
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of new bicyclo[2.1.1]hexane compact modules: an opening towards novel sp3-rich chemical space†
Loïc
Herter
 ab,
Ilias
Koutsopetras‡
b,
Lorenzo
Turelli‡
b,
Thomas
Fessard
*a and
Christophe
Salomé
*a
ab,
Ilias
Koutsopetras‡
b,
Lorenzo
Turelli‡
b,
Thomas
Fessard
*a and
Christophe
Salomé
*a
aSpiroChem AG: Rosental area, WRO-1047-3, Mattenstrasse 22, 4058 Basel, Switzerland. E-mail: christophe.salome@spirochem.com; thomas.fessard@spirochem.com
bBio-Functional Chemistry (UMR 7199), LabEx Medalis, University of Strasbourg, 74 Route du Rhin, Illkirch, 67400, France
First published on 28th October 2022
Abstract
Among the valuable saturated bicyclic structures incorporated in newly developed bio-active compounds, bicyclo[2.1.1]hexanes are playing an increasingly important role, while being still underexplored from a synthetic accessibility point of view. Here, we disclose an efficient and modular approach toward new 1,2-disubstituted bicyclo[2.1.1]hexane modules. Our strategy is based on the use of photochemistry to access new building blocks via [2 + 2] cycloaddition. The system can readily be derivatized with numerous transformations, opening the gate to sp3-rich new chemical space.
In the quest to further improve the physicochemical properties of lead compounds, medicinal chemists tend to prefer sp3-rich and strained bicyclic scaffolds as bio-isosteres.1,2 Their intrinsic properties play a fundamental role in modulating and sometimes improving the solubility, activity, and conformational restriction of candidates,3 while offering new vector’s angle opportunities. The bicyclo[1.1.1]pentane motif remains to date the most renowned example of bicycles used. Following the breakthrough in establishing its biological value as a para-substituted phenyl isostere in 2012,4 extensive research has been performed to expand the toolbox of available bicyclic structures. Bicyclo[2.1.1]hexane systems have been the highlight of numerous works.5–10 These works focused on new synthetic routes, implementation of new methodologies, and new exit vectorization. However, access to a diverse range of exit vectors is still limited and additional work is required to fully exploit the rich chemical space surrounding the [2.1.1] platform. For this reason, we were interested in exploring new atom and exit-vector arrangements for [2.1.1] scaffolds. Hence, we focused on the synthesis of 1,2-disubstituted bicyclo[2.1.1]hexanes (such as 3) as depicted in Scheme 1b.2. Only limited prior methods allowed the synthesis of such [2.1.1] hexanes by a crossed [2 + 2]-cycloaddition of 1,5-dienes using a mercury lamp (Scheme 1b.1).11 Unfortunately, the use of such a lamp is technically challenging (special equipment and glassware needed) and the reaction, therefore, is difficult to scale up. Additionally, the use of toxic stannane reagents is making the process even less amenable for large-scale synthesis. Thus, we propose a novel, robust, scalable (>10 g), and mild synthetic route toward systems such as 3 (Scheme 2).
 | ||
| Scheme 1 a) Established [2.1.1] scaffolds which are broadly applied. (b) New atom arrangement being investigated. (b.1) Prior art towards 1,2-disubstituted bicyclo[2.1.1]hexane systems bearing a ketone. (b.2) New efficient and mild route proposed towards these systems. | ||
 | ||
| Scheme 2 Synthetic route towards 8a. | ||
Our efforts started with commercially available phenylacetaldehyde 4a, which was quantitatively methylenated using a reported procedure12 with Eschenmoser's salt to access aldehyde 5a. The latter was treated with allyl magnesium chloride to yield alcohol 6a, which was further oxidized using Dess–Martin periodinane (DMP) to generate diene 7a in a gratifying yield (80%). Interestingly, only DMP oxidation gave satisfactory results for this transformation, and other methods such as MnO2, Swern, or PCC led either to degradation or slow conversion towards the desired product.
Inspired by the iridium catalyzed [2 + 2] cycloaddition designed by Kwon et al.,13 giving access to bridged benzobicycloheptanone from benzylic dienones, we tested similar conditions for our cycloaddition with dienone 7a. Gratifyingly, the use of (Ir[dF(CF3)ppy]2(dtbpy))PF6 (2 mol%) in dichloromethane under blue LED irradiation at 450 nm enabled the isolation of bicyclic scaffold 8a (Table 1, entry 1) in 55% yield. Switching the solvent to acetonitrile further improved the yield to 90% (Table 1, entry 2). We then looked at decreasing the catalyst loading to allow for a more cost-efficient synthesis on large scale. A smaller loading (Table 1, entries 3 & 4) had a modest influence on the isolated yield of 8a with for instance an 83% yield for a loading of 0.5 mol%. As a control experiment, the cycloaddition was attempted without iridium photocatalyst and as expected no reaction occurred (Table 1, entry 5).
With the optimal conditions in hand, the reaction sequence was efficiently scaled up, yielding 4.50 g of [2.1.1] 8a starting from 10 g of phenylacetaldehyde 4a, giving a global yield of 40% with only one purification by chromatography. These results are proving the scalability and robustness of our sequence. The scope of phenylacetaldehydes was evaluated (Scheme 3). Substrates 4b–e were subjected to the reaction sequence and the ones bearing bromine, fluorine, and methyl performed well towards analogs 8b–d without any noticeable issues. Ketones 8b–d were then reduced to their corresponding secondary alcohols 9b–d in excellent yields (>90%) (Scheme 3). Unfortunately, 4-methoxyphenylacetaldehyde 4e underwent the first methylenation and subsequent Grignard addition but provided a very low yield. Indeed, the methylenation of 4e (step a) did not reach completion, and the obtained substrate rapidly decomposed (dimerization), as highlighted in the works (described in the ESI) of Davies et al.14 This result guided us towards a “late-stage functionalization” approach (Scheme 4) in order to circumvent the problems inherent to available phenylacetaldehydes, multi-step preparation of intermediates and the electronic nature of substituents (Scheme 4).
 | ||
Scheme 3 Evaluation of the scope of the phenylacetaldehydes 4b–e using the designed route and reduction to the corresponding alcohols. Yields refer to isolated products after column chromatography. (a) Eschenmoser's salt, Et3N, CH2Cl2. (b) AllylMgCl, THF. (c) DMP, CH2Cl2. (d) Ir[dF(CF3)ppy]2(dtbpy)PF6 (2 mol%), CH3CN (0.075 M), blue LEDs. (e) NaBH4, MeOH. **![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Due to a low yielding first step, the synthesis was not taken further. Due to a low yielding first step, the synthesis was not taken further. | ||
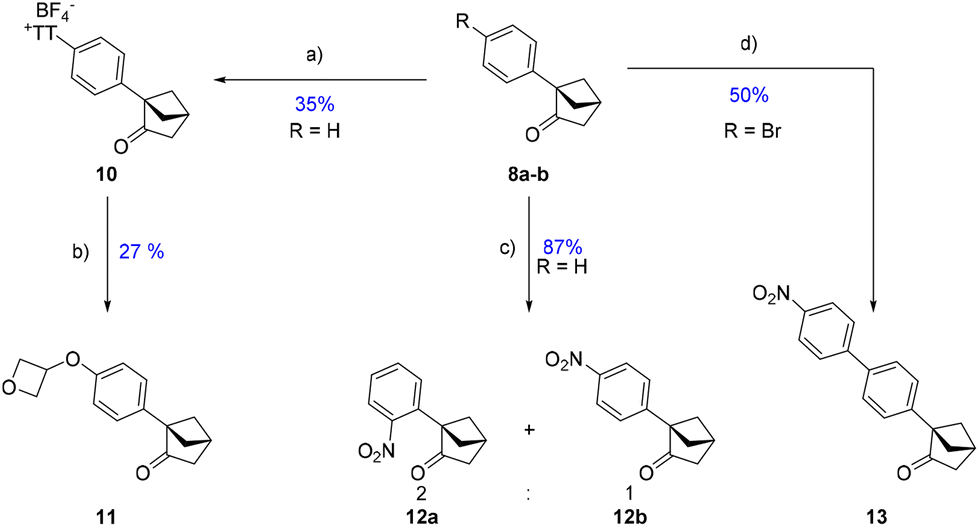 | ||
| Scheme 4 Aromatic ring functionalization of 8 derivatives. (a) Thianthrene oxide, TFAA, HBF4·Et2O, CH3CN, r.t., R = H. (b) Oxetan-3-ol, CuTC, Na2CO3, [Ir[dF(CF3)ppy]2(dtbpy)PF6], blue LED, CH3CN, r.t. (c) H2SO4, HNO3, Ac2O, AcOH, 0 °C, R = H. (d) 4-NO2-Ph-B(OH)2, Pd(PPh3)4, Cs2CO3, dioxane:H2O, R = Br. | ||
Firstly, using the “thianthrenation” approach designed by Ritter's group,15 we were able to target the para-position of the system (10). This position was further functionalized with an oxetan-3-ol,16 allowing the isolation of phenol ether 11, which demonstrates the mildness of this approach. Alternatively, nitration of the phenyl afforded a 2:1 mixture favoring, surprisingly, the ortho-substituted moiety 12a. Lastly, a Suzuki cross-coupling was tolerated and afforded 13 starting from 8b.
Subsequently, we explored various strategies to exploit the carbonyl motif of our [2.1.1] skeleton 8a as a useful point of divergence (Scheme 5). The Wittig and Horner–Wadsworth–Emmons-type reactions yielded 14 and 15, respectively. Unsaturated 15 was further hydrogenated (Pd/C, 1 bar of H2) to obtain acetate 19, while 14 was treated under acidic conditions to give the homologated aldehyde 16. The latter was quantitatively converted to carboxylic acid 17 using Pinnick's conditions, offering a handle for diversification (amides, esters, decarboxylative couplings, etc.). Nucleophiles such as TMS-CN or MeMgBr could react with the carbonyl and form compounds 18a–b (in two steps for 18a: TMSCN addition and cleavage of the TMS group formed). Bicyclo[2.1.1]hexane 8a could serve as a platform for ring-expanded compounds 22 and 23. The lactone 22 was effectively formed by the Baeyer–Villiger rearrangement with mCPBA in refluxed methylene chloride while the lactam counterpart 23 was obtained via a Schmidt reaction using benzyl azide under the conditions developed by Aubé et al.17 It is of importance to note that the regioisomers shown in Scheme 5 were the only ones formed during these ring-expansion reactions.
 | ||
| Scheme 5 Ketone derivatization of 8a. All yields refer to isolated products after column chromatography. (a) MeOCH2P(Ph3)Cl, KOtBu, THF. (b) 2 M HCl, THF. (c) NaClO2, Na2HPO4, H2O2, CH3CN:H2O. (d) (OEt)2P(O)CO2Et, NaH, THF. (e) H2, Pd/C, MeOH, r.t. o/n. (f) TMSCN, ZnI2, CH2Cl2 then HCl (2 M). (g) MeMgBr, THF. (h) Tosyl-NH2NH2, MeOH, 60 °C. (i) N-Cyclohexyl-propyl, n-BuLi, ethyl formate, THF. (j) mCPBA, CH2Cl2, 45 °C. (k) Bn-N3, TiCl4, CH2Cl2. | ||
Interestingly, any attempt to form the [3.1.1] lactam product 23 by the Beckmann rearrangement (following oxime formation) only resulted in the decomposition or recovery of the starting material. Additionally, owing to the high degree of conformational restriction and high steric energy (44.8 kcal mol−1), the ketone group of 8a seemed less prone to undergo all the “classical” reactions inherent to carbonyl reactivity. This is especially true regarding enolate chemistry, which led to unfruitful results at almost every attempt despite testing a wide array of reactions, aiming to functionalize position 3 of the system. To reinforce this statement, after the successful formation of hydrazone 20 (Scheme 5), a Shapiro reaction was attempted (with MeI as the electrophilic partner) but did not provide the endo C–C double bond, as could have been expected.18 Likewise, simple enol-reactivity with initial treatment using a base such as LDA and trapping of the supposedly formed enolate with various electrophiles such as MeI or TMS-Cl only led to the recovery of the starting material. This non-enol reactivity was already highlighted in the works of Ho et al.19 Indeed, they demonstrated that the only base/electrophile combination that effectively afforded an enolate product was N-propyl-cyclohexylamine/ethyl formate which afforded the α-formyl-ketone. This combination was attempted in our system which yielded the desired formylated product 21, albeit in a modest yield of 17%. Further studies are currently ongoing in order to improve this poor enol-reactivity. Following an extensive diversification study on both the aromatic and center cores of our newly developed system 8a, we were intrigued by the synthetic availability and feasibility of an all-aliphatic scaffold with rightly placed exit vectors. This would enable the construction of sp3-rich building blocks valuable for drug discovery. A new synthetic route had to be developed with an aliphatic group replacing the phenyl rings (Scheme 6).
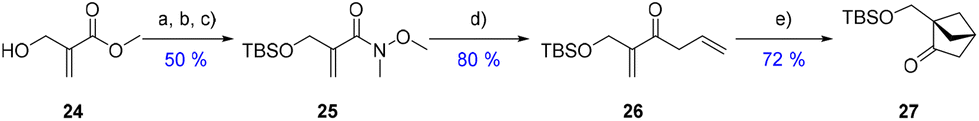 | ||
| Scheme 6 New synthetic route with only aliphatic groups. (a) LiOH·H2O, MeOH:THF, water. (b) TBS-Cl, imidazole, CH2Cl2. (c) HN(OMe)Me·HCl, EDC, DIPEA, CH2Cl2. (d) Allyl MgCl, THF. (e) ITX (10 mol%), CH3CN, 365 nm LEDs, o/n. ITX = 2-isopropyl thioxanthone. | ||
By employing a similar route to the one used in Scheme 2, the goal was to evaluate the influence of the absence of the aromatic counterpart on the radical formation events, energy transfer from the photocatalyst and therefore the cycloaddition reaction. Hydroxymethylacrylate 24 was chosen as the starting material (availability and acrylate reactivity). Compound 24 was thus saponified and TBS-protected, followed by Weinreb-amide formation to access 25. It was then subjected to allyl Grignard addition to form precursor 26 in 80% yield. Using the same conditions used previously (Schemes 2 and 3), the cycloaddition towards 27 would not reach completion, despite the extended stirring time and higher catalyst loadings. At this point, it appeared clear to us that the loss of the aromatic ring in the system affected the triplet excited state energy (ET) of diene 26 and it was therefore higher than its aromatic counterpart 7a. This change would therefore prevent a proper energy transfer from the iridium catalyst (ET = 260 kJ mol−1) under blue LED irradiation. Thus, a new photosensitizer with higher triplet excited state energy had to be selected. Following the review written by the Glorius group about energy transfer and triplet energy,20 it seemed that the thioxanthone family of photo-sensitizers could provide suitable candidates.
Due to its high energy (ET = 270 kJ mol−1),21 2-isopropyl thioxanthone (ITX) was picked for our cycloaddition towards 27. The reaction of 26 with ITX in acetonitrile under 365 nm LED (black light) irradiation in a quartz tube afforded the desired cyclized product 27 in a very rewarding 72% yield.
With a useful procedure in hand, we then focused on the diversification that this new substitution pattern offers (Scheme 7). Starting from 27, the corresponding oxime was quantitatively formed using hydroxylamine and sodium acetate. Then, the reduction of this oxime using nickel(II) and sodium borohydride in the presence of Boc anhydride afforded the Boc-protected amine 28 at position 2 of the system. The silyl-protected alcohol was effectively transformed into the very interesting non-natural amino-acid 29. A Curtius rearrangement yielded the orthogonally bis-protected-diamine building block 30. The latter was further mono-deprotected, giving 31 as a TFA salt. These transformations highlight the versatility and modularity that this scaffold offers to construct valuable sp3-rich building blocks to be incorporated into drug-like compounds.
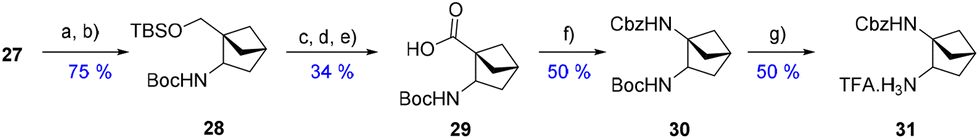 | ||
| Scheme 7 New-building blocks elaboration starting from 27. (a) HONH2·HCl, NaOAc, MeOH, 60 °C. (b) NiCl2·(H2O)6, Boc2O, NaBH4, MeOH. (c) TBAF, THF, 0 °C to r.t. (d) DMP, CH2Cl2. (e) NaClO2, Na2HPO4, H2O2, CH3CN:water (1:1) (f) BnOH, DPPA, Et3N, toluene, reflux o/n. (g) TFA, CH2Cl2. | ||
Conclusions
To conclude, we propose an efficient, scalable, and mild approach toward valuable and innovative bicyclo[2.1.1]hexane systems. The diversifications realized built a platform to efficiently introduce this scaffold into more complex structures. Ongoing studies of the dihedral angles of 1,2-substituents by X-ray analysis are underway to compare the bridged scaffold with other unsaturated cyclic linkers found in active compounds (such as 1,2-trans-cyclopentane).22Conflicts of interest
LH, CS and TF declare a conflict of interest as they are the employees and CEO of SpiroChem AG, a Swiss fine chemicals company commercializing the compounds described in this work.Acknowledgements
The authors thank Dr Guilhem Chaubet for the fruitful discussions. This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme Marie Sklodowska Curie Action ITN under Grant Agreement No. 859458.References
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- Y. P. Auberson, C. Brocklehurst, M. Furegati, T. C. Fessard, G. Koch, A. Decker, L. La Vecchia and E. Briard, ChemMedChem, 2017, 12, 590–598 CrossRef CAS PubMed.
- D. Gao, C. Penno and B. Wünsch, ChemistryOpen, 2020, 9, 874–889 CrossRef CAS PubMed.
- A. F. Stepan, C. Subramanyam, I. V. Efremov, J. K. Dutra, T. J. O'Sullivan, K. J. DiRico, W. S. McDonald, A. Won, P. H. Dorff, C. E. Nolan, S. L. Becker, L. R. Pustilnik, D. R. Riddell, G. W. Kauffman, B. L. Kormos, L. Zhang, Y. Lu, S. H. Capetta, M. E. Green, K. Karki, E. Sibley, K. P. Atchison, A. J. Hallgren, C. E. Oborski, A. E. Robshaw, B. Sneed and C. J. O'Donnell, J. Med. Chem., 2012, 55, 3414–3424 CrossRef CAS PubMed.
- J. J. Bloomfield and D. C. Owsley, Tetrahedron Lett., 1973, 14, 1795–1798 CrossRef.
- T. Gibson and W. F. Erman, J. Org. Chem., 1966, 31, 3028–3032 CrossRef CAS.
- V. V. Levterov, Y. Panasyuk, V. O. Pivnytska and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 7161–7167 CrossRef CAS PubMed.
- R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 605, 477–482 CrossRef CAS PubMed.
- B. F. Agasti S, E. Pye, N. Kaltsoyannis, G. Crisenza and D. Procter, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-v93kv.
- R. Guo, Y.-C. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988–7994 CrossRef CAS PubMed.
- R. H. Newman-Evans, R. J. Simon and B. K. Carpenter, J. Org. Chem., 1990, 55, 695–711 CrossRef CAS.
- J. Choi, H. Park, H. J. Yoo, S. Kim, E. J. Sorensen and C. Lee, J. Am. Chem. Soc., 2014, 136, 9918–9921 CrossRef CAS PubMed.
- J. Zhao, J. L. Brosmer, Q. Tang, Z. Yang, K. N. Houk, P. L. Diaconescu and O. Kwon, J. Am. Chem. Soc., 2017, 139, 9807–9810 CrossRef CAS PubMed.
- H. M. L. Davies and X. Dai, J. Org. Chem., 2005, 70, 6680–6684 CrossRef CAS PubMed.
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed.
- R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161–16166 CrossRef CAS PubMed.
- P. Desai, K. Schildknegt, K. A. Agrios, C. Mossman, G. L. Milligan and J. Aubé, J. Am. Chem. Soc., 2000, 122, 7226–7232 CrossRef CAS.
- W. Kirmse, T. Meinert, D. A. Modarelli and M. S. Platz, J. Am. Chem. Soc., 1993, 115, 8918–8927 CrossRef CAS.
- F. T. Bond and C.-Y. Ho, J. Org. Chem., 1976, 41, 1421–1425 CrossRef CAS.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- L. D. Elliott, S. Kayal, M. W. George and K. Booker-Milburn, J. Am. Chem. Soc., 2020, 142, 14947–14956 CrossRef CAS PubMed.
- A. Knight, J. L. Hemmings, I. Winfield, M. Leuenberger, E. Frattini, B. G. Frenguelli, S. J. Dowell, M. Lochner and G. Ladds, J. Med. Chem., 2016, 59, 947–964 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01669a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |