
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New classes of functionalized parylenes and poly(phenylene vinylene)s via coupling of dihaloxylyl diesters†
Jihong
Lyu
ab and
Christopher W.
Bielawski
 *ab
*ab
aCenter for Multidimensional Carbon Materials (CMCM), Institute for Basic Science (IBS), Ulsan 44919, Republic of Korea
bDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 14th January 2022
Abstract
New classes of poly(p-xylylene)s and poly(p-phenylene vinylene)s were synthesized and studied. The novel polymers differ from known variants of their kind in that the ethylene and vinylene repeat units are outfitted with ester groups, which can be readily modified through transesterification. The polymers were prepared by treating an appropriate dihalobenzene diester with either a reductant to afford the corresponding poly(p-xylylene) or a base to afford the corresponding poly(p-phenylene vinylene). Variation of the ester groups enabled a broad range of functional polymers to be prepared, including derivatives that are liquid crystalline, photoswitchable, water soluble or amenable to cycloaddition chemistry.
Introduction
Poly(p-xylylene) (PPX) is a polymer that features an alternating arrangement of arylene and ethylene units along its backbone. The unique structure affords PPX with high thermal stability, broad chemical resistance, good insulating properties, and a low dielectric constant.1,2 Although the polymer was first synthesized in the 1940s by Szwarc,3 it was later commercialized by Union Carbide and sold under the name of ‘parylene’.4 The typical synthesis of PPX entails the pyrolysis of an appropriate monomer, such as p-xylene or, more commonly, [2,2]paracyclophane. Heating the precursor to an elevated temperature (>500 °C) and under low pressure results in the deposition of a polymer film, presumably via radical intermediates that generate in situ and then combine.5,6 Although high quality films can be produced using this method, the polymers are typically intractable which challenges processing or functionalization steps.7Poly(p-phenylene vinylene) (PPV) is an unsaturated analogue of PPX that features an alternating arrangement of arylene and vinylene (carbynyl) units. Due to its conjugated electronic structure and small band gap, PPV has found utility in numerous optoelectronic applications, including organic light emitting diodes,8 solar cells,9 and sensors.10 The material can also be rendered electrically conductive upon doping.11 A variety of synthetic methods have been disclosed for synthesizing PPVs including the Heck coupling of 1,4-dibromobenzene and ethylene,12,13 Wittig or Knoevenagel condensations of terephthaldehyde and suitably functionalized xylenyl derivatives,14–16 or the ring-opening metathesis polymerization of barrelene.17–19 Variations of these methods are known and have enabled the synthesis of PPV derivatives that exhibit enhanced solubility or tailored electronic characteristics (Scheme 1).20,21
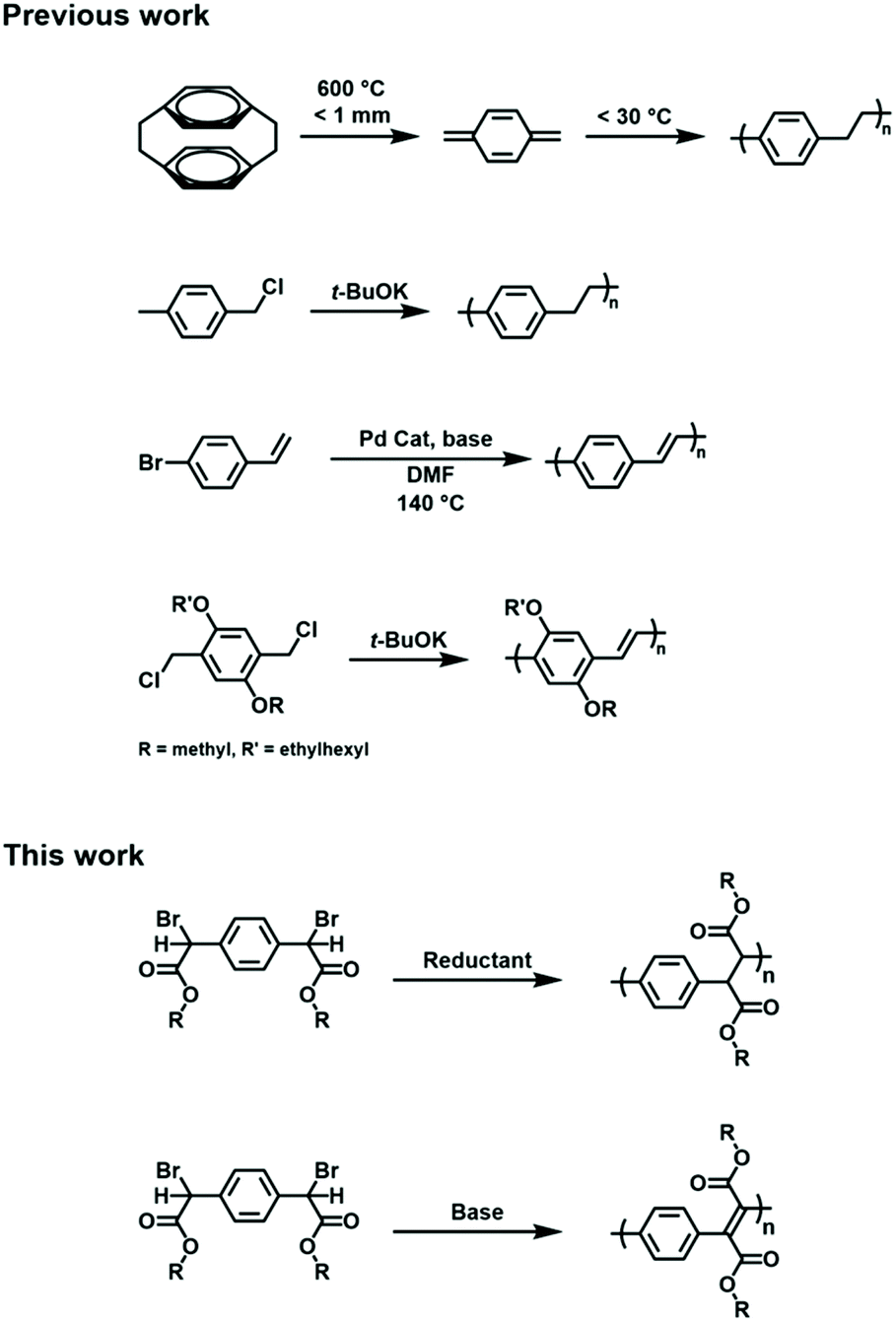 | ||
| Scheme 1 Selected examples of methods that have been used to synthesize PPX and PPV. | ||
Other methods that have been used to synthesize PPX and PPV utilize base-mediated polymerizations of α,α′-disubstituted para-xylenes and are often referred to as the ‘Gilch route’, which was named after the initial discovery of the chemistry.22 The addition of a strong base, such as potassium tert-butoxide, to α,α′-dibromo-p-xylene, for example, results in a deprotonation reaction and the formation of anionic xylyl intermediates which then self-condense. One equivalent of base affords PPX as the major product;23,24 however, excess base results in elimination (typically dehydrohalogenation) of the PPX intermediate and affords PPV.22,25 Variations of the Gilch route have been reported and used to optimize polymer molecular weight or to access functionalized derivatives, although most of these efforts have entailed modification of the arylene unit.22–24,26 For example, poly[2-methoxy-5-((2′-ethylhexyl)-oxy)-p-phenylene vinylene] (MEH-PPV) features an arylene core that has been modified to promote solubility and to exhibit a band gap needed for use in optical displays. MEH-PPV is typically prepared by condensing the corresponding functionalized para-xylene with α,α′-dibromo-p-xylene under basic conditions.20,27
We envisioned an alternative strategy to PPV and PPX derivatives that utilized a common precursor, 2,2′-(1,4-phenylene)bis(2-bromoacetic acid). Our hypothesis was that the dimerization of a reduced (i.e., dehalogenated) form of the compound should result in the formation of a PPX whereas condensation followed by elimination would afford a PPV.28,29 An intrinsic advantage of such an approach is that it enables the modification of the ethylene or vinylene repeat units and, as such, can be expected to more profoundly influence the intrinsic chemical, optoelectronic and other properties displayed by the polymers, particularly when compared to approaches that entail arene modifications or are relatively distal to the main chains. Moreover, the precursor is commercially available and can be readily adapted through esterification with various alcohols which should facilitate the integration of PPV and PPX in a broader range of applications and enable more detailed studies of the fundamental properties displayed by these polymeric materials.
Results and discussion
Model reactions
The proposed polymerization methodology requires α-bromophenylacetate derivatives to be coupled. To determine the viability of such chemistry, initial efforts were directed toward the dimerization of ethyl α-bromophenylacetate (EBPA) as a model reaction. As summarized in Table 1, a variety of bases and reductants were explored as potential reagents to effect combination and/or condensation. The addition of diisopropylethylamine (DIPEA) or triethylamine (TEA) to EBPA did not result in a significant reaction (entries 1 and 2), presumably because the amines were not sufficiently basic or reducing. As such, photoredox reagents30,31 were utilized to enhance the reducing power of the amine. Irradiating (λ = 435 nm via blue LEDs) a DMF solution of Ru(bpy)3Cl2 (5 mol%) (bpy = 2,2′-bipyridine), DIPEA (2 equiv.), and EBPA in at room temperature afforded the corresponding bibenzyl dimer (diethyl 2,3-diphenylsuccinate) (A) as a major product (entry 3). While additional DIPEA was found to be beneficial (entry 4), the use of a stronger reductant, tris[2-(2,4-difluorophenyl)pyridine]iridium(III) (fac-Ir(Fppy)3), in lieu of the aforementioned Ru catalyst (cf.: Ir: Ered = −1.46 V vs. SCE vs. Ru: −0.81 V vs. SCE)32 did not appear to significantly increase the conversion (cf., entry 5).|
|
|||
|---|---|---|---|
| Entry | Conditions (equivalents) | Product | Yieldd (%) |
| a Conditions: 0.25 mmol of EBPA, 1 mL of DMF, 18 h. b Irradiated using blue LEDs. c 0.12 mmol of EBPA was used. d Isolated yield. e Conversion of EBPA to the product shown was determined by 1H NMR spectroscopy. | |||
| 1 | DIPEA (10) | No reaction | |
| 2 | TEA (10) | No reaction | |
| 3b | Ru(bpy)3Cl2 (0.05), DIPEA (2) | A | 48e |
| 4b | Ru(bpy)3Cl2 (0.05), DIPEA (10) | A | 60e |
| 5b | Ir(fppy)3 (0.05), DIPEA (10) | A | 61e |
| 6c | TDAE (4) | A | 81 |
| 7c | TDAE (1), TEMPO (1) | A | 60 |
| 8c | TDAE (1), 2-methyl 2-nitrosopropane (1) | A | 78e |
| 9c | TDAE (1), 4-nitrobenzyl bromide (1) |

|
61 |
| 10 | K2CO3 (10) | No reaction | |
| 11 | NaOH (10) | B | 83 |
| 12 | NaOH (0.5) |

|
58 |

Efforts were also directed toward the use of organic electron donors (OEDs) to promote product formation. Tetrakis(dimethylamino)ethylene (TDAE) (Ered = −0.78 V vs. SCE),33 for example, has been shown to facilitate the reductive cleavage of carbon–halogen bonds via sequential electron transfer.34 Benzyl halides,35 bromodifluoroalkanes,33 and α-bromo ketones have been reduced with TDAE36 and, in some cases, the resulting products undergo dimerization. Treatment of EBPA with TDAE (4 equiv.) in DMF afforded the bibenzyl dimer as the major product and in the highest yield (81%; meso![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :racemic = 4:6; entry 6) among the reagents tested.
:racemic = 4:6; entry 6) among the reagents tested.
The aforementioned dimerization may involve either the coupling of radical intermediates that are generated upon reaction of EBPA with TDAE or the condensation of unreacted EBPA with carbanions that are generated upon reaction of two equivalents of TDAE with EBPA.37 To elucidate the operative mechanism, EBPA was treated with TDAE followed by various reagents designed to capture either radical or anionic intermediates. Adding either 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) or 2-methyl-2-nitrosopropane as a radical trap to an equimolar mixture of EBPA and TDAE afforded A as the major product (entries 7 and 8). Moreover, ethyl 3-(4-nitrophenyl)-2-phenylpropanoate was obtained when a stoichiometric mixture of EBPA and TDAE was treated with 4-nitrobenzyl bromide, an electrophile that is susceptible to nucleophilic substitution (entry 9). Collectively, these results indicated that TDAE may convert EBPA to an anionic intermediate which then attacks another (unreacted) unit of EBPA to afford the observed dimer via substitution.
Next, efforts were directed toward treating EBPA with inorganic bases to form the corresponding unsaturated derivatives. While the use of relatively weak inorganic bases, such as K2CO3, were ineffective (entry 10), treatment of EBPA with excess sodium hydroxide (NaOH) resulted in the formation of the stilbene dimer B as the major product (entry 11). Adding sub-stoichiometric quantities of NaOH to EBPA afforded diethyl 2-bromo-2,3-diphenylsuccinate (entry 12) which may undergo 1,2-elimination in the presence of excess base to afford its unsaturated derivative (B).
Polymer synthesis and characterization
Building on the model reactions, subsequent efforts were directed toward the synthesis of ditopic analogues of EBPA followed by their polymerization. As summarized in Scheme 2, two complementary approaches were utilized to access the requisite monomer. The strategies differed by the timing of the bromination step, which was conducted either prior to or after esterification depending on the alcohol used. In the first approach, 1,4-phenylenediacetic acid was esterified with various alkyl alcohols using catalytic quantities of H2SO4 at 80 °C. The diesters were then brominated with N-bromosuccinimide (NBS) and azobisisobutyronitrile (AIBN) as an initiator in chloroform at 60 °C to obtain the desired dihalogenated derivatives in good overall yield after purification. In the second approach, bromination preceded esterification. Due to its low solubility in chloroform, 1,4-phenylenediacetic acid was dissolved in acetonitrile, treated with NBS and AIBN, and then heated to 80 °C to afford 2,2′-(1,4-phenylene)bis(2-bromoacetic acid). The acid was then transformed via Steglich esterification, where dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine were used to activate the acid toward condensation with the added alcohol.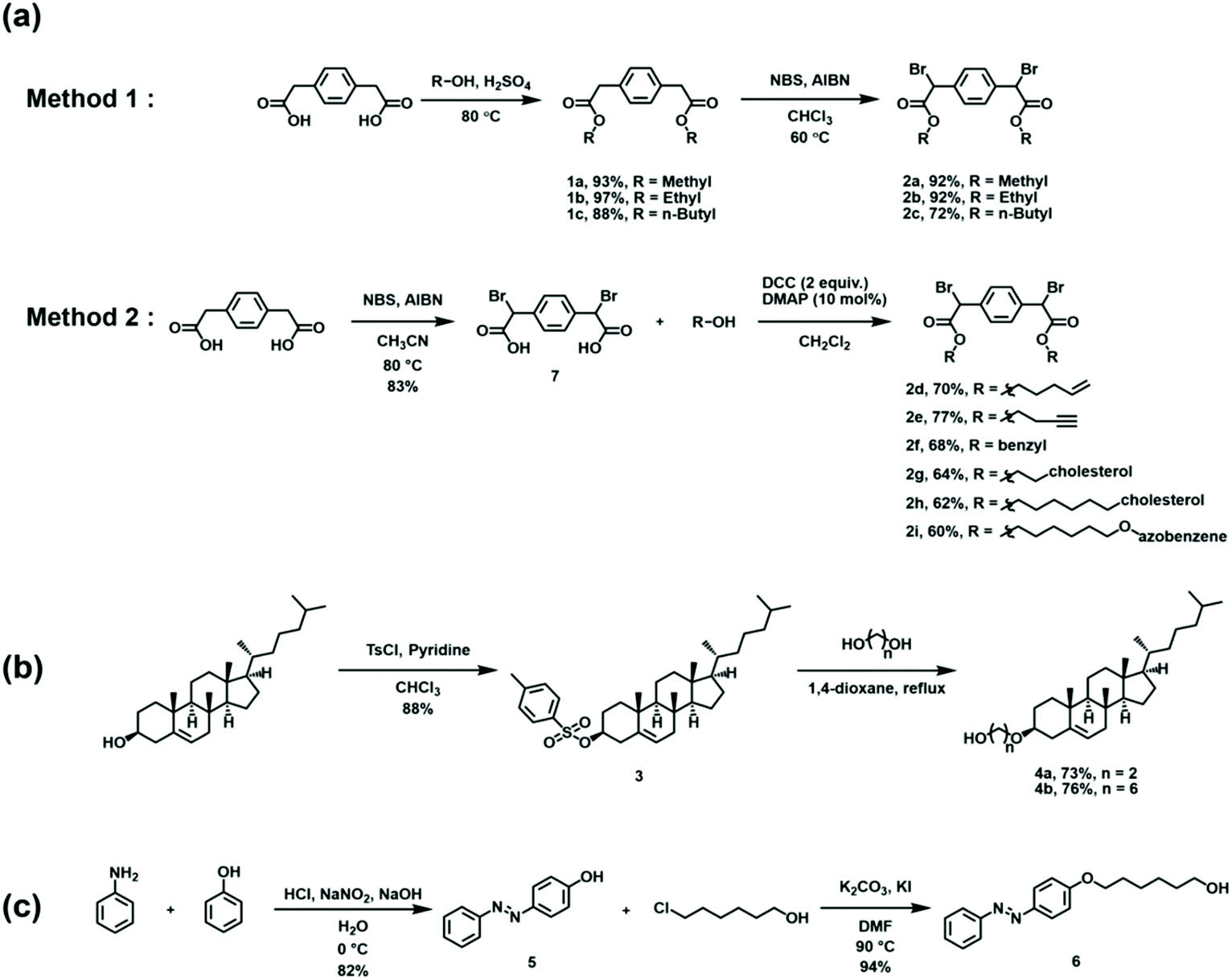 | ||
| Scheme 2 Summary of the synthetic methodology used to access key intermediates and monomers. (a) The monomers are derived from 1,4-phenylenediacetic acid. (b) Synthesis of cholesterol derivatives that feature different linker lengths. (c) Synthesis of an azobenzene derivative. | ||
With the monomers in hand, subsequent efforts were directed toward adapting the conditions developed for the model reactions to facilitate polymerization. A summary is shown in Scheme 3. The reductive polymerizations were conducted by treating DMF solutions of 2 with excess TDAE. After 24 h, the reaction mixtures were filtered to remove the TDAE+Br− salts that formed as the reaction progressed, and then the filtrate was poured into excess methanol. The precipitated solids were collected, dried, and characterized using multinuclear NMR spectroscopy, FT-IR spectroscopy, and size exclusion chromatography (SEC) (see Fig. S46–S54†). For example, the polymer obtained from 2a (i.e., PPX-2a) exhibited a diagnostic signal at δ 4.0–4.5 ppm (CDCl3) in its corresponding 1H NMR spectrum which was assigned to the ethylene moieties in the backbone. The integral of this signal was commensurate with that recorded for the signal assigned to the ester group and indicated that the two groups were present in an equimolar ratio, consistent with the structure shown. Signals at 171.5 and 173.2 ppm were also observed in the 13C NMR spectrum recorded for PPX-2a which may reflect the different stereochemistry adopted by the polymer. These signals were downfield when compared to the corresponding signal displayed by the starting material (168.6 ppm). FT-IR spectroscopic analysis of PPX-2a revealed a strong absorption at 1150 cm−1 which was assigned to the C–C backbone of the polymer chain. In contrast, the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O recorded for PPX-2a (1734 cm−1) was measured at a lower frequency when compared to that of 2a (1738 cm−1). The molecular weight of the polymer was determined by SEC to be 2.4 kDa relative to polystyrene standards and exhibited a polydispersity index (Đ) of 1.73, consistent with that expected from a step-growth polymerization. Similar analyses were performed for the other polymers derived from 2 and key results are summarized in Table 2.
O recorded for PPX-2a (1734 cm−1) was measured at a lower frequency when compared to that of 2a (1738 cm−1). The molecular weight of the polymer was determined by SEC to be 2.4 kDa relative to polystyrene standards and exhibited a polydispersity index (Đ) of 1.73, consistent with that expected from a step-growth polymerization. Similar analyses were performed for the other polymers derived from 2 and key results are summarized in Table 2.
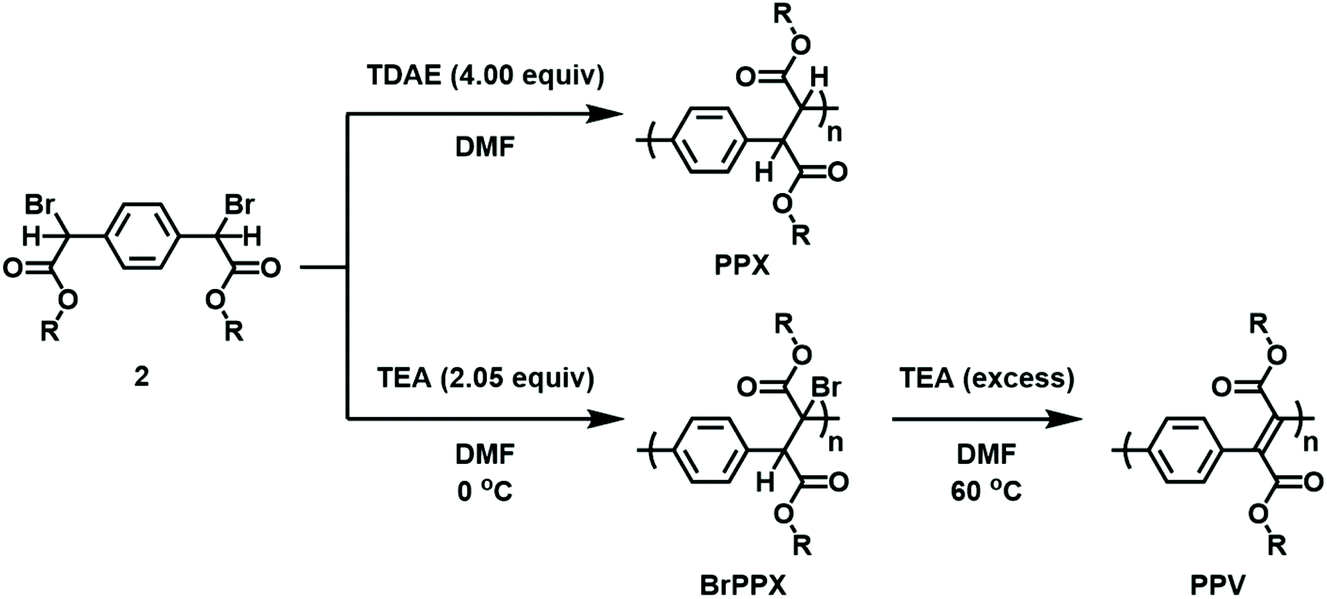 | ||
| Scheme 3 Synthetic methodology used to prepare PPX, BrPPX and PPV. | ||
| Polymer | Conc. (M) | Time (h) | Yieldb (%) |
M
nc (kDa) |
Đ |
T
d5d (°C) |
T
ge (°C) |
|---|---|---|---|---|---|---|---|
| a The polymerization reaction was performed by injecting a solution of TDAE in DMF into a prefilled DMF solution containing the corresponding monomer at room temperature. b Isolated yield. c Determined by SEC relative to polystyrene standards in THF. d The temperature at which 5 wt% loss occurred (Td5) was measured by TGA under an atmosphere of nitrogen. e The glass transition temperatures (Tg) were measured by DSC (heating rate: 20 °C min−1). f Samples were first stirred in CH2Cl2 at 35 °C for 12 h and analyzed by SEC relative to polystyrene standards in CH2Cl2. | |||||||
| PPX-2a | 0.24 | 24 | 78 | 2.4f | 1.73 | 365 | 178 |
| PPX-2b | 0.48 | 24 | 73 | 5.8 | 1.81 | 314 | 119 |
| PPX-2c | 0.48 | 24 | 60 | 9.4 | 1.71 | 353 | 79 |
| PPX-2d | 0.48 | 24 | 67 | 13.7 | 1.53 | 392 | 14 |
| PPX-2e | 0.48 | 24 | 46 | 5.2 | 1.43 | 361 | 70 |
| PPX-2f | 0.48 | 24 | 67 | 3.2 | 1.68 | 357 | 46 |
| PPX-2g | 0.12 | 24 | 95 | 5.5 | 1.34 | 338 | 84 |
| PPX-2h | 0.12 | 24 | 90 | 8.1 | 1.37 | 360 | 37 |
| PPX-2i | 0.48 | 24 | 80 | 3.9 | 1.40 | 308 | 46 |
While the conversion of EBPA to its stilbene derivative required the use of a relatively strong base (NaOH), it was discovered that 2 underwent polymerization when treated with TEA.38,39 Adding 2.05 equiv. of TEA at 0 °C to a solution of 2 resulted in the formation of a polymeric product as determined by multinuclear NMR and FT-IR spectroscopy (see Fig. S3, S4 and S55–S63†). While PPX exhibits a diagnostic 1H NMR signal at δ 4.0–4.5 ppm (CDCl3), the signal decreased in relative intensity (2H → 1H) and shifted downfield to 4.5–5.0 ppm upon conversion to BrPPX. Subsequent addition of excess TEA to BrPPX at 60 °C effectively converted the material to its PPV derivative as the 1H NMR spectrum recorded for the polymer lacked the diagnostic signal which indicated that the elimination reaction was complete. Moreover, signals that could be attributed to vinylene repeat units were also observed in the FT-IR spectra that were recorded for the PPVs (e.g., at 1610 cm−1 for PPV-2a).
The 13C NMR spectra recorded for BrPPX-2a and PPV-2a exhibited signals at 170.5 and 167.5 ppm, respectively, which provided additional support for the elimination reaction. Likewise, UV-vis spectroscopy revealed a bathochromic shift upon the addition of TEA to BrPPX-2a, presumably due to the formation of a conjugated backbone. Similar approaches were used to convert the other BrPPX derivatives described above to the corresponding PPVs, and the key results are summarized in Table 3. The λmax values displayed by the PPVs depended on their pendant substituents and ranged between 278 and 287 nm when measured in CH2Cl2 (see Fig. S6†).
| Polymer | Conc. (M) | Time (h) | Yieldb (%) |
M
nc (kDa) |
Đ |
T
d5d (°C) |
T
ge (°C) |
|---|---|---|---|---|---|---|---|
| a The dehydrohalogenation reaction was performed by adding excess TEA to a DMF solution containing the corresponding BrPPX at 60 °C. Additional characterization data for the BrPPX derivatives may be found in the ESI (see Table S1†). b Isolated yield. c Determined by SEC relative to polystyrene standards in THF. d The temperature at which 5 wt% loss occurred (Td5) was measured by TGA under an atmosphere of nitrogen. e The glass transition temperatures (Tg) were measured by DSC (heating rate: 20 °C min−1). f The polymerization reaction was conducted in THF. | |||||||
| PPV-2a | 0.30 | 18 | 96 | 17.1 | 2.15 | 228 | 142 |
| PPV-2b | 0.30 | 18 | 90 | 28.8 | 2.65 | 303 | 115 |
| PPV-2c | 0.30 | 18 | 81 | 28.2 | 2.10 | 270 | 13 |
| PPV-2d | 0.30 | 24 | 99 | 34.7 | 1.75 | 251 | 3 |
| PPV-2e | 0.30 | 24 | 67 | 16.1 | 1.89 | 303 | 52 |
| PPV-2f | 0.12 | 24 | 84 | 7.7 | 2.00 | 271 | 64 |
| PPV-2g | 0.12 | 24 | 96 | 37.9 | 1.89 | 268 | 96 |
| PPV-2h | 0.12 | 24 | 96 | 19.7 | 1.73 | 284 | 27 |
| PPV-2i | 0.12 | 24 | 88 | 3.0 | 2.39 | 254 | 52 |
Since PPXs are renowned for their thermal properties, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to evaluate the derivatives described above. For comparison, the corresponding PPVs were analyzed as well. Each of the PPXs displayed a decomposition temperature, defined as the temperature at which 5% weight loss (Td5) was observed, that exceeded 300 °C under nitrogen. The result underscored the high thermal stability exhibited by this class of polymers and demonstrated that the ester functional groups present in the ethylene linkage may be key contributors. For comparison, the Td5 values measured for the PPVs were relatively low, presumably due to the increased reactivity associated with conjugated systems. The PPXs and the PPVs displayed glass transition temperatures (Tgs) that ranged from 3 to 178 °C in a manner that depended on the structure and composition of the pendant ester groups. The broad range highlights an intrinsic advantage of the synthetic modularity as it enables access to polymers with tunable thermal properties.
Functionalized PPX and PPV derivatives: synthesis and application
Due to the modularity intrinsic to the synthetic methodology, a variety of PPX and PPV derivatives were prepared. Many of these derivatives feature functional groups that are amenable to further modification and utilization. For example, PPX-2e features pendant alkynyl groups, which can be readily converted through copper-catalyzed azide–alkyne cycloaddition chemistry.40,41 As shown in Scheme 4, the introduction of PPX-2e to benzyl azide along with copper sulfate and sodium ascorbate afforded the corresponding triazole-based product in high yield. The 1H NMR spectrum recorded for the modified polymer featured signals that were assigned to the triazole groups (5.4 ppm, CDCl3) but lacked signals that could be attributed to the alkynyl groups of the starting material (see Fig. S7†).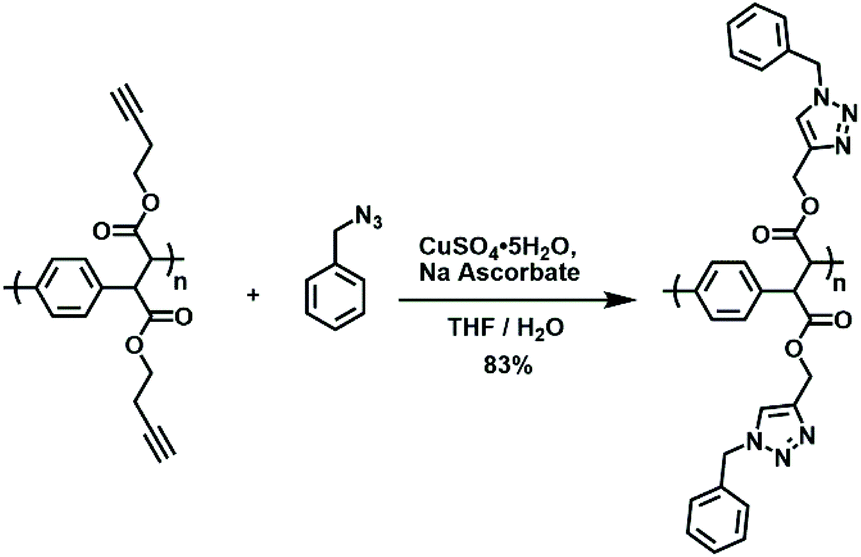 | ||
| Scheme 4 The use of copper-catalyzed azide–alkyne cycloaddition chemistry to effect post-polymerization modification. | ||
A cholesterol42 group was also attached to a PPX as well as a PPV to determine if the polymers can be bestowed with liquid crystalline properties.1,2,43–45 The syntheses of these derivatives began by condensing cholesterol with various diols followed by attachment to the aforementioned monomer precursors. Standard polymerization conditions were used, and the resulting polymers were analyzed using DSC, polarized-light optical microscopy (POM) and powder X-ray diffraction (XRD); data were also collected for the monomeric precursors to facilitate comparisons (see Fig. S97, S98 and S101–S104†). As summarized in Table 4, phase transitions were observed to occur at temperatures that depended on the length of the alkyl chains that connected the cholesterol units to the polymer backbones. For example, monomer 2g became isotropic at 117 °C whereas the corresponding transition for 2h occurred at 48 °C. A sharp signal was observed at 2Θ = 2.0° (44.2 Å) in the X-ray data recorded for 2g at 45 °C, consistent with the adoption of a smectic A phase upon melting. A broad diffraction at 2θ = 14–19° (4.7–6.3 Å) was also observed which can be attributed to the relatively short alkyl spacer that was utilized. For comparison, monomer 2h appeared to melt into a nematic phase upon heating and exhibited a broad diffraction signal due to the relatively longer linker length employed. All of the polymers appeared to adopt smectic A phases at room temperature and underwent isotropization at elevated temperatures (>120 °C) when compared to their respective monomers. These results indicated that the polymers could not only be rendered to display liquid crystalline properties but that the stabilities of the liquid crystalline phases were increased after polymerization.
| Compound | Transitiona | T (°C) | ΔHc (J g−1) |
|---|---|---|---|
| a Cr = crystalline phase; SmA = smectic A phase; N = nematic phase; Iso = isotropic liquid phase. b Onset temperature as observed by DSC at a heating rate of 10 °C min−1. c Enthalpy change. | |||
| 2g | Cr → SmA | 45 | 1.04 |
| SmA → Iso | 117 | 20.83 | |
| 2h | Cr → N | 20 | 0.74 |
| N → Iso | 48 | 3.16 | |
| PPX-2g | SmA → Iso | 128 | 1.62 |
| PPX-2h | SmA → Iso | 149 | 2.40 |
| BrPPX-2g | SmA → Iso | 164 | 2.85 |
| BrPPX-2h | SmA → Iso | 147 | 1.34 |
| PPV-2g | SmA → Iso | 160 | 2.46 |
| PPV-2h | SmA → Iso | 149 | 3.88 |
To further demonstrate the versatility and the modularity of the synthetic methodology, attention was directed toward the incorporation of azobenzene units into the polymers. Azobenzene is well known to isomerize from its thermodynamically stable trans geometry to a kinetically stable cis form upon irradiation. The process is not only reversible but is accompanied by changed in sterics and electronics. As such, azobenzenes have found utility in as photoswitches46 and have been used in a variety of patterning,47 self-healing,48 and responsive applications.49
A PPX containing pendant azobenzene units was prepared from monomer 2i. As shown in Scheme 2, aniline was first treated with NaNO2 and HCl to obtain the respective diazonium chloride, and then subjected to a Williamson ether synthesis as well as esterification to afford 2i. Treatment of 2i with TDAE afforded PPX-2i, which was subsequently characterized using the techniques described above. A CH2Cl2 solution of PPX-2i ([repeat unit]0 = 4.2 × 10−5 M) was irradiated with ultraviolet light (365 nm). As shown in Fig. 1a, the trans isomer was converted to its cis form within 2 min, as determined by a hypsochromic shift in the absorption profile recorded upon analysis of the reaction mixture. Subsequent irradiation of the solution with visible light (520 nm) returned the initial state. The reversible photoisomerization was repeated multiple times without detriment (Fig. 1b), reflective of the high fidelity of the isomerization chemistry in conjunction with the high chemical and thermal stability of the polymer. Irradiation for longer periods of time (1 h) and under more concentrated conditions ([repeat unit]0 = 0.01 M) enabled the isomerization process to reach a 94% conversion, as determined by 1H NMR spectroscopy (see Fig. S73†). In addition to electronic variation, the photoisomerization process affected the thermal properties of the polymer as well.46 After the isomerization process was complete, the solvent was removed and the residual polymer was analyzed by DSC (Fig. 1c). The trans isomer of PPX-2i exhibited a Tg at 46 °C and a phase transition at 93 °C. However, the cis isomer of PPX-2i exhibited a Tg at −22 °C as well as a broad exothermic transition upon heating to 70 °C, which was assigned to the release of heat that accompanies geometric isomerizations.
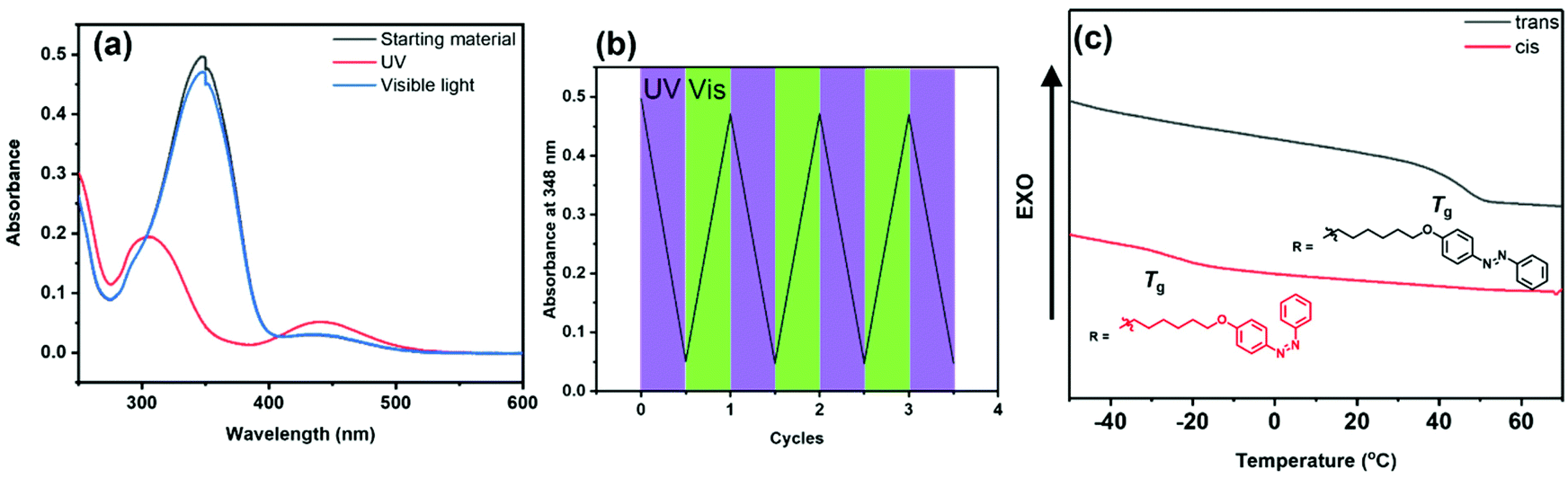 | ||
| Fig. 1 (a) UV-vis spectra recorded for PPX-2i before (black) and after irradiation with 365 nm UV light (red), and then after irradiation with 520 nm visible light (blue) in CH2Cl2. (b) Plot of the magnitude of the signal recorded at 348 nm after exposing a CH2Cl2 solution of PPX-2iversus multiple exposures to UV or visible light (indicated). (c) DSC data recorded for trans and cisPPX-2i. Additional characterization data may be found in the ESI (see Fig. S100 and S105, S106†). | ||
Finally, it was envisioned that the methodology may be adapted to access water-soluble derivatives of PPXs and PPVs, which are often desired for their processing advantages. Such types of polymers have historically been prepared through the incorporation of quaternary ammonium50 or carboxylic acid groups.13 A THF solution of the benzyl ester functionalized PPX (PPX-2f) was treated with an aqueous solution of KOH and then poured into an aqueous solution of HCl (1 M) to induce precipitation.51 The FT-IR spectrum recorded for the precipitate exhibited a vCO in the range expected for a carboxylic acid (cf., 1712 cm−1vs. 1733 cm−1 recorded for precursor; see Fig. S89†) and a broad O–H signal at 2900–3300 cm−1, consistent with the formation of the structure shown for PPX-acid, was observed. As shown in Fig. 2a, the unsaturated analogue, PPV-acid, was prepared by exposing BrPPX-2f to an aqueous solution of KOH as treatment with an aqueous solution of HCl appeared to afford anhydride side products.28,52 The carboxylic acid product exhibited a broad absorption over the range of 3000 to 3600 cm−1 and a vCO at 1566 cm−1 (Fig. 2b). Further support for the formation of the PPV-acid was obtained via UV-spectroscopy which revealed a strong bathochromic absorption when compared to polymeric starting material (Fig. 2c).
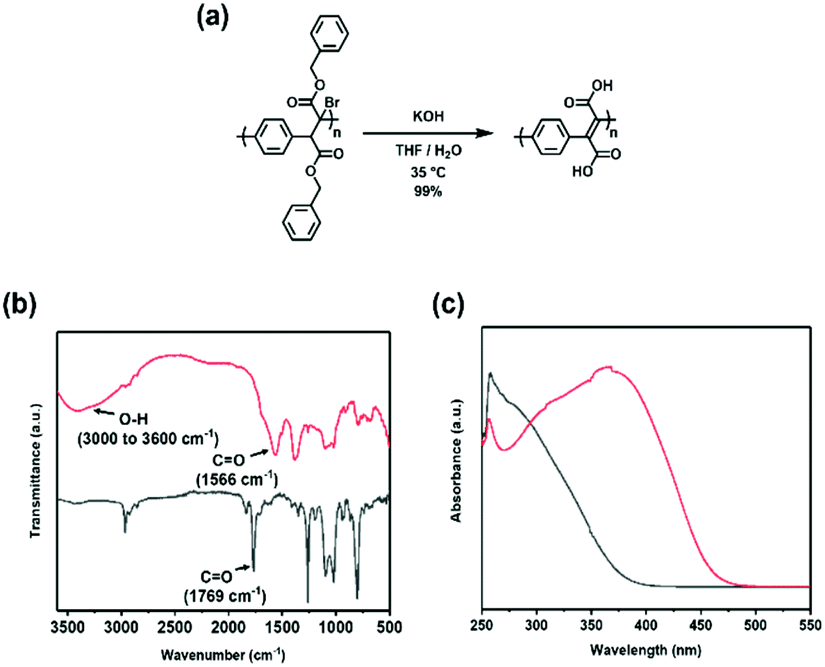 | ||
| Fig. 2 Post-polymerization modification of BrPPX-2f and corresponding characterization data. (a) Treatment of BrPPX-2f with potassium hydroxide afforded the corresponding acid, PPV-acid. (b) FT-IR spectra recorded for BrPPX-2f (black) and PPV-acid (red). (c) UV-vis spectra recorded for BrPPX-2f (black) and PPV-acid (red) in DMSO. | ||
Conclusions
New classes of PPXs and PPVs that feature functionalized ethylene and vinylene backbones were synthesized. A library of different monomers was prepared in few steps and then subsequently polymerized through exposure to a mild base or an organic reductant. The versatility of the approach enabled a broad range of derivatives PPXs and PPVs to be prepared. For example, polymers that featured pendant alkynyl groups were functionalized with azides to afford the corresponding triazoles, cholesterol to bestow the polymers with liquid crystalline characteristics, or azobenzene to enable reversible photoisomerization that manifested in distinct electronic and thermal changes. To facilitate processing, water-soluble derivatives were also synthesized. Overall, the methodology offers a new venue for accessing functional PPXs and PPVs from readily available precursors and offers a means to tune the electronic, chemical, and thermal properties of such polymers through straightforward modifications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the IBS for support (IBS-R019-D1). We thank Geon-Hui Park for assistance with the HRMS measurements and Sun Yi Lee for assistance with the XRD measurements.Notes and references
- A. Greiner, S. Mang, O. Schäfer and P. Simon, Poly(P–xylylene)s: Synthesis, polymer analogous reactions, and perspectives on structure–property relationships, Acta Polym., 1997, 48(1–2), 1–15 CrossRef CAS.
- J. Lahann and R. Langer, Novel poly(p-xylylenes): thin films with tailored chemical and optical properties, Macromolecules, 2002, 35(11), 4380–4386 CrossRef CAS.
- M. Szwarc, The C–H bond energy in toluene and xylenes, J. Chem. Phys., 1948, 16(2), 128–136 CrossRef CAS.
- W. F. Gorham, A new, general synthetic method for the preparation of linear poly–p–xylylenes, J. Polym. Sci., Part A: Polym. Chem., 1966, 4(12), 3027–3039 CrossRef CAS.
- P. Simon, S. Mang, A. Hasenhindl, W. Gronski and A. Greiner, Poly(p-xylylene) and its derivatives by chemical vapor deposition: Synthesis, mechanism, and structure, Macromolecules, 1998, 31(25), 8775–8780 CrossRef CAS.
- J. Lahann, D. Klee and H. Höcker, Chemical vapour deposition polymerization of substituted [2.2]paracyclophanes, Macromol. Rapid Commun., 1998, 19(9), 441–444 CrossRef CAS.
- T. Moss and A. Greiner, Functionalization of Poly(para–xylylene)s—Opportunities and Challenges as Coating Material, Adv. Mater. Interfaces, 2020, 7(11), 1901858 CrossRef CAS.
- B. R. Hsieh, Y. Yu, E. W. Forsythe, G. M. Schaaf and W. A. Feld, A new family of highly emissive soluble poly(p-phenylene vinylene) derivatives. A step toward fully conjugated blue-emitting poly (p-phenylene vinylenes), J. Am. Chem. Soc., 1998, 120(1), 231–232 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Synthesis of conjugated polymers for organic solar cell applications, Chem. Rev., 2009, 109(11), 5868–5923 CrossRef CAS PubMed.
- L. Chen, D. W. McBranch, H.-L. Wang, R. Helgeson, F. Wudl and D. G. Whitten, Highly sensitive biological and chemical sensors based on reversible fluorescence quenching in a conjugated polymer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(22), 12287–12292 CrossRef CAS PubMed.
- D. Gagnon, J. Capistran, F. Karasz, R. Lenz and S. Antoun, Synthesis, doping, and electrical conductivity of high molecular weight poly(p-phenylene vinylene), Polymer, 1987, 28(4), 567–573 CrossRef CAS.
- W. Heitz, W. Brügging, L. Freund, M. Gailberger, A. Greiner, H. Jung, U. Kampschulte, N. Nießner, F. Osan, H.-W. Schmidt and M. Wicker, Synthesis of monomers and polymers by the Heck reaction, Macromol. Chem. Phys., 1988, 189(1), 119–127 CrossRef CAS.
- A. R. G. Srinivas, T. E. Kerr-Phillips, H. Peng, D. Barker and J. Travas-Sejdic, Water-soluble anionic poly(p-phenylene vinylenes) with high luminescence, Polym. Chem., 2013, 4(8), 2506–2514 RSC.
- S.-A. Chen and E.-C. Chang, Structure and Properties of Cyano-Substituted Poly(2,5-dialkoxy-p-phenylene vinylene)s, Macromolecules, 1998, 31(15), 4899–4907 CrossRef CAS PubMed.
- T. Ahn, M. S. Jang, H.-K. Shim, D.-H. Hwang and T. Zyung, Blue electroluminescent polymers: control of conjugation length by kink linkages and substituents in the poly (p-phenylenevinylene)-related copolymers, Macromolecules, 1999, 32(10), 3279–3285 CrossRef CAS.
- R. M. Moslin, T. L. Andrew, S. E. Kooi and T. M. Swager, Anionic Oxidative Polymerization: The Synthesis of Poly (phenylenedicyanovinylene)(PPCN2V), J. Am. Chem. Soc., 2009, 131(1), 20–21 CrossRef CAS PubMed.
- V. P. Conticello, D. L. Gin and R. H. Grubbs, Ring-opening metathesis polymerization of substituted bicyclo [2.2.2]octadienes: a new precursor route to poly(1,4-phenylenevinylene), J. Am. Chem. Soc., 1992, 114(24), 9708–9710 CrossRef CAS.
- C. Y. Yu and M. L. Turner, Soluble Poly(p–phenylenevinylene) s through Ring–Opening Metathesis Polymerization, Angew. Chem., 2006, 118(46), 7961–7964 CrossRef.
- T. Tang, G. Ahumada and C. W. Bielawski, Direct laser writing of poly(phenylene vinylene) on poly(barrelene), Polym. Chem., 2020, 11(34), 5437–5443 RSC.
- B. R. Hsieh, Y. Yu, A. C. VanLaeken and H. Lee, General methodology toward soluble poly(p-phenylenevinylene) derivatives, Macromolecules, 1997, 30(25), 8094–8095 CrossRef CAS.
- H. Spreitzer, H. Becker, E. Kluge, W. Kreuder, H. Schenk, R. Demandt and H. Schoo, Soluble phenyl–substituted PPVs—new materials for highly efficient polymer LEDs, Adv. Mater., 1998, 10(16), 1340–1343 CrossRef CAS.
- H. Gilch and W. Wheelwright, Polymerization of α–halogenated p–xylenes with base, J. Polym. Sci., Part A: Polym. Chem., 1966, 4(6), 1337–1349 CrossRef CAS.
- O. Schäfer, F. Brink-Spalink, B. Smarsly, C. Schmidt, J. H. Wendorff, C. Witt, T. Kissel and A. Greiner, Synthesis and properties of ω–phenylalkyl–substituted poly(p–xylylene)s prepared by base–induced 1,6–dehydrohalogenation, Macromol. Chem. Phys., 1999, 200(8), 1942–1949 CrossRef.
- F. Brink-Spalink and A. Greiner, Efficient Control on Molecular Weight in the Synthesis of Poly(p-xylylene)s via Gilch Polymerization, Macromolecules, 2002, 35(9), 3315–3317 CrossRef CAS.
- W. C. Wan, H. Antoniadis, V. Choong, H. Razafitrimo, Y. Gao, W. A. Feld and B. R. Hsieh, Halogen precursor route to poly [(2,3-diphenyl-p-phenylene)vinylene] (DP-PPV): Synthesis, photoluminescence, electroluminescence, and photoconductivity, Macromolecules, 1997, 30(21), 6567–6574 CrossRef CAS.
- J. Wiesecke and M. Rehahn, Direct Observation of α–Chloro–p–quinodimethane as the Real Monomer in the Gilch Polymerization Leading to Poly(p–phenylene vinylene)s, Macromol. Rapid Commun., 2007, 28(2), 188–193 CrossRef CAS.
- C. Neef and J. Ferraris, MEH-PPV: improved synthetic procedure and molecular weight control, Macromolecules, 2000, 33(7), 2311–2314 CrossRef CAS.
- L. P. Sanow, J. Sun and C. Zhang, Photochemical stability of dicyano–substituted poly(phenylenevinylenes) with different side chains, J. Polym. Sci., Part A: Polym. Chem., 2015, 53(24), 2820–2828 CrossRef CAS.
- H. Shimomoto, T.-A. Moriya, T. Mori, T. Itoh, S. Kanehashi, K. Ogino and E. Ihara, Single-Component Polycondensation of Bis(alkoxycarbonyldiazomethyl) aromatic Compounds To Afford Poly (arylene vinylene) s with an Alkoxycarbonyl Group on Each Vinylene Carbon Atom, ACS Omega, 2020, 5(10), 4787–4797 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113(7), 5322–5363 CrossRef CAS PubMed.
- J. M. Narayanam, J. W. Tucker and C. R. Stephenson, Electron-transfer photoredox catalysis: development of a tin-free reductive dehalogenation reaction, J. Am. Chem. Soc., 2009, 131(25), 8756–8757 CrossRef CAS PubMed.
- T. Koike and M. Akita, Visible-light radical reaction designed by Ru-and Ir-based photoredox catalysis, Inorg. Chem. Front., 2014, 1(8), 562–576 RSC.
- C. Burkholder, W. R. Dolbier and M. Médebielle, Tetrakis (dimethylamino) ethylene as a useful reductant of some bromodifluoromethyl heterocycles. Application to the synthesis of new gem-difluorinated heteroarylated compounds, J. Org. Chem., 1998, 63(16), 5385–5394 CrossRef CAS.
- W. Carpenter, The Reactions of Tetrakis(dimethylamino) ethylene with Polyhalogenated Compounds, J. Org. Chem., 1965, 30(9), 3082–3084 CrossRef CAS.
- G. Giuglio-Tonolo, T. Terme, M. Médebielle and P. Vanelle, Original reaction of p-nitrobenzyl chloride with aldehydes using tetrakis(dimethylamino) ethylene (TDAE), Tetrahedron Lett., 2003, 44(34), 6433–6435 CrossRef CAS.
- Y. Nishiyama and A. Kobayashi, Synthesis of 1, 4-diketones: reaction of α-bromo ketones with tetrakis(dimethylamino) ethylene (TDAE), Tetrahedron Lett., 2006, 47(31), 5565–5567 CrossRef CAS.
- J. Broggi, T. Terme and P. Vanelle, Organic Electron Donors as Powerful Single–Electron Reducing Agents in Organic Synthesis, Angew. Chem., Int. Ed., 2014, 53(2), 384–413 CrossRef CAS PubMed.
- G. M. Brooke and M. F. Woolley, Reactions proceeding via the reactive intermediate α-vinyl-p-xylylene. Contrasting orientations in the formation of cyclic dimers and polymer, Polymer, 1993, 34(6), 1282–1288 CrossRef CAS.
- The ability to induce polymerization with the use of a relative weak base (i.e., TEA) indicates that the polymerization mechanism may differ from that of the small molecule model. For example, TEA may facilitate the conversion of 2 to a quinodimethane intermediate that then undergoes spontaneous polymerization.
- B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Frechet, Dendronized linear polymers via “click chemistry”, J. Am. Chem. Soc., 2004, 126(46), 15020–15021 CrossRef CAS PubMed.
- D. Liu and C. W. Bielawski, Direct azidation of isotactic polypropylene and synthesis of ‘grafted to'derivatives thereof using azide–alkyne cycloaddition chemistry, Polym. Int., 2017, 66(1), 70–76 CrossRef CAS.
- F. Reinitzer, Beiträge zur kenntniss des cholesterins, Monatsh. Chem. Verw. Teile Anderer Wiss., 1888, 9(1), 421–441 CrossRef.
- J. L. Pohlmann, W. Elser and P. R. Boyd, Cholesteryl ω-phenylalkanoates, Mol. Cryst. Liq. Cryst., 1973, 20(1), 87–97 CrossRef CAS.
- J.-W. Lee, D. K. Oh, C. Yelamaggad, S. A. Nagamani and J.-I. Jin, Ferroelectric liquid crystalline polyoxetanes bearing chiral dimesogenic pendants, J. Mater. Chem., 2002, 12(8), 2225–2230 RSC.
- J.-S. Hu, L.-Q. Yang, X. Zhang and Z.-W. Song, Synthesis and phase behaviour of new cholesteric monomers and side chain smectic polymers based on cholesterol, Liq. Cryst., 2010, 37(10), 1259–1268 CrossRef CAS.
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H.-J. Butt and S. Wu, Photoswitching of glass transition temperatures of azobenzene-containing polymers induces reversible solid-to-liquid transitions, Nat. Chem., 2017, 9(2), 145–151 CrossRef CAS PubMed.
- S. Sun, S. Liang, W.-C. Xu, G. Xu and S. Wu, Photoresponsive polymers with multi-azobenzene groups, Polym. Chem., 2019, 10(32), 4389–4401 RSC.
- B. Peng, H. Li, Y. Li, Z. Lv, M. Wu and C. Zhao, A photoresponsive azopyridine-based supramolecular elastomer for self-healing strain sensors, Chem. Eng. J., 2020, 395, 125079 CrossRef CAS.
- Y. Yue, Y. Norikane, R. Azumi and E. Koyama, Light-induced mechanical response in crosslinked liquid-crystalline polymers with photoswitchable glass transition temperatures, Nat. Commun., 2018, 9(1), 1–8 CrossRef.
- A. Uddin, W. Sang, Y. Gao and K. N. Plunkett, Functional Poly(p-xylylene)s via Chemical Reduction of Poly(p-phenylenevinylene)s, Macromolecules, 2019, 52(24), 9799–9803 CrossRef CAS.
- J. Seo, S. Y. Lee and C. W. Bielawski, Dewar lactone as a modular platform to a new class of substituted poly(acetylene)s, Polym. Chem., 2019, 10(47), 6401–6412 RSC.
- E. M. Beccalli, M. L. Gelmi and A. Marchesini, A New Access to Diarylmaleic Anhydrides, Eur. J. Org. Chem., 1999, 1999(6), 1421–1426 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic procedures; 1H & 13C NMR spectra; infrared spectra; thermogravimetric and differential scanning calorimetry data; GPC data; UV-vis spectra; polarized optical microscopy images; and X-ray diffraction data. See DOI: 10.1039/d1py01063h |
| This journal is © The Royal Society of Chemistry 2022 |