
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA two-site reactive platform in the synthesis of amino-functionalized amphiphilic molecules via sulfenic acids†
Paola
Bonaccorsi
 a,
Chiara Maria Antonietta
Gangemi
a,
Valentina
Greco
b,
Giuseppe
Gattuso
a and
Anna
Barattucci
*a
a,
Chiara Maria Antonietta
Gangemi
a,
Valentina
Greco
b,
Giuseppe
Gattuso
a and
Anna
Barattucci
*a
aDipartimento di Scienze Chimiche Biologiche Farmaceutiche ed Ambientali (CHIBIOFARAM), Università degli Studi di Messina, Sicily, Italy. E-mail: abarattucci@unime.it
bDipartimento di Scienze Chimiche, Università degli studi di Catania, Sicily, Italy
First published on 1st September 2022
Abstract
The synthesis of some bolaamphiphiles is described. It is a convergent approach that allows the linkage of a glucosyl derivative to a bis-functionalized platform, via a copper-free Sonogashira cross-coupling. The central core was obtained from the reaction of a suitably substituted bis-sulfoxide with diethynyl benzenes. The intermediates of such reaction are sulfenyl functions that are easily added to one triple bond of the unsaturated molecules. The functionalization at the central core, through the nucleophilic addition of ammonia or piperidine onto the two vinyl sulfonyl groups already present in the backbones of the molecules, opened the way to the preparation of more complex derivatives. The observation of the formation of new stereogenic carbons with an unexpected significantly high diastereoselectivity was justified and supported by preliminary theoretical calculations. The two ending glucosyl moieties were favourably deprotected to afford the amino-functionalized bolaamphiphilic molecules.
Introduction
Amphiphiles are molecules with at least two covalently bonded components with different affinities for the solvent, one showing a high affinity for polar solvents and another possessing strong affinity for nonpolar solvents. In particular, the term “bolaamphiphiles” (BAs) was first given to “bipolar lipid for ions and molecules which aggregate in water” by Fuhrhop and Fritsch in 19841 and 19862 papers, although already in 1951 Fuoss and Edelson3 spoke of a “bolaform electrolyte” (also “bolyte” or “bolion”) to indicate a chain of hydrophobic groups connecting two ionic functions. The prefix “bola” refers to the shape of a South American missile weapon: two balls attached to both ends of a cord. Today, the name “bolaamphiphiles” is used to indicate in general amphiphilic molecules containing a hydrophobic skeleton connected with two hydrophilic heads. The most important natural BAs are the membrane lipids of thermophilic and acidophilic archaebacteria, e.g., Sulfolobus solfataricus.4 Synthetic BAs have attracted the attention of researchers for their unique property of self-assembling both at interfaces, such as air/water or liquid/solid, and in solution, forming well defined structures, such as micelles, lamellae vesicles, nanotubes, nanorods, and nanosheets. For this reason, since the nineties, several synthetic methodologies have been developed to obtain BAs and their applications have been extensively studied, including drug delivery,5 gene delivery,6 medical imaging,7 and nanoelectronics.8 In recent years, supra-amphiphiles9 and bola-type supra-amphiphiles10 have been obtained also by the self-assembly of components bearing both hydrophilic and hydrophobic segments held together by noncovalent interactions.11 Most BAs are easily made by condensation and substitution reactions, using commercial diols or diamines or dicarboxylates as starting materials. Sulfurated molecules are required by all living organisms; therefore, they can be found in a broad range of natural products12 and pharmaceuticals.13 This is because of the various sulfur-derived functional groups that possess different properties, from the thiol function and the stable disulfide bond up to the differently oxidized sulfurated molecules. Sulfenic acids (R-SOH), for instance, are intermediates, in nature14 and in lab-chemistry,15 which cannot be usually isolated but can be transformed into other stable sulfurated functional groups such as sulfinic and sulfonic acids, sulfoxides, sulfones and disulfides.16 They have long been studied and their chemistry applied to the synthesis of several sulfur-containing molecules.17 In this paper we will discuss a nontrivial approach to the synthesis of BAs 1 (Scheme 2) and O-acetylated BAs 2 (Scheme 3) composed of a long lipophilic wire containing two vinyl sulfone moieties, and decorated at the extremities of the chain with glucose terminations (Fig. 1). The two sulfurated functions, introduced by the alkyne/sulfenic acid addition,18 can be involved in Michael-type additions, while the two carbohydrate-extremities may provide beneficial properties to the bolaamphiphilic molecules. Besides their chirality, non-toxicity and biocompatibility, sugars possess in their skeletons various hydroxyl groups leading to a good hydrophilic environment and multivalent sites that help provide structural diversity during self-assembly.19 Moreover, carbohydrates can specifically target glycoreceptors in the cellular plasma membrane and can act as ligands in the drug delivery scenario and as target molecules for molecular recognition, adding to the synthesized BAs a biomedical value. | ||
| Scheme 1 Synthesis of the “two-sites reactive platform” 7 and its functionalization with glucosyl moieties. | ||
 | ||
| Scheme 2 General procedure for amino-bolaamphiphiles 1. | ||
 | ||
| Scheme 3 General procedure for acetylated BAs 2. | ||
 | ||
| Fig. 1 Schematic representation of bolaamphiphilic molecules synthesized in this paper. | ||
Results and discussion
The synthetic procedure for obtaining BAs is described below. Commercially available 1,4-benzenedimethanthiol (3) was reacted with methyl acrylate in the presence of benzyltrimethylammonium hydroxide (TRITON B) as the base (Scheme 1). The obtained bis-thioether 4 underwent oxidation to bis-sulfoxide 5 with meta-chloroperbenzoic acid (mCPBA) at −78 °C to avoid the formation of the corresponding undesirable bis-sulfone. Thermolysis of compound 5 generated sulfenic acid functionalities that reacted with one of the two triple bonds of meta- and para-diethynylbenzenes, in a completely regiocontrolled process,20 leading to the formation of compounds 6a and 6b.21 The electronic nature of the substituents, located at the β position to the sulfinyl groups, is crucial for the generation of the sulfenyl moieties from bis-sulfoxide 5, which thermolyzes at 83 °C in 1,2-dichloroethane (DCE), has a good shelf-life and can be easily handled. Methyl acrylate was indeed recognized as the best partner for the Michael-type addition of bis-thiol 3 compared to the previously adopted diethyl isopropylidenemalonate22 that furnished unstable bis-sulfoxides and low final yield of the desired products. To favour the formation of open-chain compounds 6a and 6b, against the already reported cyclophanes (in this case isolated in very small percentages),23 a large excess of para- and meta-diethynylbenzenes was used (Scheme 1).Diastereomeric mixtures (due to the presence of two chiral sulfinyl groups) of 6a and 6b were isolated in a 70% total yield and subjected to 0 °C oxidation at the corresponding bis-vinyl sulfones 7a and 7b. With the two-site reactive central bricks in hand, the synthesis of the glucosyl arm 10 was performed using 1,4-diiodo-2,5-dimethoxybenzene249 and 1-(2-prop-1-ynyl)-2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside258 in a copper-free Sonogashira cross-coupling, with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.26
:1 ratio.26
Compounds 11a and 11b were obtained by reacting separately the central cores 7 with the glucosyl arm 10 by a Sonogashira cross-coupling, again, copper-free to avoid homo-coupling side products and decay of the sugar function (Scheme 1). Compounds 11a and 11b were purified and isolated in 49% and 46% yields, respectively.
Vinyl sulfones are known for their important role as key structural units of many biologically active compounds as well as versatile building blocks for various organic transformations.27 From a synthetic point of view, they can act as efficient Michael acceptors and 2p donors in cycloaddition reactions. The presence of two vinyl sulfonyl units in the hydrophobic core of compounds 11 allowed us to exploit further modification of their backbones, such as the introduction of new functional groups via nucleophilic addition. Ammonia was used for the functionalization, leading to the insertion of two amino groups, which caused the formation of two new stereogenic carbon atoms, in the central region of compounds 11 and the contemporary desired deprotection from acetyl functions of the two glucosyl end-moieties (Scheme 2). Noteworthily, the two amino-functionalized bolaamphiphilic compounds 1a and 1b were obtained, simply after several washing steps with MeOH, as almost unique compounds, respectively. Solubility tests were performed to verify the amphiphilic nature of compounds 1 (see page S21 in the ESI†). We could not observe any trace of diastereomeric signals in the 1H NMR spectra of each compound 1(see the ESI†).
The conformational mobility of platforms 7, due to the presence of two –CH2– moieties and three symmetrically disposed benzene rings, led us to assume a possible spatial arrangement of molecules 11 that could justify the unexpected stereochemical results. Thinking of a simpler model to study than molecules 11, we decided to explore the reactivity of platforms 7a and 7b by using an excess of piperidine (Scheme 3). Compounds 12a and 12b were obtained separately, in quantitative yields, as unique products of the Michael-type reaction (see the ESI†), requiring no further purification if not the removal of excess piperidine. The reproducibility of the stereochemical results, with a complete diastereoselectivity observed even in the case of compounds 12, prompted us to investigate by computational methods the conformations of platform 7a that could explain the preference toward one of the two distereomeric products 12a. Compound 7a was subjected to an unrestricted conformer distribution calculation. Keeping in mind that 7a possesses limited conformational flexibility,28 two relevant conformations emerged from the analysis, a ‘zig-zag’ (conformer A in Fig. 2) and a ‘folded’ one (B, Fig. 2). The geometry of these conformers was refined at the DFT level of theory, by using the hybrid B3LYP functional with a 6-311++G(d,p) basis set.
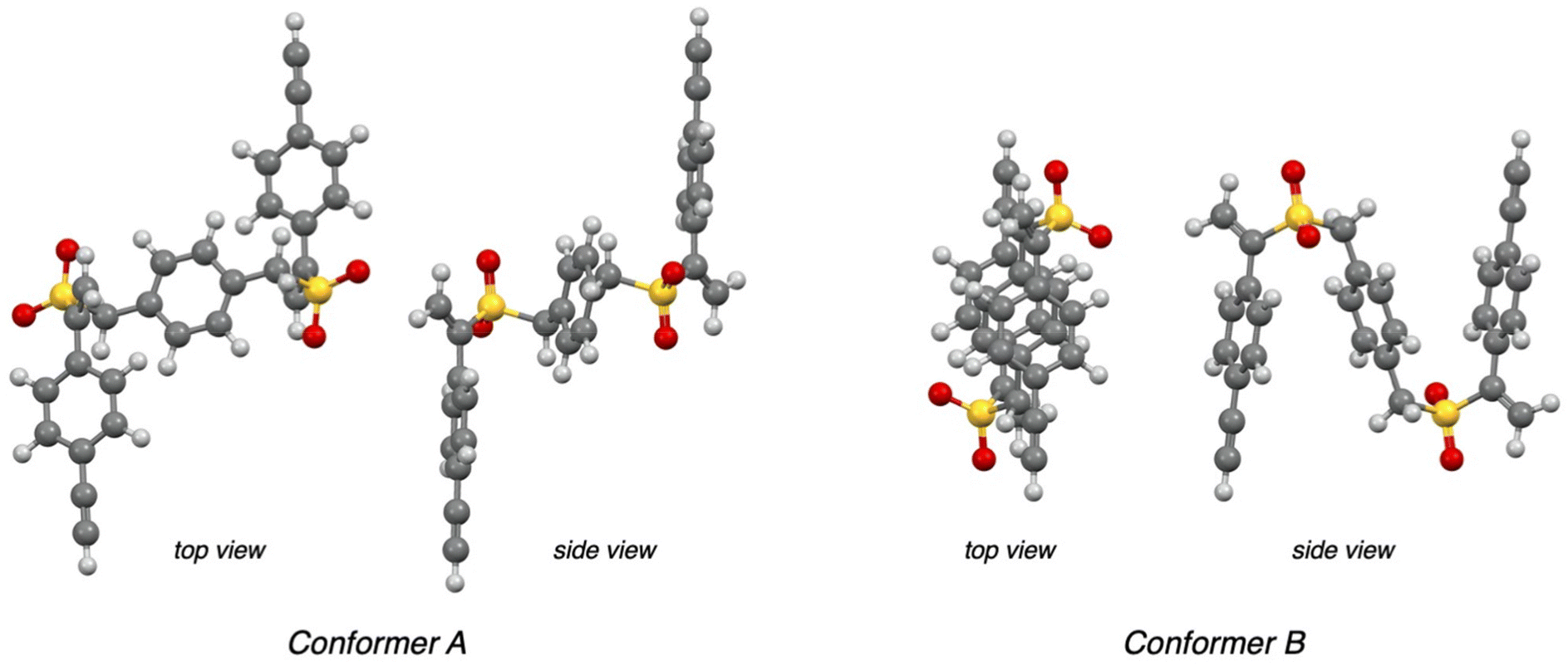 | ||
| Fig. 2 Top and side views of the optimized geometry (DFT, B3LYP/6-311++G(d,p)) of conformers A and B of compound 7a. | ||
There are some evident differences, but they share a common feature, as in both cases the geometry is driven by internal π-stacking interactions. Conformer B presents the aromatic moieties arranged to maximize – as far as the limited conformational space allows – sandwich-type Ar⋯⋯ π-stacking (4.20 Å centroid-to-centroid distance and 26.4° interplanar angle, see Fig. S1 in the ESI†). Conversely, alkene–arene π-stacking interactions29 are predominant in conformer A, with the vinyl group located at 4.3 Å from the central phenylene moiety (59° angle, Fig. S1†). Interestingly, in both conformers the vinyl moiety lies on the same plane as one of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds rather than that of the aryl ring, hinting more at a vinylsulfone Michael-type reactivity instead of a styrene-like one. Careful inspection of the two conformers suggests also that the intramolecular stacking interactions fix the position of the vinyl group practically differentiating its two faces (Fig. 3). As a consequence, the approach of a nucleophile (e.g., ammonia and piperidine) should take place preferentially to the unhindered face of the vinyl group determining, once both vinyl moieties have reacted, the exclusive formation of the meso-stereoisomer from both conformers. Remarkably, in the recent literature, the facial selectivity of a Diels–Alder reaction between the acrylic ester derivative and cyclopentadiene was demonstrated to depend on an aryl–vinyl π-stacking intramolecular interaction that locked the conformation of one of the reactants.30
O bonds rather than that of the aryl ring, hinting more at a vinylsulfone Michael-type reactivity instead of a styrene-like one. Careful inspection of the two conformers suggests also that the intramolecular stacking interactions fix the position of the vinyl group practically differentiating its two faces (Fig. 3). As a consequence, the approach of a nucleophile (e.g., ammonia and piperidine) should take place preferentially to the unhindered face of the vinyl group determining, once both vinyl moieties have reacted, the exclusive formation of the meso-stereoisomer from both conformers. Remarkably, in the recent literature, the facial selectivity of a Diels–Alder reaction between the acrylic ester derivative and cyclopentadiene was demonstrated to depend on an aryl–vinyl π-stacking intramolecular interaction that locked the conformation of one of the reactants.30
 | ||
| Fig. 3 Top and side views of the optimized geometry (DFT, B3LYP/6-311++G(d,p)) of conformers A and B of compound 7a. | ||
Pd-Catalysed copper-free Sonogashira cross-coupling was performed between the central bricks 12a and 12b and the glucosyl arm 10, as previously described, leading to compounds 2a and 2b in 55% and 62% yields, respectively, demonstrating once again the potential of the reactive platform to be transformed into multifunctionalized BAs. The multivalent nature of BAs 1 and 2, as well as the synthesis of new BAs through the exploitation of the same platforms, is now under study.
Conclusions
In this paper we have shown an easy, versatile and stereoselective synthetic procedure, to obtain bolaamphiphilic molecules, which couple one of the most applied reactions of sulfenic acids – the concerted addition to triple bonds – to the copper-free Sonogashira cross-coupling, in a convergent synthetic approach often used for the preparation of oligophenylenethynylenes (OPEs).31The synthesis of a two-site reactive central core allows its further functionalization, such as the introduction of the two polar head groups e.g. carbohydrate residues – we have used a glucosyl derivative, but many other sugars can be adopted – or the transformation of the two vinyl sulfonyl moieties – we have reacted them with ammonia and piperidine, but any other nucleophiles can be easily introduced. This second synthetic target appears particularly significant if one accounts that the introduction of structural elements, such as a lateral side chains, in the hydrophobic region of BAs can lead to bolapolyphiles (BPs) that can self-assemble into versatile 2D structures, opening the route to new molecules with diverse physical properties and self-assembly characteristics. Finally, the observed stereochemical results make the two-site reactive platforms 7 significant bricks in the synthesis of several other molecular structures. Studies on their applications are ongoing.
Experimental section
Chemicals
Solvents were purified according to standard procedures. All the syntheses were monitored by TLC on commercially available precoated plates (silica gel 60 F254), and the products were visualized with vanillin [1 g dissolved in MeOH (60 mL) and conc. H2SO4 (0.6 mL)] and a UV lamp. Silica gel 60 was used for column chromatography.Instrumentation
Proton (1H) and carbon (13C) NMR spectra were recorded on a Varian 500 spectrometer (at 500 MHz for 1H and 125 MHz for 13C) using CDCl3 or DMSO-d6 as solvents. Chemical shifts are given in parts per million (ppm) (δ relative to the residual solvent peak for 1H and 13C), coupling constants (J) are given in Hertz, and the attributions are supported by heteronuclear single-quantum coherence (HSQC) and correlation spectroscopy (COSY) experiments. Chemical shifts are reported in ppm relative to the residual CHCl3 peak (7.26 ppm) or residual DMSO peak (2.49 ppm). Combustion analyses were carried out on a FISONS EA1108 elemental analyzer, and mass analysis of final products (1a and 1b) was performed with a TSQ-Quantum access Triple Quadrupole Mass Spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), equipped with a HESI (Heated ElectroSpray Ionization) source; analyses were run in positive mode. Mass spectrometer parameters were: sheath gas flow rate, 30 (arbitrary units); aux gas flow rate, 15 (arbitrary units); spray voltage, 5.00 kV; capillary temperature, 250° C; tube lens voltage, 55 V; heater temperature, 270 °C; and scan mode: full scan.Computational studies
The conformational analysis of compound 7a was carried out with the classical molecular mechanics force field (MMFF) by using the Monte Carlo method to randomly sample the conformational space. The equilibrium geometries were then calculated at the density functional level of theory (DFT, B3LYP functional) using the 6-311++G(d,p) basis set. All quantum mechanical calculations were performed using Spartan ‘10 (Wavefunction, Inc., Irvine, CA, USA).General procedure for the synthesis of compounds 6
To a solution of 5 (1 eq.) in DCE, commercial 1,4-diethynylbenzene (for compound 6a) or commercial 1,3-diethynylbenzene (for compound 6b) (6 eq.) was added and the mixture was stirred at reflux temperature for 24 h. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (n-hexane/ethyl acetate: 80:20). The diastereomeric mixtures of compounds 6a or 6b were obtained as described in the literature.22
General procedure for the synthesis of compounds 7
To a solution of the diastereomeric mixtures of 6a or 6b (1 eq.) in DCM, a solution of m-CPBA (2.5 eq. 80 wt%) in DCM was slowly added at 0 °C and under an argon atmosphere. The reaction mixture was warmed at r.t. and monitored by TLC (n-hexane/DCM 10:90) until the disappearance of the reagent. The reaction was quenched with the addition of an equal volume of an aqueous solution of Na2S2O3 (10%w/w) and the organics washed with a sat. solution of NaHCO3 (3 times) and brine (twice). The organic phase was dried with Na2SO4 and filtered and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (n-hexane/DCM: 10:90) and 7a or 7b was obtained as a white solid.
:50). δH (500 MHz; CDCl3) 7.55 (8H, m, 2× H-2′, 2× H-3′, 2× H-5′, 2× H-6′), 7.12 (4H, s, H-2, H-3, H-5, H-6), 6.32 and 6.00 (4H, two s, 2× CH2), 4.10 (4H, s, 2× SCH2), 3.21 (2H, s, 2× ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH). δC (125 MHz; CDCl3) 147.3 (q), 132.6 (q), 132.5 and 128.6 (C-2′, C-3′, C-5′, C-6′), 131.1 (C-2, C-3, C-5, C-6), 128.7 and 123.9 (q), 128.2 (CH2), 82.6 (q), 79.4 (CH), 58.5 (SCH2). Anal. Calcd for C28H22O4S2 (486.60): C, 69.11; H, 4.56. Found: C, 69.07; H, 4.55.
:50). δH (500 MHz; CDCl3) 7.64 (2H, s, 2× H-2′), 7.59 and 7.56 (4H, dd, J4′,5′ = J5′,6′ = 7.9, 2× H-4′, 2× H-6′), 7.39 (2H, t, J2′,3′ = J3′,4′ = 7.9, 2× H-5′), 7.13 (4H, s, H-2, H-3, H-5, H-6), 6.32 and 5.99 (4H, two s, 2× CH2), 4.10 (4H, s, 2× SCH2), 3.15 (2H, s, 2× CH). δC (125 MHz; CDCl3) 147.2 (q), 133.4 and 129.0 (C-4′, C-5′, C-6′), 132.7 (q), 132.2 (C-2′), 131.1 (C-2, C-3, C-5, C-6), 129.0 and 123.0 (q), 128.2(CH2), 82.6 (q), 79.4 (CH), 58.5 (SCH2). Anal. Calcd for C28H22O4S2 (486.60): C, 69.11; H, 4.56. Found: C, 68.98; H, 4.54.
CH). δC (125 MHz; CDCl3) 147.3 (q), 132.6 (q), 132.5 and 128.6 (C-2′, C-3′, C-5′, C-6′), 131.1 (C-2, C-3, C-5, C-6), 128.7 and 123.9 (q), 128.2 (CH2), 82.6 (q), 79.4 (CH), 58.5 (SCH2). Anal. Calcd for C28H22O4S2 (486.60): C, 69.11; H, 4.56. Found: C, 69.07; H, 4.55.
:50). δH (500 MHz; CDCl3) 7.64 (2H, s, 2× H-2′), 7.59 and 7.56 (4H, dd, J4′,5′ = J5′,6′ = 7.9, 2× H-4′, 2× H-6′), 7.39 (2H, t, J2′,3′ = J3′,4′ = 7.9, 2× H-5′), 7.13 (4H, s, H-2, H-3, H-5, H-6), 6.32 and 5.99 (4H, two s, 2× CH2), 4.10 (4H, s, 2× SCH2), 3.15 (2H, s, 2× CH). δC (125 MHz; CDCl3) 147.2 (q), 133.4 and 129.0 (C-4′, C-5′, C-6′), 132.7 (q), 132.2 (C-2′), 131.1 (C-2, C-3, C-5, C-6), 129.0 and 123.0 (q), 128.2(CH2), 82.6 (q), 79.4 (CH), 58.5 (SCH2). Anal. Calcd for C28H22O4S2 (486.60): C, 69.11; H, 4.56. Found: C, 68.98; H, 4.54.
General procedure for the synthesis of compounds 11
A solution of compound 11a or 11b (1 eq.), compound 10 (2.5 eq.) and [Pd(PPh3)]4 (0.1 eq.) in dry DMF and NEt3 (1:1 ratio) was stirred at 60 °C, under an argon atmosphere for 2h, until the disappearance of the reagent by TLC (n-hexane/ethyl acetate 25:75). The solvent was removed under reduced pressure, the crude product was purified by column chromatography (DCM 100) and the desired product was obtained as a white solid.
:20). δH (500 MHz; CDCl3) 7.59 (8H, m, 2× H-2′, 2× H-3′, 2× H-5′, 2× H-6′), 7.10 (4H, s, H-2, H-3, H-5, H-6), 7.02 and 6.95 (4H, two s, 2× H-3′′, 2× H-6′′), 6.30 e 6.01 (4H, two s, 2× CH2), 5.25 (2H, t, J2′′′,3′′′ = J3′′′,4′′′ = 9.8, 2× H-3′′′), 5.11 (2H, t, J3′′′,4′′′ = J4′′′,5′′′ = 9.8, 2× H-4′′′), 5.04 (2H, dd, J1′′′,2′′′ = 8.3, J2′′′,3′′′ = 9.8, 2× H-2′′′), 4.90 (2H, d, J1′′′,2′′′ = 8.3, 2× H-1′′′), 4.65 (4H, s, 2× CH2C), 4.26 and 4.16 (2H, split AB system, J5′′′,6′′′A = 4.4, J6′′′A,6′′′B = 12.8, 2× H2-6′′′), 4.10 (4H, s, 2× SCH2), 3.89 and 3.88 (12H, two s, 4× –OCH3), 3.75 (2H, m, 2× H-5′′′), 2.07, 2.04, 2.02, and 2.01 (24H, four s, 8× CH3CO). δC (125 MHz; CDCl3) 170.6, 170.3, 169.4 and 169.3 (4× CO), 154.1 (q), 147.3 (q), 132.1 (q), 132.0 and 128.6 (C-2′, C-3′, C-5′, C-6′), 131.0 (C-2, C-3, C-5, C-6), 128.4 and 124.9 (q), 128.2 (CH2), 115.7 and 115.5 (C-3′′, C-6′′), 113.3 and 112.7 (q), 98.3 (C-1′′′), 94.0, 89.2, 87.6 and 83.3 (q), 72.8 (C-3′′′), 71.9 (C-5′′′), 71.1 (C-2′′′), 68.3 (C-4′′′), 61.8 (C-6′′′), 58.5 (SCH2), 57.0 (CH2C), 56.4 and 56.3 (–OCH3), 20.7, 20.6 and 20.5 (CH3CO). Anal. Calcd for C78H78O28S2 (1527.57): C, 61.33; H, 5.15. Found: C, 61.27; H, 5.17.
:20). δH (500 MHz; CDCl3) 7.74 (2H, s, 2× H-2′), 7.62 and 7.52 (4H, two d, J4′,5′ = J5′,6′ = 7.5, 2× H-4′, 2× H-6′), 7.40 (2H, t, J4′,5′ = J5′,6′ = 7.5, 2× H-5′), 7.13 (4H, s, H-2, H-3, H-5, H-6), 7.04 and 6.96 (4H, two s, 2× H-3′′, 2× H-6′′), 6.34 and 6.02 (4H, two s, 2× CH2), 5.27 (2H, t, J2′′′,3′′′ = J3′′′,4′′′ = 9.5, 2× H-3′′′), 5.12 (2H, t, J3′′′,4′′′ = J4′′′,5′′′ = 9.6, 2× H-4′′′), 5.05 (2H, dd, J1′′′,2′′′ = 8.0, J2′′′,3′′′ = 9.8, 2× H-2′′′), 4.91 (2H, d, J1′′′,2′′′ = 8.0, 2× H-1′′′), 4.66 (4H, s, 2× CH2C), 4.29 and 4.17 (2H, split AB system, J5′′′,6′′′A = 4.5, J6′′′A,6′′′B = 12.5, 2× H2-6′′′), 4.13 (4H, s, 2× SCH2), 3.89 (12H, s, 4× OCH3), 3.76 (2H, m, 2× H-5′′′), 2.08, 2.05, 2.03, and 2.01 (24H, four s, 8× CH3CO). δC (125 MHz; CDCl3) 170.7, 170.3, 169.5 and 169.4 (4× CO), 154.1 and 153.9 (q), 147.4 (q), 132.9, 128.9 and 128.8 (C-4′, 5′, 6′), 132.7 (q), 131.6 (C-2′), 131.1 (C-2, C-3, C-5, C-6), 128.7 and 124.0 (q), 128.2 (CH2), 115.7 and 115.6 (C-3′′, C-6′′), 113.3 and 112.6 (q), 98.3 (C-1′′′), 93.9, 89.2, 86.6 and 83.3 (q), 72.8 (C-3′′′), 71.9 (C-5′′′), 71.1 (C-2′′′), 68.3 (C-4′′′), 61.8 (C-6′′′), 58.6 (SCH2), 57.0 (CH2C), 56.5 and 56.4 (–OCH3), 20.7, 20.6 and 20.5 (CH3CO). Anal. Calcd for C78H78O28S2 (1527.57): C, 61.33; H, 5.15. Found: C, 61.55; H, 5.13.
General procedure for the synthesis of compounds 1
Compound 11a or 11b (0.2 mmol) was dissolved in THF–MeOH (1:1, 40 mL). A large excess of aqueous ammonia (12 mL) was added, and the reaction mixture was continuously stirred at r.t. overnight, until the disappearance of the starting product by TLC (n-hexane/ethyl acetate 40:60). Solvents were removed under reduced pressure and several washings with Et2O were performed to purify the product from the undesired acetamide.25
), 4.6–4.5 (6H, m, 2× CH2C, 2× CH2S; 2× –OH-6′′′), 4.44 (2H, dd, Jvic = 5.5, Jvic = 4.1, 2× –CHCH2NH2), 4.38–4.32 (4H, m, 2× –CH2S and 2× H-1′′′), 3.82 and 3.81 (12H, two s, 4× –OCH3), 3.71–2.95 (16H, m, 2× H2-6′′′, 2× H-5′′′, 2× H-4′′′, 2× H-3′′′, 2× H-2′′′, 2× –CHCH2NH2). δC (125 MHz; DMSO-d6) 154.1 and 153.9 (q), 133.3 (q), 131.9 and 130.9 (C-2′, C-3′, C-5′, C-6′), 131.7 (C-2, C-3, C-5, C-6), 128.4 and 123.0 (q), 116.3 and 116.0 (C-3′′, C-6′′), 112.9 and 112.7 (q), 101.6 (C-1′′′), 94.6, 91.7, 87.1 and 82.5 (q), 77.5, 77.1, 73.7 and 70.5 (C-2′′′, C-3′′′, C-4′′′, C-5′′′), 69.9 (–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HCH2NH2), 61.6 (C-6′′′), 57.6 (–CH2S), 56.6 and 56.2 (–OCH3), 56.2 (–CH2C), 41.8 (–CHCH2NH2). Anal. Calcd for C62H68N2O20S2 (1225.34): C, 60.77; H, 5.59; N, 2.29. Found: C, 60.82; H, 5.60; N, 2.29. ESI( + )-MS m/z calcd for C62H68N2O20S2 ([M + H]+) 1226.35, found 1226.32 and 613.23 ([M + 2H]2+).
), 4.60–4.53 (6H, m, 2× CH2C, 2× CH2S; 2× –OH-6′′′), 4.44 (2H, br t, J = 5.5; 2× –CHCH2NH2), 4.38–4.31 (4H, m, 2× –CH2S and 2× H-1′′′), 3.82 and 3.81 (two s, 12H, 4× –OCH3), 3.72–2.98 (16H, m, 2× H2-6′′′, 2× H-5′′′, 2× H-4′′′, 2× H-3′′′, 2× H-2′′′, 2× –CHCH2NH2). δC (125 MHz; DMSO-d6) 153.7 and 153.4 (q), 132.7 (q), 131.4 and 131.3 (C-2′, C-5′, C-2, C-5), 129.1 (C-4′, C-6′) 127.9 and 122.6 (q), 115.8 and 115.6 (C-3′′, C-6′′), 112.4 and 112.3 (q), 101.1 (C-1′′′) 94.1, 91.2, 86.3 and 82.0 (q), 77.0, 76.7, 73.3 and 70.1 (C-2′′′, C-3′′′, C-4′′′, C-5′′′), 69.3 (–CHCH2NH2), 61.2 (C-6′′′), 57.2 (–CH2S), 56.2 and 56.1 (–OCH3), 55.8 (–CH2C), 41.3 (–CHCH2NH2). Anal. Calcd for C62H68N2O20S2 (1225.34): C, 60.77; H, 5.59; N, 2.29. Found: C, 60.84; H, 5.57; N, 2.29. ESI(+)-MS m/z calcd for C62H68N2O20S2 ([M + H]+) 1226.35, found 1226.26 and 613.24 ([M + 2H]2+).
HCH2NH2), 61.6 (C-6′′′), 57.6 (–CH2S), 56.6 and 56.2 (–OCH3), 56.2 (–CH2C), 41.8 (–CHCH2NH2). Anal. Calcd for C62H68N2O20S2 (1225.34): C, 60.77; H, 5.59; N, 2.29. Found: C, 60.82; H, 5.60; N, 2.29. ESI( + )-MS m/z calcd for C62H68N2O20S2 ([M + H]+) 1226.35, found 1226.32 and 613.23 ([M + 2H]2+).
), 4.60–4.53 (6H, m, 2× CH2C, 2× CH2S; 2× –OH-6′′′), 4.44 (2H, br t, J = 5.5; 2× –CHCH2NH2), 4.38–4.31 (4H, m, 2× –CH2S and 2× H-1′′′), 3.82 and 3.81 (two s, 12H, 4× –OCH3), 3.72–2.98 (16H, m, 2× H2-6′′′, 2× H-5′′′, 2× H-4′′′, 2× H-3′′′, 2× H-2′′′, 2× –CHCH2NH2). δC (125 MHz; DMSO-d6) 153.7 and 153.4 (q), 132.7 (q), 131.4 and 131.3 (C-2′, C-5′, C-2, C-5), 129.1 (C-4′, C-6′) 127.9 and 122.6 (q), 115.8 and 115.6 (C-3′′, C-6′′), 112.4 and 112.3 (q), 101.1 (C-1′′′) 94.1, 91.2, 86.3 and 82.0 (q), 77.0, 76.7, 73.3 and 70.1 (C-2′′′, C-3′′′, C-4′′′, C-5′′′), 69.3 (–CHCH2NH2), 61.2 (C-6′′′), 57.2 (–CH2S), 56.2 and 56.1 (–OCH3), 55.8 (–CH2C), 41.3 (–CHCH2NH2). Anal. Calcd for C62H68N2O20S2 (1225.34): C, 60.77; H, 5.59; N, 2.29. Found: C, 60.84; H, 5.57; N, 2.29. ESI(+)-MS m/z calcd for C62H68N2O20S2 ([M + H]+) 1226.35, found 1226.26 and 613.24 ([M + 2H]2+).
General procedure for the synthesis of compounds 12
To a solution of 7a or 7b (1 eq.) in MeOH, piperidine (3.2 eq.) was added and the reaction mixture was stirred at r.t., under an argon atmosphere, until the disappearance of the starting product by TLC (n-hexane/ethyl acetate 50:50). The solvent was evaporated under reduced pressure. The desired product 12a or 12b was obtained without requiring any further purification, after removing excess piperidine under vacuum.
:50). δH (500 MHz; CDCl3) 7.48 and 7.27 (H, two d, Jorto = 7.8, 2× H-2′, 2× H-3′, 2× H-5′, 2× H-6′), 7.41 (4H, s, H-2, H-3, H-5, H-6), 4.47 and 4.38 (4H, AB system, Jgem = 13.2, 2× SCH2), 4.19 (2H, part X of ABX system, Jvic1 = 7.3, Jvic2 = 4.4, 2× –CHCH2Pi), 3.50 and 2.84 (4H, part A and B of ABX system, Jgem = 13.6 Jvic1 = 7.3 Jvic2 = 4.4, 2× –CHCH2Pi), 3.11 (2H, s, 2× CH), 2.47 (8H, m, 2× H2-2p, 2× H2-6p), 1.43 (8H, m, 2× H2-3p, 2× H2-5p), 1.26 (4H, m, 2× H2-4p). δC (125 MHz; CDCl3) 132.8 (q), 132.5 and 129.7 (C-2′, C-3′, C-5′, C-6′), 131.5 (C-2, C-3, C-5, C-6), 128.4 and 122.8 (q), 82.9 (q), 78.3 (CH), 65.2 (–CHCH2Pi), 59.2 and 59.1 (SCH2, –CHCH2Pi), 54.8 (C-2p, C-6p), 26.0 (C-3p, C-5p), 24.0 (C-4p). Anal. Calcd for C38H44N2O4S2 (656.90): C, 69.48; H, 6.75; N, 4.26. Found: C, 69.33; H, 6.73; N, 4.25.
:50). δH (500 MHz; CDCl3) δ 7.52–7.20 (8H, m, 2× H-2′, 2× H-4′, 2× H-5′, 2× H-6′), 7.42 (4H, s, H-2, H-3, H-5, H-6), 4.48 and 4.41 (4H, AB system, Jgem = 14.2, 2× SCH2), 4.19 (2H, part X of ABX system, Jvic1 = 7.8, Jvic2 = 4.6, 2× –CHCH2Pi), 3.50 and 2.83 (4H, part A and B of ABX system, Jgem = 13.2, Jvic1 = 7.8, Jvic2 = 4.6, 2× –CHCH2Pi), 3.17 (2H, s, 2× CH), 2.47 (8H, m, 2× H2-2p, 2× H2-6p), 1.43 (8H, m, 2× H2-3p, 2× H2-5p), 1.25 (4H, m, 2× H2-4p). δC (125 MHz; CDCl3) 133.3, 132.6, 130.1 and 128.8 (C-2′, C-4′, C-5′, C-6′), 132.5 (q), 131.5 (C-2, C-3, C-5, C-6), 128.5 and 122.8 (q), 82.9 (q), 78.0 (CH), 65.0 (–HCH2Pi), 59.4 and 59.2 (SCH2 and –CHH2Pi), 54.8 (C-2p, C-6p), 26.0 (C-3p, C-5p), 24.0 (C-4p). Anal. Calcd for C38H44N2O4S2 (656.90): C, 69.48; H, 6.75; N, 4.26. Found: C, 69.55; H, 6.76; N, 4.27.
General procedure for the synthesis of compounds 2
Compounds 2 were obtained following the general procedure for the synthesis of compounds 11. The mixture was reacted until the disappearance of compounds 12 by TLC (n-hexane/ethyl acetate: 40:60). The solvents were removed under reduced pressure and the reaction crude products were purified by column chromatography (n-hexane/ethyl acetate: 40:60). The desired products 2 were obtained as white solids.
:60). δH (500 MHz; CDCl3) 7.55 and 7.33 (8H, AA′XX′ system, Jorto = 8.0, 2× H-2′, 2× H-3′, 2× H-5′, 2× H-6′), 7.41 (4H, s, H-2, H-3, H-5, H-6), 7.00 and 6.93 (4H, two s, 2× H-3′′, 2× H-6′′), 5.26 (t, J2′′′, 3′′′ = J3′′′, 4′′′ = 9.0, 2× H-3′′′), 5.11 (2H, t, J3′′′, 4′′′= J4′′′, 5′′′ = 9.0, 2× H-4′′′), 5.04 (2H, t, J1′′′, 2′′′ = J2′′′, 3′′′ = 9.0, 2× H-2′′′), 4.90 (2H, d, J1′′′, 2′′′ = 9, 2× H-1′′′), 4.64 (4H, s, 2× CH2C), 4.48–4.15 (10H, m, 2× SCH2, 2× –CHCH2Pi, 2× H2-6′′′), 3.87 (12H, s, 4× OCH3), 3.76 (2H, m, 2× H-5′′′), 3.54 and 2.90 (4H, m AB of an ABX system, 2× –CHCH2Pi), 2.49 (8H, m, 2× H2-2p, 2× H2-6p), 2.07, 2.04, 2.02 and 2.00 (24H, four s, 8× CH3CO), 1.57 (8H, m, 2× H2-3p, 2× H2-5p), 1.42 (4H, m, 2× H2-4p). δC (125 MHz; CDCl3) 170.7, 170.3, 169.5 and 169.4 (4× –CO), 154.1 and 154.0 (q), 132.1 and 129.9 (C-2′, C-3′, C-5′, C-6′), 131.6 (C-2, C-3, C-5, C-6), 128.7, 128.3 and 124.1 (q), 115.7 and 115.6 (C-3′′, C-5′′), 114.6 and 112.4 (q), 98.2 (C-1′′′), 94.3, 89.1, 86.7 and 83.3 (q), 72. 8 (C-3′′′), 71.9 (C-5′′′), 71.0 (C-2′′′), 68.1 (C-4′′′), 65.3 (–CHCH2Pi), 61.8 (C-6′′′) 59.0 and 58.9 (SCH2, –CHCH2Pi), 57.1 (–CH2C), 56.5 and 56.3 (–OCH3), 54.8 (C-2p, C-6p), 25.5 (C-3p, C-5p), 23.6 (C-4p), 20.8, 20.7, 20.6 and 20.5 (CH3CO). Anal. Calcd for C88H100N2O28S2 (1697.86): C, 62.25; H, 5.94; N, 1.65. Found: C, 62.31; H, 5.93; N, 1.65.
:60). δH (500 MHz; CDCl3) 7.67–7.28 (8H, m, 2× H-2′, 2× H-4′, 2× H-5′, 2× H-6′), 7.45 (4H, s, H-2, H-3, H-5, H-6), 7.01 and 6.94 (4H, two s, 2× H-3′′, 2× H-6′′), 5.27 (2H, t, J2′′′, 3′′′ = J3′′′, 4′′′ = 9.0, 2× H-3′′′), 5.12 (2H, t, J3′′′, 4′′′ = J4′′′, 5′′′ = 9.0, 2× H-4′′′), 5.05 (2H, br t, 2× H-2′′′), 4.91 (2H, d, J1′′′, 2′′′ = 8.0, 2× H-1′′′), 4.65 (4H, s, 2× CH2C), 4.53–4.12 (10H, m, 2× SCH2, 2× –CHCH2Pi, 2× H2-6′′′), 3.87 (12H, s, 4× OCH3), 3.77 (2H, m, 2× H-5′′′), 3.57 and 3.1 (4H, m AB of an ABX system, 2× –CHCH2Pi), 2.53 (8H, m, 2× H2-2p, 2× H2-6p), 2.08, 2.05, 2.03 and 2.01 (24H, four s, 8× CH3CO), 1.66 (8H, m, 2× H2-3p, 2× H2-5p), 1.42 (4H, m, 4H, 2× H2-4p). δC (125 MHz; CDCl3) 170.8, 170.4, 169.6 and 169.5 (4× –CO), 154.1 and 153.9 (q), 132.3, 131.8, 128.7 and 128.5 (C-2′, C-3′, C-5′, C-6′, C-2, C-3, C-5, C-6), 130.6, 128.4 and 124.3 (q), 115.9 and 115.8 (C-3′′, C-6′′), 113.6 and 112.6 (q), 98.2 (C-1′′′), 94.3, 89.3, 86.6 and 83.5 (q), 72. 9 (C-3′′′), 72.0 (C-5′′′), 71.3 (C-2′′′), 68.4 (C-4′′′), 62.0 (–CHCH2Pi), 61.9 (C-6′′′), 59.9 (SCH2), 57.1 (CH2C), 56.6 and 56.5 (–OCH3), 55.1 (C-2p, C-6p), 46.1 (–CHCH2Pi), 29.6 (C-3p, C-5p), 23.5 (C-4p), 21.1 and 20.1 (CH3CO). Anal. Calcd for C88H100N2O28S2 (1697.86): C, 62.25; H, 5.94; N, 1.65. Found: C, 62.49; H, 5.92; N, 1.65.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Messina for financial support (FFABR). The publication was created with the co-financing of the European Union-FSE-REACT-EU, PON Research and Innovation 2014–2020 DM.1062/2021.References
- J.-H. Fuhrhop and D. Fritsch, Thermochromism of charge-transfer complexes between a vesiculating paraquat derivative and benzidine, J. Am. Chem. Soc., 1984, 106, 4287–4288 CrossRef CAS.
- J.-H. Fuhrhop and D. Fritsch, Bolaamphiphiles Form Ultrathin, Porous, and Unsymmetric Monolayer Lipid Membranes, Acc. Chem. Res., 1986, 19, 130–137 CrossRef CAS.
- R. M. Fuoss and D. J. Edelson, olaform Electrolytes. I. Di-(β-trimethylammonium Ethyl) Succinate Dibromide and Related Compounds, J. Am. Chem. Soc., 1951, 73, 269–273 CrossRef CAS.
- K. Rastädter, D. J. Wurm, O. Spadiut and J. Quehenberger, The Cell Membrane of Sulfolobus spp.—Homeoviscous Adaption and Biotechnological Applications, Int. J. Mol. Sci., 2020, 21, 3935–3956 CrossRef PubMed.
- M. Fariya, A. Jain, V. Dhawan, S. Shah and M. S. Nagarsenker, Bolaamphiphiles: A Pharmaceutical Review, Adv. Pharm. Bull., 2014, 4, 483–491 Search PubMed.
- J. R. Hughes, A. S. Miller, C. E. Wallace, G. N. Vemuri and P. M. Iovine, Biomedically Relevant Applications of Bolaamphiphiles and Bolaamphiphile-Containing Materials, Front. Chem., 2021, 8, 604151–604168 CrossRef.
- N. Mehwish, X. Dou, Y. Zhao and C.-L. Feng, Supramolecular fluorescent hydrogelators as bio-imaging probes, Mater. Horiz., 2019, 6, 14–44 RSC.
- N. Nuraje, H. Bai and K. Su, Bolaamphiphilic molecules: Assembly and applications, Prog. Polym. Sci., 2013, 38, 302–343 CrossRef CAS.
- G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi and F. Puntoriero, A supramolecular amphiphile from a new water-soluble calix [5] arene and n-dodecylammonium chloride, Tetrahedron Lett., 2013, 54, 188–191 CrossRef CAS.
- I. Pisagatti, L. Barbera, G. Gattuso, V. Villari, N. Micali, E. Fazio, F. Neri, M. F. Parisi and A. Notti, Tuning the aggregation of an amphiphilic anionic calix [5] arene by selective host–guest interactions with bola-type dications, New J. Chem., 2019, 43, 7628–7635 RSC.
- S. P. Wang, W. Lin, X. Wang, T. Y. Cen, H. Xie, J. Huang, B.-Y. Zhu, Z. Zhang, A. Song, J. Hao, J. Wu and S. Li, Controllable hierarchical self-assembly of porphyrin-derived supra-amphiphiles, Nat. Commun., 2019, 10, 1339 CrossRef.
- L. Minuti, E. Ballerini, A. Barattucci, P. M. Bonaccorsi, M. L. Di Gioia, A. Leggio, C. Siciliano and A. Temperini, A unified strategy for the synthesis of three conicol marine natural product, Tetrahedron, 2015, 71, 3253–3262 CrossRef CAS.
- P. M. Bonaccorsi, M. Labbozzetta, A. Barattucci, T. M. G. Salerno, P. Poma and M. Notarbartolo, Synthesis of Curcumin Derivatives and Analysis of Their Antitumor Effects in Triple Negative Breast Cancer (TNBC) Cell Lines, Pharmaceuticals, 2019, 12, 161–179 CrossRef CAS.
- A. Barattucci, M. C. Aversa, A. Mancuso, T. M. G. Salerno and P. Bonaccorsi, Transient Sulfenic Acids in the Synthesis of Biologically Relevant Products, Molecules, 2018, 23, 1030–1042 CrossRef.
- M. C. Aversa, A. Barattucci, P. Bonaccorsi, F. Marino-Merlo, A. Mastino and M. T. Sciortino, Synthesis and biological testing of thioalkane- and thioarene-spaced bis-beta-D-glucopyranosides, Bioorg. Med. Chem., 2009, 17, 1456–1463 CrossRef CAS.
- M. C. Aversa, A. Barattucci and P. Bonaccorsi, Efficient Synthesis of Unsymmetrical Disulfides through Sulfenic Acids, Eur. J. Org. Chem., 2009, 6355–6359 CrossRef CAS.
- M. C. Aversa, A. Barattucci, P. Bonaccorsi, G. Bruno, F. Caruso and P. Giannetto, Enantiopure 2-sulfinylbuta-1,3-dienes in Diels–Alder cycloadditions: a stereoselective approach to an azasteroidal skeleton, Tetrahedron: Asymmetry, 2001, 12, 2901–2908 CrossRef CAS.
- M. C. Aversa, A. Barattucci, P. Bonaccorsi and A. Contini, Addition of sulfenic acids to monosubstituted acetylenes: a theoretical and experimental study, J. Phys. Org. Chem., 2009, 22, 1048–1057 CrossRef CAS.
- B.-D. Lechner, P. Biehl, H. Ebert, S. Werner, A. Meister, G. Hause, K. Bacia, C. Tschierske and A. Blume, Controlling the Miscibility of X-Shaped Bolapolyphiles in Lipid Membranes by Varying the Chemical Structure and Size of the Polyphile Polar Headgroup, J. Phys. Chem. B, 2018, 122, 10861–10871 CrossRef CAS PubMed.
- M. C. Aversa, A. Barattucci, P. Bonaccorsi and A. Temperini, Regio- and Stereocontrolled Synthesis of (Z)-α-(Phenylseleno)sulfinyl and -sulfonyl Alkenes via Sulfenic Acids, and a Study of their Reactivity, Eur. J. Org. Chem., 2011, 5668–5673 CrossRef CAS.
- P. Bonaccorsi, F. Marino-Merlo, A. Barattucci, G. Battaglia, E. Papaianni, T. Papalia, M. C. Aversa and A. Mastino, Synthesis and biological evaluation of a new class of glycoconjugated disulfides that exhibit potential anticancer properties, Bioorg. Med. Chem., 2012, 20, 3186–3195 CrossRef CAS PubMed.
- M. C. Aversa, A. Barattucci, P. Bonaccorsi, C. Faggi and T. Papalia, Thiacyclophane cages and related bi-and tripodal molecules via transient polysulfenic acids, J. Org. Chem., 2007, 72, 4486–4496 CrossRef CAS PubMed.
- A. Barattucci, M. L. Di Gioia, A. Leggio, L. Minuti, T. Papalia, C. Siciliano, A. Temperini and P. Bonaccorsi, Stereoselective Synthesis of Dithia[3.3]cyclophane S,S’ -Dioxides with Planar and Central Chirality, Eur. J. Org. Chem., 2014, 2099–2104 CrossRef CAS.
- C. Yi, C. Blum, M. Lehmann, S. Keller, S.-X. Liu, G. Frei, A. Neels, J. Hauser, S. Schürch and S. Decurtins, Versatile strategy to access fully functionalized benzodifurans: redox-active chromophores for the construction of extended π-conjugated materials, J. Org. Chem., 2010, 75, 3350–3357 CrossRef CAS PubMed.
- G. B. Giovenzana, L. Lay, D. Monti, G. Palmisano and L. Panza, Synthesis of carboranyl derivatives of alkynyl glycosides as potential BNCT agents, Tetrahedron, 1999, 55, 14123–14136 CrossRef CAS.
- A. Barattucci, E. Deni, P. Bonaccorsi, M. G. Ceraolo, T. Papalia, A. Santoro, M. T. Sciortino and F. Puntoriero, Oligo (phenylene ethynylene) Glucosides: modulation of cellular uptake capacity preserving light ON, J. Org. Chem., 2014, 79, 5113–5120 CrossRef CAS.
- Y. Fang, Z. Luo and X. Xu, Recent advances in the synthesis of vinyl sulfones, RSC Adv., 2016, 6, 59661–59676 RSC.
- A. Barattucci, P. Bonaccorsi, T. Papalia, N. Manganaro and G. Gattuso, Kinetic control in the formation of meso-dithia [3.3]-paracyclophane S, S’-dioxide, Tetrahedron Lett., 2014, 55, 5096–5100 CrossRef CAS.
- V. Corne, A. M. Sarotti, C. Ramirez de Arellano, R. A. Spanevello and A. G. Suárez, Experimental and theoretical insights in the alkene–arene intramolecular π-stacking interaction, Beilstein J. Org. Chem., 2016, 12, 1616–1623 CrossRef CAS PubMed.
- A. M. Sarotti, I. Fernández, R. A. Spanevello, M. Á. Sierra and A. G. Suárez, π-Stacking effect on levoglucosenone derived internal chiral auxiliaries. A case of complete enantioselectivity inversion on the Diels− Alder reaction, Org. Lett., 2008, 10, 3389–3392 CrossRef CAS PubMed.
- E. Deni, A. Zamarrón, M. C. Carreño, A. Juarranz, F. Puntoriero, M. T. Sciortino, M. Ribagorda and A. Barattucci, Glucose-functionalized amino-OPEs as biocompatible photosensitizers in PDT, Eur. J. Med. Chem., 2016, 111, 58–71 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01266a |
| This journal is © The Royal Society of Chemistry 2022 |