
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the self-assembly and anti-glioblastoma efficacy of a cinnamoyl-capped dipeptide hydrogelator†
E. D.
Sitsanidis
 ab,
P. M.
Kasapidou
ac,
J. R.
Hiscock
d,
V.
Gubala
a,
H.
Castel
e,
P. I. A.
Popoola
d,
A. J.
Hall
a and
A. A.
Edwards
*a
ab,
P. M.
Kasapidou
ac,
J. R.
Hiscock
d,
V.
Gubala
a,
H.
Castel
e,
P. I. A.
Popoola
d,
A. J.
Hall
a and
A. A.
Edwards
*a
aMedway School of Pharmacy, Universities of Kent and Greenwich at Medway, Central Avenue, Chatham Maritime, Kent ME4 4TB, UK. E-mail: a.a.edwards@kent.ac.uk
bDepartment of Chemistry, Nanoscience Centre, University of Jyväskylä, P.O. Box 35, FI-40014, Finland
cMelville Laboratory for Polymer Synthesis, Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
dSupramolecular, Interfacial and Synthetic Chemistry Group, School of Physical Sciences, University of Kent, Canterbury, Kent CT2 7NZ, UK
eNormandie Univ, UNIROUEN, INSERM U1245, CBG, 76000 Rouen, France
First published on 12th September 2022
Abstract
Herein, we introduce the first diphenylalanine dipeptide hydrogelator capped with the cinnamoyl functional group (Cin-L-F-L-F). We evaluate the effects of the cinnamoyl moiety on molecular self-assembly events and resultant physical properties of the hydrogel formed. In addition, we report our preliminary results of this dipeptide's cytotoxicity against glioblastoma (GBM) cancer cells.
Introduction
Glioblastoma multiforme (GBM), a grade IV astrocytoma, is a malignant brain tumour and one of the most aggressive cancers in adults. It represents ≈80% of all primary tumours of the central nervous system (CNS) with a life expectancy of approximately 15 months from diagnosis.1 The spread of single GBM cells into the brain's parenchyma is due to the intracellular interactions of these cells with the microenvironment of the tumour.2 This explains why extracranial metastasis is rare, as GBM cells do not invade into the walls of blood vessels. The proliferation and infiltration of GBM cells into the surrounding brain tissue is triggered by signalling pathways and the expression of proteins, such as cytokines and growth factors.3,4 Such processes are stimulated by the presence of the primary components of the brain's extracellular matrix (ECM) such as collagen5 and hyaluronic acid (HA).6 In addition, there is also a link between the mechanosensing and proliferation pathways of the GBM cells as a response to the changes in the stiffness of the brain's ECM.7,8The present treatment protocol includes surgery followed by chemotherapy and radiation therapy.9 However, the success of surgical resection depends on the location, size and shape of the tumour. Furthermore, local tumour control by radiation therapy can cause necrosis and permanent neuronal damage while chemotherapy induces side effects and is limited to brain penetrant agents that can cross the blood brain barrier.10 The limited nature of GBM treatment options has led to new therapeutic approaches such as immunotherapy, antiangiogenic treatment, targeted therapies, and combination regimens.11,12 However, there is still no evidence that the use of these treatments results in an improved patient outcome, especially for recurrent GBM.11 In addition, novel anti-GBM targeted materials have been developed for use post-surgical resection such as nano-drugs13 and injectable gels.14 However, this technology has not yet successfully translated into the clinic.
Specific limitations associated with the use of injectable gels after surgical resection include the inability of the gel to adopt the shape of the resultant cavity (gel-tissue apposition), resultant changes in intracranial pressure, and issues relating to the controlled release of any drug loaded into the gel matrix.15 To help address such issues, we present herein N-protected cinnamoyl diphenylalanine dipeptide (Cin-L-F-L-F 1, Fig. 1), a novel low molecular weight hydrogelator for the preparation of soft materials for use as a GBM therapeutic hydrogel.
 | ||
| Fig. 1 Chemical structures of the phenylalanine based hydrogelators Cin-L-F-L-F 1, Fmoc-L-F-L-F 2, and Cin-L-F 3. | ||
The dipeptide diphenylalanine (L-F-L-F) and its derivatives are well-known hydrogelators.16,17 Especially, their aromatic amphiphilic analogues, which incorporate an aromatic moiety at the N-terminus, have been extensively used for the bottom up fabrication of soft materials.18–20 Amongst these low molecular weight hydrogelators, the dipeptide N-fluorenylmethoxycarbonyl diphenylalanine (Fmoc-L-F-L-F 2, Fig. 1) is the most well-studied.21–23 The mechanical properties of these hydrogels have been previously described and can be modulated.24–26 In addition, their biological activity to support or “kill” other cells has also been extensively discussed.16
To date, the only structurally similar gelator to the cinnamoyl protected dipeptide 1 is the cinnamoyl phenylalanine amino acid (Cin-L-F 3, Fig. 1) as reported by Xu et al.27 Here, the authors suggest that the cinnamoyl group is the minimum structural motif able to induce sufficient aromatic-aromatic interactions and support the gelation of a single phenylalanine amino acid. However, we now introduce the Cin-L-F-L-F dipeptide hydrogelator 1 and compare its properties to the extended aromatic gelator, Fmoc-L-F-L-F 2. In addition, we report our preliminary results on the cytotoxic effects of dipeptides 1 and 2 against GBM cancer cells for the first time.
Results and discussion
Dipeptide 1 was produced through a four step synthetic procedure, using standard amide bond formation and selective protection-deprotection reaction protocols (Fig. S1†).28 Dipeptide 1 was shown to self-assemble, via non-covalent intermolecular interactions, into helical fibres resulting in the formation of a self-supporting hydrogel. The gelation of dipeptide 1 in a phosphate buffer solution (PBS) was triggered thermally (95 °C) followed by sonication, resulting in the production of a self-supporting, semi-transparent hydrogel. However, heating or sonication alone was not sufficient to generate a gel, although the hydrogelator was completely solubilized. Instead, heating only resulted in a viscous, transparent free flowing solution that gelled only after sonication. Further tests showed that storage of this “pre-gel solution” (solution after heating only) in the fridge (4 °C) or at RT for 24 h did not result in gelation. Only after the sample was again sonicated, was a self-supporting hydrogel formed. The minimum gelation concentration (MGC) of 1 was found 2.0 mg mL−1 in PBS buffer (pH 7.4), while the gel was stable at 37 °C with a gel-to-sol phase transition temperature between 45–50 °C.The Fmoc-L-F-L-F 2 dipeptide yielded a self-supporting, semi-transparent hydrogel in PBS solution (pH 7.4) at a MGC of 2.0 mg mL−1, similar to its counterpart 1. Here gelation was induced by sonication only, with heating processes resulting in amorphous precipitation events. In contrast to the Cin-L-F-L-F 1 hydrogel, the gel-to-sol phase transition temperature of the Fmoc-L-F-L-F 2 hydrogel was higher (75–80 °C). This suggests that the replacement of the Fmoc-group by that of cinnamoyl-forms weaker hydrogels since less energy is now required to cleave the self-assembled molecules of 1, presumably due to reduced π-π stacking interactions.
To confirm the self-assembly of Cin-L-F-L-F 1, we compared the circular dichroic (CD) profile of the self-supporting gel to that of the methanolic solution of 1 (Fig. 2A and Fig. S16†). The observed CD signals of the gel sample originate from the assembly event, since in the solution phase (solution of Cin-L-F-L-F 1 in methanol) the spectrum lacks any CD signal. Interestingly, the “pre-gel” solution also gave CD signals compared to the methanolic solution indicating that self-assembly had also occurred. In both samples, the observed signals are due to the aromatic moieties of the dipeptide unit which, as expected, appear at higher wavelengths compared to the amide of the cinnamoyl group.
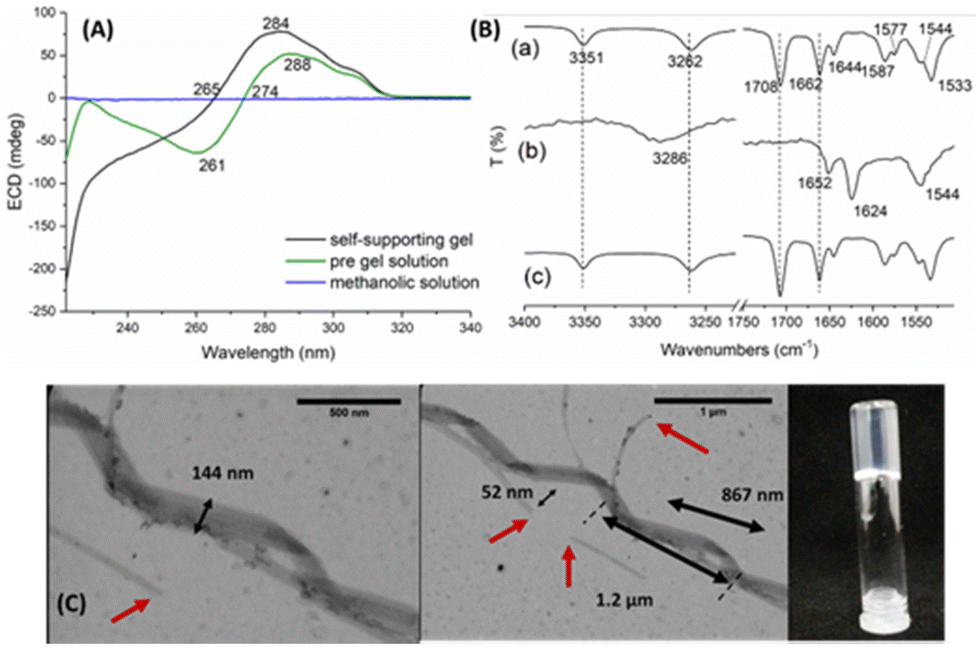 | ||
| Fig. 2 Self-assembly studies of Cin-L-F-L-F 1. (A) CD spectra of self-supporting gel, “pre-gel” solution and methanol-solvent control. A cylindrical cuvette with a path length of 0.1 mm was used. The hydrogel and “pre gel” solution formed in situ at a concentration of 2.0 mg mL−1. The methanolic solution was prepared at a concentration of 0.2 mg mL−1. All spectra were recorded at 25 °C; (B) FT-IR spectra of dipeptide 1 at the solid state (a), gel state (b) and the corresponding xerogel (c); (C) TEM imaging of the gel's fibres and vial inversion test. | ||
The “pre-gel” solution does not equal the gel spectrum, i.e., it has some assembly, but the assembly (type and/or extent of assembly) differs to that of the gel. The observed blue shift of the CD spectral features of the gel (relative to the “pre-gel” solution) presumably originates from a different orientation of the aromatic moieties or due to a combination of more than one self-assembled state. In addition, the shift could be explained by a more significant exciton coupling, resulting from the closer proximity of the aromatic moieties during self-assembly events. A tighter molecular packing leads the electronic and magnetic transition moments of the monomeric gelator units to higher energetic states and, therefore, decreased wavelength values (blue shift).29,30
Additionally, the minimum at ≈220 nm suggests that the secondary structure of the Cin-L-F-L-F 1 hydrogel may be consistent with a β-sheet arrangement akin to that observed in longer peptides. This is consistent with previously reported CD data for structurally similar systems such as dipeptide 221,31 and indole diphenylalanine.32 Indeed, their CD profiles appear qualitatively similar to those shown in Fig. 2A, with reported minimums at 218 and 220 nm, respectively.
The CD spectrum of the Fmoc-L-F-L-F 2 hydrogel that we have recorded (Fig. 3A) does not differ either to those previously published.21,31 Similarly, to the dipeptide 1 gel sample, the observed CD signals of the Fmoc-L-F-L-F 2 originate from the assembly event since in the solution phase (solution of Fmoc-L-F-L-F 2 in methanol) the spectrum lacks any CD signal. In addition, the peaks at 308 and 298 nm are due to the Fmoc- moiety, which as expected, are absent at the dipeptide 1 gel sample (Fig. 2A–3A).
 | ||
| Fig. 3 Self-assembly studies of Fmoc-L-F-L-F 2. (A) CD spectra of self-supporting gel and methanol-solvent control. A cylindrical cuvette with a path length of 0.1 mm was used. The hydrogel formed in situ at a concentration of 2.0 mg mL−1. The methanolic solution was prepared at a concentration of 0.2 mg mL−1. All spectra were recorded at 25 °C; (B) FT-IR spectra of 1 and 2 in the gel state, featuring the Amide I and II regions; (C and D) TEM imaging of the Fmoc-L-F-L-F 2 gel network. | ||
The FT-IR spectrum of Cin-L-F-L-F 1 in the gel state (Fig. 2B-b) showed a broad band at 3286 cm−1, attributed to the NH stretching vibration. This signal is significantly red-shifted (3262 cm−1) compared to the spectra obtained from the amorphous monomeric solid of 1 (Fig. 2B-a) and its corresponding xerogel (lyophilized gel – Fig. 2B-c). It should be noted that the IR spectrum of the corresponding xerogel is identical to that of the solid-state, which indicates the collapse of the supramolecular network during the freeze-drying process.33 The absence of the bands at 3351 cm−1 (–OH of the acid) and 1708 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), in the gel phase, must be related to the self-assembly. Furthermore, as observed for the gel sample, the presence of the amide II band at 1544 cm−1 and amide I bands at 1652 cm−1 and 1624 cm−1, corresponding to a combination of the NH in-plane bending, CO stretching, and CN stretching, suggests the presence of amide-amide H-bonding.
O), in the gel phase, must be related to the self-assembly. Furthermore, as observed for the gel sample, the presence of the amide II band at 1544 cm−1 and amide I bands at 1652 cm−1 and 1624 cm−1, corresponding to a combination of the NH in-plane bending, CO stretching, and CN stretching, suggests the presence of amide-amide H-bonding.
Given the peptidic nature of 1, the orientation of the amide bonds is consistent with the formation of α-helices (1652 cm−1) while the stronger peak at 1624 cm−1 refers to a β-sheet arrangement. α-Helices absorb near 1655 cm−1 with an observed downshift when are solvent-exposed (1640–1650 cm−1 in H2O and 1629–1640 cm−1 in D2O). β-Sheets usually give rise to a band around 1623–1641 cm−1 in H2O, which can be slightly red-shifted in D2O.34,35
The FT-IR spectrum of the Fmoc-L-F-L-F 2 hydrogel (Fig. 3B) showed three peaks at 1692, 1634 and 1535 cm−1, corresponding to the C-terminus CO stretch and the two amide NCO stretches respectively (amide I and II bands). These are consisted with previously reported data, indicating an antiparallel arrangement of β-sheets.21,32 In general, hydrogelators bearing the diphenylalanine (L-F-L-F) motif produce qualitatively similar FT-IR spectra. This suggests that it is not the type but rather the relative spatial orientation of the aromatic groups which is responsible for the molecular alignment of the hydrogelators (as observed by their FT-IR profile).
Transition electron microscopy (TEM) images of the Cin-L-F-L-F 1 hydrogel sample confirmed the presence of α-helical-like supramolecular assemblies (Fig. 2C). These consist of elongated flat ribbons, approximately ≈50 nm wide, which connect to each other forming wider fibres (width of 144 nm) that fold to form helical structures. The length of the major turn of the helix (two-folds) is approximately 1.2 μm, while the minor turn (one-fold) measures approximately 870 nm. Shear force tears of the fibres are also evident at various points (indicated by the red arrows), presumably as a result of disruption of the gel's matrix by dilution and/or vortexing during sample preparation. These observations are consistent with the IR interpretation. The Fmoc-L-F-L-F 2 gel sample is composed of an overlapping mesh of flat ribbons yielding a thick supramolecular network (Fig. 3C and D). The diameter of the ribbons ranges between 40–140 nm while shear force tears are also observed at various points (red arrows).
The viscoelastic properties of the Cin-L-F-L-F 1 and Fmoc-L-F-L-F 2 hydrogels were assessed by oscillatory rheology (Fig. 4). Frequency sweep measurements were performed on self-supporting gels, within the linear viscoelastic region (LVR), in which the storage (G′) and loss moduli (G′′) are independent of the strain amplitude. G′ has a higher value compared to G′′, confirming the viscoelastic nature of both materials. Cin-L-F-L-F 1 yields a hydrogel with a G′ value of approximately ≈226 Pa which is in the range of the stiffness of the brain tissue (0.1–1 kPa) and therefore could prevent potential increase of intracranial pressure upon application.7
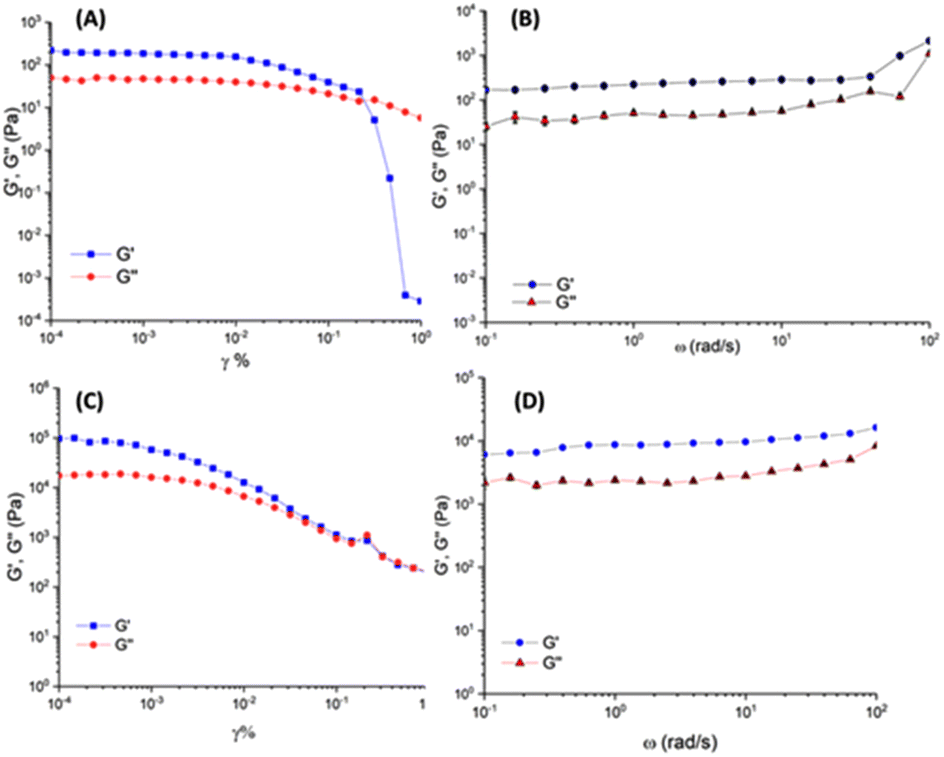 | ||
| Fig. 4 Oscillatory rheology analysis. (A) Amplitude and (B) frequency sweep measurements of the Cin-L-F-L-F 1 hydrogel (1.0 Hz frequency for amplitude sweep and 0.05% shear strain for frequency sweep). (C) Amplitude and (D) frequency sweep measurements of the Fmoc-L-F-L-F 2 hydrogel (1.0 Hz frequency for amplitude sweep and 0.03% shear strain for frequency sweep). Frequency sweep measurements were performed in triplicate. Scale bars represent standard deviation. The concentration of both gels is 2.0 mg mL−1. | ||
The Fmoc-L-F-L-F 2 hydrogel is stiffer compared to its Cin-L-F-L-F 1 counterpart with a G′ value of approximately ≈9500 Pa, almost 42-fold higher. It is of note that several parameters can modulate the stiffness of gel systems. As reported by Adams et al. the mechanosensitivity of Fmoc-L-F-L-F 2 hydrogels depend on the pH, type of buffer and the gelation protocol.26 Further to this, the indole protected diphenylalanine-based gels, as reported by Thordarson et al., show G′ values within the range of 105 to 3 × 105 Pa.32 This suggests that the type of aromatic groups incorporated to the diphenylalanine dipeptide have a significant effect on the stiffness of the hydrogels formed. However, to compare similar gel systems, the gelation conditions should be considered. Herein, the gels of 1 and 2 form under different conditions, which potentially affect the mechanic properties of the materials. Finally, although different in stiffness, both materials of 1 and 2 appear equal in terms of elasticity since no significant differences are observed at the G′–G′′ cross points of their corresponding amplitude sweep profiles (Fig. 4A/C).
As a preliminary step towards evaluation of Cin-L-F-L-F 1 for the potential treatment of GBM we assessed the cytotoxic effect of the dipeptide against model cell line U87MG, a human malignant GBM cell line. Both the gel and solution phases of 1 were evaluated. The solutions were prepared at concentrations below that of the “pre gel” solution (Table 1). In addition, we examined the cytotoxicity of both dipeptides 1 and 2 at the gel phase and we compared our data to previously reported studies.
GBM cells were seeded on the surface of both the Cin-L-F-L-F 1 and Fmoc-L-F-L-F 2 hydrogels towards 2D cell cultures. The gel specimens were prepared according to the given gelation protocols at the MGC (2.0 mg mL−1). For all samples, (gels of 1 and 2 and solutions of 1), the cell viability was assessed after 24 and 48 h of seeding by live-dead staining and MTS colorimetric assays (Fig. 5 and 6). Optical microscopy images of the cells seeded on the gels’ surface were also captured, after 24 and 48 h of incubation, to identify potential changes in the cell morphology in comparison to untreated cells and also confirm cell death.
 | ||
| Fig. 5 Cytotoxicity studies of the Cin-L-F-L-F 1 dipeptide against GBM cells at 24 and 48 h of incubation. (A) Live/dead staining images of GBM cells treated with solutions A–C; (B) Live/dead staining images of GBM cells seeded on the surface of the hydrogel; (C) MTS colorimetric assay of GBM cells treated with solutions A–C. Green and red staining indicate live and dead cells respectively. Scale bars represent 50 μm. Error bars denote the standard deviations (n = 8). Statistical analysis was performed with t-test, P < 0.0001, ****, ns: not significant. | ||
 | ||
| Fig. 6 Comparing the cytotoxicity of Cin-L-F-L-F 1 and Fmoc-L-F-L-F 2 hydrogels against GBM cells at 24 and 48 h of incubation. (A) Optical images of GBM cells seeded on the surface of the hydrogels and control; (B) MTS colorimetric assay of GBM cells of the corresponding cultures; (C) the breaking of Cin-L-F-L-F 1 hydrogel by addition of cell culture medium. Scale bars represent 50 μm. Error bars denote the standard deviations (n = 8). Statistical analysis was performed with t-test, P < 0.0001, ****. | ||
Cell viability assessment of the dipeptide 1 in the solution phase (MTS colorimetric assays) showed that solution A (0.1% w/v) was cytotoxic compared to the diluted solutions B (0.01% w/v) and C (0.001% w/v) (Fig. 5C). It was also observed that within the first 24 h, the viability of GBM cells, treated with solution A, dropped almost to 7%. The results were highly consistent with complementary results obtained from a live-dead staining assay of GBM cells treated with the corresponding solutions A–C (Fig. 5A). Interestingly, the hydrogel of 1 was also found to be cytotoxic against U87MG cells within 24 h (Fig. 5B and 6A). The viability of the seeded cells was found to be 18% at 24 h, however dropped to 5% over a 48 h period (Fig. 6B).
In addition, we confirmed changes in the cell morphology by comparing the optical microscopy images of the seeded cells on the surface of the hydrogels (of both materials of 1 and 2) with the untreated cells (positive control) (Fig. 6A). Here, the untreated cells (control) changed from an elongated “star” shape to be more spherical in shape (gels). The data revealed a typical behaviour of GBM cells when seeded on hydrogel surfaces confirming that the matrix composition, crosslinking density, biological, and biochemical properties of hydrogel systems have a profound effect on the transformation of GBM cells.36
Cin-L-F-L-F 1 was found to be cytotoxic at concentrations above 0.01% w/v (0.23 mM) in the solution phase, however the required MGC needed to prepare a self-supporting hydrogel was 2.0 mg mL−1 (4.5 mM), almost 20-fold higher. The Cin-L-F-L-F 1 hydrogel was cytotoxic as the cell viability was 18% at 24 h and then dropped to 5% at 48 h. The cytotoxicity of the Fmoc-L-F-L-F 2 hydrogel was similar, with a cell viability of 26% at 24 h and 2% at 48 h. Although the stiffness of the Cin-L-F-L-F 1 gel is within the range of that of the brain tissue, such an increase in effective gelator concentration was found to led to cell apoptosis, suggesting that the viability of GBM cells is gelator concentration dependant. However, based on previously reported studies, Fmoc-L-F-L-F 2 hydrogels were prepared at concentrations of 5.0 and 10 mg mL−1 for the 3D culture of astrocytes (two strains), MDCK and COS 7 cell lines.37 Additionally, dipeptide 2 displayed high biocompatibility towards HeLa cells at the range of concentrations 0.001–0.1% w/v, while at higher concentrations >0.1% w/v some cell toxicity was observed.32 The reported results are consistent with our findings for Cin-L-F-L-F 1 in the solution state.
To explain the cell toxicity of dipeptide 2, the authors proposed that the salts present in the cell culture media prevented the bundling that was observed in the fibres, resulting in smaller aggregates that most likely interact with the cell membrane and cause cell death.32 To assess the stability of the Cin-L-F-L-F 1 hydrogel and explain the resultant cell death, we added cell culture medium to the surface of a preformed gel and incubated the material at 37 °C/5% CO2 for 12 h. The gel was broken, and free gravitational flow was observed upon the vial inversion test (Fig. 6C). In addition, we repeated the stability test with brine, however the gel remained intact for one week. However, the Fmoc-L-F-L-F 2 hydrogel was degraded when exposed to brine. This suggests that the supramolecular network of the materials dictates their stability which varies for different gelator molecules, regardless of structural similarity. An increased ionic strength does not seem to be responsible for the degradation of the Cin-L-F-L-F 1 gel but rather the type of salts and other ingredients, present in the cell culture medium, and their interactions with the fibres.
Conclusions
In summary, the low molecular weight gelator Cin-L-F-L-F 1 produced a self-supporting hydrogel in PBS buffer at 2.0 mg mL−1. Preliminary results suggest that the material exhibits inherently cytotoxic effects against GBM tumour cells, while resembling the mechanical properties of brain tissue. Consequently, there would be no need for chemotherapeutic drugs to be encapsulated in the gel matrix if applied after resection. The present work constitutes a preliminary investigation of the cytotoxicity of dipeptide 1 on U87MG cells. We are aware that other cancer and normal cell lines need to be assessed to establish further its cytotoxicity, in addition to the haemolytic properties of the material, its stability, anticancer activity, and compatibility in animal models, which is the subject of subsequent studies.In comparison to the structurally similar gelator Fmoc-L-F-L-F 2, the hydrogel produced with Cin-L-F-L-F 1 exhibits preferential mechanical properties and similar cytotoxicity, therefore representing a step forward in the development of hydrogel systems for the treatment of GBM.
Experimental
Materials
All commercial reagents were used as supplied. tert-Butyloxycarbonyl phenylalanine and cinnamic acid were purchased from Sigma Aldrich, (S)-3-phenylalanine-t-butyl ester hydrochloride from Carbosynth and N-fluorenylmethoxycarbonyl diphenylalanine from Biogelx. Hydrogels were prepared in phosphate-buffered saline (PBS) solution (Fisher BioReagents, NaCl 137 mM, phosphate buffer 10 mM, KCl 2.7 mM, pH 7.4). Human glioblastoma cells, (U87MG, HBT-14, grade IV WHO classification, ATCC Manassas, VA, USA) were maintained in a fully supplemented (complete) medium (Dulbecco's Modified Eagle's Medium, DMEM) with 10% fetal bovine serum (FBS), 1% antibiotic/antimicotic and 1% sodium pyruvate. DMEM, antibiotic/antimicotic and FBS were purchased from Gibco by Life Technologies. Sodium pyruvate was purchased from Sigma Aldrich. MTS colorimetric assay (CellTiter 96 AQueous One Solution Proliferation Assay) was purchased from Promega and fluorescent live-dead assay from Invitrogen. All reactions were performed under inert nitrogen atmosphere unless stated otherwise. Reactions were monitored by TLC, which was performed on aluminum sheets, coated with 60 F254 silica (Merck). TLC sheets were checked for UV activity (254/325 nm) prior to staining and visualized using Hanessian's stain. Flash column chromatography was performed on silica gel (100–200 mesh, Merk).Characterization of synthetic products
High resolution mass spectra (m/z) were recorded using a Waters Synapt G2 TOF mass spectrometer (Waters, UK) with an electrospray ionization probe or a Bruker microTOF-Q mass spectrometer and spectra recorded and processed using Bruker's Compass Data Analysis software. All IR spectra were recorded in the range of 4000–650 cm−1 in attenuated total reflectance (ATR) mode, using a PerkinElmer Spectrum One FT-IR Spectrometer (given abbreviations for peak characterization: strong (s), weak (w), very weak (vw), shoulder (sh)). 1H and 13C nuclear magnetic resonance spectra (NMR) were recorded in the deuterated solvent as stated on a Jeol ECP 400 MHz FT NMR spectrometer, incorporating a tuneable H (5) 400 probe (1H: 400 MHz and 13C: 101 MHz) or on a Jeol ECA, 500 MHz FT NMR spectrometer, incorporating a NM-50TH5AT/FG2 probe, (1H: 500 MHz and 13C: 126 MHz). All chemical shifts (δ) are quoted in ppm and coupling constants (J) are averaged values (in Hz). Residual signals from the solvents or TMS signal were used as an internal reference. 1H resonances were assigned with the aid of 2D techniques such as COSY and HSQC. 13C resonances were assigned using a DEPT 135 sequence and HSQC.Hydrogel characterization and associated studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (gel:water) ratio. 5 μL of the diluted sample was then pipetted onto carbon films (400 mesh Cu, Agar Scientific) and left to dry prior imaging.
:5 (gel:water) ratio. 5 μL of the diluted sample was then pipetted onto carbon films (400 mesh Cu, Agar Scientific) and left to dry prior imaging.
Cytotoxicity assessment
Synthetic protocols (Fig. S1–S15†)
:EA 2:1, Hanessian's stain, Rf = 0.5). The solvent was then evaporated under vacuum and the residue was dissolved in DCM (10 mL) and washed with water (50 mL). The aqueous phase was back extracted twice with DCM (10 mL). The obtained organic phases were combined and washed sequentially with water (2 × 50 mL), aqueous HCl (1M, 10 mL), water (50 mL, pH of organic phase 5–6) and with a saturated aqueous solution of NaHCO3 (20 mL, pH of organic phase 6–7). The organic phase was finally dried with MgSO4, filtered and evaporated.
Off-white glassy solid, 247 mg, 70%; m.p. 134.7–135.7 °C; [α]20D +6.8° (1% w/v in DMSO); IR (vmax/cm−1): 3299 (br), 2978 (w), 2932 (vw), 1733 (sh), 1677 (sh), 1654 (s), 1523 (s), 1497 (m), 1456 (w), 1393 (sh), 1366 (s), 1252 (s), 1223 (s), 1152 (s), 1115 (sh), 1048 (w), 1024 (w), 847 (s), 740 (s), 698 (s); 1H NMR (500 MHz, D6-DMSO): δ 8.22 (d, J = 7.6 Hz, 1H), 7.32–7.15 (m, 10H), 6.86 (d, J = 8.8 Hz, 1H), 4.37 (q, J = 7.4 Hz, 1H), 4.19 (td, J = 10.6, 8.7, 4.0 Hz, 1H), 2.98 (dd, J = 7.3, 3.7 Hz, 2H), 2.92 (dd, J = 14.1, 4.0 Hz, 1H), 2.69 (dd, J = 13.8, 10.6 Hz, 1H), 1.31 (s, 9H), 1.28 (s, 9H); 13C NMR (126 MHz, D6-DMSO) δ 172.3, 171.0, 155.7, 138.7, 137.6, 129.8, 129.70, 128.71, 128.5, 127.1, 126.7, 81.2, 78.5, 56.1, 54.6, 38.0, 37.4, 28.7, 28.1; HRMS (ESI-TOF, m/z) calculated for C27H37N2O5 [M + H]+ 469.2702, found 469.270.
:EA, 3:1, Hanessian's stain) confirmed the formation of a new spot (Rf = 0.57). The reaction mixture was then neutralized (pH 6–7) using saturated aqueous NaHCO3 and extracted with EA (100 mL). The organic phase was dried with MgSO4 and evaporated to dryness. Product 5 was used without further purification or characterization other than NMR spectroscopy. The obtained spectra were in agreement with previously published data.38,39
Off-white gammy solid, 786 mg, stoichiometric yield- 100%; 1H NMR (500 MHz, D6-DMSO): δ 8.16 (d, J = 8.0 Hz, 1H), 7.29–7.12 (m, 11H), 4.44 (dt, J = 8.0, 6.9 Hz, 1H), 3.41 (dd, J = 8.4, 4.6 Hz, 1H), 2.94 (d, J = 6.9 Hz, 2H), 2.93–2.89 (m, 1H), 2.56 (dd, J = 13.5, 8.4 Hz, 1H), 1.33 (s, 9H); 13C NMR (126 MHz, D6-DMSO): δ 174.6, 170.9, 139.1, 137.5, 129.92, 129.81, 128.70, 128.63, 127.1, 126.7, 81.4, 56.3, 53.9, 41.3, 37.8, 28.1.
:EA 1:1, Hanessian's stain) confirmed the absence of dipeptide 5 (Rf = 0.10) and the appearance of a new spot (Rf = 0.66). The solvent was removed under vacuum and the obtained residue was dissolved in EA (10 mL). The solution was then extracted with water (3 × 20 mL), the organic phases were combined, dried with MgSO4 and evaporated to dryness. The obtained residue was purified by flash column chromatography (Hex:EA 3:1) to give a mixture of a white solid and a transparent oil (undissolved urea derivative). When the mixture was dissolved in a minimum amount of methanol, a white precipitate was formed which was isolated by filtration under vacuum.
Off white glassy solid, 559 mg, 80%; m.p. 90.8–92.7 °C; [α]20D −12.7° (1% w/v in DMSO); IR (vmax/cm−1): 3275 (br), 3061 (vw), 3030 (vw), 2976 (vw), 2926 (w), 1733 (s), 1649 (s), 1616 (s), 1541 (s), 1497 (sh), 1455 (w), 1367 (w), 1344 (w), 1221 (s), 1150 (s), 977(s), 847 (s), 740 (s), 698 (s) cm−1; 1H NMR (500 MHz, D6-DMSO): δ 8.48 (d, J = 7.4 Hz, 1H), 8.31 (d, J = 8.6 Hz, 1H), 7.55–7.51 (m, 2H), 7.43–7.36 (m, 3H), 7.36–7.31 (m, 1H), 7.30–7.14 (m, 10H), 6.67 (d, J = 15.9 Hz, 1H), 4.73 (td, J = 10.2, 8.6, 4.1 Hz, 1H), 4.39 (td, J = 7.8, 6.8 Hz, 1H), 3.07 (dd, J = 13.9, 4.1 Hz, 1H), 2.98 (h, J = 8.0, 7.6 Hz, 2H), 2.78 (dd, J = 13.9, 10.1 Hz, 1H), 1.32 (s, 10H); 13C NMR (126 MHz, D6-DMSO) δ 171.9, 170.9, 165.2, 139.5, 138.4, 137.6, 135.4, 130.0, 129.77, 129.71, 129.48, 128.73, 128.57, 128.06, 127.05, 126.82, 122.4, 81.2, 54.81, 54.17, 38.3, 37.4, 28.1; HRMS (ESI-TOF, m/z) calculated for C31H34N2O4 [M − H]− 497.2446, found 497.2408.
:EA 1:1, Hanessian's stain) confirmed the consumption of the starting material and the presence of a new spot (Rf = 0.24). The solvent was then evaporated, and the obtained residue was left to dry overnight under vacuum. The residue was washed three times with DCM (1.0 mL).
White solid in stoichiometric yield, 133 mg, 100%; m.p. 210.5–212.2 °C; [α]20D −14.8° (1% w/v in DMSO); IR (vmax/cm−1): 3350 (s), 3262 (s), 1707 (s), 1662 (s), 1646 (s), 1587 (s), 1533 (s), 1496 (w), 1350 (w), 1331 (w), 1307 (w), 1271 (s), 1224 (sh), 1212 (s), 1182 (s), 1116 (vw), 1054 (vw), 990 (s), 794 (w), 771 (w), 734 (s), 696 (s) cm−1; 1H NMR (500 MHz, D6-DMSO): δ 12.76 (s, 1H), 8.38 (d, J = 7.8 Hz, 1H), 8.27 (d, J = 8.6 Hz, 1H), 7.53 (d, J = 6.9 Hz, 2H), 7.39 (dt, J = 13.4, 6.9 Hz, 3H), 7.33 (d, J = 15.7 Hz, 1H), 7.29–7.14 (m, 10H), 6.67 (d, J = 15.8 Hz, 1H), 4.71 (td, J = 9.4, 4.1 Hz, 1H), 4.47 (td, J = 8.3, 5.1 Hz, 1H), 3.07 (ddd, J = 17.7, 14.0, 4.7 Hz, 2H), 2.94 (dd, J = 13.9, 8.7 Hz, 1H), 2.76 (dd, J = 14.0, 10.1 Hz, 1H); 13C NMR (126 MHz, D6-DMSO): δ 173.3, 171.8, 165.2, 139.5, 138.4, 137.9, 135.4, 130.03, 129.71, 129.49, 128.74, 128.57, 128.06, 126.98, 126.78, 122.4, 54.2, 54.0, 38.1, 37.2; HRMS (ESI-TOF, m/z) calculated for C27H27N2O4+ [M + H]+ 443.1971, found 443.1970.
Author contributions
EDS: Conceptualization, investigation, validation, writing original draft, review & editing. PMK, JRH: Investigation, validation, writing – review & editing. PIAP: Investigation, validation. VG, HC: Writing – review & editing. AJH, AAE: Project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. D. S. would like to thank the University of Kent for his PhD scholarship and the Jane and Aatos Erkko Foundation for financial support during the writing of the current manuscript. J. R. H. would like to thank the UKRI for funding (MR/T020415/1). The authors acknowledge access to/use of University of Greenwich facilities for TEM (A. P. Hurt) and CD (B. D. Alexander).References
- N. Grech, T. Dalli, S. Mizzi, L. Meilak, N. Calleja and A. Zrinzo, Cureus, 2020, 12(5), e8195 Search PubMed.
- W. Diao, X. Tong, C. Yang, F. Zhang, C. Bao, H. Chen, L. Liu, M. Li, F. Ye, Q. Fan, J. Wang and Z. C. Ou-Yang, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed.
- S. Oushy, J. E. Hellwinkel, M. Wang, G. J. Nguyen, D. Gunaydin, T. A. Harland, T. J. Anchordoquy and M. W. Graner, Philos. Trans. R. Soc., B, 2017, 373, 20160477 CrossRef PubMed.
- H. Ohgaki and P. Kleihues, Am. J. Pathol., 2007, 170, 1445–1453 CrossRef CAS PubMed.
- K. B. Pointer, P. A. Clark, A. B. Schroeder, M. Shahriar, K. W. Eliceiri and J. S. Kuo, J. Neurosurg., 2017, 126(6), 1812–1821 Search PubMed.
- B. Ananthanarayanan, Y. Kim and S. Kumar, Biomaterials, 2011, 32(31), 7913–7923 CrossRef CAS.
- C. Wang, X. Tong and F. Yang, Mol. Pharm., 2014, 11(7), 2115–2125 CrossRef CAS PubMed.
- I. E. Palamà, S. D'Amone and B. Cortese, Front. Bioeng. Biotechnol., 2018, 6, 131 CrossRef.
- M. Weller, M. van den Bent, M. Preusser, E. Le Rhun, J. C. Tonn, G. Minniti, M. Bendszus, C. Balana, O. Chinot, L. Dirven, P. French, M. E. Hegi, A. S. Jakola, M. Platten, P. Roth, R. Rudà, S. Short, M. Smits, M. J. B. Taphoorn, A. von Deimling, M. Westphal, R. Soffietti, G. Reifenberger and W. Wick, Nat. Rev. Clin. Oncol., 2021, 18(3), 170–186 CrossRef PubMed.
- Z. Banu, Int. Res. J. Pharm., 2019, 9(12), 7–12 CrossRef.
- C. Birzu, P. French, M. Caccese, G. Cerretti, A. Idbaih, V. Zagonel and G. Lombardi, Cancers, 2021, 13(1), 47 CrossRef CAS PubMed.
- M. Touat, A. Idbaih, M. Sanson and K. L. Ligon, Ann. Oncol., 2017, 28(7), 1457–1472 CrossRef CAS PubMed.
- E. Alphandéry, Cancers, 2020, 12(1), 242 CrossRef.
- G. Cirillo, U. G. Spizziri, M. Curcio, F. P. Nicoletta and F. Iemma, Pharmaceutics, 2019, 11(9), 486 CrossRef CAS PubMed.
- C. Bastiancich, P. Danhier, V. Préat and F. Danhier, J. Controlled Release, 2016, 243, 29–42 CrossRef CAS PubMed.
- S. Marchesan, A. V. Vargiu and K. E. Styan, Molecules, 2015, 20, 19775–19788 CrossRef CAS PubMed.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed.
- S. Awhida, E. R. Draper, T. O. McDonald and D. J. Adams, J. Colloid Interface Sci., 2015, 455, 24–31 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Z. Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927–935 RSC.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- J. Raeburn, G. Pont, L. Chen, Y. Cesbron, R. Lévy and D. J. Adams, Soft Matter, 2012, 8, 1168–1174 RSC.
- J. Shi, Y. Gao, Z. Yang and B. Xu, Beilstein J. Org. Chem., 2011, 7, 167–172 CrossRef CAS PubMed.
- L. S. Lin, T. L. Jr, S. E. De Laszlo, Q. Truong, T. Kamenecka and W. K. Hagmann, Tetrahedron Lett., 2000, 41, 7013–7016 CrossRef CAS.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43(15), 5211–5233 RSC.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30(13), 2523–2530 CrossRef CAS PubMed.
- A. D. Martin, A. B. Robinson, A. F. Mason, J. P. Wojciechowski and P. Thordarson, Chem. Commun., 2014, 50(98), 15541–15544 RSC.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18(11), 3531–3540 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta, 2007, 1767(9), 1073–1101 CrossRef CAS PubMed.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39(8), 549–559 CrossRef CAS PubMed.
- S. Pedron, E. Becka and B. A. C. Harley, Biomaterials, 2013, 34, 7408–7417 CrossRef CAS PubMed.
- T. Liebmann, S. Rydholm, V. Akpe and H. Brismar, BMC Biotechnol., 2007, 7, 88 CrossRef.
- J. T. Van Herpt, M. C. Stuart, W. R. Browne and B. L. Feringa, Eur. J. Chem., 2014, 20(11), 3077–3083 CrossRef CAS PubMed.
- F. Rodler, W. Sicking and C. Schmuck, Chem. Commun., 2011, 47, 7953–7955 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details and data relating to chemical and material characterisation. See DOI: https://doi.org/10.1039/d2ob01339h |
| This journal is © The Royal Society of Chemistry 2022 |