
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot ester and thioester formation mediated by pentafluoropyridine (PFP)†
Liam N. D.
Beardmore
 ,
Steven L.
Cobb
* and
William D. G.
Brittain
*
,
Steven L.
Cobb
* and
William D. G.
Brittain
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: s.l.cobb@durham.ac.uk; william.d.brittain@durham.ac.uk
First published on 23rd August 2022
Abstract
Acyl fluorides are valuable synthetic intermediates, but in some cases they can be challenging to handle and difficult to isolate given their susceptibility to degradation. In addition, many reagents utilised to prepare acyl fluorides are incompatible with in situ generation strategies and require the acyl fluoride to be isolated before any further reaction can take place. The combination of these factors has meant that acyl fluorides are currently under investigated in nucleophilic substitution processes, and often only a limited substrate scope is tolerated where they have been used. Herein, we report that pentafluoropyridine can be utilised to generate acyl fluorides in situ under mild conditions, and that they can subsequently be used to generate a range of esters and thioesters. This methodology offers a simple one-pot synthesis of esters and thioesters directly from parent carboxylic acids.
Introduction
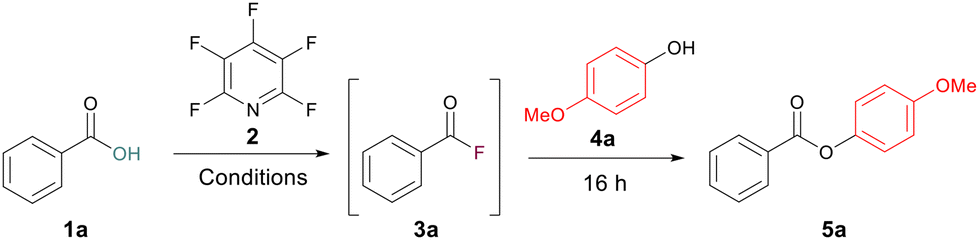
Esters are ubiquitous molecules which play important roles in medicines, fragrances, natural products and lubricants, and act as valuable synthetic intermediates for further reactions.1 The classical approach to the formation of esters on laboratory scale is through reaction of acyl chlorides or anhydrides with alcohols (Scheme 1a).2 However, acyl chlorides are often prepared using reagents such as SOCl2 or PCl3 and anhydrides are often accessed from acyl chlorides.3 The conditions utilised to access acyl chlorides can be less than ideal, especially for acid-sensitive substrates. Acyl chlorides are also prone to hydrolysis back to their parent carboxylic acids.4 Acyl fluorides are similar to acyl chlorides in their reactivity towards nucleophiles but despite this they remain overlooked as intermediates for ester formation. Acyl fluorides do have several advantages over acyl chlorides, including being less prone to hydrolysis and exhibiting a wider functional group tolerance.5 Whilst many methods to access acyl fluorides have been reported,6 being able to do so in an operationally simple and cheap manner is still challenging with many common reagents for acyl fluoride formation being highly corrosive, expensive, requiring bespoke synthesis or requiring specialist equipment to handle safely. Classical reagents such as cyanuric fluoride7 or SeF4 pyridine complexes8 pioneered by Olah are often reported in the synthesis of acyl fluorides, but these are toxic and can be difficult to handle. Although somewhat safer, reagents such as DAST or Deoxo-Fluor are often expensive and require storing at low temperatures due to their instability.9 Isolation and then reaction of acyl fluorides with nucleophiles inherently limits the substrate scope of the parent carboxylic acids which can be used. Many acyl fluorides are prone to degradation when used in their isolated forms and thus in situ generation for application in one-pot processes is desirable. The use of gaseous reagents can enable in situ acyl fluoride generation. For example, Qin and co-workers utilised SO2F2 for the generation of amides in a one pot process (Scheme 1b).10 | ||
| Scheme 1 (a) Classical approaches to ester synthesis (b) recently disclosed strategies for the synthesis of esters and amides utilising in situ acyl fluoride generation (c) this work using pentafluoropyridine for mild in situ acyl fluoride generation for the synthesis of esters and thioesters. | ||
Most recently, the use of thionyl fluoride was disclosed by Sammis and co-workers for the synthesis of N-hydroxyphthalimide esters, N-hydroxylsuccinimide esters and thioesters (Scheme 1b).11 Whilst thionyl fluoride and SO2F2 mediated reactions require little to no final purification of the generated ester, the reagents are corrosive and gaseous, requiring a specialised set-up to use effectively. Therefore, developing a one-pot acyl fluoride formation/acyl nucleophilic substitution reaction combination utilising bench-stable, commercially available, and easy-to-handle reagents would be advantageous compared to the previously reported approaches.
As part of a wider program of work within our research group to probe the properties of perfluorinated aromatics,12 we have reported the use of pentafluoropyridine (PFP) to generate acyl fluorides directly from carboxylic acids under mild conditions.13 This methodology was shown to be particularly powerful for in situ acyl fluoride formation which could then be used directly for amidation. We wished to expand the scope of acyl nucleophilic substitution reactions that could be carried out utilising in situ formation of acyl fluorides. Here we report the use of in situ PFP-mediated acyl fluoride generation for the synthesis of esters, thioesters, di-esters and amide-esters. (Scheme 1c).
Results and discussion
To begin our study, a test reaction between benzoic acid 1a and 4-methoxyphenol 4a was selected and run under our previously reported conditions for amidation.13 A 30 min acyl fluoride activation time at room temperature was used. Under these conditions a yield of 56% for the ester 5a was recorded (Table 1, entry 1). To improve this yield, we began by increasing the equivalents of the alcohol after the 30 min activation window to 1.5 equiv. and 3.75 equiv. However, both had a deleterious effect on the yield, giving 36% and 27%, respectively (Table 1, entries 2 and 3). Finally, by changing the solvent to toluene, no formation of the acyl fluoride was observed (Table 1, entry 5). This led us to postulate that the rate of acyl fluoride generation was hindering the yield during ester formation. We had previously observed this pattern of reactivity during amidation where increasing the activation period could positively affect the yield of the amide product.13 This suspicion was reinforced by the formation of a tetrafluoropyridyl bi-aryl ether as a major by-product, pointing to the PFP not being fully consumed during the acyl fluoride formation step.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Temp. | Time (h) | 4a (equiv.) | DIPEA (equiv.) | 2 (equiv.) | Solvent | Yield (%) |
| a Following formation of the acyl fluoride, 4-methoxyphenol was added and the reaction was maintained at the same temperature for 16 h. b Monitored by 19F NMR spectroscopy for acyl fluoride formation. After 4 h no sign of acyl fluoride formation was observed. | |||||||
| 1 | rt | 0.5 | 1.00 | 2.00 | 1.10 | MeCN | 56 |
| 2 | rt | 0.5 | 1.50 | 2.00 | 1.10 | MeCN | 36 |
| 3 | rt | 0.5 | 3.75 | 2.00 | 1.10 | MeCN | 27 |
| 4 | 50 °C | 4 | 1.10 | 2.00 | 1.10 | MeCN | 71 |
| 5 | 50 °C | 4 | — | 1.00 | 1.10 | Toluene | —b |
To confirm that acyl fluoride formation was the governing factor in the yield of the ester and to better understand the rate of acyl fluoride formation, 19F NMR reaction monitoring was undertaken. A series of substituted benzoic acids were treated with PFP and DIPEA in MeCN-d3 at 50 °C and 19F NMR spectra were recorded using fluorobenzene as an internal standard. The conversion to acyl fluoride was monitored at various time points. It was found that acyl fluoride generation was much quicker for electron-rich benzoic acids such as 4-methoxybenzoic acid 1b and benzoic acid 1a itself, while formation was significantly slower when the electron-poor 4-nitrobenzoic acid 1c was employed. After 1 h at 50 °C, conversion was almost complete for 1a and 1b and remained constant following 2 h of reaction time (Fig. 1).
 | ||
| Fig. 1 Graph of conversion of benzoic acids to acyl fluorides. Reactions were conducted in NMR tubes in a thermostatically controlled water bath. 19F NMR spectra were recorded at various time points using fluorobenzene as an internal standard. Integration of the internal standard vs. integration of the acyl fluoride resonance was used to determine conversion. | ||
Conversion of compound 1c, however, was only ∼20% complete after 1 h and ∼30% after 2 h (Fig. 1). The same analysis was also conducted at rt and the same trend was observed with slow conversion of 1c to 3c (see ESI, p. S26†).
It was concluded that extension of the activation period from 30 min should have a beneficial effect for most carboxylic acids. The results of the reaction monitoring also helped confirm our suspicions that unreacted PFP had remained in our reaction screening mixtures (Table 1). This unreacted PFP is extremely susceptible to nucleophilic attack and explained why a tetrafluoropyridyl bi-aryl ether had been observed as a by-product in the reaction, thus having an adverse effect on ester formation.
Following acyl fluoride reaction monitoring, it was concluded that incorporating a 4 h acyl fluoride activation window at 50 °C would allow complete conversion for most carboxylic acids, thus maximising potential ester formation. Employing these conditions to our test reaction increased the yield of 5a to 71% (Table 1, entry 4).
With optimised reaction conditions in hand, we set about exploring the scope of the ester formation. We began with the synthesis of a range of esters with benzoic acid 1a as the carboxylic acid component which would allow us to test the reactions tolerance to various alcohols. Across a wide range of alcohols, good to excellent yields of esters were obtained. Aliphatic (5b, 5k and 5l), benzylic (5e, 5h) and aromatic alcohols (5a, 5d, 5f, 5g and 5i) were all well tolerated. High yielding reactions occurred when phenol or 3-hydroxypyridine were employed as the alcohol components, giving compounds 5d and 5j in 80% overall yields. In general, the reaction proceeded well across all alcohols tested, even with electron-deficient (5g) or sterically hindered (5i) phenols, for which acceptable to excellent yields of 58% and 92% respectively were recorded. Tertiary butanol was the only alcohol where no conversion was observed (5c) presumably due to the high degree of steric hinderance around the nucleophile. In order to test the developed methodology on the gram scale we remade compound 5l starting from 1.00 g of benzoic acid whilst the yield of the reaction decreased at this larger scale the product was successfully formed in 61% yield to give 1.03 g of the ester (Scheme 2).
 | ||
| Scheme 2 Scope of benzoic acid derived esters. Note: isolated yields following purification. aNo conversion to the ester was observed by 1H NMR. bReaction was carried out with 1.00 g of benzoic acid. | ||
After investigating the alcoholic component, we next turned our attention to the carboxylic acid component. Aromatic carboxylic acids worked well with compounds 5m, 5n, 5o, 5q and 5r, all leading to greater than 50% yield of the corresponding esters. Examples 5p and 5s were produced in 49% and 18% yields, respectively. This is likely due to the electron-deficient nature of the alcohol component in these cases, as electron-deficient acids gave good yields for other compounds such as 5n (55% yield) and 5r (69% yield). To explore if increasing the reaction temperature could have a positive effect on reaction yield for examples which had given lower yields at 50 °C compound 5s was resynthesised this time inside of a sealed tube at 100 °C. Increasing the reaction temperature increased the yield of 5s to 77%. Long-chained aliphatic acids (5t and 5u) proceeded smoothly, giving good yields of the esters (79% and 75%). When short-chained propanoic acid was employed, yields were lower (5v, 48%; 5x, 46%). These lower yields were attributed to the volatile nature of the generated acyl fluoride. To minimise the complication from the volatility we ran these reactions at 35 °C to limit boil off of the generated acyl fluoride. We tested the developed methodology on acids with known biological activity, and thus we prepared the analogues of naproxen (5y) and ibuprofen (5z) in 69% and 63% yield, respectively. Finally, 1,4-dibenzoic acid was employed to determine whether it were possible to generate di-acyl fluorides using the same approach. The reaction proceeded to generate the di-ester 5aa in 40% yield (Scheme 3).
 | ||
| Scheme 3 Scope of one-pot esterification reaction. Note: isolated yields following purification. aReaction was conducted in a sealed tube at 100 °C. bReactions were conducted at 35 °C. | ||
We hypothesised that the reaction protocol could also be employed to rapidly build molecular complexity from diols or amino alcohols. 2,2′-Biphenol gave an excellent yield (85%) of the bis-ester 7a whilst diethylene glycol and BINOL also gave good yields of their corresponding bis-esters 7b (64%) and 7c (72%). Finally, we wished to probe if an esterification and amidation could be carried out at the same time to generate a product containing both ester and amide linkages. By using 4-aminophenol as the nucleophilic component, the amide-ester 7d was successfully generated in 31% yield (Scheme 4).
 | ||
| Scheme 4 Multiple substitution processes carried out simultaneously. Note: isolated yields following purification. | ||
Thioesters are another class of highly useful compound which have widespread use in medicinal chemistry and are a cornerstone of native chemical ligation.14 We postulated that our ester forming reaction would also be applicable to thioesters by simply switching to a thiol as the nucleophilic component of the substitution. To investigate this hypothesis, we conducted the generation of benzoyl fluoride 3a using the same reaction conditions as for esterification before adding 1 equiv. of benzyl mercaptan and letting the reaction stir for 16 h at 50 °C. The corresponding thioester 9a was generated in high yield (89%). Changing the identity of the thiol to aliphatic examples (9b, 9c and 9g) delivered good to excellent yields of thioesters. Thiophenol was also amenable to the reaction to give the thioester 9d in 52% yield. The reaction was also found to be applicable to tertiary thiols with 9e being generated in 35% yield. Altering the carboxylic acid to, picolinic acid gave 9f in 59% yield, 4-methoxy benzoic acid gave a thioester (9h) with benzyl mercaptan in 49% yield, and with naproxen as the carboxylic acid a yield of 68% for the thioester 9i was recorded. Finally, to confirm that the developed methodology could be used with di-thiols, ethane di-thiol was employed to successfully generate the di-thioester 9j in 37% yield (Scheme 5).
 | ||
| Scheme 5 Scope of one-pot thioester formation. Note: isolated yields following purification. | ||
Conclusions
We have demonstrated that pentafluoropyridine (PFP) can be utilised to couple carboxylic acids and nucleophiles to generate esters, thioesters, di-esters and amide-esters under mild conditions. The one-pot reaction proceeds via the in situ generation of an acyl fluoride. Preparation of the acyl fluoride in situ allows any potential issues with acyl fluoride instability to be overcome and thus a wider range of carboxylic acids to be used. Using 19F NMR reaction monitoring we were able to optimise our reaction protocol to generate high yields of esters across a range of alcohol and carboxylic acid partners. Expanding the scope further, we demonstrated that we could rapidly build molecular complexity through the use of di-carboxylic acids or bis-nucleophiles to generate di-esters, di-thioesters or amide-esters. Finally, we explored the application of the methodology to deliver thioesters, again showing good to excellent yields of products. We believe that this methodology represents a manner to utilise acyl fluorides for the synthesis of esters and thioesters under mild conditions without the need to handle corrosive or gaseous reagents. This reaction manifold offers an alternative to traditional acyl chloride or acid anhydride esterification for substrates which may not be suitable for these approaches.Experimental
General procedure for the synthesis of esters
To an oven dried glass vial or Radley's carousel tube equipped with a stirrer bar was added carboxylic acid (1 equiv.), acetonitrile dried over 4 Å molecular sieves (3 mL), diisopropylethylamine (DIPEA) (2 equiv.) and pentafluoropyridine (PFP) (1.1 equiv.). This mixture was allowed to stir at 50 °C for 4 h, at which point the desired alcohol (1 equiv.) was added. The mixture was then stirred at 50 °C for 16 h. Following this time, the mixture was concentrated under reduced pressure, the resulting residue was dissolved in a minimum amount of dichloromethane (DCM) and the recovered crude material was purified directly by flash column chromatography which yielded the desired compounds.General procedure for the synthesis of thioesters
To an oven dried glass vial or Radley's carousel tube equipped with a stirrer bar was added carboxylic acid (1 equiv.), acetonitrile dried over 4 Å molecular sieves (3 mL), diisopropylethylamine (DIPEA) (2 equiv.) and pentafluoropyridine (1.1 equiv.). This mixture was allowed to stir for a period of 4 h, after which thiol (1 equiv.) was added. The mixture was then allowed to stir at 50 °C for 16 h after this time, the mixture was allowed to cool, was concentrated under reduced pressure, and the resulting residue was dissolved in a minimum amount of dichloromethane (DCM) and purified directly by flash column chromatography which yielded the desired compounds.Author contributions
L. N. D. B. conducted the reported reactions, wrote sections of the manuscript and prepared the ESI.† S. L. C. co-conceived and co-supervised the study. W. D. G. B. co-conceived and co-supervised the study, wrote the manuscript and contributed towards the ESI.† All co-authors were involved in data analysis, contributed ideas and contributed towards the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. N. D. B. would like to thank the James Pantyfedwen Foundation for a studentship grant. W. D. G. B would like to acknowledge the Leverhulme Trust for an Early Career Fellowship (ECF-2020-454 and Durham University), Durham University for a Grant Seedcorn Award and the Royal Society of Chemistry for a Research Enablement Grant (E21-9509829332) for financial support. Dr Juan A. Aguilar is thanked for NMR spectroscopy support. Mr Peter Stokes is thanked for mass spectrometry support.References
- (a) M. Tsakos, E. S. Schaffert, L. L. Clement, N. L. Villadsen and T. B. Poulsen, Nat. Prod. Rep., 2015, 32, 605–632 RSC; (b) M. Sedighi and M. A. Lipton, Org. Lett., 2005, 7, 1473–1475 CrossRef CAS PubMed; (c) M. Zabadal, A. P. Pelliccioli, P. Klán and J. Wirz, J. Phys. Chem. A, 2001, 105, 10329–10333 CrossRef CAS; (d) J. V. McCullagh and S. P. Hirakis, J. Chem. Educ., 2017, 94, 1347–1351 CrossRef CAS; (e) S. Rayne and K. Forest, Flavour Fragrance J., 2016, 31, 385–394 CrossRef CAS; (f) I. Bassanini, K. Hult and S. Riva, Beilstein J. Org. Chem., 2015, 11, 1583–1595 CrossRef CAS PubMed.
-
(a) S. Gaspa, A. Porcheddu and L. De
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Luca, Adv. Synth. Catal., 2016, 358, 154–158 CrossRef CAS;
(b) F. E. A. Van Waes, J. Drabowicz, A. Cukalovic and C. V. Stevens, Green Chem., 2012, 14, 2776–2779 RSC;
(c) S. Takimoto, J. Inanaga, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1976, 49, 2335–2336 CrossRef CAS;
(d) J.-Q. Chen, X. Tu, Q. Tang, K. Li, L. Xu, S. Wang, M. Ji, Z. Li and J. Wu, Nat. Commun., 2021, 12, 5328 CrossRef CAS PubMed.
Luca, Adv. Synth. Catal., 2016, 358, 154–158 CrossRef CAS;
(b) F. E. A. Van Waes, J. Drabowicz, A. Cukalovic and C. V. Stevens, Green Chem., 2012, 14, 2776–2779 RSC;
(c) S. Takimoto, J. Inanaga, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1976, 49, 2335–2336 CrossRef CAS;
(d) J.-Q. Chen, X. Tu, Q. Tang, K. Li, L. Xu, S. Wang, M. Ji, Z. Li and J. Wu, Nat. Commun., 2021, 12, 5328 CrossRef CAS PubMed. - (a) B. D. Hosangadi and R. H. Dave, Tetrahedron Lett., 1996, 37, 6375–6378 CrossRef CAS; (b) J. A. Cade and W. Gerrard, Nature, 1953, 172, 29–29 CrossRef CAS PubMed; (c) J. A. Greenberg and T. Sammakia, J. Org. Chem., 2017, 82, 3245–3251 CrossRef CAS PubMed; (d) I. A. El-Sakka and N. A. Hassan, J. Sulfur Chem., 2005, 26, 33–97 CrossRef CAS; (e) H. Chen, X. Xu, L. L. Liu, G. Tang and Y. Zhao, RSC Adv., 2013, 3, 16247–16250 RSC; (f) X. Wu, L. Zhou, R. Yang, F. Guo, Z.-L. Tang and J. Xiao, J. Chem. Res., 2020, 44, 301–304 CrossRef CAS; (g) F. Li, X. Wu, F. Guo, Z.-L. Tang and J. Xiao, Eur. J. Org. Chem., 2021, 4314–4317 CrossRef CAS; (h) I. Shiina, M. Kubota, H. Oshiumi and M. Hashizume, J. Org. Chem., 2004, 69, 1822–1830 CrossRef CAS PubMed.
- (a) H. K. Hall, J. Am. Chem. Soc., 1956, 78, 1450–1454 CrossRef CAS; (b) G. E. K. Branch and A. C. Nixon, J. Am. Chem. Soc., 1936, 58, 2499–2504 CrossRef CAS; (c) R. F. Hudson and J. E. Wardill, J. Chem. Soc., 1950, 1729–1733 RSC.
- (a) Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574–594 CrossRef CAS PubMed; (b) C. G. Swain and C. B. Scott, J. Am. Chem. Soc., 1953, 75, 246–248 CrossRef CAS; (c) D. P. N. Satchell, J. Chem. Soc., 1963, 555–557 RSC; (d) B. D. Song and W. P. Jencks, J. Am. Chem. Soc., 1989, 111, 8470–8479 CrossRef CAS; (e) D. N. Kevill and M. J. D'Souza, J. Org. Chem., 2004, 69, 7044–7050 CrossRef CAS PubMed; (f) B. D. Song and W. P. Jencks, J. Am. Chem. Soc., 1989, 111, 8479–8484 CrossRef CAS.
- (a) M. Gonay, C. Batisse and J.-F. Paquin, Synthesis, 2021, 653–665 CrossRef CAS; (b) M. Gonay, C. Batisse and J.-F. Paquin, J. Org. Chem., 2020, 85, 10253–10260 CrossRef CAS PubMed; (c) P. J. Foth, T. C. Malig, H. Yu, T. G. Bolduc, J. E. Hein and G. M. Sammis, Org. Lett., 2020, 22, 6682–6686 CrossRef CAS PubMed; (d) T. Scattolin, K. Deckers and F. Schoenebeck, Org. Lett., 2017, 19, 5740–5743 CrossRef CAS PubMed; (e) Y. Liang, Z. Zhao, A. Taya and N. Shibata, Org. Lett., 2021, 23, 847–852 CrossRef CAS PubMed; (f) M. Meanwell, J. Lehmann, M. Eichenberger, R. E. Martin and R. Britton, Chem. Commun., 2018, 54, 9985–9988 RSC; (g) H.-X. Song, Z.-Y. Tian, J.-C. Xiao and C.-P. Zhang, Chem. – Eur. J., 2020, 26, 16261–16265 CrossRef CAS PubMed; (h) X. Wang, F. Wang, F. Huang, C. Ni and J. Hu, Org. Lett., 2021, 23, 1764–1768 CrossRef CAS PubMed; (i) S. B. Munoz, H. Dang, X. Ispizua-Rodriguez, T. Mathew and G. K. S. Prakash, Org. Lett., 2019, 21, 1659–1663 CrossRef CAS PubMed; (j) A. L'Heureux, F. Beaulieu, C. Bennett, D. R. Bill, S. Clayton, F. LaFlamme, M. Mirmehrabi, S. Tadayon, D. Tovell and M. Couturier, J. Org. Chem., 2010, 75, 3401–3411 CrossRef PubMed.
- G. A. Olah, M. Nojima and I. Kerekes, Synthesis, 1973, 487–488 CAS.
- G. A. Olah, M. Nojima and I. Kerekes, J. Am. Chem. Soc., 1974, 96, 925–927 CrossRef CAS.
- (a) M. Kaźmierczak and M. Bilska-Markowska, Eur. J. Org. Chem., 2021, 5585–5604 CrossRef; (b) R. P. Singh and J. N. M. Shreeve, Synthesis, 2002, 2561–2578 CAS.
- S.-M. Wang, C. Zhao, X. Zhang and H.-L. Qin, Org. Biomol. Chem., 2019, 17, 4087–4101 RSC.
- (a) T. G. Bolduc, C. Lee, W. P. Chappell and G. M. Sammis, J. Org. Chem., 2022, 87, 7308–7318 CrossRef CAS PubMed; (b) C. Lee, B. J. Thomson and G. M. Sammis, Chem. Sci., 2022, 13, 188–194 RSC.
- (a) W. D. G. Brittain and S. L. Cobb, Org. Biomol. Chem., 2019, 17, 2110–2115 RSC; (b) W. D. G. Brittain and S. L. Cobb, J. Org. Chem., 2020, 85, 6862–6871 CrossRef CAS PubMed; (c) W. D. G. Brittain and C. R. Coxon, Chem. – Eur. J., 2022, 28, e202103305 CAS; (d) D. Gimenez, C. A. Mooney, A. Dose, G. Sandford, C. R. Coxon and S. L. Cobb, Org. Biomol. Chem., 2017, 15, 4086–4095 RSC.
- W. D. G. Brittain and S. L. Cobb, Org. Lett., 2021, 23, 5793–5798 CrossRef CAS PubMed.
- (a) V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed; (b) S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Nat. Rev. Chem., 2018, 2, 0122 CrossRef CAS; (c) V. Diemer, O. Firstova, V. Agouridas and O. Melnyk, Chem. – Eur. J., 2022, 28, e202104229 CAS; (d) Y. Asahina, T. Kawakami and H. Hojo, Chem. Commun., 2017, 53, 2114–2117 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01268e |
| This journal is © The Royal Society of Chemistry 2022 |