We attributed this to the electronic differences between the electron-rich polystyryl radical (PSt˙) and the electron-poor acrylate-based radicals (i.e. PMA˙ and PBA˙). PSt˙ cross-propagates to the electron-rich norbornene end-group more slowly than do either of the acrylate-based radicals; this observation is in agreement with the indicative copolymerization reactivity ratios.32,33¶ It appears that electronics plays a less significant role in the DB% in the oxanorbornene case, which provides further indirect evidence for the contribution of retro Diels–Alder to branching. Indicative copolymerization reactivity ratios, between St and maleimide, suggest a tendency towards alternation which would lead to the consumption of a maleimide at the chain end. On the other hand an unreacted, electron-rich oxanorbornene would be expected to behave in almost the same way as norbornene in St polymerization (i.e. display conversion via cross-propagation to the chain end). Similar polymerization outcomes were obtained when targeting PSt of Xn = 250 (i.e. [St]:[RAFT] = 250:1) with 7-ONb delivering materials with higher Mn and Đ, and significantly higher DB% than those with 8-Nb (see Table S1, entries 5 and 6, and Fig. S1(e) and (f)†).
 | ||
| Fig. 3 Pseudo-first order kinetics plots (a and c) for the conversion of St (red circles) and the (oxa)norbornene end-group (black squares), and SEC chromatograms illustrating the evolution of the molar mass distribution with the reaction time (b and d) for polymerizations of St with the RAFT agents 7-ONb (a and b) or 8-Nb (c and d). All polymerizations were performed with [St]:[RAFT] = 50:1. | ||
In summary, cross propagation of the propagating species (PMA˙, PBA˙ and PSt˙) to the olefinic RAFT chain-end was found to occur in all cases discussed, albeit in varying degrees.
This results in a proportion of branched structures, a generally undesired topological ‘impurity’ in the final polymer sample.
This is also expected to adversely impact the preparation of the targeted bottlebrush polymers due to the (partial) consumption of the ROMP polymerizable end-group. It is clear from the results discussed above that 8-Nb is the preferred RAFT agent for preparing macromonomers from monosubstituted monomers via RAFT. Significantly more ‘defects’ in the macromonomer structure are due to the trade-off of using the more easily prepared 7-ONb instead of 8-Nb. It should be noted that even when using the norbornene-based RAFT agent 8-Nb in the polymerization of the acrylates MA and BA significant end-group consumption was observed (up to 20%, see Table 1, entries 2 and 4). To decrease the incidence of these ‘defects’ in the synthesis of MMs we recommend targeting higher molar masses and quenching the reaction at a lower conversion; from a copolymerization standpoint this effectively decreases the monomer feed ratio of the norbornene chain-end reducing the rate of cross-propagation.
Macromonomer synthesis via RAFT polymerization
Following on from the kinetic investigations described above, we successfully prepared three macromonomers based on MA, BA, and St with low molar masses (∼2000–4000 g mol−1) by RAFT polymerization using the RAFT agents (7-ONb or 8-Nb). The reactions used to prepare the MMs were quenched after the desired time and purified by precipitation three times in methanol to completely remove any unreacted monomers present. 1H-NMR spectroscopy and size exclusion chromatography (SEC) were used to characterize the resulting MMs. The properties of the prepared MMs are summarized in Table 2 and the SEC chromatograms are shown in Fig. 4 (black traces). | ||
| Fig. 4 SEC chromatograms of macromonomers (black) and bottlebrush polymers (red) for the polymerizations of (a) PMA-7-ONb, (b) PMA-8-Nb, (c) PBA-7-ONb, (d) PBA-8-Nb, (e) PSt-7-ONb and (f) PSt-8-Nb. Polymerizations were performed with [MM]:[G3] = 25:1. | ||
| Entry | Macromonomer | Time (h). | M n (NMR) (g mol−1). | M n(SEC) (g mol−1). | Đ . | L (ω-end)c. | DB % (SEC)d,e. |
|---|---|---|---|---|---|---|---|
| a Calculated from 1H NMR. b SEC THF eluent, T = 40 °C (data reported in polystyrene equivalents). c L% = ([CTA]0/([CTA]0 + df × [I]0 × 1 − ekdt) × 100%,27,28 where f is the initiator efficiency ( =0.7),29d is the number of chains formed by radical–radical termination ( =1),30 and kd = 9.67 × 10–6 s−1 at 60 °C31 or kd = 1.95 × 10–5 s−1 at 65 °C (calculated from Arrhenius parameters).31 d DB % = percentage degree of branching. e Calculated following the deconvolution of SEC chromatograms. | |||||||
| 1 | PMA-7-ONb | 1 | 2700 | 3300 | 1.24 | 99.8 | 15 |
| 2 | PMA-8-Nb | 1.5 | 3100 | 3600 | 1.15 | 99.6 | 6 |
| 3 | PBA-7-ONb | 1 | 4300 | 5600 | 1.28 | 99.8 | 20 |
| 4 | PBA-8-Nb | 1 | 3400 | 3900 | 1.14 | 99.8 | 2 |
| 5 | PSt-7-ONb | 10 | 3100 | 2900 | 1.19 | 96.6 | 1 |
| 6 | PSt-8-Nb | 10 | 2800 | 2500 | 1.08 | 96.6 | 0 |
Akin to the data described above, the norbornenyl RAFT agent 8-Nb delivered MMs with the lowest Đ and DB% values in the case of each monomer (see Table 2, entries 2, 4 and 6.). The oxanorbornenyl RAFT agent 7-ONb resulted in higher dispersities (and bimodality in the molar mass distribution) (see entries 1, 3 and 5, Table 2). Compared to styrene MMs, the acrylate MMs exhibit slightly higher dispersities and DB% (see Table 2 and Fig. 4-black traces).
ROMP of macromonomers
Grafting-through polymerization via ROMP of the (oxa)norbornene macromonomers (see Table 2) was carried out in THF using a MM to catalyst ratio of 25:1 with a Grubbs third generation catalyst, G3 (see Scheme 4 and Table 3). Exhibiting rapid initiation kinetics and high functional-group tolerance, G3 is well known to successfully polymerize sterically hindered substrates, allowing for the synthesis of polymers with narrow molar mass distributions.
 | ||
| Scheme 4 Grafting-through ROMP of RAFT Macromonomers (PMA-7-ONb, PBA-7-ONb, PSt-7-ONb, PMA-8-Nb, PBA-8-Nb, and PSt-8-Nb). | ||
| Entrya | Macromonomer (MM) | MM Mnb (g mol−1) | Residual MM % | M n (SEC) (g mol−1) | Đ |
|---|---|---|---|---|---|
| a [MM] = 0.33 M in THF, [MM]/[G3] = 25, t = 3 h. b Calculated from 1H NMR. c From SEC THF eluent, T = 40 °C (data reported in polystyrene equivalents). | |||||
| 1 | PMA-7-ONb | 2700 | 11 | 117000 |
1.29 |
| 2 | PMA-8-Nb | 3100 | 3 | 125000 |
1.35 |
| 3 | PBA-7-ONb | 4300 | 37 | 162000 |
1.27 |
| 4 | PBA-8-Nb | 3200 | 5 | 117000 |
1.23 |
| 5 | PSt-7-ONb | 2900 | 20 | 53000 |
1.40 |
| 6 | PSt-8-Nb | 2800 | 4 | 90000 |
1.58 |
Due to the difference in thermal stability between the RAFT agents 7-ONb and 8-Nb and their behaviour in RAFT polymerization (i.e. higher DB%) as described above, it was found that the bottlebrush polymers based on 7-ONb have higher levels of residual MMs than those prepared using 8-Nb; ROMP of MMs based on 8-Nb gives a bottlebrush polymer with a low amount of MM residue (≤5%).
Bottlebrush polymers with a [MM]/[I] ratio of 50:1 were prepared and full characterization was attempted, however, the polymers contained fractions that were larger than the exclusion limit of our SEC columns, limiting the ability to assess their molar masses and dispersity accurately (see Table S2 and Fig. S2 in the ESI†). Keeping this drawback aside these materials displayed similar MM incorporation for the [MM]/[I] 25:1 samples.
From these experiments it is clear that using less effective 7-ONb in the RAFT synthesis of MMs also leads to less desirable outcomes (e.g. higher residual MM%) in the sequential RAFT/ROMP process for the preparation of bottlebrush polymers than the use of 8-Nb. We postulate that the altered architecture of the MMs derived from 7-ONb and the related decrease in the amount of “ROMP-able” end-groups per unit mass of MMs (both brought about by the higher incidence of cross-propagation to the olefin chain end during the RAFT syntheses) contribute to the poorer performance of oxanorbornene-based MMs in the preparation of bottlebrush polymers.
Conclusions
Two new RAFT agents with either an oxanorbornenyl or norbornenyl moiety were successfully prepared. The design of the RAFT agent has been found to affect the RAFT polymerization and hence the sequential ROMP process. Three different monomers (MA, BA, and St) were selected for RAFT polymerizations. The thermal stability of RAFT agents and the monomer electronics and sterics were found to affect the resultant MM structure and molar mass distribution.In the case of the oxanorbornenyl RAFT agent 7-ONb, the polymerization analysis revealed the increase of branching as a function of conversion, due to copolymerization to a maleimide end-group following thermal extrusion of furan. On the other hand, the RAFT agent 8-Nb with the norbornenyl moiety is more thermally stable and yields MMs with low branching, due to the limited copolymerization reactivity of norbornenyl alkenes towards acrylate or styrene propagating species. The more significant “imperfections” in the 7-ONb derived MMs (in comparison to those prepared using 8-Nb) were found to affect the molecular properties of the bottlebrush polymers prepared via ROMP; NMR and SEC analyses revealed significant differences in the residual MM concentrations following polymerization.
Author contributions
MN: investigation, data analysis, drafted sections of the original manuscript, and review and editing of the manuscript; KN: data analysis, peak fitting and deconvolution of SEC data, review and editing of the manuscript; DJK: conceptualization, investigation, data analysis, drafted sections of the original manuscript, supervision, and review and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Naguib would like to thank the Cultural Affairs & Missions Sector of Egypt for financial support. D.J.K. acknowledges the Royal Society of Chemistry and the Faculty of Science and Engineering at the University of Wolverhampton for funding. The authors would also like to thank Materia Inc. (USA) for the kind donation of Grubbs catalysts.Notes and references
- Z. Li, K. Zhang, J. Ma, C. Cheng and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5557–5563 CrossRef CAS PubMed.
- S. Jha, S. Dutta and N. B. Bowden, Macromolecules, 2004, 37, 4365–4374 CrossRef CAS.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- Y. Xu, W. Wang, Y. Wang, J. Zhu, D. Uhrig, X. Lu, J. K. Keum, J. W. Mays and K. Hong, Polym. Chem., 2016, 7, 680–688 RSC.
- X. Li, S. L. Prukop, S. L. Biswal and R. Verduzco, Macromolecules, 2012, 45, 7118–7127 CrossRef CAS.
- G. Xie, M. R. Martinez, M. Olszewski, S. S. Sheiko and K. Matyjaszewski, Biomacromolecules, 2019, 20, 27–54 CrossRef CAS PubMed.
- I. Choinopoulos, Polymers, 2019, 11, 298/291–298/231 CrossRef PubMed.
- J. C. Foster, S. Varlas, B. Couturaud, Z. Coe and R. K. O'Reilly, J. Am. Chem. Soc., 2019, 141, 2742–2753 CrossRef CAS PubMed.
- J. Rzayev, ACS Macro Lett., 2012, 1, 1146–1149 CrossRef CAS.
- S. C. Radzinski, J. C. Foster and J. B. Matson, Polym. Chem., 2015, 6, 5643–5652 RSC.
- X. Tian, J. Ding, B. Zhang, F. Qiu, X. Zhuang and Y. Chen, Polymers, 2018, 10, 318 CrossRef PubMed.
- D. L. Patton and R. C. Advincula, Macromolecules, 2006, 39, 8674–8683 CrossRef CAS.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS PubMed.
- A. Li, J. Ma, G. Sun, Z. Li, S. Cho, C. Clark and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1681–1688 CrossRef CAS.
- S. Laroque, M. Reifarth, M. Sperling, S. Kersting, S. Kloepzig, P. Budach, J. Storsberg and M. Hartlieb, ACS Appl. Mater. Interfaces, 2020, 12, 30052–30065 CrossRef CAS PubMed.
- J. C. Foster, S. C. Radzinski, S. E. Lewis, M. B. Slutzker and J. B. Matson, Polymer, 2015, 79, 205–211 CrossRef CAS.
- Y. Zhou, Y. Qu, Q. Yu, H. Chen, Z. Zhang and X. Zhu, Polym. Chem., 2018, 9, 3238–3247 RSC.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- J. Kötteritzsch, S. Stumpf, S. Hoeppener, J. Vitz, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2013, 214, 1636–1649 CrossRef.
- M. B. Runge and N. B. Bowden, J. Am. Chem. Soc., 2007, 129, 10551–10560 CrossRef CAS PubMed.
- F. Yu, A. M. Spring, L. Li, F. Qiu, K. Yamamoto, D. Maeda, M. Ozawa, K. Odoi and S. Yokoyama, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1278–1284 CrossRef CAS.
- J. B. Matson and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 6731–6733 CrossRef CAS PubMed.
- D. J. Keddie, C. Guerrero-Sanchez, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2011, 44, 6738–6745 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- K. J. Sykes, S. Harrisson and D. J. Keddie, Macromol. Chem. Phys., 2016, 217, 2310–2320 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 639–649 CrossRef CAS.
- J. Gardiner, I. Martinez-Botella, J. Tsanaktsidis and G. Moad, Polym. Chem., 2016, 7, 481–492 RSC.
- S. Houshyar, D. Keddie, G. Moad, R. Mulder, S. Saubern and J. Tsanaktsidis, Polym. Chem., 2012, 3, 1879–1889 RSC.
- G. Moad and D. H. Solomon, in The Chemistry of Radical Polymerization, Elsevier Science Ltd, Amsterdam, 2nd edn, 2005, pp. 233–278 Search PubMed.
- G. Moad and D. H. Solomon, in The Chemistry of Radical Polymerization, Elsevier Science Ltd, Amsterdam, 2nd edn, 2005, pp. 49–166 Search PubMed.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley, New York, 1999 Search PubMed.
- Z.-M. Dong, X.-H. Liu, X.-L. Tang and Y.-S. Li, Macromolecules, 2009, 42, 4596–4603 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of additional polymers prepared by RAFT and ROMP targeting higher degrees of polymerization, including additional pseudo-first order kinetic plots and SEC data. See DOI: 10.1039/d1py01599k |
| ‡ The conversion of the (oxa)norbornene end-group is calculated in the same standard manner in which conversion of the vinyl monomer is achieved; the resonances from vinylic end-groups are well resolved in all cases. A representative example can be found in the ESI (Fig. S1†). |
| § No categorical evidence of in situ maleimide end-group formation could be observed in NMR analysis of the kinetic samples or of the final product polymers. We believe this is due to its rapid consumption upon its formation, in addition to the already relatively low concentration of RAFT groups in the polymerization mixture, rendering analysis of the molecular microstructure difficult. Additionally, the furan by-product was also not observed, presumably due to its volatility; all kinetic samples were taken directly from the polymerization reaction mixtures that were all at higher temperatures than the boiling point of furan (31 °C). |
| ¶ Indicative copolymerization reactivity ratios, calculated from Q–e values, for norbornene systems: methyl acrylate (MA)/norbornene (Nor): rMA = 3.90, rNor = 0.181; n-butyl acrylate (BA)/norbornene (Nor): rBA = 5.83, rNor = 0.146; styrene (St)/norbornene (Nor): rSt = 7.25, rNor = 0.045. |
| || Indicative copolymerization reactivity ratios, calculated from Q–e values, for maleimide systems: methyl acrylate (MA)/maleimide (MI): rMA = 0.36, rMl = 2.62; n-butyl acrylate (BA)/maleimide (MI): rBA = 0.47, rMl = 1.81; styrene (St)/maleimide (MI): rSt = 0.178, rMl = 0.170. |
| This journal is © The Royal Society of Chemistry 2022 |