
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel electrochemically-mediated peptide dethiylation in processes relevant to native chemical ligation†
Charles M. G.
Lamb
,
Jian
Shi
,
Jonathan D.
Wilden
 and
Derek
Macmillan
*
and
Derek
Macmillan
*
Department of Chemistry, UCL, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: d.macmillan@ucl.ac.uk
First published on 30th August 2022
Abstract
Here we explore electrochemical dethiylation in processes relevant to Native Chemical Ligation (NCL). NCL's reliance on the redox active amino acid cysteine and β-mercaptamine derivatives suggests a potential role for electrochemistry. We show that the application of a 1 V potential to platinum electrodes in 0.15 M TCEP solution is sufficient to convert Cys to Ala in cyclic peptides, and to cleave the 2-mercapto-2-phenethyl class of acyl transfer auxiliary.
Introduction
In recent years electrochemical processes have made a significant impact on organic synthesis, delivering novel, efficient, and environmentally benign reactions.1 In the context of peptide chemistry, electrochemistry is less explored aside from well-known redox processes such as oxidation and reduction of disulfide bonds, reactions at aromatic sidechains and C-terminal decarboxylation/functionalization.2 Owing to the heavy reliance on desulfurization chemistry in NCL-type processes, we became interested in exploring electrochemistry for this purpose.Native chemical ligation is a powerful reaction for the synthesis of proteins,3 and in its simplest guise unites one peptide component bearing a C-terminal thioester, with another containing an N-terminal cysteine. Occasionally a large target protein has cysteine residues suitably positioned along its backbone to allow ligation between synthetically accessible components of similar size. However, this situation is relatively rare. It can be overcome, in many cases, using synthetic β-mercapto analogues of naturally occurring amino acids,4 and acyl transfer auxiliaries.5 Whilst extremely powerful tools for protein total synthesis, disadvantages of these approaches include lengthy asymmetric syntheses of amino acid analogues, and acyl transfer auxiliaries often perform poorly at ligation junctions where neither amino acid (thioester, or auxiliary linked) are glycine.
A further popular way of solving this problem is to protect all naturally occurring cysteines in the protein throughout the synthesis and replace alanine, a far more frequently occurring amino acid and more likely to be suitably positioned in the sequence, with newly introduced cysteines. This allows ligations to take place under typical NCL conditions using a readily available naturally occurring amino acid. Once complete, all free cysteines are desulfurized to alanine and naturally occurring cysteine residues are then deprotected prior to protein folding. Ultimately desulfurization removes all traces of the ligation process (Scheme 1A).6 This approach is equally suited for the removal of the 2-mercapto-2-phenethyl class of acyl-transfer auxiliary (Scheme 1B) where desulfurization gives rise to a radical intermediate that initiates auxiliary cleavage.6d,7
 | ||
| Scheme 1 Typical products of (A) NCL and (B) 2-mercapto-2-phenethyl acyl transfer auxiliary-mediated NCL, where initial desulfurization and radical formation reduces Cys to Ala and facilitates cleavage of the auxiliary. TCEP = tris-carboxyethyl phosphine. Here we explore the use of electricity for this purpose. | ||
Desulfurization often involves exposing peptides to highly flammable, noxious or malodorous chemicals and we were keen to investigate whether, using electrochemistry, we could discover a process that may remove or reduce the dependence on such reagents.
Results and discussion
We first examined reduction of available Ac-Cys-OMe (1) and a 1.0 V potential was employed as this extends beyond the redox potentials typically associated with oxidation and reduction of disulfide bonds. Initially, employing graphite electrodes in a divided electrochemical cell with NaCl as the electrolyte, various conditions were investigated. Notably, in an unbuffered reaction, the pH would drift over time to pH 14 leading to sample decomposition. Replacing water with 0.1 M Na phosphate buffer (pH 5.8) stabilized the reaction pH and replaced NaCl as the electrolyte but no conversion to Ac-Ala-OMe (2) was observed (Table 1, entries 1–3) after 18 h, prompting an investigation of alterative electrode materials (Table 1, entries 4–21). Successful desulfurization was clearly dependent on the application of the 1.0 V potential (Fig. 1). | ||
| Fig. 1 1H NMR spectra of crude products obtained in the presence (upper trace) and absence (lower trace) of an applied potential. Only 5–10% conversion to the reduced product is observed in the control reactions where no electricity is applied. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Electrode material | Cell type | Potential/V | [TCEP]/M | Ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 :2 |
|
a All reactions employed a silver reference electrode and 10 mg 1 at a final concentration of 0.011 M in 4:1 H2O/MeCN containing 0.1 M Na phosphate buffer; pH 5.8.
|
|||||
| 1 | Graphite | Divided | ±1 | 0 | 1:0 |
| 2 | Graphite | Undivided | 1 | 0 | 1:0 |
| 3 | Graphite | Undivided | 1 | 0.2 | 1:0 |
| 4 | Pt | Undivided | 1 | 0 | 1:0 |
| 5 | Pt | Divided | ±1 | 0.05 | 1:0 |
| 6 | Pt | Undivided | 1 | 0.05 | 9:10 |
| 7 | Pt | Undivided | 1 | 0.1 | 6:13 |
| 8 | Pt | Undivided | 1 | 0.15 |
0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 9 | Pt | Undivided | 1 | 0.2 |
0:1
|
| 10 | Pt | Undivided | 0 | 0.2 | 7:1 |
| 11 | Stainless steel | Divided | +1 | 0 | 1:0 |
| 12 | Stainless steel | Undivided | 1 | 0 | 1:0 |
| 13 | Stainless steel | Undivided | 1 | 0.1 | 1:0 |
| 14 | Cu | Divided | +1 | 0 | 1:0 |
| 15 | Cu | Undivided | 1 | 0 | 1:0 |
| 16 | Cu | Undivided | 1 | 0.1 | 1:0 |
| 17 | Ti | Divided | +1 | 0 | 1:0 |
| 18 | Ti | Undivided | 1 | 0 | 1:0 |
| 19 | Ti | Undivided | 1 | 0.1 | 7:2 |
| 20 | Ti | Undivided | 1 | 0.2 |
6:7
|
| 21 | Ti | Undivided | 0 | 0.2 | 19:1 |

Reaction progress was monitored by comparing the integration of CHα protons in the crude reaction mixture (see ESI†). No reduction was observed when utilizing stainless steel or copper electrodes. Notably, in the case of platinum and titanium, reduction was taking place in the absence of the usual hydrogen donor (t-BuSH) and free radical initiator (VA-044). Overall, regardless of electrode material employed, or to which half-cell 1 was added to, no reduction was observed in a divided electrochemical cell. Comparing entries for platinum (8 and 10) and titanium (20 and 21), reduction was dependent on the application of a potential, with platinum proving to be the more effective material, showing complete reduction in 4:1 H2O/MeCN in the presence of 0.15 M tris-carboxyethyl phosphine (TCEP). The progress of the reaction also appeared dependent on the presence of TCEP (entries 4 and 18), although at significantly lower concentrations than those often reported in the literature (0.4–0.5 M). The reaction was performed on a larger scale such that Ac-Ala-OMe could be isolated (90% yield) and the optical rotation was determined to be −91.2, consistent with the literature value,8 confirming that the stereochemical integrity of the starting material had been preserved. For subsequent reactions we employed platinum electrodes, a silver reference electrode, and conducted reactions in 4:1 H2O/MeCN with 0.1 M and 0.15 M final concentrations of sodium phosphate buffer (pH 5.8) and TCEP respectively.
Due to an ongoing interest in peptide cyclisation via N → S acyl transfer we investigated desulfurization of short cyclic peptides including an Agardhipeptin A: cyclo(–His1–Gly2–Trp3–Pro4–Trp5–Gly6–Leu7–)9 analogue and crotogossamide: cyclo(–Gly1–Ala2–Ser3–Gly4–Leu5–Asn6–Gly7–Ile8–Phe9–).10 The parent peptide sequences were circularly permuted to afford linear precursors 3 and 4, suitable for head-to-tail cyclisation via N → S acyl transfer (Fig. 2).11 Cyclisation to afford 5 and 6 proceeded under typical reaction conditions. However 4 tended to precipitate upon heating to 60 °C, so the cyclisation was conducted in 3 M guanidine hydrochloride. Cyclic peptides 5 and 6 were isolated in yields of 55% and 57% respectively. In order to obtain a standard compound for comparison 5 was subjected to typical metal-free desulfurization reaction conditions utilizing VA-044, 0.2 M TCEP, and tBuSH at 37 °C. After 22 h the reduced peptide 7 was isolated in 54% yield. Next 5 and 6 were subjected to electrochemical reduction under identical conditions to reduction of 1 and the reduced peptides 7 and 8 accumulated over a period of 6 h at room temperature, and were isolated in yields of 33% and 27% respectively. Whilst yields were moderate for this class of molecule, it was encouraging that the reactions appeared to proceed smoothly, as determined by HPLC and LC-MS. However, there was some evidence of side-products accumulating over time during dethiylation of 5. Based on significant literature precedent,12 and the absence of this modification upon dethiylation of 6, oxidation was presumed to have taken place on the indole ring of tryptophan. Mindful that peptide Cα-decarboxylation reactions are well known to electrochemistry and, to further explore the scope and limitations of this reaction we examined reduction of two further model peptides. The first was simply the linear precursor 3, containing two cysteine residues. The fully-reduced peptide was isolated in 13% yield but Trp oxidation (presumed) and Cα-decarboxylation were also observed during the reaction by LC-MS. The second model peptide contained sulfur in the form of methionine in place of leucine in a further cyclic Agardhipeptin analogue. The reduced peptide was isolated in 25% yield, and once again possible oxidation of Trp was observed as the only significant byproduct (see ESI†).
 | ||
| Fig. 2 Synthesis and desulfurization of cyclic peptides. Reagents and conditions: (i) 0.1 M Na phosphate buffer (pH 5.8), 10% w/v MESNa, 0.5% TCEP, 60 °C, 24 h. 55% (5), 57% (6) (ii) 0.2 M TCEP, t-BuSH, VA-044, pH 6, 37 °C, 22 h, 54%. (iii) Pt electrodes, undivided cell, 0.1 M sodium phosphate buffer; pH 5.8/MeCN (4:1), 0.15 M TCEP, 1.0 V, 6 h room temp. 33% (7), 27% (8). HPLC and LC-MS data confirm the identical nature of 7 produced by different methods and the identity of 8. | ||
Having demonstrated electrochemically induced dethiylation in peptides we were also keen to explore its application to cleavage of the 2-mercapto-2-phenethyl acyl transfer auxiliary.8 We considered this reaction had potential to be more efficient than cysteine reduction as a consequence of the benzylic radical intermediate formed upon dethiylation. To prepare a model peptide for reaction first the auxiliary was prepared from phenyl acetic acid (9). In a procedure modified from the literature 9 was α-brominated using N-bromosuccinimide (NBS) and benzoyl peroxide to afford the α-bromo acid as the major product. The resulting mixture 9 and α-bromo acid was coupled to N,O-dimethylhydroxylamine hydrochloride to afford Weinreb amide 10, which was isolated by column chromatography in 38% yield over two steps. The bromide was displaced by trityl thiol, and then the desired aldehyde 12 was obtained in 66% yield after reduction with LiAlH4. A model peptide (sequence: GRAFS) was assembled on Rink amide resin using Fmoc-based solid phase peptide synthesis (SPPS) and the auxiliary was installed on the resin-bound peptide by reductive amination. The final auxiliary-linked peptide 13 was cleaved from the resin and obtained in 27% yield after HPLC purification. A thioester precursor peptide 14 was also readily assembled on solid support, isolated in 40% yield and converted to the 2-mercaptoethanesulfonic acid (MESNA) thioester 15 by N → S acyl transfer11 in 22% yield (43% based on recovered starting material). 15 was ligated to 13 under typical NCL conditions and the product H–LYRAG(Aux)GRAFS–NH216 was isolated in 40% yield. Under our previously developed electrochemical conditions, we were then delighted to observe complete cleavage of the auxiliary within 2 h at room temperature to afford 17 in 60% yield (Fig. 3). The only minor byproduct observed in the reaction likely corresponded to the N-formylated peptide7b with an observed mass of 1124.5 Da (M +28 Da).
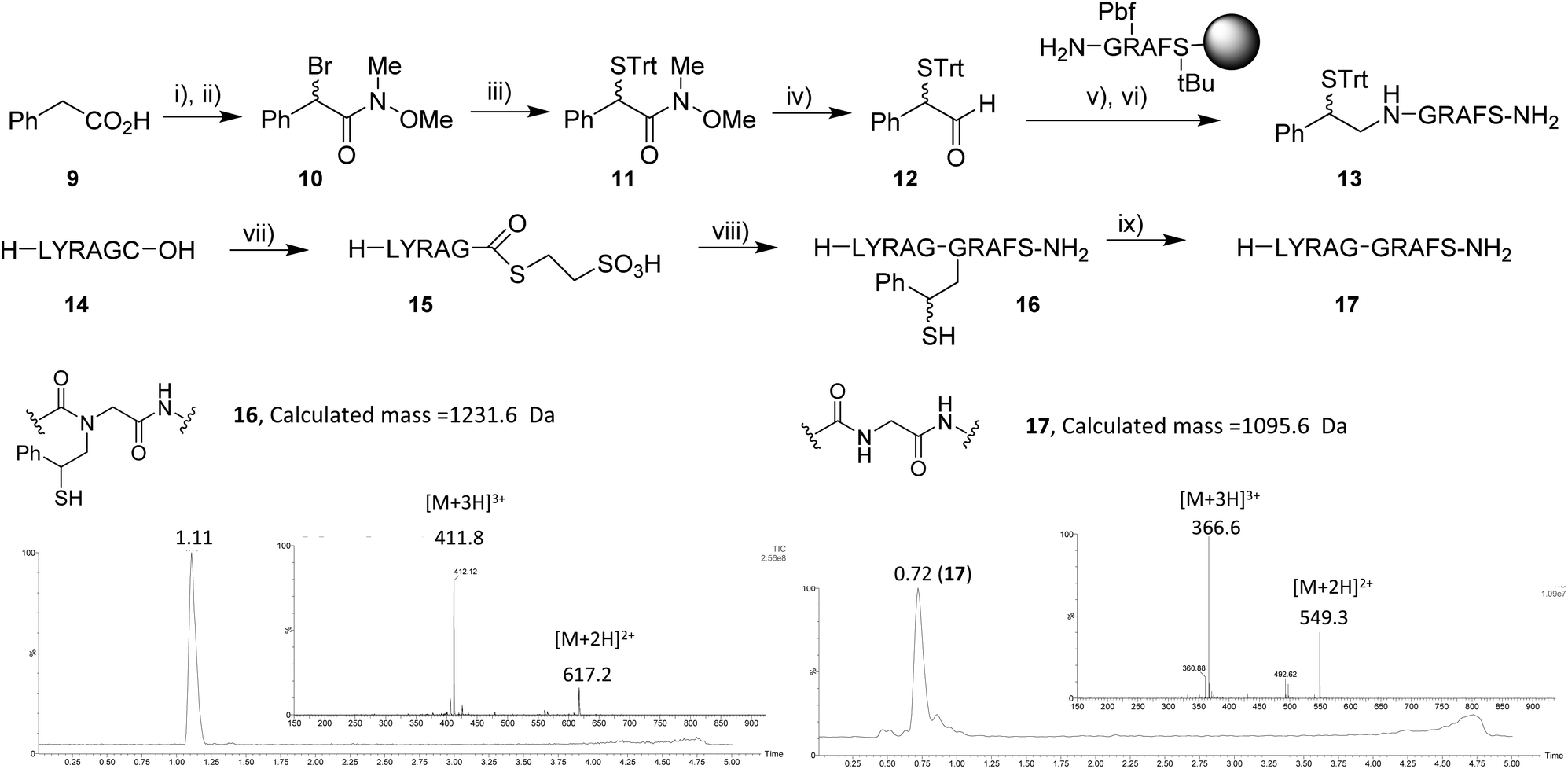 | ||
| Fig. 3 Synthesis of model peptide and auxiliary cleavage investigation. Reagents and conditions: (i) NBS, (PhCO2)2, 1,2-DCE, reflux, 2 h. (ii) N,O-Dimethylhydroxylamine·HCl, EDCI, NEt3, DCM, 0 °C–rt, 24 h (38% over 2 steps). (iii) TrtSH, K2CO3, DMF, 60 °C, 5 h, 21%. (iv) LiAlH4, THF, −78 °C, 1 h, 66%. (v) Resin bound peptide, NaCNBH3, 3:1 NMP/iPrOH, 5% AcOH, rt, 16 h. (vi) 95% TFA, 2.5% H2O, 2.5% EDT, rt, 4 h, 27%. (vii) 0.1 M Na phosphate (pH 5.8), 10% w/v MESNA, 0.5% TCEP, 60 °C, 24 h, 22%. (viii) 13, 50 mM sodium phosphate buffer (pH 8.0), 20 mM TCEP, 50 mM MPAA, rt, 2 h, 44%. (ix) Pt electrodes, undivided cell, 0.1 M sodium phosphate; pH 8.5/MeCN (4:1), 0.15 M TCEP, 1.0 V, 2 h rt, 60%. | ||
Conclusions
In summary, we have demonstrated a simple protocol for electrochemical dethiylation of peptides, and acyl transfer auxiliary cleavage. To the best of our knowledge this is the first time electrochemistry has been applied in this context. By reducing the number of required reagents, we have also raised the green credentials of the process. Initially we considered that application of a reducing potential may reduce the C–S bond directly. In fact, the reaction retains the need for TCEP and suggests initiation by anodic oxidation of the thiol to radical cation 18 (Scheme 2), which goes on to form sulfur centered radical 19 after deprotonation.13 Platinum is known to demonstrate a greater tendency for one electron oxidation and radical desorption relative to other electrode materials e.g. carbon.14 Interaction of the thiyl radical with TCEP to form intermediate 20, followed by β-scission6d likely produces the desired carbon-centered radical which can abstract hydrogen from thiol starting material or from molecular hydrogen produced at the cathode. | ||
| Scheme 2 Possible mechanism for electrochemical dethiylation. | ||
Auxiliary cleavage is likely to follow a similar initial pathway to the benzylic radical. It is possible that β-scission of the benzylic radical to the amido radical precedes hydrogen abstraction. However, the likely presence of N-formylated material during the reaction, and in the absence of morpholine, agrees with recent mechanistic studies where the benzylic radical is first trapped by molecular oxygen prior to fragmentation.7b
Whilst we observe some of the known side-reactions that occur in densely functionalized peptides, further experiments employing lower potentials, rapid alternating polarity,15 flow electrochemistry16 and electrochemical mediators,17 might all further increase chemoselectivity and product recovery. Interestingly, intercepting the carbon centred radical intermediates may also provide access to site specifically modified proteins. This was previously accomplished by addition of radicals to dehydroalanine (Dha)-containing peptides and proteins18 yet this “reversed” protocol has the advantage that it can retain amino acid stereochemistry.
Experimental section
General experimental details
Chemicals were purchased from Sigma-Aldrich, Acros, Fischer Scientific and Nova Biochem, and were used without further purification. All nuclear magnetic resonance (NMR) experiments were recorded at room temperature on a Bruker AMX 600 MHz instrument. Preparative reversed-phase high performance liquid chromatography (RP-HPLC) was performed using a Dionex Ultimate 3000 system equipped with a Phenomenex Jupiter 10μ Proteo 90A, C12, 250 × 21.2 mm column. A mobile phase of 0.1% TFA (v/v) in water (solvent A)/acetonitrile (solvent B) over a 5–60% acetonitrile gradient over 60 min, and were monitored at wavelengths 230 nm, 254 nm, and 280 nm. Analytical reversed-phase high performance liquid chromatography (RP-HPLC) was performed using a Dionex Ultimate 3000 equipped with a Phenomenex SphereClone 5μ ODS, C18 250 × 4.6 mm column. Separations involved a mobile phase of 0.1% TFA (v/v) in water (solvent A)/acetonitrile (solvent B) over a 5–95% acetonitrile gradient, and were monitored at wavelengths 230 nm, 254 nm, and 280 nm. Analytical LC-MS was carried out on Waters uPLC/SQD-LC mass spectrometer instrument equipped with a C18, 2.1 × 50 mm column. Separations were conducted with a linear gradient of 5–95% acetonitrile containing 0.1% formic acid over 10 min using a flow rate of 0.6 ml min−1. High resolution mass spectrometry was obtained from a Q-Exactive Orbitrap mass spectrometer.Electrochemical desulfurization of N-acetylcysteine methyl ester
N-Acetyl-L-cysteine methyl ester 1 (25 mg, 0.14 mmol) was dissolved in 4 mL of a solution comprised of 0.15 M TCEP (adjusted to pH 5.8 with 2 M NaOH) and 0.1 M pH 5.8 sodium phosphate buffer. Acetonitrile (1.0 mL) was added to ensure 1 was fully dissolved. The solution was transferred to a 25 mL three necked flask and platinum wire electrodes submerged 1 cm into the solution. A silver reference electrode was also inserted into the middle neck of the flask and all electrodes were connected to a Ivium Technologies Vertex model potentiostat operating in chronoamperometry mode. The solution was stirred vigorously at room temperature and a constant potential of 1.0 V was supplied. After 18 hours the electrical current was disconnected and the solution extracted with DCM (3× 5 mL). The organic fractions were combined, dried over MgSO4, and concentrated under vacuum to produce a grey solid of N-acetyl-L-alanine methyl ester (18.5 mg, 0.13 mmol, 90%). 1H NMR (600 MHz, CDCl3) δH/ppm 6.28 (1H, bs, NH), 4.56 (1H, quint, J = 6.8 Hz, MeCH), 3.72 (3H, s, OCH3), 1.99 (3H, s, NCOCH3), 1.37 (3H, d, J = 7.0 Hz, CHCH3). 13C NMR (400 MHz, CDCl3) δC/ppm 173.8, 169.7, 52.6, 48.1, 23.3, 18.6. [α]298KD = −91.2 deg cm3 g−1 dm−1 (c = 1.0 g dcl−1 in H2O).9ESI-MS: calculated m/z for C6H11NO3 [M]+: 145.07, observed [M + Na]+ 168.20.General peptide synthesis procedure
Model peptides were synthesised on a 0.05 mmol scale using either Novasyn TGT resin pre-loaded with Fmoc–Cys(Trt)–OH or Rink amide MBHA resin, and an ABI 433A automated peptide synthesiser following the Fastmoc protocol: 10 equivalents of Fmoc–Xaa–OH, HBTU/HOBt as the coupling reagents and DIPEA as base. After peptide synthesis the dry resin was suspended in a trifluoroacetic acid cleavage cocktail (5 mL, 95% TFA, 2.5% EDT, 2.5% water) then stirred for 4 h at room temperature. The solution was then filtered to obtain a yellow solution to which diethyl ether (20 mL) was added to induce peptide precipitation. The sample was then centrifuged at 3500 rpm (4 °C) for 15 minutes and the supernatant discarded. The pellet was washed with diethyl ether (20 mL) then centrifuged again at 3500 rpm (4 °C). The supernatant was again discarded and the precipitated peptide dissolved in distilled water and acetonitrile (5 mL, 5–10% MeCN as required for solubility) then purified by preparative HPLC. The eluted fractions containing the peptide (determined by UV absorption and LC-MS) were combined and the acetonitrile removed in vacuo. Lyophilization produced a white fluffy solid.Desulfurization of 5 was complete after 6 h where LC-MS confirmed full consumption of the starting material and the electrical current was disconnected. The desulfurized peptide was purified directly from the solution via preparative HPLC and the eluted fractions freeze-dried to produce 7 (1.6 mg, 33%) as a fluffy white solid. ESI-HRMS: calculated m/z for C44H54N11O7S [M + H]+ 848.4208, observed [M + H]+ 848.4200.
:1) as eluent to afford the Weinreb amide 10 as a pale-yellow oil (3.64 g, 14.1 mmol, 38% over 2 steps). 1H NMR (600 MHz, CDCl3) δH/ppm 7.56 (1H, d, J = 8 Hz, ArH), 7.50 (1H, d, J = 8 Hz, ArH), 7.38–7.29 (3H, m, ArH), 5.99 (1H, s, BrCH), 3.57 (3H, s, NOCH3), 3.21 (3H, s, NCH3). 13C NMR (126 MHz, CDCl3) δC/ppm 171.0, 136.7, 129.1, 129.0, 128.8, 128.4, 60.6, 35.3, 34.1. ESI-MS: calculated m/z for C10H12BrNO2 [M]+ 257.01, observed [M + H]+ 258.39.
Bromide 10 (3.64 g, 14.1 mmol), trityl thiol (8.96 g, 32.4 mmol) and potassium carbonate (4.48 g, 32.4 mmol) were dissolved in dry DMF (30 mL) and stirred at 60 °C for 5 h. The reaction mixture was allowed to cool to room temperature and diluted with DCM (100 mL) and water (100 mL), then the organic phase separated. The water was extracted twice more with DCM (2× 50 mL) and the combined organic layers washed with water (4× 50 mL) and saturated NaCl (50 mL). The solution was then dried over MgSO4, filtered, and concentration under vacuum gave the crude product as a yellow powder. This was purified via flash chromatography over silica with 4:1 pet. ether/EtOAc as the eluant to produce a yellow solid which was washed with diethyl ether and dried under vacuum, yielding 11 as a fluffy white powder (1.32 g, 2.91 mmol, 21%). 1H NMR (600 MHz, CDCl3) δH/ppm 7.43–7.39 (m, 5H, ArH), 7.25–7.16 (m, 13H, ArH), 7.10–7.06 (m, 2H, ArH), 4.58 (bs, 1H, TrtSCH), 3.15 (bs, 3H, OCH3), 2.97 (bs, 3H, NCH3). 13C NMR (126 MHz, CDCl3) δC/ppm 171.5, 144.5, 130.1, 129.4, 128.6, 128.5, 127.9, 127.5, 127.5, 126.8, 69.5, 61.2, 50.1, 32.5. ESI-MS: calculated m/z for C29H27NO2S [M]+ 453.18, observed [M + H]+ 454.31.
11 (1.32 g, 2.91 mmol) was dissolved in dry THF (50 mL) under nitrogen. The solution was then cooled to −78 °C in an acetone-dry ice bath. LiAlH4 (1 M in THF, 3.5 mL, 3.5 mmol) was added dropwise over 5 min and the mixture stirred for 60 min. After this time KHSO4 (4–5 drops, 5% w/v) was added dropwise and the mixture allowed to warm to room temperature. The mixture was diluted with DCM (100 mL) and washed with aqueous KHSO4 (2× 100 mL, 5% w/v), dried over MgSO4 and concentrated in vacuo. The crude product was purified via flash chromatography with petroleum ether/ethyl acetate (1:2) as the eluant, to afford aldehyde 12 as a viscous yellow oil (756 mg, 1.92 mmol, 66%). 1H NMR (500 MHz, CDCl3) δH/ppm 9.01 (d, 1H, J = 3.4 Hz, CHO), 7.45–7.41 (m, 5H, ArH), 7.34–7.25 (m, 10H, ArH), 7.24–7.20 (m, 3H, ArH), 7.12–7.10 (m, 2H, ArH), 4.00 (d, 1H, J = 3.1 Hz, TrtSCH). 13C NMR (126 MHz, CDCl3) 193.8, 147.0, 144.2, 134.8, 132.6, 130.2, 130.0, 129.9, 129.8, 129.6, 128.4, 128.3, 128.2, 128.1, 128.1, 127.9, 127.5, 127.4, 127.3, 127.2, 127.1, 69.5, 58.7. ESI-MS: calculated m/z for C27H22OS [M]+ 394.14, observed [M + H]+ 395.15.
:1 NMP/iPrOH containing 5% glacial AcOH. This solution was added to the resin followed by NaCNBH3 (31.4 mg, 0.5 mmol) and the reaction was shaken at room temperature for 16 h. The reaction was drained, the resin was washed, and the peptide cleaved as described above and purified via preparative HPLC and lyophilized to produce a white fluffy solid (8.9 mg, 27%), tR = 24.2 min. ESI-MS: calculated m/z for C31H45N9O6S [M]+: 671.3, observed [M + H]+ 672.61.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from UCL and EPSRC.References
- (a) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415 CrossRef CAS; (b) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386 RSC; (c) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed.
- (a) A. S. Mackay, R. J. Payne and L. R. Malins, J. Am. Chem. Soc., 2022, 144, 23 CrossRef CAS PubMed; (b) Y. Lin and L. R. Malins, J. Am. Chem. Soc., 2021, 143, 11811 CrossRef CAS PubMed; (c) D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mével, D. Deniaud, M. Boujtita and S. G. Gouin, J. Am. Chem. Soc., 2018, 140, 17120 CrossRef CAS; (d) S. L. S. Leite, V. L. Pardini and H. Viertler, Synth. Commun., 1990, 20, 393 CrossRef CAS.
- (a) S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338 RSC; (b) P. Dawson, T. Muir, I. Clark-Lewis and S. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- L. R. Malins, N. J. Mitchell and R. J. Payne, J. Pept. Sci., 2014, 20, 64 CrossRef CAS PubMed.
- J. Offer, Pept. Sci., 2010, 94, 530 CrossRef CAS PubMed.
- (a) S. Tsuda, M. Mochizuki, H. Nishio and T. Yoshiya, ChemBioChem, 2016, 17, 2133 CrossRef CAS PubMed; (b) Y.-C. Huang, C.-C. Chen, S. Gao, Y.-H. Wang, H. Xiao, F. Wang, C.-L. Tian and Y.-M. Li, Chem. – Eur. J., 2016, 22, 7623 CrossRef CAS PubMed; (c) N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049 CrossRef CAS PubMed; (d) Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248 CrossRef CAS PubMed; (e) L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526 CrossRef CAS PubMed.
- (a) F. Loibl, Z. Harpaz and O. Seitz, Angew. Chem., Int. Ed., 2015, 54, 15055 CrossRef PubMed; (b) S. F. Loibl, A. Dallmann, K. Hennig, C. Juds and O. Seitz, Chem. – Eur. J., 2018, 24, 3623 CrossRef CAS PubMed.
- B. Zupančič, B. Mohar and M. Stephan, Adv. Synth. Catal., 2008, 350, 2024 CrossRef.
- H. J. Shin, H. Matsuda, M. Murakami and K. Yamaguchi, Tetrahedron, 1996, 52, 13129 CrossRef CAS.
- S. Quintyne-Walcott, A. R. Maxwell and W. F. Reynolds, J. Nat. Prod., 2007, 70, 1374 CrossRef CAS.
- (a) D. Macmillan, Synlett, 2017, 1517 CrossRef CAS; (b) W. Chen, V. A. Kinsler, D. Macmillan and W.-L. Di, PLoS One, 2016, 11, e0166268 CrossRef PubMed; (c) B. Cowper, L. Shariff, W. Chen, S. M. Gibson, W.-L. Di and D. Macmillan, Org. Biomol. Chem., 2015, 13, 7469 RSC; (d) B. Cowper, D. J. Craik and D. Macmillan, ChemBioChem, 2013, 14, 809 CrossRef CAS; (e) D. Macmillan, M. De Cecco, N. L. Reynolds, L. F. A. Santos, P. E. Barran and J. R. Dorin, ChemBioChem, 2011, 12, 2133 CrossRef CAS PubMed.
- J. Roeser, H. P. Permentier, A. P. Bruins and R. Bischoff, Anal. Chem., 2010, 82, 7556 CrossRef CAS PubMed.
- Y. Yuan, Y. Chen, S. Tang, Z. Huang and A. Lei, Sci. Adv., 2018, 4, eaat5312 CrossRef CAS PubMed.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866 CrossRef CAS PubMed.
- Y. Kawamata, K. Hayashi, E. Carlson, S. Shaji, D. Waldmann, B. J. Simmons, J. T. Edwards, C. W. Zapf, M. Saito and P. S. Baran, J. Am. Chem. Soc., 2021, 143, 16580 CrossRef CAS PubMed.
- T. Noël, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858 CrossRef.
- (a) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941 RSC; (b) R. Francke, Curr. Opin. Electrochem., 2019, 15, 83 CrossRef CAS.
- T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W.-L. Ng, R. Raj, S. Faulkner, M. R. J. Vallée, A. Phanumartwiwath, O. D. Coleman, M.-L. Thézénas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, 1465 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic methods and spectra for all synthesised materials. See DOI: https://doi.org/10.1039/d2ob01499h |
| This journal is © The Royal Society of Chemistry 2022 |