
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Meyer–Schuster rearrangement of propargylic alcohols mediated by phosphorus-containing Brønsted acid catalysts†
Lalita
Radtanajiravong
 ,
Jake
Peters
,
Jake
Hummell
and
Silvia
Díez-González
*
,
Jake
Peters
,
Jake
Hummell
and
Silvia
Díez-González
*
Imperial College London, Department of Chemistry, MSRH, 82 Wood Lane, London W12 0BZ, UK. E-mail: s.diez-gonzalez@imperial.ac.uk
First published on 31st August 2022
Abstract
Commercially available (aqueous) hypophosphorus acid is an efficient catalyst for the synthesis of α,β-unsaturated carbonyl compounds from their corresponding propargylic alcohols. Reactions were carried out in technical toluene in the presence of air and in several instances the desired products were isolated analytically pure after a simple work-up.
Introduction
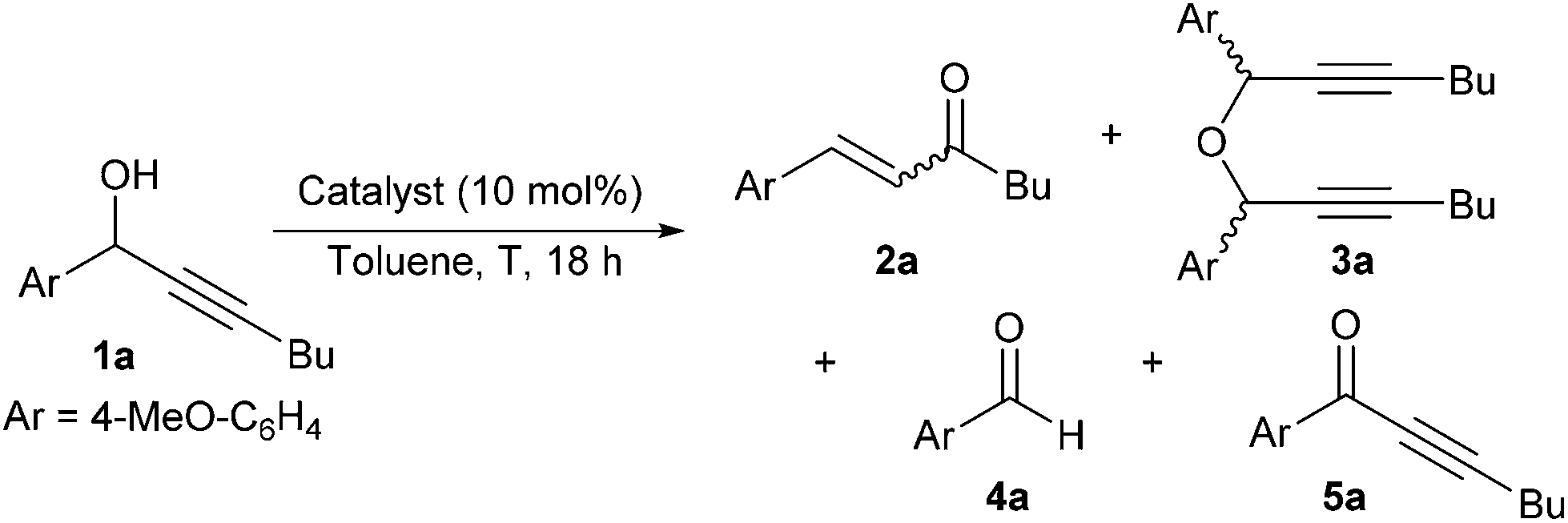
Propargylic alcohols are popular intermediates due to their easy preparation and versatile reactivity. They are the obvious precursors for a range of propargylic moieties occurring in natural products and synthetic pharmaceuticals, and their rearrangement produces α,β-unsaturated carbonyl compounds, equally desirable compounds. This rich reactivity is not without its caveats since often promoting a specific transformation over other available pathways remains a challenge.The Meyer–Schuster rearrangement was originally reported in a range of acidic media such as acetic acid, acetyl chloride and concentrated sulfuric acid, and related Rupe isomerisation is competitive when the starting alcohol contains any beta-hydrogens, leading to an alternative enone as the final product (Scheme 1).1,2 The use of strong acids and high temperatures for accessing the key carbocationic intermediates hampered the early widespread application of these powerful rearrangements. In consequence, the alternative activation of the alkyne by a range of soft Lewis acids/transition metal catalysts is now well established for Meyer–Schuster rearrangement.3,4
 | ||
| Scheme 1 The Meyer–Schuster and Rupe rearrangements. | ||
These recent developments typically involve mild reaction conditions, but some issues remain, including cost effectiveness since ruthenium and gold-based catalysts are arguably the most popular options. Therefore, it is not surprising that efforts towards reliable Brønsted acid-based catalytic systems for Meyer–Schuster rearrangements are still ongoing.5,6 For instance, the level of stereocontrol of the desired unsaturated products by either heteropoly acids,7 or tailored boronic acids8 is truly remarkable.9 Alternatively, recent reports on simple and economical Brønsted acids such as phosphorous acid or p-toluenesulfonic acid address some of the classic limitations of these isomerisations, still they present clear limitations. PTSA can stereoselectively promote the Meyer–Schuster rearrangement of secondary propargylic alcohols but the reaction is limited to terminal alkynes and 30 mol% acid in hot dichloroethane (cancerogenic) are required.10 On the other hand, 1.5 equivalents of phosphorous acid (OH)2P(O)H were needed to isomerise a range of mono- and disubstituted hydroxyalkynes in an overpressured vessel under inert atmosphere.11 In our previous studies on nucleophilic substitutions of propargylic alcohols with either HBF4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 or diethylphosphite13 as catalysts we occasionally noticed the formation of rearrangement by-products. Capitalising on these observations, we herein report our efforts to develop an economical, readily accessible and user friendly methodology for Meyer–Schuster reactions.
12 or diethylphosphite13 as catalysts we occasionally noticed the formation of rearrangement by-products. Capitalising on these observations, we herein report our efforts to develop an economical, readily accessible and user friendly methodology for Meyer–Schuster reactions.
Results and discussion
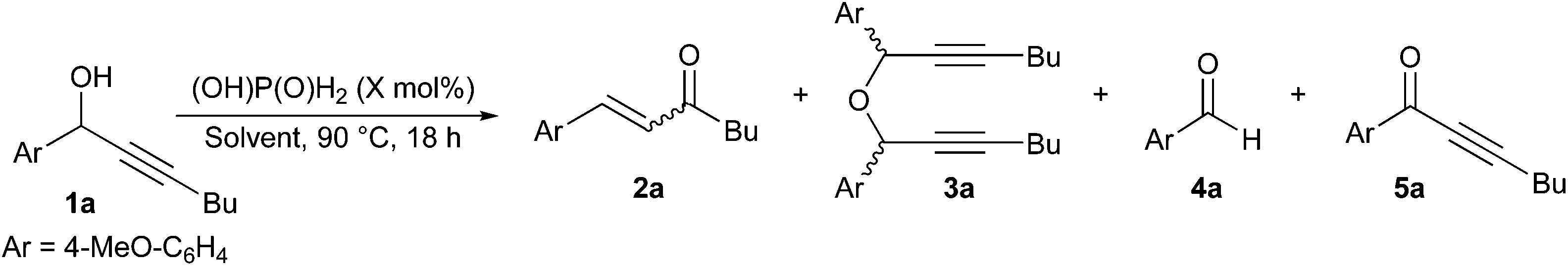
Commercially available phosphorus-based acids were tested in technical toluene at 90 °C in the presence of air with 1a as model substrate to obtain enone 2a (Table 1). No starting alcohol was recovered in any of these reactions and while trace amounts of aldehyde 4a and ketone 5a were formed in most tests, the main by-product in these reactions was ether 3a, issue of a condensation reaction of the starting propargylic alcohol. While a reasonable NMR yield of 60% was achieved with phosphorous acid (Table 1, entry 2), diethyl phosphite and aqueous hypophosphorous acid gave the highest conversions in enone 2a with high and complete selectivity, respectively (Table 1, entries 3 and 5). In comparison, significant decomposition was observed with polyprotic acids and diphenyl phosphate (Table 1, entries 1, 2 and 4). Overall, there was no correlation between the catalyst pKa and the yield in enone 2a, and aqueous hypophosphorous acid was chosen as the optimal catalyst due to its lower price per mol.14 Temperatures lower than 90 °C reduced the conversion and E/Z selectivity for enone 2a, as well as increased product decomposition and formation of ether 3a (Table 1, entries 5–7).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | T (°C) | 2a (%) |
E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :Z :Z
|
3a (%) | 4a (%) | 5a (%) |
| a Reaction conditions: 1a (1 mmol), catalyst (10 mol%) in technical toluene (1 mL), T (°C), 18 h. b 1H NMR yields and ratios were calculated using dibromomethane as internal standard and are the average of at least two independent experiments. c 85 wt% aqueous solution. d 50 wt% aqueous solution. | |||||||
| 1c | aq. (OH)3P(O) | 90 | 31 | 94:6 |
<5 | <5 | 5 |
| 2 | (OH)2P(O)H | 90 | 60 | 97:3 |
<5 | <5 | <5 |
| 3 | (OH)P(OEt)2 | 90 | 84 | 95:5 |
— | <5 | 5 |
| 4 | (OH)P(O)(OPh)2 | 90 | 19 | 83:17 |
17 | <5 | — |
| 5d | aq. (OH)P(O)H2 | 90 | 90 | E only | <5 | <5 | 5 |
| 6d | aq. (OH)P(O)H2 | 70 | 63 | 64:36 |
<5 | <5 | <5 |
| 7d | aq. (OH)P(O)H2 | 50 | 11 | 30:70 |
53 | <5 | <5 |
Alternative solvents were then screened with the model substrate but none of the apolar or polar solvents tested could match toluene in terms of reactivity or selectivity (Table 2, entries 1–5). No by-products issue of a Friedel-Craft reaction of toluene were observed in any of these reactions, while lower acid loadings reduced the NMR yields for 2a even if no starting alcohol was recovered. On the other hand, the stereoselectivity remained very high in all cases (Table 2, entries 1 and 6–8).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Cat. (mol%) | 2a (%) |
E:Z
|
3a (%) | 4a (%) | 5a (%) |
| a Reaction conditions: 1a (1 mmol), aq. (OH)P(O)H2 (X mol%) in technical solvent (1 mL), 90 °C, 18 h. b 1H NMR yields and ratios were calculated using dibromomethane as internal standard and are the average of at least two independent experiments. c Isolated yield. | |||||||
| 1 | Toluene | 10 | 90 (95)c | E only | <5 | <5 | 5 |
| 2 | Cyclohexane | 10 | 68 | 97:3 |
— | <5 | — |
| 3 | 1,2-Dioxane | 10 | 67 | 86:14 |
— | <5 | 5 |
| 4 | 2-Me-THF | 10 | 52 | 80:20 |
— | <5 | 5 |
| 5 | Acetonitrile | 10 | 47 | 88:12 |
— | <5 | <5 |
| 6 | Toluene | 5 | 71 | 97:3 |
<5 | <5 | <5 |
| 7 | Toluene | 2.5 | 68 | 97:3 |
<5 | <5 | <5 |
| 8 | Toluene | 1 | 65 | 96:4 |
<5 | <5 | <5 |
The 31P NMR of the commercial aqueous solution used as catalyst in CDCl3 displays two triplets, a sharp one at 12.1 ppm and a broad one at 12.5 ppm with respect to H3PO4 as external reference (1JPH = 568 Hz). This supports tetrahedral hypophosphorous acid (or phosphinic acid) as the major tautomer with respect to less stable trigonal pyramidal phosphonous acid as well as a significant dimerisation in solution through hydrogen bonding (Scheme 2).15 Similar spectra were obtained in toluene-d8.16 When enone 2a was prepared under the optimised conditions no hypophosphorous acid was detected at the end of the reaction, but phosphorous acid (ca. 12%)17 was identified in the reaction crude. This indicates that while phosphorous acid can catalyse this rearrangement, hypohosphorous acid eventually disproportionates into phosphorous acid at the reaction temperature,18 which would then increase the percentage of decomposition of the reaction mixture (see Table 1, entry 2).15 Nevertheless, the bulk of the signals in 31P NMR appeared in the 25–70 ppm region, which is characteristic of R3PO derivatives. Either (OH)P(O)H2 and/or (OH)2P(O)H might act as nucleophiles producing either propargylic or allenic H-phosphinic acids,19,20 however, the instability of the formed species prevented further characterisation. Comparable results were obtained when the model reaction was carried out in the presence of diethyl phosphite, the other efficient catalyst identified in the original screening (see Table 1, entry 3).
 | ||
| Scheme 2 Phosphorous species in Meyer–Schuster rearrangement reactions. | ||
With an optimised system in hand, we next explored the scope of the reaction (Table 3). A range of enones 2 was prepared in moderate to excellent yields as well as α,β-unsaturated esters and aldehydes (Table 3, entries 4–6). The reaction conditions had to be slightly modified for several substrates to minimise undesired decomposition, or formation of by-products 3. In several instances diethyl phosphite was used as alternative catalyst in an attempt to improve the overall yield in the desired α,β-unsaturated carbonyl compounds. While similar results were obtained in most cases, no overall improvement was achieved. Only a trimethylsilyl substituent at the acetylenic position precluded the Meyer–Schuster rearrangement from taking place with ether 3f as the only identifiable reaction product (Table 3, entry 6). Gratifyingly, many of the desired rearrangement products were isolated analytically pure after a simple aqueous work-up, and purification by column chromatography was only required for problematic substrates such as 2g, 2j or 2l. Unsurprisingly, no reaction was observed with alkyl (propyl or cyclohexyl) propargylic alcohols.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Rearrangement | Product | T (°C) | 2 (%) |
E:Zb |
3 (%) |
| a 1H NMR yields and ratios were calculated using dibromomethane as internal standard and are the average of at least two independent experiments. Isolated yields are provided in brackets. b E/Z ratios were calculated from the crude 1H NMR spectra. c aq. (OH)P(O)H2 (5 mol%). | ||||||
| 1 |
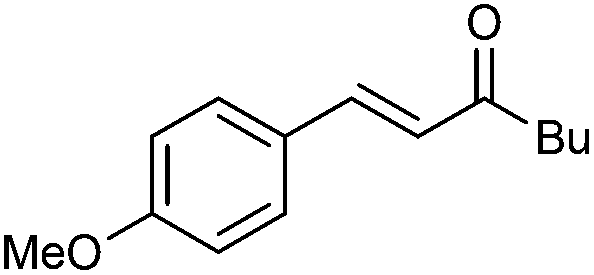
|
2a | 90 | 90 (95) | E only | <5 |
| 2 |

|
2b | 90 | 91 (81) | E only | — |
| 3 |
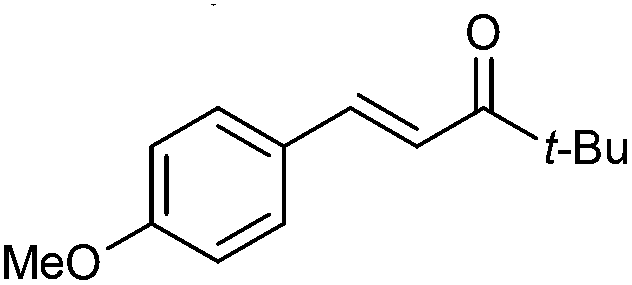
|
2c | 90 | >95 (99) | E only | — |
| 4 |
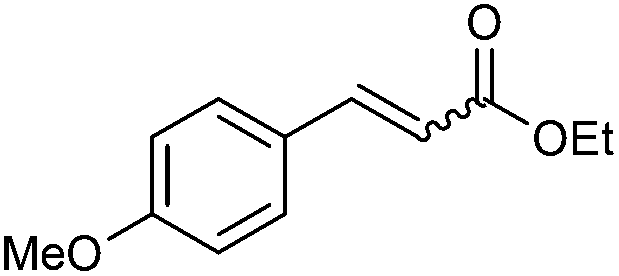
|
2d | 90 | 91 (91) | 1:1 |
— |
| 5 |

|
2e | 90 | 55 (51) | 2:1 |
— |
| 6 |

|
2f | 90 | — | — | 50 |
| 110 | — | — | 8 | |||
| 7 |

|
2g | 90 | 41 (34) | E only | — |
| 8 |
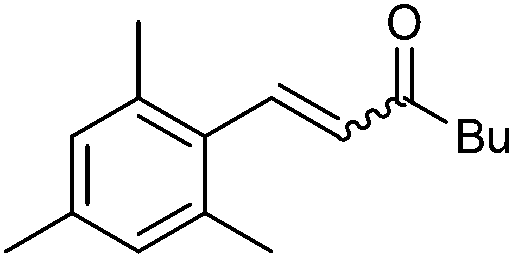
|
2h | 90 | >95 (93) | 53:47 |
— |
| 9 |
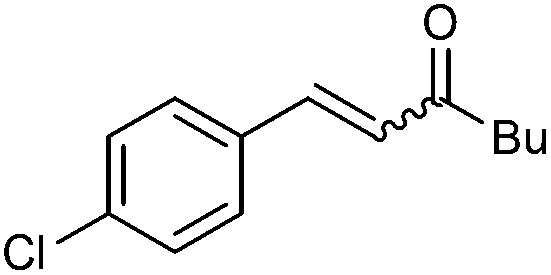
|
2i | 90 | 59 | 88:12 |
27 |
| 110 | 68 (58) | E only | — | |||
| 10 |
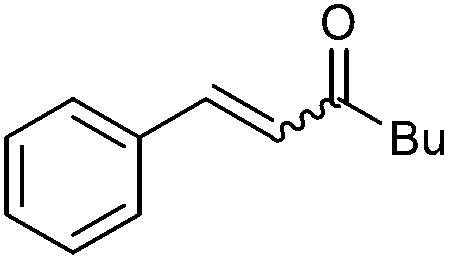
|
2j | 90 | 44 | 76:24 |
32 |
| 110c | 64 (69) | E only | — | |||
| 11 |

|
2k | 90 | 90 (85) | — | — |
| 12 |

|
2l | 90 | 11 | — | — |

While bis-aryl propargylic alcohol 1k formed the corresponding enone in good yields, the reaction of a tertiary alcohol bearing a methyl group at the Cα position suffered from severe decomposition and the desired enone 2l was only observed in low conversion together with traces of enyne 6l (Table 3, entries 11, 12 and Scheme 3A). Lowering the reaction temperature to 50 °C improved the formation of 6l so it could be isolated and fully characterised. This enyne is a known intermediate in Rupe rearrangements but the hydration of 6l was never observed under the tested conditions, even after extended reaction times.
 | ||
| Scheme 3 Reactions of tertiary alcohol 1l. 1H NMR yields were calculated using dibromomethane as internal standard and are the average of at least two independent experiments. Isolated yields are provided in brackets. aReaction carried out in the presence of 1.5 equiv. of 4-cyanoaniline. | ||
We were surprised by these results since we had previously isolated enone 2l in fair yield during our studies on nucleophilic substitution reactions with diethylphosphite as catalyst, instead of the expected propargylic amine (Scheme 3B).13 However, only traces of 2l, if any, were observed in the absence of 4-cyanoaniline, which indicates that the basicity of this nucleophile is key to tame the reactivity of phosphorous acid, the most acidic phosphorous by-product observed either from diethylphosphite or hypophosphorous acid under our conditions. Indeed, the use of a suitable base might be key to avoid decomposition, particularly for substrates susceptible of undergoing competitive Rupe rearrangement.
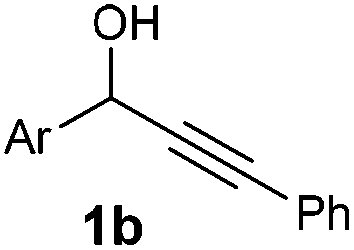
Finally, we looked into the role of ethers 3 in these rearrangement reactions since the formation of this by-product was systematically favoured by lower reaction temperatures with our catalytic system. Indeed, ether 3b was the only observed product from the reaction of alcohol 1b at temperatures up to 50 °C, whereas a mixture of 2b and 3b was formed at 60 °C (Table 4, entries 1–3). Interestingly, similar results were obtained at 60 °C when ether 3b was used as starting material, while no reaction was observed in the absence of acid. As expected, lower stereoselectivities were obtained at lower reaction temperatures (see Table 3, entry 2). These results confirm that ethers 3 can act as intermediates in the formation of α,β-unsaturated carbonyl compounds,7 probably through the protonation of ether 3b followed by the thermal cleavage into the corresponding propargylic alcohol 1b and carbocation derivative that might then engage in a Meyer–Schuster rearrangement.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Starting material | Conditions | 1b (%) | 2b (%) |
E:Z
|
3b (%) |
| a Reaction conditions: 1b (1 mmol), aq. (OH)P(O)H2 (X mol%) in technical toluene (1 mL), T (°C), 24 h. b 1H NMR yields/recoveries and ratios were calculated using dibromomethane as internal standard and are the average of at least two independent experiments. Isolated yield is provided in brackets. | ||||||
| 1 |
|
Cat. (10 mol%), RT | 22 | — | — | 78 |
| 2 | Cat. (10 mol%), 40 °C | <5 | — | — | >95 | |
| 3 | Cat. (10 mol%), 60 °C | <5 | 26 | 65:35 |
69 (64) | |
| 4 |

|
Cat. (10 mol%), 60 °C | — | 31 | 65:35 |
51 |
| 5 | 60 °C | — | — | — | >95 | |

Conclusions
The preparation of α,β-unsaturated carbonyl compounds from propargylic alcohols assisted by commercially available aqueous hypophosphorous acid does not require costly metals and delivers the desired products in technical solvent without the requirement of inert atmosphere, or (in many cases) further purification after a work-up. While Brønsted acid catalysts are intrinsically limited to substrates able to form sufficiently stable carbocations, different substitution patterns on the propargylic aromatic substituent and at the acetylenic position allowed the formation of unsaturated ketones, esters and aldehydes.These reactions are deceptively simple. While they are distinctly easy to carry out, the effects of the reaction temperature and speciation of the phosphorous catalyst are not that straightforward. Higher temperatures promote the conversion of ethers 3 into the desired unsaturated products 2 and improve the overall stereoselectivity of the reaction, nevertheless, they also lead to significant decomposition of the reaction mixture. Prolonged periods of heating in the presence of moisture and oxygen, while user-friendly, inevitably leads to the formation of different phosphorous species throughout the reaction, which might be effective catalysts as well and/or promote decomposition. A better understanding of the different roles could lead to an improved control and reactivity in this and other transformations mediated by Brønsted acids.
Experimental section
General considerations
All reactions were carried out in air using technical solvents without any particular precautions to exclude moisture or oxygen, unless stated otherwise. Commercially available reagents were used as received without further purification. Column chromatography and TLC were performed on silica gel (Kieselgel 60), using UV light and a phosphomolybdic acid dip to visualize the products.General procedure for the Meyer–Schuster rearrangement reactions
The chosen propargylic alcohol (1.0 mmol) was added to a solution of aq. (OH)P(O)H2 (50 wt% aq. solution, 5–10 mol%) in technical toluene (1.0 mL). The reaction mixture was stirred at 90–110 °C on a heating block for 18 h, before being cooled to room temperature. The reaction mixture was quenched with a saturated aqueous solution of NaHCO3 and then extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. If necessary, the obtained residue was then purified by column chromatography (reaction crude was dry-loaded onto stationary phase).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The Royal Thai Government is acknowledge for a PhD studentship to L.R.References
- S. Swaminathan and K. V. Narayanan, Chem. Rev., 1971, 71, 429–438 CrossRef CAS.
- Z. Wang, Rupe Rearrengement in Comprehensive Organic Name Reactions and Reagents, 2010, DOI:10.1002/9780470638859.conrr553.
- D. A. Engel and G. B. Dudley, Org. Biomol. Chem., 2009, 7, 4149–4158 RSC.
- V. Cadernio, P. Crochet, S. E. García-Garrido and J. Gimeno, Dalton Trans., 2010, 39, 4015–4031 RSC.
- For a comprehensive review on recent Bonsted-acid mediated Meyer-Schuster rearrangements, see: F. Justaud, A. Hachem and R. Grée, Eur. J. Org. Chem., 2021, 514–542 CrossRef CAS.
- For a review on related intercepted Meyer–Schuster rearrangements, see: D. Roy, P. Tharra and B. Baire, Asian J. Org. Chem., 2018, 7, 1015–1032 CrossRef CAS.
- M. Egi, M. Umemura, T. Kawai and S. Akai, Angew. Chem., Int. Ed., 2011, 50, 12197–12200 CrossRef CAS PubMed.
- H. Zheng, M. Lejkowski and D. G. Hall, Chem. Sci., 2011, 2, 1305–1310 RSC.
- For the diastereoselective formation of β-enaminoes, see: Y.-W. Kang, Y. J. Cho, S. J. Han and H.-Y. Jang, Org. Lett., 2016, 18, 272–275 CrossRef CAS PubMed.
- J. Park, J. Yun, J. Kim, D.-J. Jang, C. H. Park and K. Lee, Synth. Commun., 2014, 44, 1924–1929 CrossRef CAS.
- X. Gan, Z. Fu, L. Liu, Y. Yan, C. Chen, Y. Zhou and J. Dong, Tetrahedron Lett., 2019, 60, 150906 CrossRef CAS.
- E. Barreiro, A. Sanz-Vidal, E. Tan, S.-H. Lau, T. D. Sheppard and S. Díez-González, Eur. J. Org. Chem., 2015, 7544–7549 CrossRef CAS.
- L. Radtanajiravong and S. Díez-González, ACS Omega, 2019, 4, 12300–12307 CrossRef CAS PubMed.
- Around £15 per mol for hypophosphorous acid versus £30 per mol for diethyl phosphite at Sigma-Aldrich as of July 2022.
- N. S. Golubev, R. E. Asfin, S. N. Smirnov and P. M. Tolsoi, Russ. J. Gen. Chem., 2006, 76, 915–924 CrossRef CAS.
- See ESI† for further details.
- H. C. Fisher, L. Prost and J.-L. Montchamp, Eur. J. Org. Chem., 2013, 7973–7978 CrossRef CAS.
- G. T. Shechkov, I. A. Pevneva and O. A. Meshkova, Russ. J. Appl. Chem., 2003, 76, 1354–1355 CrossRef CAS.
- Y. Belabassi, A. F. Gushwa, A. F. Richards and J. L. Montchamp, Phosphorus, Sulfur, Silicon Relat. Elem., 2008, 183, 2214–2228 CrossRef CAS.
- G. Hu, C. Shan, W. Chen, P. Xu, Y. Gao and Y. Zhao, Org. Lett., 2016, 18, 6066–6069 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01259f. For FAIR data for NMR spectra, see Imperial College Research Services Data Repository, 2022, DOI: 10.14469/hpc/11111. |
| This journal is © The Royal Society of Chemistry 2022 |