
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic, asymmetric azidations at carbonyls: achiral and meso-anhydride desymmetrisation affords enantioenriched γ-lactams†
Simon N.
Smith
 ,
Cristina
Trujillo
and
Stephen J.
Connon
*
,
Cristina
Trujillo
and
Stephen J.
Connon
*
Trinity Biomedical Sciences Institute, School of Chemistry, The University of Dublin, Trinity College, Dublin 2, Ireland. E-mail: connons@tcd.ie
First published on 21st July 2022
Abstract
An unprecedented organocatalytic process involving the asymmetric addition of azide to meso-anhydrides has been developed, promoted by novel sulfamide-substituted Cinchona alkaloid-based catalysts. Readily available glutaric anhydrides can be smoothly converted to enantioenriched hemi-acyl azides and from there to either γ-amino acids or γ-lactams.
Since the first preparation of phenyl azide by Griess in 1864,1 the reactivity of organic azides (R-N3) as [1,3]-dipoles, electrophiles, nucleophiles and radical acceptors have been widely exploited.2,3 In addition, the capacity of azide-containing compounds to liberate molecular nitrogen facilitates an array of reaction pathways with the capacity to yield complex products from relatively-simple precursors.
In nucleophilic substitution reactions, azide can either be a useful N1 synthon for the introduction of functional groups (primary amine, amide) or used to install a particular structural motif (1,2,3-triazoles, tetrazoles). Although the high reactivity of azide can be beneficial – with a Mayr nucleophilicity parameter4 exceeding that of some α-effect nucleophiles – the utilisation of organic azides in a catalytic asymmetric context is challenging. Both organometallic and organocatalytic approaches to asymmetric azidations have been explored.5 Jacobsen and co-workers6 used the privileged ‘salen’ ligand in the Cr-catalysed silylazidation of meso-epoxides with trimethylsilyl azide (TMSN3, Fig. 1A) – later expanded to the kinetic resolution of epoxides,7 the desymmetrisation of meso-aziridines8 and the first asymmetric β-azidation of α,β-unsaturated imides with excess hydrazoic acid (HN3).9 As the intrinsic properties of HN3 (toxic, volatile and explosive) prevent its practical use,10 the pursuit of organocatalytic, asymmetric strategies to obviate the direct use of HN3 has been of interest.11–16
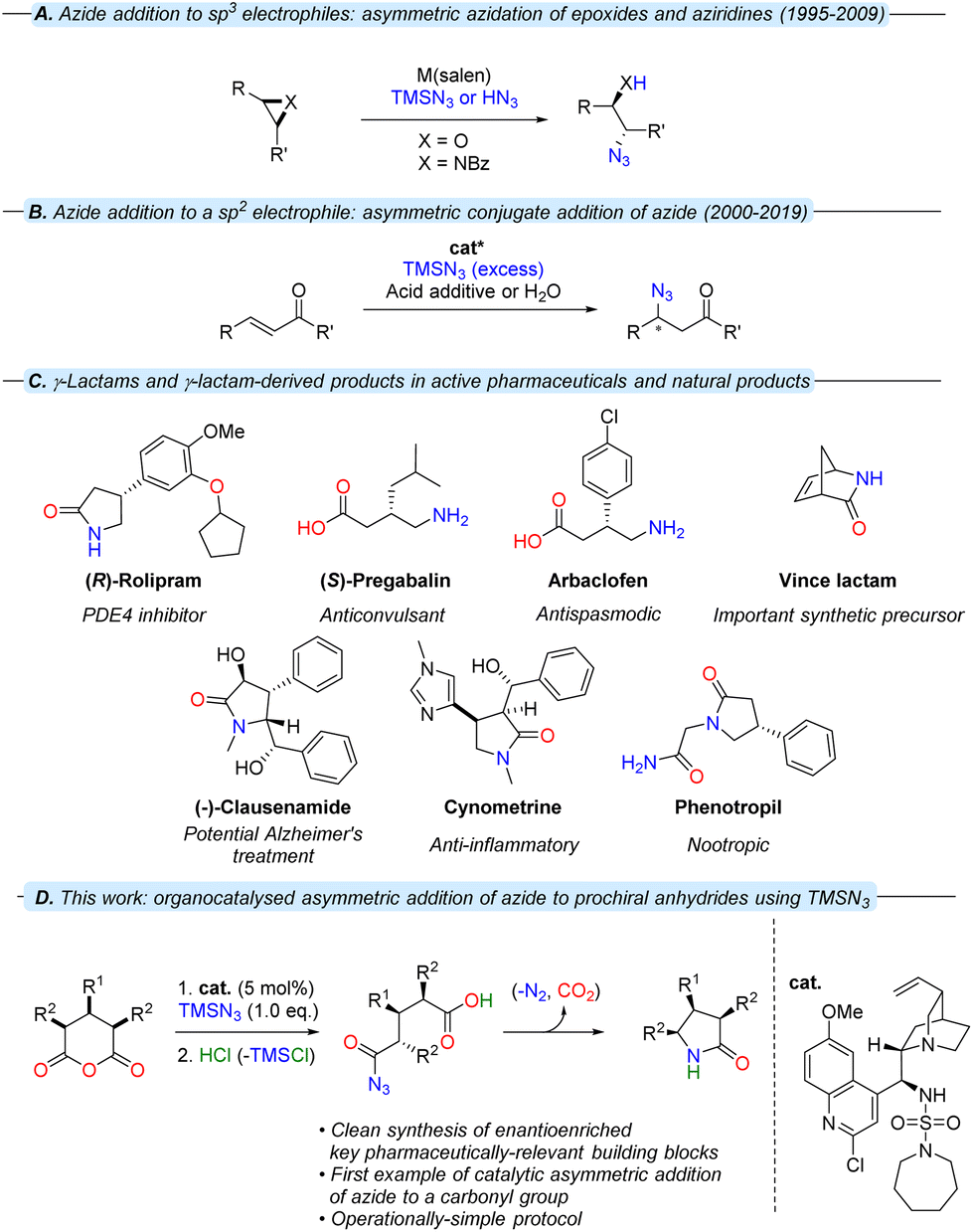 | ||
| Fig. 1 Asymmetric nucleophilic azidations; the prevalence of chiral γ-lactams in the synthesis of biologically-active compounds and a summary of this work. | ||
The first organocatalytic, asymmetric β-azidation, reported by Miller and co-workers in 2000,11 relied on safer generation of HN3in situ through the use of TMSN3 and an organic acid additive (Fig. 1B). Asymmetric β-azidations were further developed thereafter by other research groups,16a culminating recently in the first organic acid-free β-azidation of α,β-unsaturated ketones.16b Recently, we reported the first organocatalytic reactions between cyclic anhydrides and TMSN3, which allowed controlled access to a variety of pharmaceutically-active γ-amino acid and γ-lactam derivatives from anhydrides.17
This bioactive class of compounds that contains the γ-aminobutyric acid (GABA) motif are potent central nervous system-active agents (Fig. 1C),18–24 and are present in a wide variety of natural products and numerous APIs. The advantage of this reaction cascade lies in rapid access to valuable scaffolds from uncomplicated substrates in a robust manner. In a similar fashion, enantioselective desymmetrisations of achiral or meso-anhydrides via existing organocatalytic methodologies (alcoholysis, thiolysis, cycloaddition, inter alia) can offer a powerful strategy to access molecular complexity from inexpensive, accessible precursors.25–28
Though great advances have been made in the catalytic enantioselective azidations;29,30 an analogous catalytic, asymmetric transformation involving the reaction of azide at a carbonyl centre has not yet been reported. In this report, we have expanded upon a study involving a racemic variant of this process17 and demonstrate the first examples of the enantioselective desymmetrisation of prochiral cyclic anhydrides via azidolysis (Fig. 1D).
At the outset, 1 was chosen as the model substrate. After considerable experimentation (see ESI†), suitable reaction conditions were developed in order to facilitate an initial catalyst screen (Table 1). As in the racemic process,17 tertiary amines were effective promoters of the silylazidation of 1 with equimolar TMSN3 to produce the intermediate acyl azide 2 in CHCl3 at −50 °C.31 In order to separate any confounding factors that could alter the enantioselectivity in the desymmetrisation step, the intermediate acyl azide 2 was efficiently quenched with excess pyrrolidine to provide amido ester 3 after desilylation, extraction of the acid and methylation with TMSCHN2.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | t (h) | Conversiona (azide 2, %) | eeb (%) |
| a Determined by 1H NMR spectroscopic analysis. b Determined by CSP-UHPLC, see ESI.† | ||||
| 1 | — | 24 | <5 | — |
| 2 | 4 | 16 | 67 | −3 |
| 3 | 5 | 16 | 99 | rac |
| 4 | 6 | 16 | 99 | 16 |
| 5 | 7 | 16 | 90 | 8 |
| 6 | 8 | 16 | 90 | rac |
| 7 | 9 | 16 | 81 | 14 |
| 8 | 10 | 16 | 86 | −28 |
| 9 | 11 | 16 | 72 | 24 |
| 10 | 12 | 16 | 72 | 34 |
| 11 | 13 | 16 | 90 | 55 |
| 12 | 14 | 16 | 90 | 54 |
| 13 | 15 | 24 | 90 | 47 |
| 14 | 16 | 18 | 89 | 40 |
| 15 | 17 | 18 | 90 | 48 |
| 16 | 18 | 18 | 99 | 58 |
| 17 | 19 | 18 | 99 | 40 |
| 18 | 20 | 24 | 80 | 56 |
| 19 | 21 | 18 | 90 | 33 |
| 20 | 22 | 24 | 99 | 29 |
| 21 | 23 | 24 | 90 | 23 |
| 22 | 24 | 18 | 99 | 57 |
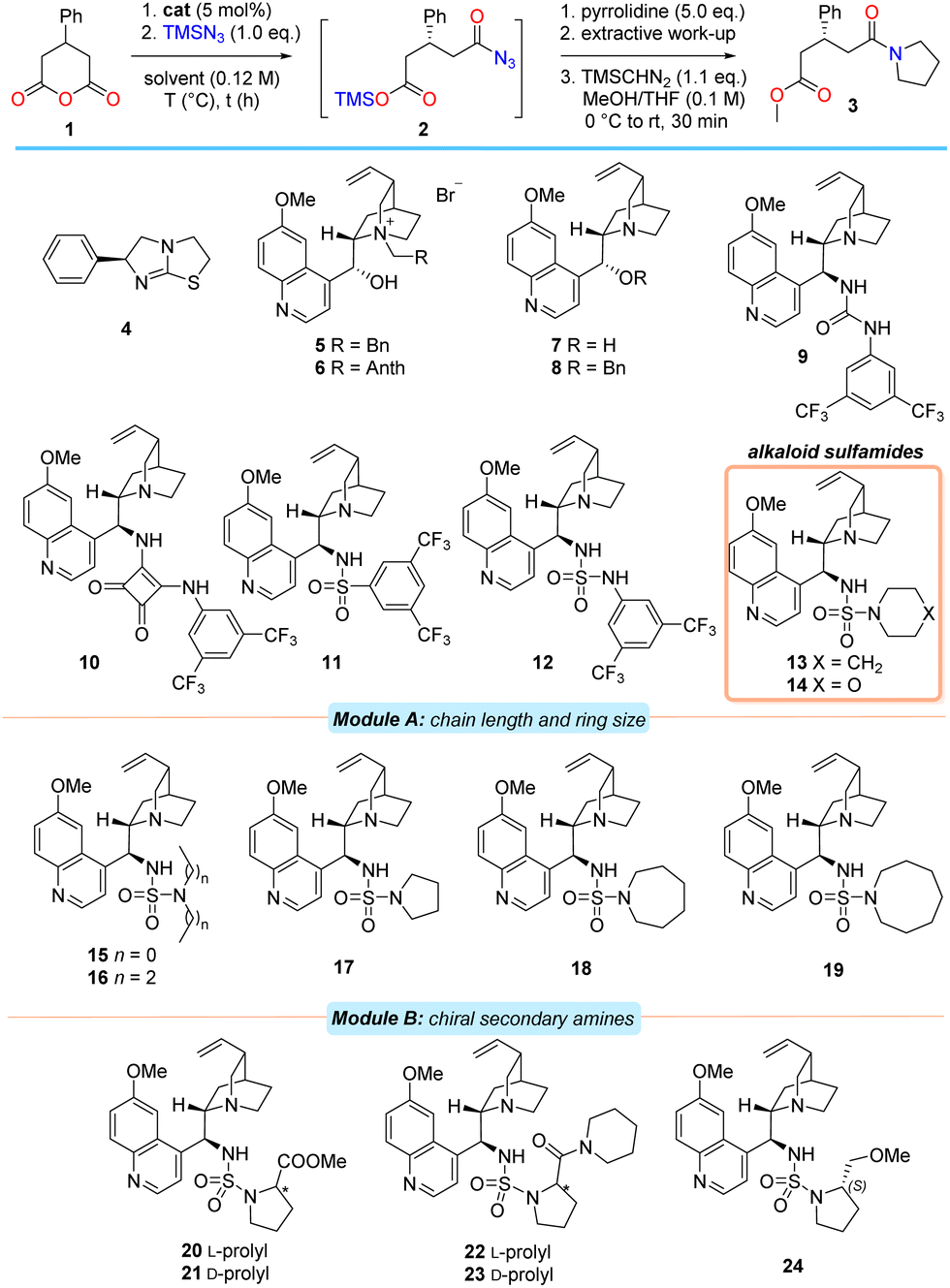
Commercial (S)-benzotetramisole32 (4), natural configuration Cinchona alkaloid-based phase-transfer agents 5–6 and bifunctional free-base alkaloid catalysts 7–8 were found to promote the reaction efficiently but with an almost complete absence of enantiocontrol (entries 1–6). Examination of the 9-epi-quinine-derived urea, -squaramide and -sulfonamide catalysts 9–11 with superior hydrogen bond donor (HBD) units provided amido acid 3 in only modest ee and curiously, with a preference for the formation of the opposite enantiomer in the case of squaramide 10 (entries 7–9).16 Incorporation of the sulfamide motif as the HBD into the Cinchona alkaloid scaffold (i.e., alkaloid 12) proved advantageous and provided 3 in marginally improved ee (entry 10). Further modest improvement in enantioselectivity was observed after exchange of the aniline moiety for an aliphatic, secondary amine (i.e., catalyst 13, entry 11). This was somewhat surprising in view of both literature precedent33 and the loss of the catalyst's ability to participate in efficient bifurcated hydrogen bond donation. However, substitution of the piperidine unit for morpholine did little to influence the enantioselectivity of the process, suggesting that the electronic characteristics at the secondary amine substituent of the sulfamide (i.e., 14) are unimportant (entry 12). After establishing the class of HBD most suitable, further modifications of both the tertiary sulfamide unit and alkaloid core were undertaken.
A modular catalyst design strategy was adopted (Table 1). Module A examined the effect of either incorporating acyclic amines or modifying amine ring size on enantioselectivity. Module B involved an additional peripheral chirality element. Module C represents the later combination of the optimal structural features from modules A and B and the finalisation of catalyst development (Table 2).
Alteration of the secondary amine to either an acyclic amine or a reduction in ring size from 6 to 5 resulted in poorer enantioselectivities relative to 13 (i.e., 15–17, entries 13–15). While increasing the heterocycle ring size from 6 to 7 atoms was beneficial (i.e., catalyst 18, entry 16), further expansion to an azocane system resulted in substantially poorer ee (i.e., 19, entry 17).
Evaluation of 20 and 21, prepared from proline methyl ester antipodes, revealed stark differences between the diastereomers with respect to reactivity and selectivity. In the case where the stereochemistry on the prolyl unit matched that at C9 of the alkaloid core (i.e., ‘matched’ centres), the reaction required marginally extended reaction times, but provided the product in significantly higher ee compared to the ‘mismatched’ case (entries 18 and 19). The same (albeit less pronounced) effect was also observed in the case of the diastereomeric prolinamide-derived sulfamides 22 and 23 (entries 20 and 21). Separately, evaluation of the ‘matched’ case of methyl ether 24 provided the amido acid 3 in a slightly more selective process than obtained using 20 (entry 22).
Examination of model systems (see ESI†) based on fragments of catalyst 13 revealed that the quinoline endocyclic nitrogen atom (located far from the catalyst's stereochemical information) could independently participate in the activation of TMSN3, thereby competing with catalysis at the quinuclidine moiety. This could be obviated in the model system through the installation of a chlorine atom at C2. In a similar vein, methoxy-quinolines were expected to be more active catalysts than quinoline itself.
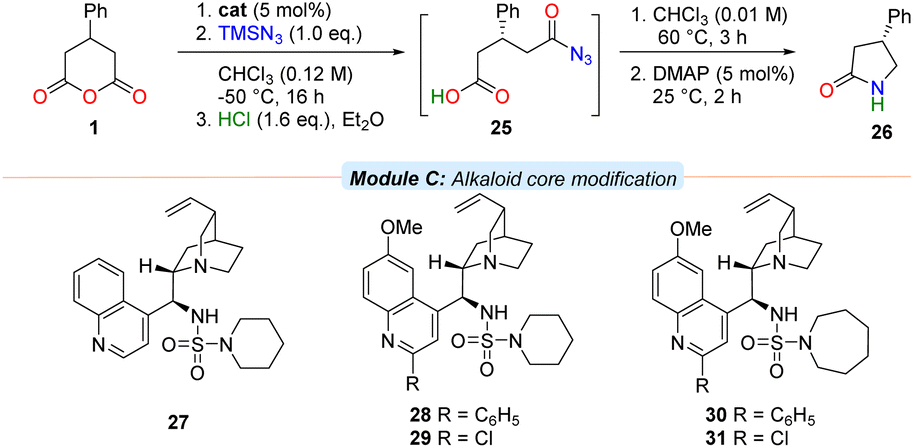
To test the hypothesis that the quinoline moiety negatively contributes to enantioselective reaction in bifunctional systems, the cinchonidine-derived piperidine sulfamide 27, along with C2′-substituted analogues of 13 and 18 (i.e., 28–29 and 30–31 respectively) were prepared and evaluated (Table 2). Anhydride 1 was subjected to the azidolysis conditions promoted by core-modified sulfamide catalysts 27–31, and the intermediate silyl ester 2 then cleaved with anhydrous HCl to isolate the acyl azide 25. Facile Curtius rearrangement and subsequent lactamisation provided the more potent enantiomer of (R)-phenibut lactam (26).
Gratifyingly, the cinchonidine-derived sulfamide 27 outperformed the analogous quinine-derived catalyst 13 (entry 1). A further increase in selectivity was observed on substitution of the C2′ position of the quinoline unit of the catalyst to incorporate either a phenyl group or a chlorine atom (entries 2 and 3). A consistent trend in enantioselectivity was observed upon examination of both the C2′-phenyl azepane sulfamide 30 (entry 4), and its C2′-chloro derivative 31; the latter proved a marginally more selective promoter of the desymmetrisation process (entry 5, 70% ee).
With conditions in hand for the enantioselective azidolysis, a range of cyclic anhydrides 32 were subjected to the azidative desymmetrisation procedure to provide acyl azides 33, promoted by sulfamide 31. These intermediates could be telescoped into Curtius rearrangement and ring-contractive lactamisation steps (vide supra) to provide enantioenriched γ-lactams 34 in one-pot (Table 3).

|
Both electron-donating and electron-withdrawing substituents on the aromatic ring were well-tolerated; providing access to β-aryl-γ-lactams 26 and 35–40 in uniformly high yields and good ee, most notably arbaclofen lactam (36) and the PDE4 inhibitor rolipram (37). Regarding aliphatic substitution patterns: while methyl and isopropyl substituents were compatible when placed at the 3-position (i.e., lactams 42 and 43), the presence of larger silyl ether and isobutyl groups led to a small loss in enantioselectivity, although reactivity in the subsequent lactamisation process was maintained (i.e., lactam 41 and pregabalin lactam (44), respectively). Interestingly, comparable enantioselectivities were obtained when conformationally-locked norcamphoric anhydride was examined, providing access to the Vince lactam derivative 45.
As is the case in the analogous sulfonamides,34Cinchona alkaloid-derived sulfamides are found to exist as a pair of two rotational isomers (rotamers) in a ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at room temperature on the 1H NMR spectroscopic timescale, which interconvert by rotation about the C9–C4′ bond axis (Fig. 2). Although it could be postulated that one rotamer could be contributing negatively to stereoselection; variable temperature-NMR spectroscopy of piperidine sulfamide 13 revealed temperature-dependent convergence of rotamer populations, with the major rotamer at room temperature (i.e., rotamer A) present almost exclusively at low temperature when observed in situ during catalysis (see ESI†).
:1 ratio at room temperature on the 1H NMR spectroscopic timescale, which interconvert by rotation about the C9–C4′ bond axis (Fig. 2). Although it could be postulated that one rotamer could be contributing negatively to stereoselection; variable temperature-NMR spectroscopy of piperidine sulfamide 13 revealed temperature-dependent convergence of rotamer populations, with the major rotamer at room temperature (i.e., rotamer A) present almost exclusively at low temperature when observed in situ during catalysis (see ESI†).
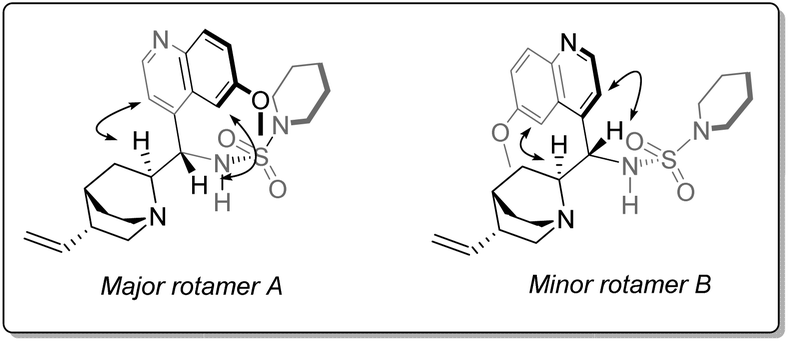 | ||
| Fig. 2 The two rotamers associated with 13, showing key NOE interactions involved in corroborating in silico-derived data. | ||
Although the isolated catalyst does not display this temperature-dependent behaviour as the free base form, the monoprotic acetate salt of 13 exhibited similar behaviour to that observed in situ (Fig. 3). Attempts to isolate the analogous hydrogen azide salt by several methods were unsuccessful. This can be adequately rationalised in the context of similar studies;35 poor room temperature association has been observed in other amine complexes with HN3, resulting in its dissociation on irreversible loss of HN3(g) by evaporation. However, as the pKa (AcOH) ≅ pKa (HN3) at 25 °C, and given the similarities regarding the temperature-dependent behaviour (with respect to rotamer ratios and 1H NMR spectroscopic chemical shifts) displayed by the catalyst species in situ and the isolated AcOH salt of 13 were found, the evidence suggests that the catalytically-active species in solution is the structurally-related HN3 complex with 13.
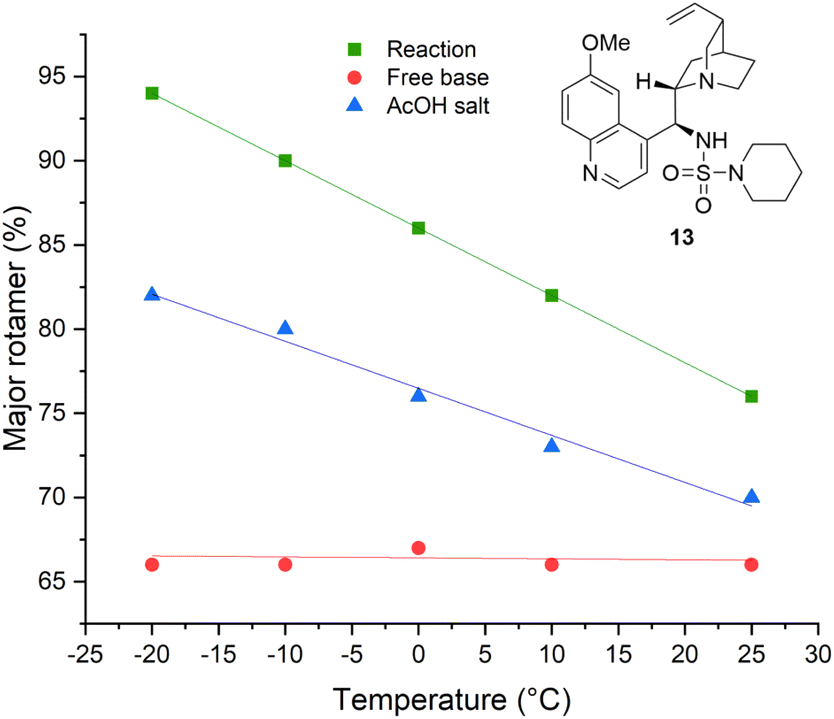 | ||
| Fig. 3 Temperature-dependent populations of rotameric states of catalyst 13 as the free base, the acetic acid salt and the catalyst observed in situ by variable-temperature 1H NMR spectroscopy. | ||
Furthermore, it can be proposed that the free base form of the model sulfamide catalyst 13 is first converted to the active hydrazoate complex 13a by trapping of adventitious HN3, which is present in small amounts in commercial samples of TMSN3 (Fig. 4). This nucleophilic species then facilitates transfer of azide to the anhydride via a stereodetermining addition–elimination reaction at the prochiral carbonyl centre of 1. The resulting carboxylate 13b is then silylated by TMSN3 to liberate the product 25 and regenerate the active catalyst 13a.
 | ||
| Fig. 4 Proposed catalytic cycle for the desymmetrisation of cyclic anhydrides with equimolar TMSN3 promoted by Cinchona alkaloid sulfamide organocatalysts. | ||
A DFT conformational analysis exploring the low-energy chemical space associated with 13 was performed. Two predominant conformers differing by the rotation of the C9–C4′ bond were identified (Fig. 5). The Boltzmann population ratio (66:34) predicted by the calculation in CHCl3 is in good agreement with those obtained from 1H NMR spectroscopic analysis. In addition, a repeat of the calculations at 223 K yielded a very similar population ratio of 61:39.
 | ||
| Fig. 5 DFT calculations: structures, relative stabilities and QTAIM analysis of the major rotamers of catalyst 13. | ||
A characterisation of the different intramolecular non-covalent interactions was also performed by means of the quantum theory of atoms in molecules (QTAIM) methodology (Fig. 5). An intramolecular hydrogen-bond between the quinuclidine N-atom and the sulfamide unit is a discernible rigidifying feature of both conformations. The other interactions identified appear to be weak in nature.
As is the case in the analogous sulfonamides,34 the Cinchona alkaloid-derived sulfamide catalysts exist as a pair of rotamers in a ca. 2:1 ratio at 25 °C on the 1H NMR spectroscopic timescale, which interconvert by rotation about the C9–C4′ bond axis. Variable temperature-NMR spectroscopy revealed a convergence of rotamer populations, with the major rotamer at room temperature present almost exclusively at low temperature when observed in situ during catalysis (see ESI†). Although the isolated catalyst does not display this temperature-dependent behaviour as the free base form, the AcOH salt of 13 exhibited similar behaviour to that observed in situ. For spectroscopic evidence supporting a hydrazoic acid salt of 13 catalyst resting state, DFT calculations on the catalyst rotamers and a proposed reaction mechanism, see the ESI.†
In a demonstration of the potential synthetic utility of the desymmetrisation, lactam 26 could be prepared on a larger scale under the developed conditions and then converted to enantiopure form in 61% overall yield in a single recrystallisation, which can be transformed to either the anxiolytic phenibut35 or the nootropic phenotropil36 (Scheme 1).
 | ||
| Scheme 1 Recrystallisation of (R)-phenibut lactam 26. | ||
In summary, the first catalytic asymmetric addition of azide to a carbonyl electrophile has been developed. In the presence of novel bifunctional Cinchona alkaloid-derived sulfamide catalysts, prochiral glutaric anhydride derivatives undergo desymmetrisation via addition of TMSN3. The resulting enantioenriched acyl azide derivatives can be readily converted to either γ-amino acid derivatives or a wide range of γ-lactams of considerable medicinal/pharmaceutical interest. Further studies on the scope, utility and mechanism are underway.
Experimental section
NMR spectral data were obtained from a Bruker DPX (400 MHz) or Bruker Avance II (600 MHz) using CDCl3, DMSO-d6 or D2O with chemical shift data referenced relative to residual protic resonances of the deuterated solvent, (δH = 7.26, 2.50, and 4.79 ppm respectively). 13C (100.9 or 150.9 MHz) spectra were recorded on the same instruments with total proton decoupling. Additional 2D spectral acquisitions (HSQC-ME, HMBC, TOCSY, NOESY/EXSY) were obtained in order to assist in the assignment of resonances where required. Conventional abbreviations for describing peak morphologies in NMR spectroscopic analysis are observed (i.e. s, singlet; br s, broad singlet; d, doublet; dd, doublet of doublets, etc.). All coupling constants (J) are reported in hertz (Hz). Infrared spectra were obtained as neat solids or liquids unless otherwise stated on a Perkin-Elmer Spectrum100 FT-IR instrument fitted with an attenuated-total reflectance (ATR) accessory. Abbreviations used for descriptions of transmission band intensities are as follows: w, weak; m, medium; s, strong; vs, very strong; br., broad.Thin-layer chromatography (TLC) analyses were performed using Merck-F254 silica gel plates and were visualised under ultraviolet (UV) irradiation, potassium permanganate, ninhydrin, ammonium molybdate or bromocresol green staining methods. Column and flash chromatography was performed using Sigma-Aldrich 60 Å, 230–400 mesh particle silica gel. Melting point data were recorded on a Griffin Melting Point Apparatus; readings were obtained in triplicate and are reported uncorrected. High-resolution mass spectrometry experiments were carried out in the Mass Spectrometry Unit, School of Chemistry, TCD.
Anhydrous CHCl3 (amylene-stabilised) and HCl (as 2 M solution in Et2O) were obtained from Sigma-Aldrich Ireland and transferred to reaction vessels using Schlenk techniques. Hünig's base on polystyrene (DIPEA@PS, product ID: 38343) was purchased from Sigma-Aldrich Ireland and all other chemicals were of regent-grade, obtained from commercial suppliers and used without further purification unless otherwise noted.
Safety considerations
While we have not experienced any issues surrounding the use of TMSN3 in these studies, it is imperative that the appropriate safety precautions are taken, especially when working on reaction scales >1 mmol. In the following preparations, TMSN3 has the potential to liberate toxic and explosive HN3 on contact with H2O or in acidic media. Any volatiles removed should be carried out in a well-ventilated fume hood and reactions performed with a blast shield in large-scale preparations. It is advised that all azide-containing waste should be quenched cautiously, and in an appropriate manner.37General procedure A: preparation of 9-epi-9-amino Cinchona alkaloids from native configuration alkaloids
To an oven-dried 250 mL round bottomed flask containing a stirrer bar, Cinchona alkaloid derivative (18.50 mmol) and PPh3 (5.82 g, 22.19 mmol) under Ar atmosphere, anhydrous THF (125 mL, 0.15 M) was added via syringe. The solution was cooled to 0 °C before DIAD (4.40 mL, 22.19 mmol) and DPPA (4.77 mL, 22.19 mmol), were added sequentially dropwise via syringe. The resulting yellow solution was warmed to room temperature and stirred at 20 °C for 24 h. The flask was fitted with a reflux condenser and the solution stirred at 50 °C for a further 2 h. PPh3 (5.82 g, 22.19 mmol) was added portionwise with stirring and the solution heated at 50 °C for 2 h or until nitrogen evolution had ceased. H2O (26.4 mL, 0.7 M) was added and the solution stirred at room temperature for 16 h. The resulting mixture was concentrated as far as possible in vacuo and the residue partitioned between 2 M HCl and CH2Cl2 (100 mL each). The aqueous phase was removed and the organic layer extracted with 2 M HCl (3 × 50 mL). The combined aqueous extracts were washed with CH2Cl2 (5 × 50 mL) and concentrated as far as possible. The viscous residue was stirred in EtOH and the resulting precipitate filtered and dried in vacuo. The precipitate can be purified by reprecipitation from boiling MeOH using EtOAc as antisolvent to give the alkaloid hydrochloride as a powder.

General procedure B: sulfamoyl chloride synthesis
SO2Cl2 (1.5 eq.) in anhydrous CH2Cl2 (1.00 M) was cooled to −20 °C under Ar atmosphere before a solution of NEt3 (1.5 eq.) and the appropriate secondary amine (1.0 eq.) in anhydrous CH2Cl2 (2 M with respect to amine) was added dropwise via syringe (>30 min, exothermic). The resulting solution was stirred at −20 °C for 30 min before warming to room temperature over 1.5 h. The resulting yellow mixture was slowly poured into ice-H2O using CH2Cl2 to effect the transfer. The biphasic mixture was partitioned and the organic layer washed with H2O and brine before being dried over anhydrous MgSO4, filtered and the filtrate concentrated in vacuo. The residue was dissolved in the minimum CH2Cl2 and passed through a short plug of silica, eluted with CH2Cl2 to provide the analytically-pure sulfamoyl chloride product after drying in vacuo.General procedure C: Cinchona alkaloid sulfamide preparation from sulfamoyl chlorides
To a 25 mL round bottomed flask containing a magnetic stirrer bar and the appropriate alkaloid hydrochloride salt (1.00 mmol) was added anhydrous CH2Cl2 (5.00 mL, 0.20 M). To the resulting suspension, NEt3 (4.20 mmol) was added dropwise at −5 °C and the resulting suspension stirred vigorously for 30 min before sulfamoyl chloride (1.20 mmol) was added dropwise via syringe. The resulting solution was stirred at room temperature for 24–48 h until consumption of the sulfamoyl chloride was observed by TLC analysis. The solution was diluted with CH2Cl2 (25 mL) and washed sequentially with half-saturated NaHCO3(aq.), H2O and brine (2 × 10 mL each). The solution was dried over anhydrous MgSO4, filtered and concentrated in vacuo to give a yellow oil which was purified by flash chromatography as appropriate.

Azepane-1-sulfonyl chloride
Prepared according to general procedure B using azepane (376 μL, 3.33 mmol) and purified by passing through a short plug of silica, eluting with CH2Cl2 to give the product as a colourless oil (394.4 mg, 60%). TLC (CH2Cl2, ninhydrin): Rf = 0.83. δH (400 MHz, CDCl3): 3.46–3.53 (4H, m, H-1), 1.80–1.86 (4H, m, H-2) and 1.63–1.69 (4H, m, H-3) ppm. δC (101 MHz, CDCl3): 50.1 (C-1), 27.5 (C-3) and 27.0 (C-2) ppm. νmax (neat)/cm−1: 2932 (m), 2860 (m), 1462 (w), 1386 (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, s), 1367 (s), 1172 (s), 1144 (m), 1042 (m), 888 (m) and 693 (s) cm−1.
O, s), 1367 (s), 1172 (s), 1144 (m), 1042 (m), 888 (m) and 693 (s) cm−1.
2′-Chloro-9-amino-(9-deoxy)-epi-quininium trihydrochloride (precursor to 31)
Prepared according to general procedure A using C2′-chloroquinine38 (1.39 g, 3.88 mmol) and precipitated after co-evaporation of residual H2O with EtOH to give the product as a bright yellow powder (1.09 g, 80%), m.p. 198–204 °C (decomp.); [α]24D = +2.5 (c = 0.20, H2O). 1H, 13C NMR and EXSY spectroscopic analyses in DMSO-d6 revealed rotameric species in the ratio 93:7 at 25 °C. 13C resonances are clearly observable for the major rotamer only. Major rotamer: δH (400 MHz, DMSO-d6): 11.12, 9.48 (3H, br s), 8.15 (1H, s), 7.97 (1H, d, J 9.2), 7.83 (1H, d, J 1.9), 7.58 (1H, dd, J 9.2, 1.9), 5.87–5.96 (1H, m), 5.82 (1H, d, J 10.4), 5.26 (1H, d, J 17.3), 5.16 (1H, d, J 10.5), 4.61–4.68 (1H, app. q), 4.09–4.18 (1H, m), 4.01 (3H, s), 3.70–3.76 (1H, m), 3.28–3.38 (2H, m), 2.76 (1H, br s), 1.80–1.92 (3H, m), 1.57–1.63 (1H, m) and 0.87 (1H, dd, J 13.3, 8.4) ppm. δC (151 MHz, DMSO-d6): 158.7, 146.9, 143.6, 141.6, 138.3, 130.4, 126.7, 123.7, 122.0, 116.7, 103.0, 58.7, 56.4, 52.1, 47.7, 41.6, 35.9, 25.5, 23.6 and 23.4 ppm. Minor rotamer: δH (400 MHz, DMSO-d6): 11.12, 9.48 (3H, br s), 8.11 (1H, s), 7.98 (1H, d, J 9.0), 7.54 (1H, dd, J 9.0, 2.0), 7.49 (1H, d, J 2.0), 5.77–5.86 (1H, m), 5.41 (1H, d, J 17.5), 5.22–5.25 (1H, m, J 10.4), 5.16 (1H, d, J 10.5), 4.96 (1H, app. q.), 4.06 (3H, s), 3.93–3.95 (1H, m), 3.70–3.76 (1H, m), 3.28–3.38 (2H, m), 2.76 (1H, br s), 1.99 (1H, br s), 1.80–1.92 (2H, m), 1.21–1.29 (1H, m) and 1.06–1.15 (1H, dd, J 13.3, 8.4) ppm. νmax (neat)/cm−1: 3478 (m, NH st.), 2560 (w), 1617 (s), 1510 (m), 1460 (m), 1395 (m), 1320 (w), 1279 (m), 1235 (s), 1140 (s), 1019 (m), 920 (s), 831 (s), 774 (s), 728 (w) and 681 (s) cm−1. HRMS (APCI+) m/z: Found: 358.1685 ([M + H]+ C20H25ClN3O; requires 358.1680).
C2′-Chloroquinine azepane sulfamide 31
Prepared according to general procedure C using 2′-chloro-9-amino-(9-deoxy)-epi-quininium trihydrochloride (573 mg, 1.33 mmol) and azepane-1-sulfonyl chloride (291.3 mg, 1.47 mmol), purified by flash chromatography (7:3 CH2Cl2/EtOAc) to give the title product as a white, crystalline powder (271 mg, 39%), m.p. 58–60 °C. TLC (98:2 CH2Cl2/MeOH): Rf = 0.44. [α]22D = +2.1 (c = 0.13, CHCl3). 1H, 13C NMR and EXSY spectroscopic analyses in CDCl3 revealed rotameric species in the ratio 70:30 at 25 °C. Major rotamer: δH (600 MHz, CDCl3): 7.95 (1H, d, J 9.2), 7.56 (1H, s), 7.48 (1H, d, J 2.7), 7.42 (1H, dd, J 9.2, 2.7), 6.12 (1H, br. s), 5.68–5.74 (1H, m), 5.03 (1H), 4.94–4.99 (2H, m), 3.98 (3H, s), 3.19–3.26 (2H, m), 2.75–2.83 (2H, m), 2.55–2.72 (5H, m), 2.28–2.33 (1H, m), 1.57–1.68 (3H, m), 1.37–1.41 (1H, m), 1.27–1.38 (6H, m), 0.86–0.92 (1H, m) ppm. δC (151 MHz, CDCl3): 158.4, 148.5, 148.0, 144.1, 141.0, 130.9, 127.5, 122.7, 121.2, 114.9, 101.5, 61.3, 55.8, 55.75, 53.0, 48.2, 40.4, 39.4, 28.6, 27.9, 27.4, 26.7, 25.2 ppm. Minor rotamer: δH (600 MHz, CDCl3): 7.96 (1H, d, J 9.2), 7.87 (1H, d, J 2.8), 7.40 (1H, dd, J 9.2, 2.8), 7.30 (1H, s), 6.29 (1H, br. s), 5.60–5.66 (1H, m), 4.89–4.95 (2H, m), 4.34 (1H, d, J 10.9), 3.93 (3H, s), 3.35–3.40 (1H, m), 3.19–3.25 (1H, m), 3.05–3.12 (1H, m), 2.72–2.76 (1H, m), 2.55–2.72 (5H, m), 2.28–2.33 (1H, m), 1.73–1.76 (1H, m), 1.58–1.61 (2H, m), 1.27–1.38 (7H, m) and 0.94–0.99 (1H, m) ppm. δC (151 MHz, CDCl3): 157.3, 147.5, 145.1, 144.7, 141.0, 131.0, 125.9, 124.0, 122.5, 114.8, 104.0, 62.6, 56.0, 55.7, 48.3, 40.0, 39.6, 28.6, 27.6, 27.4, 26.6, 26.5 ppm. νmax (neat)/cm−1: 3189 (w, br, N–H st.), 3073 (w, N–H st.), 2926 (m, C–H st.), 2862 (w), 1620 (s), 1581 (m), 1505 (s), 1455 (s), 1394 (m), 1234 (m), 1228 (m), 1143 (vs, br), 1101 (w), 1044 (w), 1030 (w), 987 (m), 941 (s), 880 (m), 828 (m), 768 (w), 692 (vs), 669 (m), 617 (w) and 576 (s) cm−1. HRMS (APCI+) m/z: Found: 519.2195 ([M + H]+ C26H36ClN4O3S; requires 519.2192).
General procedure D: organocatalytic, enantioselective synthesis of chiral γ-lactams from prochiral anhydrides
To a 5 mL round bottomed flask containing a magnetic stirrer bar, sulfamide 31 (6.4 mg, 0.012 mmol) and achiral or meso-anhydride 32 (0.246 mmol) under Ar atmosphere, anhydrous CHCl3 (2.00 mL, 0.12 M) was added via syringe before the solution was cooled to −50 °C for 30 min. TMSN3 (32.4 μL, 0.246 mmol) was then added in one portion and the resulting solution stirred at −50 °C for 16 h. HCl in Et2O (200 μL, 0.400 mmol) was added in one portion and the resulting solution stirred for 15 min at −50 °C. The solution was filtered, using anhydrous CHCl3 (1 mL) to effect the transfer and volatiles removed expediently in vacuo to give the analytically-pure acyl azide 33. The solid was placed under Ar atmosphere and anhydrous CHCl3 (25.0 mL, 0.01 M) added via syringe. The resulting solution was heated gently (vigorous gas evolution observed at ca. 40 °C) to 60 °C for 3 h under Ar atmosphere. The solution of isocyanate was then cooled to 25 °C before DMAP (1.5 mg, 0.012 mmol) was added in one portion and the resulting solution stirred vigorously at 25 °C for 2 h. The solution was concentrated in vacuo and the residue purified by flash column chromatography to give the γ-lactam product.:2 CH2Cl2/MeOH) to give the product as a white powder (35.7 mg, 90%, 69% ee), m.p. 75–76 °C (lit.,39 m.p. 73–75 °C). TLC (CH2Cl2/MeOH, 98:2): Rf = 0.42. A larger scale preparation using anhydride 1 (190.2 mg, 1.00 mmol) afforded the title product by the same method (148.3 mg, 92%, 70% ee) which was recrystallised from hot Hex/EtOAc to provide large, colourless plate crystals (100.6 mg, 62%, >99% ee) with [α]22D = −39.6 (c = 0.91, CHCl3), (lit.,40 [α]25D = −39.4 (c = 0.90, CHCl3) for 99% ee of the (R)-enantiomer). Spectroscopic data correlates well to that in the literature.7 CSP-SFC analysis: step 3 was employed with UV detection at 254 nm; RT: 3.45 min (minor enantiomer) and 3.56 min (major enantiomer). δH (400 MHz, CDCl3): 7.33–7.36 (2H, m), 7.25–7.28 (3H, m), 6.09 (1H, br s), 3.79 (1H, dd, J 9.4, 8.3), 3.71 (1H, app. quin.), 3.43 (1H, dd, J 9.4, 7.3), 2.75 (1H, dd, J 17.0, 9.0) and 2.52 (1H, dd, J 17.0, 8.9) ppm. δC (100 MHz, CDCl3): 177.7, 142.1, 129.1, 127.4, 126.9, 49.7, 40.5 and 38.1 ppm. HRMS (APCI+) m/z: Found: 162.0912 ([M + H]+; C10H12NO requires: 162.0913).
:2 CH2Cl2/MeOH) to give the product as a white powder (40.5 mg, 94%, 65% ee), m.p. 110–112 °C (lit.,41 m.p. 108–110 °C). TLC (EtOAc): Rf = 0.40. [α]22D = −9.6 (c = 0.15, CHCl3), (lit.,40 [α]20D = −33.7 (c = 0.95, CHCl3) for 99% ee). CSP-SFC analysis (see ESI†); RT: 5.50 min (minor enantiomer) and 5.88 min (major enantiomer). δH (400 MHz, CDCl3): 7.14 (4H, app. s), 6.71 (1H, br. s), 3.77 (1H, dd, J 9.4, 8.3), 3.61–3.70 (1H, m), 3.40 (1H, dd, J 9.4, 7.4), 2.77 (1H, dd, J 16.9, 8.8), 2.49 (1H, dd, J 16.9, 8.9) and 2.33 (3H, s) ppm. δC (100 MHz, CDCl3): 177.9, 139.0, 136.8, 129.5, 126.7, 49.8, 40.0, 38.2 and 21.0 ppm.
:2 CH2Cl2/MeOH): Rf = 0.31. [α]22D = −16.5 (c = 0.15, CHCl3), (lit.,39 [α]20D = −39.0 (c = 1.00, CHCl3) for 99% ee). CSP-SFC analysis (see ESI†): RT: 3.47 min (major enantiomer) and 3.70 min (minor enantiomer). δH (600 MHz, CDCl3): 7.30–7.32 (2H, m), 7.17–7.20 (2H, app. d.), 6.14 (1H, br. s, H-1), 3.78 (1H, dd, J 9.5, 8.3), 3.65–3.70 (1H, m), 3.38 (1H, dd, J 9.5, 7.1), 2.74 (1H, dd, J 16.9, 9.0) and 2.46 (1H, dd, J 16.9, 8.6) ppm. δC (151 MHz, CDCl3): 177.2, 140.6, 133.0, 129.0, 128.1, 49.3, 39.7 and 37.7 ppm.
:2 CH2Cl2/MeOH) to give the title product as an off-white crystalline powder (64.3 mg, 95%, 70% ee), m.p. 132–133 °C (lit.,43 m.p. 131–133 °C). TLC (98:2 CH2Cl2/MeOH): Rf = 0.30. [α]22D = −12.1 (c = 0.15, MeOH), (lit.,39 [α]27D = −33.0 (c = 1.00, MeOH) for 99.3% ee). CSP-SFC analysis: RT: 4.01 min (minor enantiomer) and 4.22 min (major enantiomer). δH (400 MHz, CDCl3): 6.83–6.84 (1H, m), 6.76–6.79 (2H, m), 6.06 (1H, br s.), 4.74–4.79 (1H, m), 3.83 (3H, s), 3.75 (1H, dd, J 9.3, 8.2), 3.38 (1H, dd, J 9.3, 7.4), 2.71 (1H, dd, J 16.9, 8.8), 2.47 (1H, dd, J 16.9, 8.9)1.78–1.97 (6H, m) and 1.56–1.66 (2H, m) ppm. δC (100 MHz, CDCl3): 177.6, 149.3, 148.0, 134.6, 118.9, 113.9, 112.3, 80.8, 56.2, 49.8, 40.1, 38.1, 32.9 and 24.1 ppm.
:2 CH2Cl2/MeOH) to give the title product as a white crystalline powder (37.4 mg, 91%, 65% ee), m.p. 86–88 °C. TLC (98:2 CH2Cl2/MeOH): Rf = 0.28. [α]22D = −14.0 (c = 0.15, CHCl3). CSP-SFC analysis (see ESI†): RT: 3.35 min (major enantiomer) and 3.52 min (minor enantiomer). δH (400 MHz, CDCl3): 7.32 (1H, dd, J 5.0, 2.9), 7.02 (1H, dd, J 2.9, 1.3), 6.99 (1H, dd, J 5.0, 1.3), 6.58 (1H, br. s), 3.73–3.82 (2H, m), 3.39–3.45 (1H, m), 2.70–2.77 (1H, m,) and 2.44–2.54 (1H, m, H-2b) ppm. δC (100 MHz, CDCl3): 177.8, 142.7, 126.7, 126.2, 120.4, 49.2, 37.9 and 35.9 ppm.
:1 EtOAc/CH2Cl2) to give the product as a white, crystalline powder (39.5 mg, 90%, 66% ee), m.p. 97–99 °C (lit.,44 m.p. 98–99 °C). TLC (1:1 EtOAc/CH2Cl2): Rf = 0.15. [α]22D = −7.8 (c = 0.15, MeOH), (lit.,45 [α]25D = −26.2 (c = 1.00, MeOH) for 96% ee). CSP-SFC analysis (see ESI†): RT: 3.01 min (major enantiomer) and 3.14 min (minor enantiomer). δH (400 MHz, CDCl3): 7.18–7.23 (2H, m), 6.99–7.05 (2H, m), 6.70 (1H, br s), 3.77 (1H, dd, J 9.3, 8.3), 6.62–3.71 (1H, m), 3.37 (1H, dd, J 9.3, 7.2), 2.72 (1H, dd, J 16.9, 8.9) and 2.44 (1H, dd, J 16.9, 8.7) ppm. δF (376 MHz, CDCl3): −115.53 (s) ppm. δC (100 MHz, CDCl3): 177.8, 162.0 (d, 1JC–F 245.5), 138.0 (d, 4JC–F 3.1), 128.4 (d, 2JC–F 8.0), 115.8 (d, 3JC–F 21.2), 49.8, 39.8 and 38.2 ppm.
:2 CH2Cl2/MeOH) to give the product as a white, crystalline powder (45.2 mg, 94%, 65% ee), m.p. 112–114 °C (lit.,41 m.p. 112–115 °C). TLC (EtOAc): Rf = 0.49. [α]22D = −9.4 (c = 0.10, CHCl3). CSP-SFC analysis (see ESI†): RT: 6.77 min (minor enantiomer) and 7.13 min (major enantiomer). δH (400 MHz, CDCl3): 7.39 (1H, dd, J 7.8, 1.4), 7.33 (1H, dd, J 7.7, 1.6), 7.25–7.29 (1H, m), 7.18–7.23 (1H, m), 6.45 (1H, br. s), 4.12–4.20 (1H, m), 3.86 (1H, dd, J 9.7, 8.2), 3.42 (1H, dd, J 9.7, 6.0), 2.79 (1H, dd, J 17.0, 9.1) and 2.53 (1H, dd, J 17.0, 7.3) ppm. δC (101 MHz, CDCl3): 177.5, 139.3, 133.8, 130.0, 128.4, 127.4, 127.2, 48.3, 36.68 and 36.66 (C-3) ppm.
:1 Hex/EtOAc) to give the title product as a white powder (42.4 mg, 80%, 56% ee), m.p. 78–80 °C (lit.,46 m.p. (from PE/EtOAc) 84–86 °C). TLC (1:1 Hex/EtOAc, ninhydrin): Rf = 0.19. [α]22D = −2.4 (c = 0.15, CHCl3), (lit.,47 [α]22D = −7.4 (c = 1.30, CHCl3)). CSP-SFC analysis (see ESI†): RT: 2.61 min (minor enantiomer) and 2.73 min (major enantiomer). δH (400 MHz, CDCl3): 5.98 (1H, br s), 4.53–4.58 (1H, m), 3.58 (1H, dd, J 10.0, 6.0), 3.24 (1H, dd, J 10.0, 3.4), 2.54 (1H, dd, J 17.0, 6.8) and 2.26 (1H, dd, J 17.0, 4.2) ppm. δC (100 MHz, CDCl3): 176.2, 68.0, 51.6, 40.5, 25.8, 18.0, −4.7, and −4.8 ppm.
:2 CH2Cl2/MeOH) to give the product as a white powder (22.2 mg, 91%, 70% ee), m.p. 54–55 °C (lit.,48 m.p. (from Hex) 53–55 °C). TLC (95:5 CH2Cl2/MeOH, KMnO4): Rf = 0.50. [α]22D = −4.0 (c = 0.10, CHCl3), (lit.,49 [α]25D = −20.3 (c = 1.20, CHCl3) for 99% ee). CSP-SFC analysis (see ESI†): RT: 5.26 min (minor enantiomer) and 5.43 min (major enantiomer). δH (400 MHz, CDCl3): 6.25 (1H, br s), 3.53 (1H, dd, J 9.4, 7.6), 2.99 (1H, dd, J 9.4), 2.51–2.64 (1H, m), 2.48 (1H, dd, J 16.5, 8.5), 1.97 (1H, dd, J 16.5, 7.1) and 1.16 (3H, d, J 6.7) ppm. δC (100 MHz, CDCl3): 178.6, 49.6, 38.5, 29.6 and 19.7 ppm.
:2 CH2Cl2/MeOH) to give the product as a white powder (29.0 mg, 92%, 70% ee), m.p. 90–92 °C (lit.,50 m.p. 96–97 °C). TLC (97:3 CH2Cl2/MeOH, ninhydrin): Rf = 0.25. [α]22D = +1.8 (c = 0.10, CHCl3), (lit.,50 [α]25D = +16.9 (c = 1.05, CHCl3) for 99% ee). CSP-SFC analysis (see ESI†): RT: 5.12 min (minor enantiomer) and 5.30 min (major enantiomer). δH (600 MHz, CDCl3): 5.89 (1H br. s), 3.46 (1H, dd, J 9.3, 8.3), 3.09 (1H, dd, J 9.2, 8.3), 2.39 (1H, dd, J 16.7, 8.7), 2.17–2.26 (1H, m), 2.07 (1H, dd, J 16.7, 9.6), 1.56–1.64 (1H, m), 0.93 (3H, d, J 6.7) and 0.90 (3H, d, J 6.6) ppm. δC (151 MHz, CDCl3): 178.3, 46.2, 42.3, 35.2, 32.5, 20.6 and 20.0 ppm.
:5 CH2Cl2/MeOH): Rf = 0.8. [α]22D = −0.81 (c = 0.16, CHCl3), (lit.,51 [α]20D = −2.42 (c = 1.00, CHCl3) for 99% ee). CSP-SFC analysis (see ESI†): RT: 1.88 min (minor enantiomer) and 2.00 min (major enantiomer). δH (600 MHz, CDCl3): 6.28 (1H, br s), 3.47 (1H, dd, J 9.3, 7.9), 2.98 (1H, dd, J 9.3, 7.1), 2.53 (1H, app. sept.), 2.40 (1H, dd, J 16.7, 8.6), 1.97 (1H, dd, J 16.7, 8.5), 1.52–1.61 (1H, m), 1.30–1.37 (2H, m) and 0.89 (6H, app. t, J 6.5) ppm. δC (151 MHz, CDCl3): 178.5, 48.3, 43.9, 37.1, 33.0, 26.2, 22.7 and 22.5 ppm. HRMS (ESI+) m/z: Found: 164.1047 ([M + Na]+; C8H15NNaO requires: 164.1046).
:2 CH2Cl2/MeOH) to give title compound as a white powder (21.8 mg, 80%, 72% ee), m.p. 79–81 °C (lit.,52 m.p. (from iPrOH) 78–81 °C. TLC (98:2 CH2Cl2/MeOH, ninhydrin): Rf = 0.26. [α]22D = −48.6 (c = 0.15, CHCl3), (lit.,53 [α]22D = −160.0 (c = 1.00, CHCl3)). CSP-SFC analysis (see ESI†): RT: 2.67 min (major enantiomer) and 2.78 min (minor enantiomer). δH (400 MHz, CDCl3): 6.02 (1H, br s), 3.86–3.89 (1H, m), 2.71–2.74 (1H, m), 1.77–1.93 (3H, m), 1.54–1.66 (2H, m) and 1.41 (1H, dt, J 9.3, 1.4) ppm. δC (100 MHz, CDCl3): 181.2, 55.4, 45.1, 41.3, 30.2 and 23.7 ppm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Irish Research Council (IRC GOIPG/2017/972) is gratefully acknowledged.Notes and references
- J. P. Griess, Proc. R. Soc. London, 1864, 13, 375 CrossRef.
- E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297 CrossRef CAS.
- J. Aubé, C. Fehl, R. Liu, M. C. McLeod and H. F. Motiwala, Compr. Org. Synth., 2014, 6, 598 Search PubMed.
- T. B. Phan and H. Mayr, J. Phys. Org. Chem., 2006, 19, 706 CrossRef CAS.
- Selected reviews: (a) L. W. Xu and C. G. Xia, Eur. J. Org. Chem., 2005, 633 CrossRef CAS; (b) M. Goswami and B. de Bruin, Eur. J. Org. Chem., 2017, 1152 CrossRef CAS PubMed; (c) P.-G. Ding, X.-S. Hu, F. Zhou and J. Zhou, Org. Chem. Front., 2018, 5, 1542 RSC.
- (a) L. E. Martínez, J. L. Leighton, D. H. Carsten and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5897 CrossRef; (b) K. B. Hansen, J. L. Leighton and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 10924 CrossRef CAS.
- (a) J. F. Larrow, S. E. Schaus, E. N. Jacobsen, V. Har, V. Uni and R. V. May, J. Am. Chem. Soc., 1996, 14, 7420 CrossRef; (b) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421 CrossRef CAS PubMed.
- (a) B. Wu, J. R. Parquette and T. V. RajanBabu, Science, 2009, 326, 1662 CrossRef CAS PubMed; (b) B. Wu, J. C. Galluci, J. R. Parquette and T. V. RajanBabu, Chem. Sci., 2014, 5, 1102 RSC.
- J. K. Myers and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 8959 CrossRef CAS.
- F. González-Bobes, N. Kopp, L. Li, J. Deerberg, P. Sharma, S. Leung, M. Davies, J. Bush, J. Hamm and M. Hrytsak, Org. Process Res. Dev., 2012, 16, 2051 CrossRef.
- T. E. Horstmann, D. J. Guerin and S. J. Miller, Angew. Chem., Int. Ed., 2000, 39, 3635 CrossRef PubMed.
- D. J. Guerin and S. J. Miller, J. Am. Chem. Soc., 2002, 124, 2134 CrossRef CAS PubMed.
- M. Nielsen, W. Zhuang and K. A. Jørgensen, Tetrahedron, 2007, 63, 5849 CrossRef CAS.
- E. B. Rowland, G. B. Rowland, E. Rivera-Otero and J. C. Antilla, J. Am. Chem. Soc., 2007, 129, 12084 CrossRef CAS PubMed.
- T. Bellavista, S. Meninno, A. Lattanzi and G. Della Sala, Adv. Synth. Catal., 2015, 357, 3365 CrossRef CAS.
- (a) M. S. Taylor, D. N. Zalatan, A. M. Lerchner and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 1313 CrossRef CAS PubMed; (b) J. Humbrías-Martín, M. C. Pérez-Aguilar, R. Mas-Ballesté, A. Dentoni Litta, A. Lattanzi, G. Della Sala, J. A. Fernández-Salas and J. Alemán, Adv. Synth. Catal., 2019, 361, 4790 CrossRef.
- (a) S. N. Smith, R. Craig and S. J. Connon, Chem. – Eur. J., 2020, 26, 13378 CrossRef CAS PubMed; (b) S. N. Smith and S. J. Connon, Eur. J. Org. Chem., 2021, 5540 CrossRef CAS.
- (a) J. S. Bryans and D. J. Wustrow, Med. Res. Rev., 1999, 19, 149 CrossRef CAS PubMed; (b) T. Ivšić and Z. Hameršak, Tetrahedron: Asymmetry, 2013, 24, 21 CrossRef.
- O. A. C. Petroff, Neuroscientist, 2002, 8, 562 CrossRef CAS PubMed.
- A. V. Kalueff and D. J. Nutt, Depression Anxiety, 2007, 24, 495 CrossRef CAS PubMed.
- W. Froestl, Future Med. Chem., 2011, 3, 163 CrossRef CAS PubMed.
- P. Nuss, Neuropsychiatr. Dis. Treat., 2015, 1, 165 Search PubMed.
- R. Vardanyan, V. Hruby, R. Vardanyan and V. Hruby, in Synth. Best-Seller Drugs, Academic Press, 2016, p. 155 Search PubMed.
- A. G. Malykh and M. R. Sadaie, Drugs, 2010, 70, 287 CrossRef CAS PubMed.
- For selected reviews detailing anhydride desymmetrisation by alcoholysis see: (a) A. C. Spivey and B. I. Andrews, Angew. Chem., Int. Ed., 2001, 40, 3131 CrossRef CAS; (b) Y. Chen, P. McDaid and L. Deng, Chem. Rev., 2003, 103, 2965 CrossRef CAS PubMed; (c) I. Atodiresei, I. Schiffers and C. Bolm, Chem. Rev., 2007, 107, 5683 CrossRef CAS PubMed; (d) Z. Rodriguez-Docampo and S. J. Connon, ChemCatChem, 2012, 4, 151 CrossRef CAS; (e) A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474 RSC.
- Thiolysis: (a) T. Honjo, S. Saisno, M. Shiro and Y. Nagao, Angew. Chem., Int. Ed., 2005, 44, 5838 CrossRef CAS PubMed; (b) A. Peschiulli, C. Quigley, S. Tallon, Y. K. Gun'ko and S. J. Connon, J. Org. Chem., 2008, 73, 6409 CrossRef CAS PubMed; (c) A. Peschiulli, B. Procuranti, C. J. O'Connor and S. J. Connon, Nat. Chem., 2010, 2, 380 CrossRef CAS PubMed.
- Reductive and alkylative desymmetrisation: (a) H. Fernández-Pérez, P. Etayo, J. R. Lao, J. L. Núñez-Rico and A. Vidal-Ferran, Chem. Commun., 2013, 49, 10666 RSC; (b) E. A. Bercot and T. Rovis, J. Am. Chem. Soc., 2005, 127, 247 CrossRef CAS PubMed; (c) E. E. Stache, T. Rovis and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 3679 CrossRef CAS PubMed.
- Anhydride desymmetrisation by fluoridation: R. Craig, M. Litvajova, S. A. Cronin and S. J. Connon, Chem. Commun., 2018, 54, 10108 RSC.
- Catalytic (enantioselective) haloazidation is also possible: (a) P. K. Shyam and H.-Y. Jang, Eur. J. Org. Chem., 2014, 1817 CrossRef CAS; (b) P. Zhou, L. Lin, L. Chen, X. Zhong, X. Liu and X. Feng, J. Am. Chem. Soc., 2017, 139, 13414 CrossRef CAS PubMed; (c) P. Zhou, X. Liu, W. Wu, C. Xu and X. Feng, Org. Lett., 2019, 21, 1170 CrossRef CAS PubMed.
- Recent radical-mediated carboazidation and diazidation reactions of α,β-unsaturated ketones: W. Liu, M. Pu, J. He, T. Zhang, S. Dong, X. Liu, Y.-D. Wu and X. Feng, J. Am. Chem. Soc., 2021, 143, 11856 CrossRef CAS PubMed.
- The superiority of chloroform as a solvent was surprising, as we have found ethereal solvents to be optimal – without exception – in multiple different reactions involving chiral bifunctional catalysts where the product is a carboxylic acid. We would suggest that this is due to the intermediacy of a silylated carboxylic acid derivative. For examples see: (a) A. Peschiulli, Y. Gun'ko and S. J. Connon, J. Org. Chem., 2008, 73, 2454 CrossRef CAS PubMed; (b) C. Cornaggia, F. Manoni, E. Torrente, S. Tallon and S. J. Connon, Org. Lett., 2012, 14, 1850 CrossRef CAS PubMed; (c) F. Manoni, C. Cornaggia, J. Murray, S. Tallon and S. J. Connon, Chem. Commun., 2012, 48, 6502 RSC; (d) F. Manoni and S. J. Connon, Angew. Chem., Int. Ed., 2014, 53, 2628 CrossRef CAS PubMed; (e) C. Cornaggia, S. Gundala, F. Manoni, N. Gopalasetty and S. J. Connon, Org. Biomol. Chem., 2016, 14, 3040 RSC; (f) S. A. Cronin, A. Gutiérrez Collar, S. Gundala, C. Cornaggia, E. Torrente, F. Manoni, A. Botte, B. Twamley and S. J. Connon, Org. Biomol. Chem., 2016, 14, 6955 RSC; (g) F. Manoni, U. Farid, C. Trujillo and S. J. Connon, Org. Biomol. Chem., 2017, 15, 1463 RSC; (h) R. Claveau, B. Twamley and S. J. Connon, Chem. Commun., 2018, 54, 3231 RSC; (i) M. L. Aiello, U. Farid, C. Trujillo, B. Twamley and S. J. Connon, J. Org. Chem., 2018, 83, 15499 CrossRef CAS PubMed; (j) U. Farid, M. L. Aiello and S. J. Connon, Chem. – Eur. J., 2019, 25, 10074 CrossRef CAS PubMed; (k) A. Gutiérrez Collar, C. Trujillo and S. J. Connon, Chem. – Eur. J., 2019, 25, 7270 CrossRef PubMed; (l) B. Lockett-Walters, C. Trujillo, B. Twamley and S. J. Connon, Chem. Commun., 2019, 55, 11283 RSC.
- V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351 CrossRef CAS PubMed.
- R. Claveau, B. Twamley and S. J. Connon, Org. Biomol. Chem., 2018, 16, 7574 RSC.
- K. Blise, M. W. Cvitkovic, N. J. Gibbs, S. F. Roberts, R. M. Whitaker, G. E. Hofmeister and D. Kohen, J. Org. Chem., 2017, 82, 1347 CrossRef CAS PubMed.
- X. T. Guo, J. Shen, F. Sha and X. Y. Wu, Synthesis, 2015, 47, 2063 CrossRef CAS.
- A. N. Reznikov, E. V. Golovin and Y. N. Klimochkin, Russ. J. Org. Chem., 2013, 49, 663 CrossRef CAS.
- F. González-Bobes, N. Kopp, L. Li, J. Deerberg, P. Sharma, S. Leung, M. Davies, J. Bush, J. Hamm and M. Hrytsak, Org. Process Res. Dev., 2012, 16, 2051 CrossRef.
- M. Litvajova, E. Sorrentino, B. Twamley and S. J. Connon, Beilstein J. Org. Chem., 2021, 17, 2287 CrossRef CAS PubMed.
- C. Shao, H. J. Yu, N. Y. Wu, P. Tian, R. Wang, C. G. Feng and G. Q. Lin, Org. Lett., 2011, 13, 788 CrossRef CAS PubMed.
- J. P. Hilton-Proctor, O. Ilyichova, Z. Zheng, I. G. Jennings, R. W. Johnstone, J. Shortt, S. J. Mountford, M. J. Scanlon and P. E. Thompson, Eur. J. Med. Chem., 2020, 191, 112120 CrossRef CAS PubMed.
- I. J. Montoya-Balbás, B. Valentín-Guevara, E. López-Mendoza, I. Linzaga-Elizalde, M. Ordoñez and P. Román-Bravo, Molecules, 2015, 20, 22028 CrossRef PubMed.
- T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed.
- P. L. Feldman, M. F. Brackeen, D. J. Cowan, B. E. Marron, F. J. Schoenen, J. A. Stafford, E. M. Suh, P. L. Domanico, D. Rose, M. A. Leesnitzer, E. S. Brawley, A. B. Strickland, M. W. Verghese, K. M. Connolly, R. Bateman-Fite, L. S. Noel, L. Sekut and S. A. Stimpson, J. Med. Chem., 1995, 38, 1505 CrossRef CAS PubMed.
- A. S. Paraskar and A. Sudalai, Tetrahedron, 2006, 62, 4907 CrossRef CAS.
- Q. Lang, G. Gu, Y. Cheng, Q. Yin and X. Zhang, ACS Catal., 2018, 8, 4824 CrossRef CAS.
- M. Yar, M. G. Unthank, E. M. McGarrigle and V. K. Aggarwal, Chem. – Asian J., 2011, 6, 372 CrossRef CAS PubMed.
- A. G. H. Wee, G. J. Fan and H. M. Bayirinoba, J. Org. Chem., 2009, 74, 8261 CrossRef CAS PubMed.
- R. M. Wiedhopf, E. R. Trumbull and J. R. Cole, J. Pharm. Sci., 1973, 62, 1206 CrossRef CAS PubMed.
- V. Rodríguez, M. Sánchez, L. Quintero and F. Sartillo-Piscil, Tetrahedron, 2004, 60, 10809 CrossRef.
- D. Enders and O. Niemier, Heterocycles, 2005, 66, 385 CrossRef CAS.
- J. Sietmann, M. Ong, C. Mück-Lichtenfeld, C. G. Daniliuc and J. M. Wiest, Angew. Chem., Int. Ed., 2021, 60, 9719 CrossRef CAS PubMed.
- Z. Galla, E. Forró and F. Fülöp, Tetrahedron: Asymmetry, 2016, 27, 729 CrossRef CAS.
- M. L. Cardoso do Vale, J. E. Rodríguez-Borges, O. Caamaño, F. Fernández and X. García-Mera, Tetrahedron, 2006, 62, 9475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01040b |
| This journal is © The Royal Society of Chemistry 2022 |